
Abstract
Antidepressants demonstrate modest response rates in the treatment of major depressive disorder (MDD). Despite previous genome-wide association studies (GWAS) of antidepressant treatment response, the underlying genetic factors are unknown. Using prescription data in a population and family-based cohort (Generation Scotland: Scottish Family Health Study; GS:SFHS), we sought to define a measure of (a) antidepressant treatment resistance and (b) stages of antidepressant resistance by inferring antidepressant switching as non-response to treatment. GWAS were conducted separately for antidepressant treatment resistance in GS:SFHS and the Genome-based Therapeutic Drugs for Depression (GENDEP) study and then meta-analysed (meta-analysis n = 4213, cases = 358). For stages of antidepressant resistance, a GWAS on GS:SFHS only was performed (n = 3452). Additionally, we conducted gene-set enrichment, polygenic risk scoring (PRS) and genetic correlation analysis. We did not identify any significant loci, genes or gene sets associated with antidepressant treatment resistance or stages of resistance. Significant positive genetic correlations of antidepressant treatment resistance and stages of resistance with neuroticism, psychological distress, schizotypy and mood disorder traits were identified. These findings suggest that larger sample sizes are needed to identify the genetic architecture of antidepressant treatment response, and that population-based observational studies may provide a tractable approach to achieving the necessary statistical power.
Similar content being viewed by others

Introduction
Major depressive disorder (MDD) is a disabling condition with a high global impact [1, 2]. Antidepressants are the first-line treatment for MDD patients but response is modest with only approximately 50% achieving remission after completing two treatments [3]. The mechanisms underlying antidepressant resistance remain elusive but are of key value if more effective therapies are to be identified and developed.
Genome-wide association studies (GWAS) of antidepressant treatment response have yet to establish any replicated genetic variants [4,5,6,7,8,9,10]. Two large meta-analyses similarly reported no genome-wide significant associated variants. The first, a meta-analysis of the GENDEP (Genome-based Therapeutic Drugs for Depression), MARS (Munich Antidepressant Response Study) and the STAR*D (Sequenced Treatment Alternatives to Relieve Depression) [11] studies comprised of 2256 MDD cases, and the second, between the NEWMEDS (Novel Methods Leading to New Medications in Depression and Schizophrenia) and STAR*D [7] projects comprised of 2897 MDD cases. An additional analysis in the first meta-analysis restricted to citalopram or escitalopram did, however, identify an intergenic variant (5q.15.1) [11]. The largest GWAS to date examining treatment resistance (n = 1311) versus responders (n = 7795) was conducted by Li et al. utilising self-report information from 23andMe and found no significantly associated genetic variants, although found one variant (4.q22.1) associated with bupropion response [12]. Numerous candidate genes have also been investigated but the results are inconsistent [13]. Furthermore, the largest polygenic risk score (PRS) analysis in antidepressant response to date, (which utilised GENDEP/STAR*D data) yielded no significant associations for response itself, MDD or schizophrenia [14].
Discovering genomic variants associated with resistance to antidepressants could advance personal treatment, help identify resistant individuals earlier and inform our understanding of MDD. A recent systematic review reported non-response was associated with illness severity including higher suicide risk, number of hospitalisations and antidepressant dosage, but not cognitive ability [15]. In fact, several phenotypic associations have been found in treatment resistant individuals; more comorbidities and suicide attempts [16], increased neuroticism and decreased extraversion, openness and conscientiousness [17, 18]. Identifying genetic loci may therefore help to identify resistant individuals earlier and enable timelier intervention.
Currently pharmocogenetic studies are limited by small sample sizes [19] and low statistical power. Numerous studies have indicated the need for large sample sizes in genetic studies [20, 21]. The recent Li et al study maximised sample size by utilising self-report questionnaires [12], whilst other groups have examined treatment resistance in both MDD or schizophrenia by using prescription data [22,23,24].
