
Abstract
Previous observational studies reported associations between non-steroidal anti-inflammatory drugs (NSAIDs) and major depressive disorder (MDD), however, these associations are often inconsistent and underlying biological mechanisms are still poorly understood. We conducted a two-sample Mendelian randomisation (MR) study to examine relationships between genetic variants and NSAID target gene expression or DNA methylation (DNAm) using publicly available expression, methylation quantitative trait loci (eQTL or mQTL) data and genetic variant-disease associations from genome-wide association studies (GWAS of MDD). We also assessed drug exposure using gene expression and DNAm levels of NSAID targets as proxies. Genetic variants were robustly adjusted for multiple comparisons related to gene expression, DNAm was used as MR instrumental variables and GWAS statistics of MDD as the outcome. A 1-standard deviation (SD) lower expression of NEU1 in blood was related to lower C-reactive protein (CRP) levels of −0.215 mg/L (95% confidence interval (CI): 0.128–0.426) and a decreased risk of MDD (odds ratio [OR] = 0.806; 95% CI: 0.735–0.885; p = 5.36 × 10−6). A concordant direction of association was also observed for NEU1 DNAm levels in blood and a risk of MDD (OR = 0.886; 95% CI: 0.836–0.939; p = 4.71 × 10−5). Further, the genetic variants associated with MDD were mediated by NEU1 expression via DNAm (β = −0.519; 95% CI: −0.717 to −0.320256; p = 3.16 × 10−7). We did not observe causal relationships between inflammatory genetic marker estimations and MDD risk. Yet, we identified a concordant association of NEU1 messenger RNA and an adverse direction of association of higher NEU1 DNAm with MDD risk. These results warrant increased pharmacovigilance and further in vivo or in vitro studies to investigate NEU1 inhibitors or supplements for MDD.
Similar content being viewed by others


Introduction
From the literature, associations between cell inflammation and depression are highly credible [1]. Patients with MDD exhibit increased inflammatory factor levels that are typically related to chronic inflammation, including inflammatory cytokines such as tumor necrosis factor (TNF), interleukin-1β (IL-1β) and interleukin-6 (IL-6) and acute phase proteins such as C-reactive protein (CRP) [2]. Increased inflammatory markers have also been associated with several depressive symptoms and suicidal MDD [3]. A study measuring cytokines in ‘never-treated’ relatively homogeneous patients with MDD reported that the majority of cytokines were elevated [4].
Elevated inflammatory factor levels in individuals with MDD may be attributable to ineffective clinical management [5]. For example, chronic depression and elevated depression severity at follow-up could be predicted by higher IL-6 levels [6]. CRP and TNF have also been associated with symptom severity in MDD patients [7]. Stimulation-like inflammation and infection may occur via activation of the microglia, which are the brain’s immune cells, to secrete proinflammatory cytokines that act on MDD related pathways, such hypothalamic-pituitary-adrenal (HPA) axis activation and increased indoleamine-2,3-deoxygenase activity [8]. Recent evidence has also suggested that persistent low-level inflammation is related to treatment-resistant depression and low responses to antidepressant treatments [8, 9].
Based on these evidence, recent studies examined the effects of modulating the immune system during MDD [9,10,11]; they used NSAIDs as add-ons to conventional antidepressant therapy, however, data also exists for NSAIDs monotherapy as antidepressants [10,11,12]. A recent meta-analysis summarised 36 randomised controlled trials (RCTs), including 10,000 patients, and reported that both monotherapy, add-on NSAIDs therapy and other add-on therapies, such as cytokine-inhibitor monotherapy and statin add-on therapy, generated antidepressant efficacy [12]. However, in contrast, previous studies also showed that NSAID efficacy for depressive symptoms was negligible [11, 13].
Over the past 20 years, studies have proposed the notion that inflammation and brain-immune interactions are involved in MDD pathogenesis [14, 15]. However, due to a lack of data consistency in terms of add-ons to conventional anti-depression drugs and high treatment resistance rates, other mechanisms such as genetics are proposed to affect NSAIDs efficacy towards depression. Thus, while many studies have explored the effects of NSAIDs on MDD, the biological mechanisms underpinning NSAIDs function in depressed patients remain poorly understood. Given the higher prevalence of inflammation in patients with MDD, understanding the mechanistic links between NSAIDs and MDD may identify more targeted anti-depressive therapies and facilitate better-informed prescription decisions by clinicians for patients with comorbidity [16].
Typically, RCTs are highly structured investigations used to identify the causal effects of drugs in disease [17]. However, RCTs are expensive and time-consuming, which could explain why there has been a lack of large, high-quality RCTs exploring the effects of NSAIDs in MDD. RCTs typically evaluate the relatively short-term effects of an intervention on intermediate biomarkers or populations, but without estimating the effects on genetic variants [18]. MR uses genetic variants which are robustly associated with exposure as instrumental variables to explore causal associations between the exposure and outcome [18]. Additionally, the effects of genetic variants used to instrument variables in the MR design are present at conception [19], thus MR studies can be used to estimate the long-term effects of exposure of interest on the risk of an outcome [17]. Genetic variants related to NSAIDs target messenger RNA expression or DNA methylation (DNAm), also called expression or methylation quantitative trait loci (eQTLs or mQTLs, respectively), and can be viewed as instrumental variables representing NSAIDs exposure [20, 21]. In this study, we used eQTL and mQTL data to identify suitable genetic instruments to investigate the expression of NSAIDs targets. Using this strategy, MR analysis could overcome the limitations of observational studies, e.g., by limiting potential confounders, inferring causality and using existing study samples.
Recent studies investigated CRP [21] and IL-6 [22] associations with MDD risk and suggested a potential causal relationship between inflammatory factors and a subset of depressive symptoms. However, it is unknown if NSAIDs affect MDD and if these putative affects towards MDD are mediated by inflammatory factors.
