
Abstract
Neurodegenerative diseases likely share common underlying pathobiology. Although prior work has identified susceptibility loci associated with various dementias, few, if any, studies have systematically evaluated shared genetic risk across several neurodegenerative diseases. Using genome-wide association data from large studies (total n = 82,337 cases and controls), we utilized a previously validated approach to identify genetic overlap and reveal common pathways between progressive supranuclear palsy (PSP), frontotemporal dementia (FTD), Parkinson’s disease (PD) and Alzheimer’s disease (AD). In addition to the MAPT H1 haplotype, we identified a variant near the chemokine receptor CXCR4 that was jointly associated with increased risk for PSP and PD. Using bioinformatics tools, we found strong physical interactions between CXCR4 and four microglia related genes, namely CXCL12, TLR2, RALB, and CCR5. Evaluating gene expression from post-mortem brain tissue, we found that expression of CXCR4 and microglial genes functionally related to CXCR4 was dysregulated across a number of neurodegenerative diseases. Furthermore, in a mouse model of tauopathy, expression of CXCR4 and functionally associated genes was significantly altered in regions of the mouse brain that accumulate neurofibrillary tangles most robustly. Beyond MAPT, we show dysregulation of CXCR4 expression in PSP, PD, and FTD brains, and mouse models of tau pathology. Our multi-modal findings suggest that abnormal signaling across a ‘network’ of microglial genes may contribute to neurodegeneration and may have potential implications for clinical trials targeting immune dysfunction in patients with neurodegenerative diseases.
Similar content being viewed by others

Introduction
Uncovering the shared genetic architecture across neurodegenerative diseases may elucidate underlying common disease mechanisms and promote early disease detection and intervention strategies. Progressive supranuclear palsy (PSP), frontotemporal dementia (FTD), Parkinson’s disease (PD) and Alzheimer’s disease (AD) are age-associated neurodegenerative disorders placing a large emotional and financial impact on patients and society. Despite variable clinical presentation, PSP, AD and FTD are characterized by abnormal deposition of tau protein in neurons and/or glia in the frontal, temporal, and insular cortical gray matter and hindbrain1,2. PSP is associated with 4-repeat (4R) tau inclusions1,3; FTD is characterized by 3-repeat (3R)-only, 4R-only, or mixed (3R and 4R) tau inclusions or TAR DNA-binding protein 43 aggregates4; and AD is characterized by extracellular amyloid plaques and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau (mixed 3R and 4R tau inclusions)5,6. While PD is classically characterized by alpha-synuclein deposits, recent studies support the role of tau and NFTs in modifying PD clinical sympotomatology and disease risk7,8,9. Prior work has shown that PSP shares overlapping pathobiology with FTD, AD, and PD10,11,12,13.
Genome-wide association studies (GWAS) and candidate gene studies have identified single nucleotide polymorphisms (SNPs) within the H1 haplotype in MAPT locus (which encodes tau) that increase risk for PSP, FTD, AD, and PD14,15,16,17,18,19. However, beyond MAPT, the extent of genetic overlap across these diseases and its relationship with common pathogenic processes observed in PSP, FTD, AD, and PD remain poorly understood.
Genomic studies evaluating shared risk among numerous phenotypes or diseases suggest genetic pleiotropy, where a single gene or genetic variant may impact different traits17,18,19,20. The recent proliferation of GWAS data for rare disorders like PSP paired with large studies of more common diseases such as AD, PD, and FTD provides unique opportunities to gain statistical power and identify risk loci which may not have otherwise been identified in the original GWAS study. Further, it allows for the systematic evaluation of genetic overlap across different disorders, thereby informing shared biological pathways and processes commonly altered in both conditions. Here, using previously validated methods20,21,22,23, we assessed shared genetic risk across PSP, PD, FTD, and AD. We then applied molecular and bioinformatic tools to elucidate the role of these shared risk genes in neurodegenerative diseases.