In the present study, we employed a complementary approach utilising prescription data in a population and family-based cohort (Generation Scotland: Scottish Family Health Study; GS:SFHS) to define a dichotomous and a semi-quantitative measure of antidepressant resistance; treatment resistance and stages of resistance, respectively. We conducted a GWAS of antidepressant treatment resistance with meta-analysis with the GENDEP cohort and stages of antidepressant resistance in GS:SFHS only and calculated narrow-sense heritability estimates. Gene and gene-set enrichment analysis on both traits were also conducted and we further examined genetic correlations. We also utilised PRS techniques to examine pleiotropy between the genetic liability of MDD, schizophrenia and bipolar disorder in antidepressant treatment resistance and stages of resistance.
Methods
Cohort description
Generation Scotland: Scottish Family Health Study
GS:SFHS is a family and population-based cohort of 23,960 individuals (mean age = 47.6, s.d. = 15.4) within Scotland. Participants were eligible if they were aged above 18 years and had a first-degree relative also willing to participate in the study. Recruitment has been described in detail elsewhere [25, 26]. Genotype data were available for 20,032 participants and data on mood, cognitive function and personality traits were obtained through interview (see Supplementary Materials). Briefly, four cognitive tests (digit symbol coding, vocabulary, verbal fluency and logical memory), neuroticism and extraversion (measured by the Eysenck Personality Questionnaire), schizotypal personality questionnaire (SPQ), mood disorder questionnaire (MDQ), Scottish Index of Multiple Deprivation (SIMD) and number of years in education were all assessed. MDD was measured by structured clinical interview for DSM-IV (SCID) given a positive screening during the original interview (further details in Supplementary Materials).
Prescription data were available through data linkage to the Prescribing Information System administered by National Health Service (NHS) Scotland Information Services Division. Written informed consent for linkage was obtained for 98% of GS:SFHS and only those individuals that provided informed consent were analysed. Further information regarding the prescription records are found in the Supplementary Materials (see Supplemental Table S1 and S2). To define all MDD antidepressant users, records were excluded if the daily dose was below the minimum recommendations given by the British National Formulary (BNF) for MDD [27] and the duration was below 6 weeks of continuous treatment (as this is considered adequate duration [28, 29]). Following this pruning, we totalled the number of different antidepressants prescribed to each individual. This was then used as a measure of non-response, assuming that switching to a different antidepressant reflected failure or lack of clinical response. Drug switching due to side effects is expected to take place before the 6th week of treatment. Individuals with schizophrenia, schizoaffective disorder and bipolar disorder were excluded (n = 164). Additionally, antidepressants per individual were each defined as a “prescription episode” whereby uninterrupted prescriptions for the same antidepressant are considered one prescription episode. This was done in order to differentiate a repeat prescription for one episode from multiple depression episodes where the same antidepressant was given on more than one occasion (more information in the Supplementary Materials).
Defining treatment resistance and stages of resistance
Within the antidepressant users (as defined above), treatment resistance was assessed in GS:SFHS using only individuals that had been prescribed at least one antidepressant at an adequate dose and duration (as above, n = 3452). Case status for treatment resistance was defined as those individuals who had been prescribed more than two antidepressants providing 250 treatment resistant cases and 3202 non-treatment resistant controls. There have been significant difficulties defining treatment resistant depression in research but the general consensus is that it should be defined as non-response to more than two antidepressants [30].
Individual response to antidepressants decreases with more unsuccessful trials [3], it has therefore been suggested that a semi-quantitative stages of resistance phenotype might be more informative than a dichotomous approach [31]. Stages of antidepressant resistance were defined as the number of different antidepressants prescribed given an adequate dose and duration (as above). It was coded 1–4 with all individuals receiving more than four different antidepressants assigned a value of 4. This definition included 3452 individuals on antidepressants (2557, 645, 186 and 64 on 1, 2, 3 and 4+ antidepressants, respectively). Whilst this definition takes into account the first stage of the Massachusetts General Hospital definition [29], it does not account for augmentation or electroconvulsive therapy (ECT) as these data were not available. This definition therefore provides a semi-quantitative measure of resistance based on the number of treatments taken, providing additional incremental information beyond our binary measure.
Genome-based therapeutic drugs for depression
GENDEP is a 12-week study that examined antidepressant response in 867 individuals (mean age = 42.7, s.d. = 11.6) taking escitalopram and noritriptyline. Response was measured by the Montgomery–Åsberg Depression Rating Scale [32]. Antidepressant treatment resistance was defined as those who did not respond to more than 2 antidepressant therapies including GENDEP treatments and previous treatments (cases: 109, controls: 668), as described in a previous study [33]. A full description of the cohort is provided in Supplemental Table S3.