In this study, we sought to identify NSAIDs associations with MDD using a two-sample MR approach and multi-omics datasets. We used publicly available eQTL and mQTL datasets to identify suitable genetic instruments for gene expression and DNAm (drug exposure proxies) analyses of NSAIDs targets. We also used the most recent and largest MDD GWAS to explore and characterise associations between NSAIDs, inflammatory factors and MDD.
Subjects and methods
Identifying NSAID target genes
We identified different NSAIDs classes using the Anatomical Therapeutic Chemical classification system using the World Health Organisation Collaborating Centre for Drug Statistics Methodology. Genes whose protein products were targeted by any of NSAIDs active ingredients were identified using DrugBank [23] (https://go.drugbank.com/) and ChEMBL [24] (https://www.ebi.ac.uk/chembl/) databases. We only included genes that were named as NASIDs targets in both databases.
Genetic instruments for NSAID target gene expression
Using a publicly available dataset from the eQTLGen consortium [25], we identified genetic variants with a minor allele frequency > 0.01 which were related to the expression of NSAIDs targets (n = 31,684). Only cis regions were accessed with a distance of single nucleotide polymorphisms (SNPs). The eQTL dataset was limited to a 1-SD change in gene expression levels for each affected allele. The cis region was defined as within 2 Mb of a probe in either direction. We used Consortium for the Architecture of Gene Expression (CAGE) eQTL summary data [26] (n = 2765; predominantly European heritage) to validate genetic instrument of inflammation identification (eTable 1, Supplementary Tables).
We explored DNAm levels of significant NSAIDs target genes in blood using a meta-analysis of blood mQTL [27] data from the Brisbane Systems Genetics Study (n = 614) and the Lothian Birth Cohorts of 1921 and 1936 (n = 1366). Participants were all of European ancestry (eTable 1, Supplementary Tables). We annotated the closest genes to DNAm probes using annotation files from Price et al. [28]. We also used whole blood mQTL data from the UKHLS study (n = 1193; European heritage) to validate our results [29].
Outcome data
Publicly available GWAS summary data for MDD (170,756 cases and 329,443 controls of European ancestry) were downloaded from the website, and excluded UK Biobank participants [30] (eTable 2, Supplementary Tables). Moreover, we also used GWAS MDD summary data from the UK Biobank (UKB) (113,769 cases and 208,811 controls of European ancestry) to validate our analyses [31] (eTable 2, Supplementary Tables).
Using summary-based MR (SMR) and independent heterogeneity instruments (HEIDI) to detect pleiotropic associations
MR analysis of NSAIDs target gene expression in blood and inflammation
We used the SMR method [32] to perform two-sample MR analyses (Supplementary Methods). To identify associations between changes in gene expression and inflammatory factor levels with drug exposure, SMR analyses were conducted using blood gene expression as the exposure and inflammatory factor levels as the outcome. Inflammatory factor levels were assessed using GWAS summary data for CRP [33] (n = 204,402), TNF [34] (n = 21,758), IL-6 [35] (n = 1301) and IL-1β [36] (n = 4910) in European ancestry (eTable 2, Supplementary Tables). We only included potential genes whose expression in blood was related to inflammatory factor levels at a nominal p value (p < 0.05) in analyses. Estimates from SMR for assessing associations between gene expression and inflammatory factors represented changes in inflammatory factor levels per 1-SD increase in gene expression.
MR analysis of NSAIDs target gene expression and DNAm in blood and MDD
SMR analyses were performed to estimate associations of a 1-SD change in gene expression (eQTL data) and DNAm (mQTL data) in MDD (GWAS summary data). Main results were presented as binary variables, such as OR for MDD per 1-SD change in gene and DNAm expression, where the direction of expression for the gene or DNAm change was harmonised to show inflammatory factor level decreasing associations. To control the genome-wide type I error rate, Bonferroni corrections were used to account for multiple testing.
MR analysis integrate multi-omics data in blood
We conducted an SMR analysis using blood DNAm as the exposure and gene expression as the outcome. This was to evaluate the relationship of a 1-SD change in DNAm with transcript levels, which were significantly associated with inflammatory factor levels and MDD. For SMR and HEIDI evaluations of associations between DNAm and transcripts, we only tested for relationships between single DNAm sites and single gene expression probes within a 2 Mb distance of cis regions. Bonferroni correlations were used to adjust for multiple comparisons.
Two-sample MR analyses evaluating associations between inflammatory factor levels and MDD
We evaluated association estimates between inflammatory factor levels and MDD using the generalised summary data-based MR (GSMR) method—we used multiple genetic variants, which were related to risk factors, as instruments to identify potential causality [37]. Genome-wide Complex Trait Analysis (version 1.91.7β1) was used to perform GSMR. HEIDI excludes linkage SNPs (i.e., two distant genetic variants in linkage disequilibrium (LD) independently affected gene expression and phenotype). For sensitivity analyses, a two-sample MR analysis was performed using random-effects inverse-variance weighted, weighted median, simple mode, weight mode and MR-Egger methods [38].
Sensitivity analysis
Additional sensitivity analyses, such as horizontal pleiotropy and colocalisation analysis, were performed for significant MR associations. Analyses using additional summary statistics were also performed to validate the main outcomes.
Colocalisation analysis
For statistically significant MR associations, Bayesian colonisation analyses were performed to evaluate the posterior probability of the same causal variants; the genetic variants with statistically significant MR associations for gene expression and outcomes were assessed in the coloc (v3.1) R package [39]. Default parameters were used for analysis.