Methods
Participant samples
We obtained publicly available PSP-GWAS summary statistic data from the NIA Genetics of Alzheimer’s Disease Storage Site, which consisted of 1114 individuals with PSP (cases) and 3247 controls (stage 1) at 531,451 SNPs (Table 1, for additional details see18). In this study, we focused on stage 1 of the PSP GWAS dataset. Individuals were diagnosed with PSP according to NINDS criteria24. We evaluated complete summary statistic GWAS data from clinically diagnosed FTD, AD, and PD. The International Parkinson’s Disease Genetics Consortium (IPDGC) provided PD-GWAS summary statistic data. The IPDGC cohort consists of 5333 cases and 12,019 controls with genotype or imputed data at 7,689,524 SNPs (Table 1, for additional details see9). The International FTD GWAS Consortium (IFGC) provided phase 1 FTD-GWAS summary statistic data, which consisted of 2154 FTD cases and 4308 controls with genotypes or imputed data at 6,026,384 SNPs (Table 1, for additional details see25 and Supplemental Information). The FTD dataset included multiple subtypes within the FTD spectrum: bvFTD, semantic dementia, progressive non-fluent aphasia, and FTD overlapping with motor neuron disease. We obtained publicly available, AD-GWAS summary statistic data from the International Genomics of Alzheimer’s Disease Project (IGAP Stage 1) (see Supplemental Information). The IGAP Stage 1 cohort consists of 17,008 AD cases and 37,154 controls with genotyped or imputed data at 7,055,881 SNPs (Table 1, for additional details see26 and Supplemental Information). All four cohorts were primarily of European ancestry and the studies’ authors controlled for population stratification using a principal components analysis approach. Inclusion/exclusion criteria were established by each study’s authors; please see the publications provided in Table 1 for additional details. In each study, informed consent was obtained from all subjects. Institutional approval was provided by each study’s respective committee.
Identification of shared risk loci—conjunction FDR
We evaluated SNPs associating with PSP (Phase 1), FTD (IFGC Phase 1), AD (IGAP Stage 1) and PD (IPDGC Phase 1) using techniques for evaluating genetic pleiotropy (for additional details see Supplemental Information and21,23). Briefly, for two phenotypes A and B, pleiotropic ‘enrichment’ of phenotype A with phenotype B exists if the effect sizes of the genetic associations with phenotype A become larger as a function of increased association with phenotype B. For each phenotype, Z scores were derived for each SNP association given the Wald statistics. We then corrected the Z scores for potential genomic inflation(for additional methodological details used in our study, see Supplemental Information and see27 for additional information on genomic inflation). To assess enrichment, we constructed fold-enrichment plots of nominal –log10(p) values for all PSP SNPs and a subset of SNPs determined by the significance of their association with AD, FTD, and PD. Under expected null, the average effect sizes given a group of SNPs would be the same when the conditioned effect increases. Therefore, in fold-enrichment plots, enrichment is indicated by an upward deflection of the curve for phenotype A if it shares genetic effects with phenotype B. To assess for polygenic effects below the standard GWAS significance threshold, we focused the fold-enrichment plots on SNPs with nominal –log10(p) < 7.3 (corresponding to p-value > 5 × 10−8). The enrichment can be interpreted in terms of true discovery rate (TDR = 1−False Discovery Rate [FDR]) (for additional details see Supplemental Information and28).
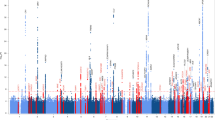
To identify specific loci involved in both PSP and AD, FTD or PD, we computed conjunction FDR21,22,23. Conjunction FDR, denoted by FDRtrait1& trait2 is defined as the posterior probability that a SNP is null for either phenotype or both simultaneously, given the p-values for both traits are as small, or smaller, than the observed p-values. A conservative estimate of the conjunction FDR is given by the maximum statistic in taking the maximum of FDRtrait1|trait2 and FDR trait2|trait1 (for additional details see Supplemental Information and21). We used an overall FDR threshold of <0.05. To visualize the results of our conjunction FDR analysis, we constructed Manhattan plots based on the ranking of conjunction FDR to illustrate the genomic location of the pleiotropic loci. Rather than representing novel risk variants where replication is needed in independent datasets, conjunction FDR pinpoints genetic variants jointly associated with two or more phenotypes/diseases and in this context, ‘replication’ may not be meaningful21.
Functional evaluation of shared risk loci
To assess whether the PSP, FTD, AD, and PD overlapping SNPs modify gene expression, we evaluated cis-expression quantitative trait loci (eQTLs, DNA sequence variants that influence the expression level of one or more genes) in a publicly available dataset from 134 neuropathologically confirmed normal control brains (UKBEC, http://braineac.org/)29 and validated these eQTLs in the GTex dataset30. We also evaluated eQTLs using a blood-based dataset31. We applied an analysis of covariance (ANCOVA) to test for association between genotypes and gene expression. SNPs were tested using an additive model.
Network-based functional association analyses
To evaluate potential protein and genetic interactions, co-expression, co-localization and protein domain similarity for the pleiotropic genes, we used GeneMANIA (www.genemania.org), an online web-portal for bioinformatic assessment of gene networks32. In addition to visualizing the composite gene network, we also assessed the weights of individual components within the network33.