Genotyping, imputation and quality control procedures
Generation Scotland: Scottish Family Health Study
Blood samples were stored and genotyped at the Wellcome Trust Clinical Research Facility, Edinburgh (www.wtcrf.ed.ac.uk). Details of the DNA extraction and genotyping have been given elsewhere [34]. Imputation to a combined reference panel of 1000 Genomes Phase 1 Version 3 and the UK10K haplotype reference panels was completed using Minimac3 and phasing was conducted utilising SHAPEIT2 [35]. All individuals were white British and multidimensional scaling (MDS) components were also used to identify and remove people who were relative outliers for these measures of population ancestry. Quality Control (QC) inclusion criteria were INFO > 0.9, missingness per single nucleotide polymorphism (SNP) or individual < 1%, Hardy-Weinberg equilibrium (HWE) P-value cut-off of > 1 × 10–6, minor allele frequency (MAF) > 1%. 7,395,460 SNPs and 3452 individuals (and 250 cases for treatment resistance) passed QC criteria.
Genome-based therapeutic drugs for depression
DNA was extracted from blood samples and genotyped using the Illumina Human610quad bead chip (Illumina, Inc., San Diego). Imputation to the Haplotype Reference Consortium (HRC) data version 1 reference panel [36] was completed using Minimac3. QC exclusion criteria were poor imputation quality (r2 < 0.3 (using the Markov Chain method [37])), missingness per SNP > 5%, missingness per individual > 3%, MAF < 1%, related individuals (identity-by-descent > 0.188). Individuals with gender discrepancies, abnormal heterozygosity and population outliers were excluded. 7,518,836 SNPs and 761 individuals (108 cases) passed QC criteria.
Statistical analysis
Bivariate analysis for all antidepressant users and MDD
Bivariate analysis was completed in GCTA v1.91.4 [38] between all antidepressant users (as defined above, n = 3452), and Structured Clinical Interview for DSM Disorders (SCID)-diagnosed MDD (details in Supplementary Materials) to assess the degree of shared genetic architecture between these traits. Bivariate values were taken between two variance component measures; genetics (G) and kinship (K), which represent common genetics and pedigree-associated genetics (including rare variants) respectively. The G component is equivalent to methods used to measure SNP heritability and K is an altered G with a pairwise relatedness threshold less than 0.05 set to zero, this technique has been previously published [39]. The model was controlled for age, sex and the first four MDS components fitted to control for population stratification. Statistical significance was estimated using the likelihood-ratio test (LRT).
Genome-wide association study
GWAS in GS:SFHS on antidepressant treatment resistance and stages of resistance were completed utilising linear mixed model analysis in GCTA (Genome-wide Complex Trait Analysis) [38]. Age, sex and the first four MDS components were fitted as covariates and, to account for the family structure in GS:SFHS, genetic relationship matrices (GRMs) were fitted as random effects (see Supplementary Materials). To counter the loss of power that is caused by inclusion of a candidate SNP as both a random effect (in the GRM) and a fixed effect, the leave-one-chromosome-out method was utilised [40]. Due to the use of linear mixed models on a binary trait, treatment resistance, Taylor series transformation [41] was used to convert beta and standard error values from the linear scale to the liability scale (see Supplementary Materials).
GWAS in GENDEP was completed on unrelated individuals utilising logistic regression in PLINK [42]. Models were corrected for age, centre, baseline severity and the first four principal components, to control for population stratification.
Meta-analysis between GS:SFHS and GENDEP in antidepressant treatment resistance was completed in METAL [43] with the inverse variance weighted method. A total of 7,120,598 SNPs were in common across both samples.