Horizontal pleiotropy analysis
One genetic variant may be associated with the expression of more than one gene, which could thus violate the MR assumption that the instrumental variable only has an association with an outcome via drug target gene expression changes. We extracted associations with nearby genes (within a 2 Mb window) to evaluate horizontal pleiotropy. SMR analyses were used to test if the expression of these nearby genes was associated with nominal significance (p < 0.05), and associated or not, with the outcome. We reported genes that passed nominal significance as we were interested in identifying specific genes which were related to the trait rather than all genes or the study as a whole.
MR analysis of gene expression in the brain and MDD
We performed SMR analyses on gene expression in brain tissues and MDD using the PsychENCODE resource, with a sample size of 1387 unique donors. The effects estimate from SMR represented the effect on the risk (OR) of disease per 1-SD change in gene expression. We also performed SMR analysis on gene expression data in the GTEx (v8) dataset at 13 different brain regions, with sample sizes ranging between 130 and 250 unique donors. All association analyses were performed within a 2 Mb window around each gene or transcription start site.
Target gene expression profiles in tissue
To analyse the expression of genes and DNAm profiles, we used two whole blood sample datasets (GSE98793 and GSE125105 datasets) downloaded from the Gene Expression Omnibus website (https://www.ncbi.nlm.nih.gov/gds). The GSE98793 dataset used an Affymetrix Human Genome Array which included 64 MDD patients and 64 healthy control whole blood samples. Expression parameters comprised fragmented per kilobase of transcript per million mapped reads with a log2 transformation, which were transformed to transcripts per kilobase million. Moreover, genes with a zero SD in expression were removed (they were irrelevant to further analysis). The GSE125105 dataset used Illumina Infinium 450k Human DNAm bead-chip technology and included 489 MDD patients and 210 healthy controls. Before analyses, datasets were normalised using z-scores. t-tests were used to identify differences in gene expression levels between MDD patients and healthy controls.
To explore the biological mechanisms of significant genes using analytical strategies, the GTEx Portal (https://gtexportal.org/home/) was used to identify target gene expression levels in 13 brain regions and whole blood samples. We also explored exon expression levels in 54 tissues (GTEx V7).
MR power calculations
We performed power calculations using an online tool [40] (http://sb452.shinyapps.io/power/) to estimate causal effects at 80% power detection.
Weak instrumental bias in MR analyses
The F value from exposure regression (DNAm or gene expression) in instrumental variables (eQTL or mQTL SNP) was used in one-sample MR studies to assess instrumental variable strength. As a general rule, instrumental variables with an F value > 10 were not considered ‘weak instruments’ [41]. For two-sample MR analysis, the F value was generated using the approximation report by Bowden et al. [42] (information is provided in Supplementary Methods).
Results
Genetic instrument selection
A study design summary and primary results are shown (Fig. 1). In total, 91 gene encoding proteins were identified, which were experimentally shown to be modified by one or more NSAIDs (eTable 3, Supplementary Tables). Of these, 61 genes were expressed in blood as annotated by eQTLGen. Of these, the expression of 19 genes was associated with inflammatory factor levels at P_SMR < 0.05 (eTable 4; Supplementary Tables) and these were included in the main analysis. For selected genes, independent cis-eQTL SNPs were included that reached GWAS significance (p < 5 × 10−8). Of these, the most significant cis-eQTL SNPs were selected as genetic instruments, with F values > 10 (eTable 4, Supplementary Tables).

bxy estimated effect of exposure on outcome; eQTL expression quantitative trait loci, mQTL methylation quantitative trait loci, GWAS genome-wide association study, HEIDI heterogeneity in dependent instrument, MR Mendelian randomisation, SMR summary-based MR, SNP single-nucleotide variant, and WHOCC World Health Organization Collaborating Centre, MDD major depressive disorder.
MR analysis of gene expression in blood and MDD
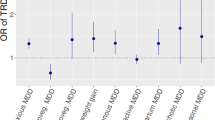
Using a significant threshold of p < 6.58 × 10−4 and a HEIDI test p ≥ 0.05 value to exclude linkage associations, we identified a 1-SD decrease in blood for neuraminidase 1 (NEU1) expression, which was associated with decreased CRP levels of 0.277 mg/l (95% CI: 0.128–0.426) (eTable 4, Supplementary Tables) and a lower risk of MDD (OR = 0.806; 95% CI: 0.735–0.885; p = 5.36 × 10−6) (Fig. 2 and eTable 5, Supplementary Tables). The genetic mutations of top associated SNP (rs367364) regulated gene expression probes (ENSG00000204386) to change the degree of the NEU1 expression. Decreased NEU1 expression could lead to decreased CRP levels and a lower risk of MDD. Colocalisation analysis identified a > 85% posterior probability for the same variant (rs367364) regulating NEU1 (OMIM 608272) expression and affecting MDD risk (eTable 6, Supplementary Tables).

Forest plot of the association between a 1-SD change in expression of 16 inflammatory factor levels lowering drug target genes in blood with risk for MDD. Data are represented as odds ratios (ORs) with 95% CI (error bars). The direction of gene expression change reflects the inflammatory factor levels lowering association. Therefore, an OR of greater than 1.00 suggests an decreased risk of MDD associated with NSAIDs treatment. Associations are statistically significant after correcting for multiple testing (16 genes × 3 inflammatory factors) and have a heterogeneity in dependent instrument (HEIDI) p ≥ 0.05, statistically significant after correcting for multiple testing but have a HEIDI p < 0.05 (indicating association likely due to linkage) or did not pass the multiple testing correction. SMR indicates summary-based Mendelian randomisation.
Using CAGE eQTL and MDD (UKB) GWAS datasets, these results indicated a concordant association between NEU1 expression and MDD risk. The results also indicated that a 1-SD decrease in blood NEU1 expression was associated with decreased inflammatory factor levels (CRP) of 0.168 mg/l (95% CI: 0.045–0.290) (eTable 7, Supplementary Tables) and a lower risk of MDD-UKB (OR = 0.966; 95% CI: 0.948–0.985, p = 5.19 × 10−4) (eFig. 1 and eTable 8, Supplementary Tables). From primary analysis, the top SNP (rs3130063) in ENSG00000204386 validation analysis was 46 kilobase pairs (kbp) distant from the top associated SNP (rs367364). The r2 and D’ values for rs3130063 and rs367364 were 0.25 and 0.55, respectively.