Gene expression alterations in PSP, PD, and FTD brains
To determine whether pleiotropic genes were differentially expressed in PSP, PD, and FTD brain tissue, we analyzed gene expression of pleiotropic genes in publically available datasets. We analyzed gene expression data from: (1) the temporal cortex and cerebellum of 80 control and 84 PSP brains (syn5550404); (2) the frontal cortex; hippocampus and cerebellum of 11 controls and 17 FTLD-U (7 brains with or 10 brains without progranulin (GRN) mutations) (Gene Expression Omnibus (GEO) dataset GSE13162)34; and (3) the substantia nigra of 23 control and 22 PD brains (GEO dataset GSE7621)35.
Evaluating gene expression of pleiotropic loci in tau transgenic mouse models
We evaluated gene expression profiles for the nearest genes associated with our shared risk loci using publicly available P301L-tau transgenic mouse model (mutant human MAPT gene) from Mouseac (www.mouseac.org)36. Briefly, microarray gene expression data was collected from three brain regions (cortex, hippocampus and cerebellum) from wild-type and P301L-tau transgenic mice. Gene expression levels were log transformed and expressed as a function of age. The presence of NFT pathology was evaluated and scored as previously reported by immunohistochemistry36. Using repeated measures ANOVAs within the hippocampus, cortex and cerebellum, we examined whether gene expression levels of PSP, FTD, AD, and PD variants are significantly different between the P301L-tau transgenic and wild-type mice, across 2, 4, 8 and 18 months of age. To maximize our ability to detect an effect, we used all expression data available for each line of mice and age grouping. As our data was publicly available, there was no randomization or blinding of the data. Please see (www.mouseac.org)36 for exact sample sizes used in each analysis and additional information on the mouse data used in this study.
Code availability
The code used to conduct pleiotropy analyses is not yet publicly available. Please contact the authors with any inquiries related to the code.
Results
Selective shared genetic risk between PSP, PD, and FTD
We observed SNP enrichment for PSP SNPs across different levels of significance of association with FTD (Fig. 1a). Using progressively stringent p-value thresholds for PSP SNPs (i.e., increasing values of nominal –log10(p)), we found up to 150-fold genetic enrichment using FTD and lower, but still notable, enrichment in PD (Figs. 1a, b). In contrast, we found minimal or no enrichment in PSP SNPs as a function of AD (Figs. 1a, b).

a Fold enrichment plots of enrichment vs. nominal −log10 p-values (corrected for inflation) in progressive supranuclear palsy (PSP) below the standard GWAS threshold of p < 5 × 10-8 as a function of significance of association with Alzheimer’s disease (AD, panel A), frontotemporal dementia (FTD, panel B) and Parkinson’s disease (PD, panel C) at the level of −log10(p) ≥ 0, −log10(p) ≥ 1, −log10(p) ≥ 2 corresponding to p ≤ 1, p ≤ 0.1, p ≤ 0.01, respectively. Blue line indicates all SNPs. b “Conjunction” Manhattan plot of conjunction and conditional –log10 (FDR) values for progressive supranuclear palsy (PSP) given Alzheimer’s disease (AD; PSP|AD, teal), frontotemporal dementia (PSP; PSP|FTD, orange) and Parkinson’s disease (PD; PSP|PD, red). SNPs with conditional and conjunction –log10 FDR > 1.3 (i.e., FDR < 0.05) are shown with large points. A black line around the large points indicates the most significant SNP in each LD block and this SNP was annotated with the closest gene, which is listed above the symbols in each locus. c Network interaction graph illustrating physical interactions, co-expression, predicted, pathway, co-localization, gene interactions and shared protein domains for CXCR4 and MAPT. Network plot was generated from GeneMANIA (www.genemania.org)
At a conjunction FDR < 0.05, we identified 2 SNPs that were associated with increased risk for both PSP and PD (Table 2, Fig. 1b): rs749873 (intergenic; closest gene = CXCR4, on chr2, minimum conjunction FDR = 0.01; Supplemental Fig. 1a) and rs11012 (UTR-3; closest gene = PLEKHM1 within MAPT region on chr 17, PSP p-value = 4.9 × 10−39, minimum conjunction FDR = 9.3 × 10−9). CXCR4 is a chemokine receptor implicated in immune processes, microglia recruitment, neuronal guidance, neural stem cell proliferation, and neurodevelopmental processes37,38,39,40. At a conjunction FDR < 0.05, we identified 1 SNP that was associated with increased risk for both PSP and FTD (Table 2, Fig. 1b): rs199533 (exonic; closest gene = NSF within MAPT region on chr 17, PSP p-value = 3.5 × 10−41, minimum conjunction FDR = 9.3 × 10−9; Supplemental Fig. 1b). Notably, rs11012 and rs199533 is in strong linkage disequilibrium (LD) with SNP rs1800547, which tags the H1 haplotype of MAPT (rs11012 pairwise D’ = 0.96, r2 = 0.71; rs199533 pairwise D’ = 1, r2 = 0.94). The H1 haplotype of MAPT has been implicated in risk for both PSP, PD, and FTD18,19. In contrast to PSP and PD as well as PSP and FTD, we found no significant overlapping loci between PSP and AD.