Gene and gene-set enrichment analysis
Gene and gene-set analysis were completed using MAGMA (v1.04) [44] (further details in Supplementary Materials). Individual level data were utilised for analysis of both antidepressant treatment resistance and stages of resistance in GS:SFHS and summary statistics data used for analysis in GENDEP treatment resistance. Antidepressant treatment resistance in GS:SFHS and GENDEP was then meta-analysed in MAGMA using fixed effect meta-analysis. To map SNPs to gene and biologically-meaningful gene sets, SNPs were annotated using NCBI 37.3 and, for the gene-set analysis, gene-annotation files from the Gene Ontology (GO) Consortium (http://geneontology.org/) were taken from the Molecular Signatures Database (MSigDB) v5.2. GO is an inclusive set of 5917 gene pathways that cover a wide variety of functions, including molecular functions, cellular components and biological processes. Gene sets were corrected for multiple testing using the MAGMA default setting correcting for 10,000 permutations.
Pedigree-based heritability
Pedigree-based heritability of antidepressant treatment resistance and stages of resistance was calculated in R using MCMCglmm [45]. This was achieved by constructing a variance component matrix that takes into account all pedigree information and then fitting it into a univariate model as a random effect. MCMCglmm uses a Bayesian framework to estimate heritability. For treatment resistance, the logit link function was used to account for the binary nature of the phenotype.
Genetic correlation analysis
Genetic correlations were calculated using a bivariate analysis in ASReml-R (http://www.vsni.co.uk/software/asreml/). Correlations between antidepressant treatment resistance and stages of resistance were examined with eight personality and cognitive variables; neuroticism, extraversion, schizotypal personality questionnaire (SPQ), mood disorder questionnaire (MDQ), general cognitive ability (‘g’, formed from four varied cognitive test scores), Scottish Index of Multiple Deprivation (SIMD), education and the general health questionnaire (GHQ). The ASReml-R method was utilised as it can account for the family structure in GS:SFHS. Genetic correlation measurements were calculated between pedigree-based heritabilities as the sample sizes were too small to conduct SNP-based correlations. More information on the variables and methods used can be found in the Supplementary Materials.
Polygenic risk scoring analysis
PRS were constructed utilising PLINK [42]. This method has been previously described [46] and further information is available in the Supplementary Materials. Summary statistics taken from the Psychiatric Genomics Consortium (PGC) were used to construct PRS for MDD (unpublished data, see Supplementary Materials), schizophrenia [47] and bipolar disorder [48] in the GS:SFHS cohort to examine genetic liability to the disorders in a treatment resistant population. PRS were reported across five P-value thresholds (<0.01, <0.05, <0.1, <0.5 and <1).
Association of PRS to the trait was analysed by linear mixed model analysis in ASReml-R (http://www.vsni.co.uk/software/asreml/) with antidepressant treatment resistance or stages of resistance as the dependent variable and PRS as the independent variable. All models were adjusted for age, sex, and the first four MDS components and, to account for related individuals, an additive relationship matrix (expected relatedness derived from pedigree information) was fitted as a random effect. Wald’s conditional F-test was used to derive P-values for all fixed effects (see Supplementary Materials). Taylor series approximation [41] was used for the treatment resistance variable, as above.
AVENGEME [49] was used to calculate power in the PRS analysis assuming 5% of SNPs had an effect in the training sample and all markers were independent. Two theoretical covariances were tested at 0.5 and 0.25.
Results
Genetic correlation of all antidepressant users and MDD
Antidepressant use and MDD demonstrated significantly overlapping genetic architectures for both common (rg = 1.0, P = 0.026) and pedigree-related genetics (rk = 0.88, P = 1.7 × 10−16, Supplemental Table S4). Both correlations indicate that ‘any antidepressant use’ defined from prescription records using dose and duration filters is a valid means of identifying a genetically representative MDD population. Common genetics accounted for 0.10 (CI= 0.02 – 0.19) of the variance in antidepressant use (i.e. SNP heritability) and pedigree-related genetics 0.43 (CI= 0.32 – 0.54), SCID MDD measurements in this cohort have been previously reported (Zeng et al, [50], PMID= 27838479).
Genome-wide association study
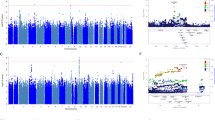
In the antidepressant treatment resistance meta-analysis of 4213 individuals (cases = 358, controls = 3855), no SNP reached genome-wide significance (P > 5 × 10−8). The most significant SNP identified was an intergenic variant located at 10p26.13 (lead SNP rs188352979, P = 3.25 × 10−7, OR = 2.87, CI = 2.47–3.28; Fig. 1).