MR analysis of DNAm in MDD
We used a significant threshold of p < 3.54 × 10−5 and a HEIDI p ≥ 0.05 value to exclude linkage associations and found that a 1-SD increase in blood NEU1 DNAm was associated with a lower risk of MDD (OR = 0.886; 95% CI: 0.836–0.939; p = 4.71 × 10−5) (eTable 9, Supplementary Tables). The genetic mutations of top associated SNP (rs693906) change the degree of NEU1 DNAm by regulating DNAm probes (cg00397479). Decreased NEU1 expression could lead to decreased CRP levels and a lower risk of MDD. We observed a reverse association between NEU1 DNAm and MDD risk using validated mQTL and GWAS datasets (OR = 0.969; 95% CI: 0.954–0.986; p = 3.01 × 10−4) (eTable 10, Supplementary Tables). The top SNP (rs3130063) in the validation analysis of cg00397479 was 31 kbp distant from the top associated SNP (rs693906) in primary analysis.
MR associations across multi-omics platforms
We integrated two omics results to identify NEU1 pleiotropy between DNAm and MDD, and between gene expression and MDD. Then, possible associations were explored between DNAm and gene expression using DNAm as the exposure and the transcript as the outcome. The results for these pairs are shown in the median model (Fig. 3A) and indicated that the effects of genetic variants in the MDD regulating the expression of gene mediates loci through DNAm.

A The effects of DNA methylation (DNAm) on trait, DNAm on gene expression, and gene expression on trait are evaluated using the SMR and HEIDI method and integrated to identify potential mediation mechanisms in which an SNP exerts an effect on the trait by altering the DNAm level, which then regulates the expression levels of a functional gene. The detailed steps were (1) Use SMR to determine association between DNAm and gene expression; (2) Use SMR to determine associations between DNAm and major depressive disorder (MDD); (3) Use SMR to determine associations between gene expression and MDD. B When DNAm in the promoter is higher, the binding of transcription factor is disrupted, thereby suppressing the expression of NEU1. When DNAm is low, transcription factors usually bind to the promoter, and the expression level of NEU1 increase.
Most DNAm sites in DNAm-transcript associations are located in promoter or enhancer regions, making it possible to infer the regulation the degree of DNAm and gene expression by genetic variant. A notable example was the cg00397479-NEU1-MDD axis (Fig. 4). β values for the causal association between DNAm to MDD, DNAm to gene and gene to MDD were −0.120, −0.519 and 0.215, respectively (eTable 11, Supplementary Tables). Indeed, the DNAm probe (cg00397479) resided in the transcriptional start sites (TSS) region of NEU1 (ENSG00000204386) across multiple tissues and cells, as identified in the Epigenome Integration Across Multiple Annotation Project (EpiMap) database. The corresponding DNAm locus belonged to the TSS functional region, suggesting a possible biological pathway for disease risk regulation. The genetic mutation (rs693906) regulated the methylation probe (cg00397479) to alter DNAm levels. When these levels at the NEU1 TSS were high, transcription factor binding was disrupted and suppressed NEU1 expression (β = −0.519; 95% CI: −0.717 to −0.320256; p = 3.16 × 10−7) and decreased MDD risk (β = −0.120; 95% CI: 0.062 to −0.178; p = 4.71 × 10−5). Furthermore, the top SNP in mQTL and GWAS analysis of the cg00397479 probe was 180 kbp distant from the top associated SNP in the eQTL and GWAS analysis of ENSG00000204386. The r2 and D’ values for rs693906 and rs367364 were 0.35 and 0.65, respectively.

The top plot shows −log10 (p values) of SNPs from the major depressive disorder (MDD) GWAS. The red diamond and blue circle represent −log10 (p values) for probes from the SMR tests for associations of gene expression and DNA methylation (DNAm) probes, respectively. The second plot shows eQTL results for the probe ENSG00000204386 (tagging NEU1). The third plot shows mQTL results for the DNAm probe cg00397479. The bottom plot shows 18 chromatins (indicated by colours) of 833 samples from EpiMap for different primary cells and tissue types (rows).
From this evidence, we hypothesised that a genetic variant in the TSS functional region of NEU1 led to DNAm, then down-regulated NEU1 expression (Fig. 3B). Decreased NEU1 expression could theoretically reduce MDD risks.
From validation analysis, a concordant association was observed between NEU1 DNAm, gene expression and MDD risk (eTable 12, Supplementary Tables). β values for DNAm to MDD, DNAm to gene and gene to MDD were −0.031, −0.841 and 0.033, respectively. The genetic mutation (rs3130063) regulated the methylation probe (cg00397479) to alter the degree of DNAm. The top SNP (rs3130063) in mQTL and GWAS was the same as the top SNP in eQTL and GWAS analysis of ENSG00000204386.
Sensitivity analyses
MR analyses of NEU1 brain expression
We failed to observe that NEU1 expression in all 14 brain regions was associated with MDD risk (eTable 13, Supplementary Tables).
Assessing the NEU1 eQTL for horizontal pleiotropy
The eQTL SNP for NEU1 expression in blood (rs367364) was related to the expression of 42 adjacent genes (p < 0.05), including three genes at p < 1 × 10−3 (APOM [OMIM 606907], TNXB [OMIM 600985] and HSPA1A [OMIM 140550] (eTable 6, Supplementary Tables). However, using SMR and colocalisation analyses, we failed to identify associations between the expression of these adjacent genes with MDD (eTable 14, Supplementary Tables), and we failed to identify horizontal pleiotropy. Possible reasons for the observed associations between NEU1 expression and MDD are shown (eFig. 2, Supplementary Figs.).