Expression quantitative trait loci (eQTL) and gene expression analyses
Consistent with our previous findings, rs749873 was associated with expression of MCM6 within the CXCR4 locus in human brains and with CXCR4 expression in whole blood41. In the human CNS, rs749873 modifies MCM6 expression in the tibial nerve41. Interestingly, rs749873 is in high LD with rs2011946 (r2 = 0.91; D’ = 1), a SNP we previously reported to be shared across PSP and corticobasal degeneration (CBD)41. As previously reported, rs199533 was significantly associated with MAPT expression in human brains (p = 2 × 10−12)41.
Protein-protein and co-expression networks for MAPT and CXCR4
Using GeneMANIA, we examined the proteins that physically interact with and/or are co-expressed with MAPT or CXCR4 (Fig. 1c). As previously reported41, CXCR4 demonstrated the strongest physical interaction with chemokine motif ligand 12 (CXCL12), toll-like receptor 2 (TLR2), Ras-related protein-B (RALB) and C–C chemokine receptor 5 (CCR5) (Fig. 1c) (Supplemental Table 1). CXCL12 is the ligand for CXCR442,43. TLR2 encodes a toll-like receptor which is utilized by the innate immune system to detect pathogenic material—it is expressed on microglia as well as astrocytes and has been proposed as an inhibitor of neural progenitor cell proliferation44. RALB is a small GTPase protein that interacts with CXCL12 and is implicated in B-cell migration45. CCR5 is a chemokine structurally related to CXCR4 and modulates the migration of microglia and blood brain barrier integrity46,47.
We found that MAPT showed robust physical interactions with FKBP4 and STUB1 (Fig. 1c; Supplemental Table 1). FKBP4 (also known as FKBP52) is a peptidylprolyl isomerase which is involved in dynein interaction and glucocorticoid receptor movement to the nucleus48. STUB1 is a ubiquitin ligase with diverse functions that has been implicated in AD49.
CXCR4 gene expression alterations in PSP, PD, and other neurodegenerative disease brains
We next sought to determine whether CXCR4 is differentially expressed in PSP, PD, and FTD brains. We included FTD brain tissues in our analyses because no SNP data for rs749873 was available for primary pleiotropy analysis in FTD, leaving open the possibility that CXCR4 expression may be altered in FTD. Compared with control brains, we found that CXCR4 was significantly upregulated in brains with a neuropathological diagnosis of PSP and FTD, especially within the cerebellum and hippocampus (Figs. 2a, b, Table 3). Further, we found CXCR4 was also significantly upregulated in PD cases (Fig. 2c; Table 3). We also evaluated expression levels in PSP, FTD, and PD brains of the four genes that showed strong physical interactions with CXCR4 in our network analyses (CXCL12, TLR2, RALB and CCR5; weighted connection with CXCR4 > 0.25, see above). We found that CXCL12 expression was significantly dysregulated in PSP, and PD, but not in FTD (Table 3). TLR2 levels were significantly altered specifically in FTD. Of note, CCR5 levels were not available for analysis. Neither MAPT or RALB expression was significantly altered in PSP, FTD or PD brains relative to controls (Table 3). Thus, our data suggest that expression of CXCR4 and functionally associated genes are significantly altered in regions of the brain susceptible to different forms of neurodegenerative disease pathology.