Manhattan and Q-Q plots of the GWAS of antidepressant treatment resistance in (a) Generation Scotland: Scottish Family Health Study, (b) Genome-based Therapeutic Drugs for Depression and (c) the meta-analysis between the two cohorts. Genome-wide significance level (P < 5 × 10−8) is represented by a red line and suggestive threshold (P < 1 × 10-5) is represented by a blue line
In the GWAS of stages of antidepressant resistance in GS:SFHS (n = 3452), no SNP reached genome-wide significance (P > 5 × 10−8). The most significant SNP identified was an intergenic variant located at 10q22.1 (lead SNP rs116902282, P = 1.5 × 10−7, beta = 0.49, s.e. = 0.076; Fig. 2).

Manhattan and Q-Q plots of the GWAS of antidepressant stages of resistance. Genome-wide significance level (P < 5 × 10−8) is represented by a red line and suggestive threshold (P < 1 × 10−5) is represented by a blue line
The top four loci for each GWAS below a P-value of 5 × 10−7 for the antidepressant treatment resistance meta-analysis and stages of antidepressant resistance can be found in Table 1. Heterogeneity statistics for the treatment resistance meta-analysis are also reported, we found either no evidence or nominal evidence of heterogeneity between GS:SFHS and GENDEP that was not significant after adjustment for multiple correction at a genome-wide level.
Gene and gene-set enrichment analysis
Gene-based analysis with MAGMA did not identify any genes significantly associated after false discovery rate (FDR) multiple testing correction. Similarly, in the gene-set analysis no gene-set passed multiple testing correction over 10,000 permutations. The most significant genes and gene sets are listed in Supplemental Table S5.
Pedigree-based heritability and genetic correlations
Pedigree-based heritability was calculated in GS:SFHS at 0.60 (CI = 0.22–0.87) for antidepressant treatment resistance and 0.27 (CI = 0.24–0.31) for stages of antidepressant resistance.
Significant positive genetic correlations were found between antidepressant treatment resistance and neuroticism (rg = 0.66, PFDR = 0.0091), MDQ (rg = 0.86, PFDR = 0.0072) and GHQ (rg = 0.96,PFDR = 8.8 × 10–5). For stages of antidepressant resistance, they were identified between neuroticism (rg = 0.51, PFDR = 0.023), SPQ (rg = 0.44, PFDR = 0.036), MDQ (rg = 0.69,PFDR = 0.027) and GHQ (rg = 0.71,PFDR = 0.0011). All these correlations survived correction for multiple testing with FDR (Table 2).
Genetic liability of psychiatric traits with antidepressant treatment resistance and stages of resistance using polygenic risk scores techniques
Antidepressant treatment resistance and stages of antidepressant resistance were positively and nominally associated with MDD PRS at P-value thresholds (PT) 0.1, 0.5 and 1, and antidepressant treatment resistance only with schizophrenia PRS at PT < 0.01. There were no significant associations between antidepressant treatment resistance or stages of antidepressant resistance with bipolar disorder PRS (Table 3). No result survived FDR correction and power analyses indicated we were underpowered to detect an association between MDD and bipolar disorder PRS with antidepressant treatment resistance and stages of antidepressant resistance. Schizophrenia PRS was powered to detect an association at all thresholds in both antidepressant treatment resistance and stages of resistance given a genetic correlation of 0.5. At a genetic correlation of 0.25, stages of antidepressant resistance had adequate power at all thresholds whilst antidepressant treatment resistance was only powered atPT 0.01 (Table 3).