MR analysis of inflammatory factor levels in MDD
We identified no evidence to support an association between genetically estimated CRP (OR = 0.1.01, 95% CI: 0.987–1.033; p = 0.40), IL-6 (OR = 0.97, 95% CI: 0.942–1.008; p = 0.14) and TNF (OR = 1.01, 95% CI: 0.987–1.027; p = 0.48) levels in MDD using 56, 9 and 5 SNPs, which were respectively associated with GWAS significance (p = 5 × 10−8) as MR instruments (after HEIDI outlier filtering to remove pleiotropic SNPs) (eTable 15, Supplementary Tables).
NEU1 expression and DNAm profiles and tissue enrichment analysis
We integrated NEU1 expression and DNAm profiles from GSE98793 and GSE125105 datasets and observed no differences in profiles between MDD patients and healthy controls (eFig. 3, Supplementary Figs.).
The eQTL SNP (rs367364) affected NEU1 expression by affecting of transcription regulation combinations. The T allele (eFig. 4A, Supplementary Figs.) was associated with lower gene expression (p = 2.12 × 10−3). Assuming that differences in gene expression were related to differences in protein activity, patients carrying a T allele at this eQTL SNP (genotype TT) of NEU1 were similar to the treatment group of the NSAIDs .
Using GTEx project data, NEU1 expression was higher in cerebellar and cerebellum regions when compared with other brain regions (eFig. 4B, Supplementary Figs.). Also, exon 5 of NEU1 was highly expressed in adrenal gland tissue (eFig. 4C, Supplementary Figs.).
Discussion
Using QTL and GWAS summary statistics, we used a two-sample MR approach to infer the potential effects of NSAIDs towards MDD. Lower NEU1 expression and higher DNAm NEU1 expression levels were associated with decreased inflammatory factor (CRP) levels and decreased MDD risk. Our integrative analyses, based on multi-omics datasets, showed that higher DNAm levels were associated with lower NEU1 expression. Combined results indicated that NSAIDs targeting gene expression or DNAm could decrease MDD risk. We did not observe any association between genetic inflammatory factor level estimates and MDD. Our results indicated that even minor potential effects of inflammatory factor levels (CRP, IL-6, TNF and IL-1) on MDD are not likely and reveal that any association between NEU1 and MDD is independent of inflammatory factors (CRP, IL-6, TNF and IL-1).
NEU1 is one of four mammalian neuraminidase isoenzymes and is predominately located in the lysosome [43, 44]. The protein is encoded by the sialidase gene (NEU1) and plays a crucial role in the lysosomal catabolism of sialylated glycoconjugates [45]. In addition to this crucial role, growing evidence now suggests that NEU1 is vital for the immune system and is mechanistically involved in cell signalling during immune responses [44,45,46]. Recently, it was shown that NEU1 regulated toll-like receptor (TLR) activation on macrophages; specifically, ligand binding to TLRs induced NEU1 activity and led to receptor activation, nitric oxide and proinflammatory cytokine production [47]. Importantly, these NEU1 roles in macrophages are confirmed by several in vitro studies [47,48,49,50,51].
Animal studies [14, 52, 53] also confirmed that NEU1 deficiency diminished lymphocyte and macrophage stimulation, thereby decreasing immune cell availability for cytokine and antibody production [54]. Critically, global or macrophage-specific NEU1 knockout mice were associated with improved vascular inflammation, lowered apoptosis, decreased reactive oxygen species production, mitigated extracellular matrix degradation and improved M2 macrophage polarisation [52]. As NEU1 appears to be required for inflammatory signalling in microglia, the gene could function as a novel target to reduce central nervous system inflammation-related neurodysfunction disorders, including MDD [5, 48]. Abnormal activation of microglia, which are immunological guardians of the brain, and increased microglial cell numbers were observed in depression and anxiety disorders, although it was unclear how this related to psychopathological conditions [55]. Recently, a study described depression as a microglia-associated disorder, and apart from excessive microglia activation and increased cell numbers, microglial decline and senescence were observed in patients with depression [56]. Therefore, microglial activation may suppress neurogenesis and neuroplasticity, further promoting the incidence of depression-like symptoms [57]. From our conclusions, we speculate that NEU1 could participate in MDD pathogenesis by regulating microglia or macrophages, which in turn regulate cytokine levels, glutamate secretion and HPA axis activity. However, how NEU1 directly affects MDD pathogenesis remains unknown as limited studies have focused on NEU1 activities.
Ikeda et al. recently conducted a behavioural analysis using NEU1-knockout (NEU1-KO) zebrafish [58] and reported that fish showed regular swimming capabilities similar to wild-types but they exhibited lower social interactions, aggression and abnormal social preferences [58], indicating a potential effect of NEU1 on emotional activities. Notwithstanding this, the extent to which brain function is conserved between fish and mammals is controversial; the distribution and structure of sialoglycans and associated enzymes differ between fish and humans [58]. Thus, elucidating abnormal behaviour and emotional reaction mechanisms in NEU1-KO mammalian models and cell lines is clinically warranted. Herein, our results indicated that NSAIDs decreased NEU1 expression or increased NEU1 DNAm which could reduce MDD risk. Given this evidence, we hypothesise that decreased NEU1 may act in a negative feedback loop to reduce inflammation by monocytes/macrophages and potentially relieve MDD development.