a CXCR4 expression in neuropathologically normal tissue compared to PSP. b CXCR4 expression in neuropathologically normal tissue compared to sporadic FTD and FTD due to GRN mutations. (c) CXCR4 expression in neuropathologically normal tissue compared to PD. d-e Line plots illustrating CXCR4 gene expression in tau transgenic (red line) and wild-type mice (black line) from 2 to 18 months of age in the d hippocampus. e Total tau pathology over time is also illustrated
Given the role of CXCR4 in astroglial signaling and microglial activation50, we asked whether the observed upregulation of CXCR4 in neurodegenerative disease was driven by proliferation and activation of microglia in response to neurodegenerative pathology. To assess this, we evaluated whether commonly used markers of microglial activation, namely TMEM119 and AIF151,52, are also upregulated across neurodegenerative diseases. Unlike CXCR4, across all evaluated brain regions within PSP, FTD, and PD brains, we found no evidence for altered TMEM119 and AIF1 expression (Table 3).
We next examined whether CXCR4 and AIF1 or TMEM119 were significantly correlated in PSP, FTD, or PD brains. We observed a significant correlation between CXCR4 and AIF1 in FTD brains (combined FTD-GRN + and FTD-GRN-: r2 = 0.35 and p < 1 × 10−4). As indicated in Table 3, TMEM119 was not available within the FTD dataset. In PSP brains, we found a marginally significant correlation between CXCR4 and TMEM119 (PSP AIF1: r2 = 5.7 × 10−4 and p = 0.83; PSP TMEM119: r2 = 0.05 and p = 0.05). However, CXCR4 and AIF1 or TMEM119 were not correlated in PD brains (AIF: r2 = 9.15 × 10−5 and p = 0.96 and TMEM119: r2 = 0.10 and p = 0.10). Thus, these findings provide evidence suggesting that CXCR4 could contribute to neurodegeneration through microglial activation as well as pathways beyond microglial dysfunction.
CXCR4 expression is elevated in tau transgenic mouse model
To assess whether CXCR4 expression is associated with NFT pathology, we examined CXCR4 expression in brains from a transgenic mouse model of tau aggregation (Tau-P301L transgenic mice). Tau-P301L mice develop NFT pathology in the hippocampus and neocortical regions by 6 months of age, while the cerebellum fails to develop NFT pathology36,53. CXCR4 expression was significantly elevated in the hippocampus (F = 10.3, p = 0.0023) in Tau-P301L mice compared with wild-type mice (Fig. 2d). In contrast, within the cerebellum (F = 0.001, p = 0.98) and cortex (F = 1.06, p = 0.308), we found no difference in CXCR4 expression between the Tau-P301L and wild-type mice across any of the time points. MAPT expression, however, was not significantly altered in Tau-P301L mice compared with wild-type in both the hippocampus and cerebellum (Supplemental Fig. 2).
We additionally assessed expression levels of the four genes (CXCL12, TLR2, RALB and CCR5) that showed strong physical interactions with CXCR4 in our network analyses (see Fig. 1c). Within the hippocampus and cortex, TLR2 was significantly elevated over time (F = 7.07, p = 1.35 × 10−7 and F = 2.9, p = 8.19 × 10−6, respectively) and CCR5 expression was significantly decreased over time (F = 0.3, p = 0.002) in Tau-P301L mice but not in wild-type mice. Expression of CXCL12 (F = 0.19, p = 0.06) and RALB (F = 0.02, p = 0.09) were not significantly altered in Tau-P301L mice (Supplemental Figs. 3–6). Within the cerebellum, which remains free of tau aggregates, we found no evidence for gene expression alterations in CXCL12, TLR2, RALB, and CCR5 in Tau-P301L mice compared with wild-type mice. Thus, expression of CXCR4 and functionally associated genes is significantly altered in regions of the mouse brain that accumulate NFTs most robustly.
Finally, we evaluated whether TMEM119 and AIF1 expression is associated with CXCR4 expression and significantly elevated within the hippocampus. Across all evaluated time points (2, 4, 6, and 18 months) and within wild-type and Tau-P301L transgenic mice, we found that TMEM119 and AIF1 levels were significantly correlated with CXCR4 expression specifically within the hippocampus (Supplemental Results). Similarly, predominantly within the hippocampus, we found that TMEM119 and AIF1 expression was elevated over time in Tau-P301L mice compared with wild-type mice (Supplemental Results). Evaluating CXCR4 associated genes, we found a strong association between microglia and inflammation related TLR2 and CCR5 expression and TMEM119 and AIF1 expression, within the hippocampus, cortex and cerebellum (Supplemental Results). In contrast, we found no correlation between levels of CXCL12 and RALB with TMEM119 or AIF1 (Supplemental Results). Together, these findings suggest that innate immune system signaling and microglial activation markers are associated with neurodegeneration and tauopathy.