Although, our study indicated a protective effect of NEU1, the use of acetylsalicylic acid to target MDD and its impact on MDD remains controversial in population-based studies [59,60,61]. Recently, an RCT involving 19,114 older adults showed that taking low-dose aspirin (acetylsalicylic acid) at 100 mg/day did not lower depression rates when compared with placebo [59]. Moreover, a meta-analysis of 12 observational, 5 case-control and 7 prospective cohort studies showed a significant association between aspirin use and depression (OR/Risk ratio = 1.10; 95% CI: 1.05–1.16) [60]. Another systematic review of six clinical studies indicated that the low-dose aspirin (80–100 mg/day) was safe, well-tolerated and potentially efficacious for improving depressive symptoms in patients with unipolar and bipolar depression [61]. These studies indicated that other important information, including drug dose and exposure duration, inter-individual variation with respect to drug metabolism and genetics exerted modifying roles in terms of drug efficacy and toxicity. Thus, conducting specific RCTs for different dose subgroups and drug exposure durations could help characterise the cumulative exposure effects on outcomes. Performing MR analysis does have a well-defined interpretation of causal relationship as an intervention on the genetic code occurs at conception. Thus, MR estimates could represent a life-long difference in exposure between genetic subgroups [62]. Furthermore, most RCTs are performed on mature individuals, who may in a stage of disease progression is irreversible. There may be no intervention on the exposure in a mature cohort which can imitate the genetic effect [62]. Hence, causation assessments using MR investigations and clinical interventions could identify true causal exposure associations for MDD and provide more evidence for MDD prevention.
No causal association between inflammatory factor levels and MDD was identified by using MR method, our results suggested that any associations between NEU1 and MDD were not likely to be mediated by the changes of inflammatory factors (CRP, IL-6 and TNF) levels. Other two-sample MR analyses focused on causal relationships between cytokines; MDD did not consistently demonstrate CRP or IL-6 associations with other depressive symptoms, except for an association between IL-6 signalling and suicidality [21]. The association between inflammation and MDD is unequivocal; over the past two decades, growing evidence has suggested that MDD is associated with systemic immune activation, comprising abnormalities in inflammatory makers, immune cell numbers and antibody titres [5]. Our results do not show a deniable association between immune system activity and MDD, thus, it is plausible that other inflammatory profiles, besides CRP, IL-6 and TNF, may be associated with different depression subtypes [63, 64]. Conducting more comprehensive and high-quality studies could facilitate a better understanding of immune-neuropsychological interactions. By further exploring the relationships arising between immunity and MDD, we can improve our understanding of disease conditions and help design better therapies to promote the quality of life of individuals with MDD.
Besides NSAIDs, we also identified NEU1 as a target for oseltamivir. Oseltamivir was developed for prophylactic and therapeutic use against influenza, specifically targeting the viral enzyme’s highly-conserved active site [65]. It has been reported that some oseltamivir-treated children had experienced adverse neuropsychiatric events [66,67,68]. To estimate the incidence rate of adverse neuropsychiatric events in patients given oseltamivir, Toovey et al. conducted an analysis covering Japan, the US, and other countries and finding such neuropsychiatric events generally fell into the categories of abnormal behaviours, delusions, and perceptual disturbances [66]. In addition, there were cases of delirium and delirium-like events, depressed consciousness levels, parasomnia, suicidal events, accidents, and injuries [66]. One 15-year-old girl in Korea was diagnosed with depressive episode after taking oseltamivir [68]. Reportedly, the neuropsychiatric adverse events were more common in children than in adults [67]. Based on the neuropsychiatric reverse effect of oseltamivir in children, the implication of NEU1 targeted drugs, including NSAIDs and oseltamivir, for the effect of treatment for depression should be explained with caution in both children and teenagers.
Study limitations and strengths
Firstly, our assumption that alterations in gene expression exerted changes in protein levels and activity may not always be the case. Therefore, our failure to detect associations may not indicate a lack of biological effects of drug therapy. Secondly, our study did not detect associations between expression changes in drug target genes and DNAm in specific tissues with MDD. For example, we did not detect significant associations between NEU1 and MDD in brain tissue, which may have been restricted by eQTL dataset sample size. Given this and varying instrument strength in different brain regions (eTable 13), our study did not identify any smaller but clinically relevant effects of NEU1 expression in any brain regions. Moreover, a lack of statistical power in identifying outcomes could also be another viable explanation for some of our study limitations (for power calculations, eTable 16 Supplementary Tables). Thirdly, we did not estimate any associations between NSAIDs and other inflammatory factors or MDD sub-symptoms—factors worth exploring in the future. Lastly, estimates from our MR analyses may not have reflected the actual effects of drug exposure. Other important information, such as doses and drug exposure duration, individual drug metabolism variants, the ability to reach target tissue and drug-binding affinity could have exerted modifying effects on drug efficacy and toxicity, thus making it difficult to extrapolate the effects between drug exposure and our analyses.
However, our study had some unique strengths. Human genetics continuously provides evidence for new drug discovery and drug safety. Loss-of-function mutations in drug target genes are ideal MR instrumental variables, however, as these variants are rare, related MR studies are largely unfeasible. Alternatively, we used eQTL and mQTL genetic variants as MR instrumental variables. Available GWAS, eQTL and mQTL datasets were exploited to investigate causality, and we avoided observational study and RCT limitations by using a two-sample MR design, e.g., limited sample size and reduced confounding bias, while using cis-SNPs to represent messenger RNA or DNAm levels could help to minimise the potential for horizontal pleiotropy. We also explored associations between DNAm and gene expression, which could help unravel biological mechanisms underpinning inflammation in MDD.
Conclusions
We identified a concordant association for lower NEU1 messenger RNA and higher DNAm levels with MDD risk, with sensitivity analyses suggesting that any NEU1 association with MDD were likely to be independent of inflammatory factor associations. We speculate that the further lowering of NEU1 activity or increasing NEU1 DNAm expression in patients with MDD could improve symptoms or limit disease episodes. Our findings warrant further investigations into the functional role of NEU1 in MDD and the potential exploitation of drug-repurposing for this condition.
Data availability
All the data used in this study could be accessed online. The links for data used in this study can be found in the Mendeley data (https://doi.org/10.17632/xj67ctfpkw.1).
References
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Black C, Miller BJ. Meta-analysis of cytokines and chemokines in suicidality: distinguishing suicidal versus nonsuicidal patients. Biol Psychiatry. 2015;78:28–37.
Syed SA, Beurel E, Loewenstein DA, Lowell JA, Craighead WE, Dunlop BW, et al. Defective inflammatory pathways in never-treated depressed patients are associated with poor treatment response. Neuron. 2018;99:914–24.e3.
Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107:234–56.
Lamers F, Milaneschi Y, Smit JH, Schoevers RA, Wittenberg G, Penninx BWJH. Longitudinal association between depression and inflammatory markers: results from the Netherlands study of depression and anxiety. Biol Psychiatry. 2019;85:829–37.
Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–15.
Uher R, Tansey KE, Dew T, Maier W, Mors O, Hauser J, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry. 2014;171:1278–86.
Chamberlain SR, Cavanagh J, Boer P, de, Mondelli V, Jones DNC, Drevets WC, et al. Treatment-resistant depression and peripheral C-reactive protein. Br J Psychiatry. 2019;214:11–19.
Andrade C. Anti-inflammatory treatments for depression: perspectives on how to read a meta-analysis critically. J Clin Psychiatry. 2019;80. https://doi.org/10.4088/jcp.19f12907.
Eyre HA, Air T, Proctor S, Rositano S, Baune BT. A critical review of the efficacy of non-steroidal anti-inflammatory drugs in depression. Prog NeuroPsychopharmacol Biol Psychiatry. 2015;57:11–16.
Köhler‐Forsberg O, Lydholm CN, Hjorthøj C, Nordentoft M, Mors O, Benros ME. Efficacy of anti‐inflammatory treatment on major depressive disorder or depressive symptoms: meta‐analysis of clinical trials. Acta Psychiatr Scand. 2019;139:404–19.
Gallagher PJ, Castro V, Fava M, Weilburg JB, Murphy SN, Gainer VS, et al. Antidepressant response in patients with major depression exposed to NSAIDs: a pharmacovigilance study. Am J Psychiatry. 2012;169:1065–72.
Song C, Wang H. Cytokines mediated inflammation and decreased neurogenesis in animal models of depression. Prog NeuroPsychopharmacol Biol Psychiatry. 2011;35:760–8.
Feltes PK, Doorduin J, Klein HC, Juárez-Orozco LE, Dierckx RA, Moriguchi-Jeckel CM, et al. Anti-inflammatory treatment for major depressive disorder: implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J Psychopharmacol. 2017;31:1149–65.
Miller AH. Beyond depression: the expanding role of inflammation in psychiatric disorders. World Psychiatry. 2020;19:108–9.
Chauquet S, Zhu Z, O’Donovan MC, Walters JTR, Wray NR, Shah S. Association of antihypertensive drug target genes with psychiatric disorders. JAMA Psychiatry. 2021;78:623–31.
Ference BA, Holmes MV, Smith GD. Using Mendelian randomization to improve the design of randomized trials. Cold Spring Harb Perspect Med. 2021;11:a040980.
Gill D, Walker VM, Martin RM, Davies NM, Tzoulaki I. Comparison with randomized controlled trials as a strategy for evaluating instruments in Mendelian randomization. Int J Epidemiol. 2019;49:1404–6.
Lee YH. Overview of Mendelian randomization analysis. J Rheum Dis. 2020;27:241–6.
Kappelmann N, Arloth J, Georgakis MK, Czamara D, Rost N, Ligthart S, et al. Dissecting the association between inflammation, metabolic dysregulation, and specific depressive symptoms. JAMA Psychiatry. 2021;78:161–70.
Kelly KM, Smith JA, Mezuk B. Depression and interleukin-6 signaling: a Mendelian RANDOMIZATION STudy. Brain Behav Immun. 2021;95:106–14.
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72.
Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45:D945–54.
Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. Biorxiv. 2018;10:447367.
Lloyd-Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet. 2017;100:371.
McRae AF, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, et al. Identification of 55,000 replicated DNA methylation QTL. Sci Rep. 2018;8:17605.
Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenet Chromatin. 2013;6:4.
Hannon E, Gorrie-Stone TJ, Smart MC, Burrage J, Hughes A, Bao Y, et al. Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am J Hum Genet. 2018;103:654–65.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun. 2018;9:1470.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
Ligthart S, Vaez A, Võsa U, Stathopoulou MG, Vries PS, de, Prins BP, et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. 2018;103:691–706.
Gilly A, Park Y-C, Png G, Barysenka A, Fischer I, Bjornland T, et al. Whole genome sequencing analysis of the cardiometabolic proteome. Nat Commun. 2020;11:854752.
Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020;2:1135–48.
Offenbacher S, Jiao Y, Kim SJ, Marchesan J, Moss KL, Jing L, et al. GWAS for Interleukin-1β levels in gingival crevicular fluid identifies IL37 variants in periodontal inflammation. Nat Commun. 2018;9:3686.
Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9:224.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Wang G, Sarkar A, Carbonetto P, Stephens M. A simple new approach to variable selection in regression, with application to genetic fine-mapping. J. R. Stat. Soc. 2020;82:1273–300.
Burgess S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol. 2014;43:922–9.
Staiger D, Stock JH. Instrumental variables regression with weak instruments. Econometrica. 1997;65:557.
Bowden J, FDG M, Minelli C, Smith GD, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45:1961–74.
Bonten EJ, Annunziata I, d’Azzo A. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci. 2014;71:2017–32.