Discussion
We identified CXCR4 as a novel locus associated with increased risk for both PSP and PD. Building on extensive prior work, we also confirmed the role of variants within MAPT in driving PSP, PD, and FTD risk. We found that CXCR4 and functionally associated genes exhibit altered expression across a number of neurodegenerative diseases. In a mouse model of tauopathy, CXCR4 and functionally associated genes were altered in the presence of tau pathology. Together, our findings suggest that alterations in expression of CXCR4 and associated microglial genes may contribute to age-associated neurodegeneration.
Utilizing GWAS summary statistics from multiple neurodegenerative diseases, our results suggest that shared genetic risk factors may underlie the pathobiological processes occurring in PSP and PD. We found up to a 150-fold enrichment in PSP as a function of FTD, lower enrichment in PD, and no enrichment in AD. These findings were unexpected given the established role of the MAPT H1 haplotype in AD26. Despite the lack of strong genetic association across these three neurodegenerative diseases, we found that CXCR4 expression was altered in brains that are pathologically confirmed for PSP, PD, and FTD. Thus, these findings support our hypothesis that these three neurodegenerative disorders share common pathobiological pathways.
CXCR4 is a chemokine receptor protein with broad regulatory functions in the immune system and neurodevelopment39,40,43,54,55,56. CXCR4 has been shown to regulate neuronal guidance and apoptosis through astroglial signaling and microglial activation50. Furthermore, it has been shown that CXCR4 is involved in cell cycle regulation through p53 and Rb57,58. Importantly, small molecular agonists and antagonists to CXCR4 have been described59. AMD3100 is an FDA-approved CXCR4 antagonist and a CXCR7 agonist that is commonly used to enhance hematopoietic stem cell proliferation60.
Using a bioinformatics approach, we identified four genes, namely CXCL12, TLR2, RALB and CCR5 that showed a strong association with CXCR4. We additionally found that expression of CXCR4 and functionally associated genes were altered in multiple neurodegenerative diseases and associated with hippocampal tau pathology in transgenic mouse models. These findings suggest that a network of CXCR4 and associated genes may act in concert to influence neurodegeneration.
Given that PSP, PD, and FTD are neuropathologically characterized by degeneration in the midbrain, cerebellum, and (to a lesser extent) neocortical regions1,2,61, our findings may suggest that subtle alterations of CXCR4 and MAPT may predispose to regionally specific brain degeneration in later life. Though we found no evidence of altered expression of known microglial markers within human neurodegenerative brains, we found that CXCR4 expression was significantly upregulated in PSP, FTD, and PD brains. Furthermore, within the tauopathy mouse model, we found a strong association between CXCR4 and TMEM119 and AIF1 within the hippocampus. Additionally, across all evaluated regions, we found a strong relationship between microglia and inflammation related genes, namely CCR5 and TLR2 expression and TMEM119 and AIF1 expression. Finally, expression of TMEM119 and AIF1 was markedly elevated predominantly within the hippocampus in Tau-P301L mouse model expression data. Thus, results from the Tau-P301L mouse model data suggest that upregulated CXCR4 expression observed during age-associated neurodegeneration may be related to inflammatory mechanisms.
Our study benefits from its use of multiple well-validated GWAS datasets and its integration of multiple information modalities ranging from population level genetic data to RNA expression in mouse models of tauopathy. Our study used GWAS data composed of common SNPs and thereby cannot inform the potential role that rare variation may play as a risk factor for PSP, PD, and FTD. A limitation of our study is that we do not have records indicating which GWAS SNPs were directly genotyped vs. imputed, limiting our ability to assess the quality of SNP ascertainment in our cohorts. Further, while we demonstrated that rs749873 modified CXCR4 expression in human blood, we were unable to test for an eQTL in a large dataset of control human brains. Given differences in sample size between the two tissue types, this could be a function of statistical power. Indeed, previous work has shown that many eQTLs are shared across tissues and that ability to detect an eQTL varies with sample size30.
In conclusion, by integrating large neurodegenerative GWAS data with gene expression data from neurodegenerative diseases and transgenic mouse models, our multi-modal findings indicate that CXCR4 is associated with PSP and PD neurodegeneration. Clinically, our results provide additional evidence that immune and microglial dysfunction contribute to the pathophysiology in PSP, PD, and FTD. These findings have important implications for future work focused on monitoring microglial activation as a marker of disease progression and on developing anti-inflammatory therapies to modify disease outcomes in patients with neurodegenerative diseases.
References
Dickson, D. W., Rademakers, R. & Hutton, M. L. Progressive supranuclear palsy: pathology and genetics. Brain. Pathol. 17, 74–82 (2007).
Kovacs, G. G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23 (2015).
Boxer, A. L. et al New diagnostics and therapeutics for progressive supranuclear palsy. Lancet Neurol. 16, 552–563, https://doi.org/10.1016/S1474-4422(17)30157-6 (2017).
Bang, J., Spina, S. & Miller, B. L. Frontotemporal dementia. Lancet 386, 1672–1682 (2015).
Dubois, B. et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 6, 734–746 (2007).
Hyman, B. T. et al. National Insitute on Aging- Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement 8, 1–13 (2013).
Simón-Sánchez, J. et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312 (2009).
Irwin, D. J., Lee, V. M.-Y. M.-Y. & Trojanowski, J. Q. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636 (2013).
Nalls, M. A. et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 377, 641–649 (2011).
Bronner I. F. et al. Comprehensive mRNA expression profiling distinguishes tauopathies and identifies shared molecular pathways. PLoS ONE 4, (2009). https://doi.org/10.1371/journal.pone.0006826.
Ahmed, R. M. et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatr. 87, 1234–1241, https://doi.org/10.1136/jnnp-2014-308350 (2016).
Moussaud, S. et al. Alpha-synuclein and tau: teammates in neurodegeneration? Mol. Neurodegener. 9, 43 (2014).
Hauser, M. A. et al. Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch. Neurol. 62, 917–921 (2005).
Allen, M. et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res. Ther. 6, 39 (2014).
Verpillat, P. et al. Association between the extended tau haplotype and frontotemporal dementia. Arch. Neurol. 59, 935–939 (2002).
Baker, M. et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715 (1999).
Zabetian, C. P. et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson’s disease. Ann. Neurol. 62, 137–144 (2007).
Höglinger, G. U. et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705 (2011).
Ferrari, R. et al Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatr. 88, 152–164, https://doi.org/10.1136/jnnp-2016-314411 (2016).
Desikan, R. S. et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation 131, 2061–2069 (2015).
Yokoyama, J. S. et al. Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol. 94158, 1–7 (2016).
Andreassen, O. A. et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am. J. Hum. Genet. 92, 197–209 (2013).
Andreassen, O. A. et al Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS. Genet. 9, e1003455 (2013).
Hauw, J. J. et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44, 2015–2019 (1994).
Ferrari, R. et al. Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurol. 13, 686–699 (2014).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Devlin, B. & Roeder, K. Genomic control for association studies. Biometrics 55, 997–1004 (1999).
Desikan, R. S. et al. Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol. Psychiatry 12, 1–8, https://doi.org/10.1038/mp.2015.6 (2015).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science (80-) 348, 648–660 (2015).
Westra, H.-J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Warde-Farley D. et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, (2010) https://doi.org/10.1093/nar/gkq537.
Mostafavi, S., Ray, D., Warde-Farley, D., Grouios, C. & Morris, Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9, S4 (2008). Suppl 1.
Chen-Plotkin, A. S. et al. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum. Mol. Genet. 17, 1349–1362 (2008).
Lesnick, T. G. et al. A genomic pathway approach to a complex disease: Axon guidance and Parkinson disease. PLoS. Genet. 3, 0984–0995 (2007).
Matarin, M. et al. A Genome-wide Gene-Expression Analysis and Database in Transgenic Mice during Development of Amyloid or Tau Pathology. Cell. Rep. 10, 633–644 (2015).
Arnò, B. et al. Neural progenitor cells orchestrate microglia migration and positioning into the developing cortex. Nat. Commun. 5, 5611 (2014).
Albright, A. V. et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J. Virol. 73, 205–213 (1999).
Zou, Y. R., Kottmann, aH., Kuroda, M., Taniuchi, I. & Littman, D. R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393, 595–599 (1998).
Lu, M., Grove, E. A. & Miller, R. J. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc. Natl. Acad. Sci. USA 99, 7090–7095 (2002).
Yokoyama, J. S. et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 99(10), 1–13, https://doi.org/10.1073/pnas.092013799 (2017).
Yang, S. et al. Cxcl12/Cxcr4 signaling controls the migration and process orientation of A9-A10 dopaminergic neurons. Development 140, 4554–4564 (2013).
Kokovay, E. et al. Adult SVZ Lineage Cells Home to and Leave the Vascular Niche via Differential Responses to SDF1/CXCR4 Signaling. Cell. Stem. Cell. 7, 163–173 (2017).