Sieve I, Münster-Kühnel AK, Hilfiker-Kleiner D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc Pharm. 2018;100:26–33.
Seyrantepe V, Iannello A, Liang F, Kanshin E, Jayanth P, Samarani S, et al. Regulation of phagocytosis in macrophages by neuraminidase 1*. J Biol Chem. 2010;285:206–15.
Sieve I, Ricke-Hoch M, Kasten M, Battmer K, Stapel B, Falk CS, et al. A positive feedback loop between IL-1β, LPS and NEU1 may promote atherosclerosis by enhancing a pro-inflammatory state in monocytes and macrophages. Vasc Pharm. 2018;103:16–28.
Abdulkhalek S, Amith SR, Franchuk SL, Jayanth P, Guo M, Finlay T, et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for toll-like receptor activation and cellular signaling*. J Biol Chem. 2011;286:36532–49.
Wang Q, Chen Z, Peng X, Zheng Z, Le A, Guo J, et al. Neuraminidase 1 exacerbating aortic dissection by governing a pro-inflammatory program in macrophages. Front Cardiovasc Med. 2021;8:788645.
Allendorf DH, Puigdellívol M, Brown GC. Activated microglia desialylate their surface, stimulating complement receptor 3‐mediated phagocytosis of neurons. Glia. 2020;68:989–98.
Amith SR, Jayanth P, Franchuk S, Finlay T, Seyrantepe V, Beyaert R, et al. Neu1 desialylation of sialyl α-2,3-linked β-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell Signal. 2010;22:314–24.
Allendorf DH, Franssen EH, Brown GC. Lipopolysaccharide activates microglia via neuraminidase 1 desialylation of toll‐like receptor 4. J Neurochem. 2020;155:403–16.
Khan A, Das S, Sergi C. Therapeutic potential of Neu1 in Alzheimer’s disease via the immune system. Am J Alzheimers Dis Other Dement. 2021;36:1533317521996147.
Walker DG, Lue L-F. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7:56.
El‐Etr M, Rame M, Boucher C, Ghoumari Abdel M, Kumar N, Liere P, et al. Progesterone and nestorone promote myelin regeneration in chronic demyelinating lesions of corpus callosum and cerebral cortex. Glia. 2015;63:104–17.
Serafini G, Amore M, Rihmer Z. The role of glutamate excitotoxicity and neuroinflammation in depression and suicidal behavior: focus on microglia cells. Neuroimmunol Neuroinflamm. 2015;2:127.
Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci. 2015;38:637–58.
Brites D, Fernandes A. Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci. 2015;9:476.
Ikeda A, Komamizu M, Hayashi A, Yamasaki C, Okada K, Kawabe M, et al. Neu1 deficiency induces abnormal emotional behavior in zebrafish. Sci Rep. 2021;11:13477.
Berk M, Woods RL, Nelson MR, Shah RC, Reid CM, Storey E, et al. Effect of aspirin vs placebo on the prevention of depression in older people. JAMA Psychiatry. 2020;77:1012–20.
Kim H, Kim J, Jung J. The association between aspirin use and depression: a systematic review and meta‐analysis of observational studies. Pharmacoepidemiol Drug Saf. 2020;29:613–22.
Ng QX, Ramamoorthy K, Loke W, Lee MWL, Yeo WS, Lim DY, et al. Clinical role of aspirin in mood disorders: a systematic review. Brain Sci. 2019;9:296.
Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377–89.
Dunjic-Kostic B, Ivkovic M, Radonjic NV, Petronijevic ND, Pantovic M, Damjanovic A, et al. Melancholic and atypical major depression—connection between cytokines, psychopathology and treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2013;43:1–6.
Kaestner F, Hettich M, Peters M, Sibrowski W, Hetzel G, Ponath G, et al. Different activation patterns of proinflammatory cytokines in melancholic and non-melancholic major depression are associated with HPA axis activity. J Affect Disord. 2005;87:305–11.
Jamieson B, Jain R, Carleton B, Goldman RD. Use of oseltamivir in children. Can Fam Physician. 2009;55:1199–201.
Toovey S, Rayner C, Prinssen E, Chu T, Donner B, Thakrar B, et al. Assessment of neuropsychiatric adverse events in influenza patients treated with oseltamivir: a comprehensive review. Drug Saf. 2008;31:1097–14.
Fuyuno I. Tamiflu side effects come under scrutiny. Nature. 2007;446:358–9.
Chung S, Joung YS. Oseltamivir (tamiflu) induced depressive episode in a female adolescent. Psychiatry Investig. 2010;7:302–4. https://doi.org/10.4306/pi.2010.7.4.302.
Acknowledgements
This study was possible thanks to our access to publicly available GWAS and eQTL (methylation) summary statistics, including data from the Psychiatric Genetics Consortium (including GWAS summary statistics for major depression from PGC and UK Biobanks, GWAS summary statistics on inflammatory factors from the IEU open GWAS project and GWAS Catalogue, the eQTLGen Consortium, the GTEx Project, the PsychENCODE Project data and data resources in the SMR pipeline).
Funding
This work is supported by City University of Hong Kong New Research Initiatives/Infrastructure Support from Central (APRC) (Grant No. 9610401).
Author information
Authors and Affiliations
Contributions
QH participated in the study design, analysed the data and wrote the paper. ANB, JL, KCHW and RH participated in the discussion of the study design and data analysis, and edited the manuscript. BF, APSK, XT and MKMK provided insightful comments. KHKC conceived the study, design and coordinated the study and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, Q., Wu, K.C.H., Bennett, A.N. et al. Non-steroidal anti-inflammatory drug target gene associations with major depressive disorders: a Mendelian randomisation study integrating GWAS, eQTL and mQTL Data. Pharmacogenomics J 23, 95–104 (2023). https://doi.org/10.1038/s41397-023-00302-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-023-00302-1