Hanke, M. L. & Kielian, T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin. Sci. 121, 367–387 (2011).
de Gorter, D. J. J. et al. The small GTPase Ral mediates SDF-1-induced migration of B cells and multiple myeloma cells. Blood 111, 3364–3372 (2008).
Carbonell, W. S., Murase, S. S.-I., Horwitz, A. A. F. & Mandell, J. W. J. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J. Neurosci. 25, 7040–7047 (2005).
Alkhatib, G. The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 4, 96–103 (2009).
Galigniana, M. D., Radanyi, C., Renoir, J. M., Housley, P. R. & Pratt, W. B. Evidence that the Peptidylprolyl Isomerase Domain of the hsp90-binding Immunophilin FKBP52 is Involved in Both Dynein Interaction and Glucocorticoid Receptor Movement to the Nucleus. J. Biol. Chem. 276, 14884–14889 (2001).
Tsvetkov, P., Adamovich, Y., Elliott, E. & Shaul, Y. E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J. Biol. Chem. 286, 8839–8845 (2011).
Bezzi, P. et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710 (2001).
Bennett, M. L. et al New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. 113, E1738–E1746 (2016).
Karch, C. M. et al. Expression of Novel Alzheimer’s Disease Risk Genes in Control and Alzheimer’s Disease Brains. PLoS ONE 7, (2012). https://doi.org/10.1371/journal.pone.0050976.
Lewis, J. et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405 (2000).
Borrell, V. & Marín, O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat. Neurosci. 9, 1284–1293 (2006).
Klein, R. S. & Rubin, J. B. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 25, 306–314 (2004).
Wong, M., Xin, W. & Duman, R. S. Rat LCR1: cloning and cellular distribution of a putative chemokine receptor in brain. Mol. Psychiatr. 1, 133–140 (1996).
Khan, M. Z. et al. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell. Death. Differ. 15, 1663–1672 (2008).
Khan, M. Z. et al. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J. Neurovirol. 9, 300–314 (2003).
Mishra, S. K., Tawani, A., Mishra, A. & Kumar, A. G4IPDB: A database for G-quadruplex structure forming nucleic acid interacting proteins. Sci. Rep. 6, 38144 (2016).
Kalatskaya, I. et al. AMD3100 Is a CXCR7 Ligand with Allosteric Agonist Properties. Mol. Pharmacol. 75, 1240–1247 (2009).
Wu, T. & Hallett, M. The cerebellum in Parkinson’s disease. Brain 136, 696–709 (2013).
Acknowledgements
We thank the International FTD-GWAS Consortium (IFGC), International Parkinson’s Disease Genomic Consortium (IPDGC) and International Genomics of Alzheimer’s Project (IGAP) for providing summary statistics data for these analyses. Further acknowledgments for IFGC, IPDGC and IGAP are found in the Supplemental material. This research was supported by grants from the National Institutes of Health (NIH-AG046374 [CMK] and K01 AG049152 [JSY]), Larry J. Hillblom Foundation (2016-A-005-SUP [JSY]), Research Council of Norway (#213837, #225989, #223273, and #237250/EU JPND [OAA]), South East Norway Health Authority (2013-123), Norwegian Health Association, the Radiological Society of North America (RMS1741 [LWB]) (RSD), ASNR Foundation AD Imaging Award (RSD), National Alzheimer’s Coordinating Center (NACC) Junior Investigator (JI) Award (RSD), the Tau Consortium (JSY), and Alzheimer's Society grant 284 (RF).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The IFGC and IPDGC members are listed in Supplementary Acknowledgments
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bonham, L.W., Karch, C.M., Fan, C.C. et al. CXCR4 involvement in neurodegenerative diseases. Transl Psychiatry 8, 73 (2018). https://doi.org/10.1038/s41398-017-0049-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-017-0049-7
This article is cited by
-
Structural basis of dimerization of chemokine receptors CCR5 and CXCR4
Nature Communications (2023)
-
Up-regulation of microglial chemokine CXCL12 in anterior cingulate cortex mediates neuropathic pain in diabetic mice
Acta Pharmacologica Sinica (2023)
-
Neuroinflammatory transcriptional programs induced in rhesus pre-frontal cortex white matter during acute SHIV infection
Journal of Neuroinflammation (2022)
-
Neural transcriptomic signature of chronic wasting disease in white-tailed deer
BMC Genomics (2022)
-
The CXCL12/CXCR4/ACKR3 Response Axis in Chronic Neurodegenerative Disorders of the Central Nervous System: Therapeutic Target and Biomarker
Cellular and Molecular Neurobiology (2022)