Discussion
We utilised antidepressant prescription records to explore common genetic factors in antidepressant treatment resistance and stages of resistance in a population and family-based cohort of 3452 individuals. In the treatment resistance GWAS meta-analysis, the most significant locus was located at 10q26.13 at P = 3.3 × 10−7; lead SNP rs188352979. This SNP is intergenic and lies in between genes ACADSB and HMX3.ACADSB encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD) which is an enzyme involved in the metabolism of fatty acids [51]. Of interest, differences in mitochondrial fatty acid metabolism have been found between ketamine responders and non-responders in bipolar disorder [52]. HMX3 is a transcription factor that is involved in the specification of neuronal cells needed for hypothalamus development and hypothalamic–pituitary–adrenal (HPA) axis [53]. Disruptions in the HPA axis are known to be associated with MDD, MDD severity and antidepressant response [55, 56]. A single locus at 10q22.1 was associated with stages of antidepressant resistance at P = 1.71 × 10−7, lead SNP rs116902282. This is an intergenic variant that lies between functional genes C10orf35 and COL13A1. C10orf35 is a protein coding gene that has previously been associated with uterine leiomyoma [56]. COL13A1 encodes the alpha chain of one of the nonfibrillar collagens. No variant in either analysis reached the required threshold for genome-wide statistical significance (P > 5 × 10−8). Gene and gene-set enrichment did not identify any significant associations with either antidepressant treatment resistance or stages of resistance. Nonetheless, modest to high pedigree-based heritability estimates indicate that 60% of the variance in antidepressant treatment resistance and 27% of the variance in stages of resistance can be explained by genetics, although these estimates had large confidence intervals. This indicates that further exploration into genetic contributions in antidepressant resistance is warranted.
Antidepressant treatment resistance was significantly and positively genetically correlated with neuroticism (rg = 0.66), MDQ (rg = 0.86) and GHQ (rg = 0.96) indicating overlapping genetic architecture between these traits. In stages of antidepressant resistance, these same traits also demonstrated significant genetic correlation; neuroticism (rg = 0.51), MDQ (rg = 0.69) and GHQ (rg = 0.71), and we additionally identified a significant genetic correlation with SPQ (rg = 0.44). We consider all correlations over 0.5 to be high genetic correlation (as defined previously [57]) and therefore only identify one correlation not considered to be so. The MDQ and SPQ correlation indicates that more resistant individuals may share genetic components with schizotypy and mood disorder personality traits. Genetic overlap between psychological distress and antidepressant resistance is indicative that individuals susceptible to distress are associated with a poorer outcome in antidepressants and should be further investigated. Nevertheless, only a modest correlation was identified between stages of antidepressant resistance and SPQ and it is difficult to ascertain causal inferences with any of these correlations. Moreover, we did not find any correlation in antidepressant treatment resistance or stages of resistance with general intelligence, education or social deprivation traits, however, a lack of power due to small sample size (n = 3452 with only 250 treatment resistant cases) may have been major contributing factor.
Using PRS, we investigated whether poor response to antidepressants indicate a higher liability to other mental disorders (schizophrenia or bipolar disorder) as well as higher genetic loading of MDD itself. We did not find any significant association with antidepressant treatment resistance or stages of resistance, however, power with the current sample size was only adequate in analysis with schizophrenia PRS at a genetic correlation of 0.5 and additionally 0.25 for stages of antidepressant resistance. Results for MDD and bipolar disorder PRS should therefore be treated with caution, although the nominal associations between MDD PRS and stages of antidepressant resistance may be worth further investigation with larger sample sizes.
Interindividual variability in drug therapies can often be attributed to genetic variability in cytochrome P450s (CYP450) and metabolic transporters. In this study, we did not identify any associations with metabolic variants and response but it has been widely explored in previous literature, although inconsistent results have been reported for both CYP450s [58] and metabolic transporters [59]. For instance, nortriptyline has been widely demonstrated to show differential serum level per CYP2D6 metaboliser type (reviewed previously [58]), however a GENDEP study of 223 individuals taking escitalopram and 161 individuals taking nortriptyline found that whilst serum levels of the drugs varied per CYP450 genotype, treatment response was not affected [60]. Nevertheless, it has been found that CYP2D6 poor metabolisers have a higher proportion of side effects [61]. Venlafaxine, however, has been more consistently shown to have lower response and remission rates amongst CYP2D6 poor metabolisers [62, 63]. This indicates that, given access to larger samples, studies exploring individual antidepressants may be beneficial to identify metabolic profiles that may or may not be affecting response. Nonetheless, no GWAS to our knowledge has so far implicated CYP450 in association with treatment response.
One of the strengths of this study was that it used data from a population-based cohort and is a good representation of antidepressant users in a MDD sample in the general Scottish population. Nevertheless, certain limitations of this study should be noted. Although currently diagnosed schizophrenia and bipolar patients were removed prior to this analysis and minimum dose was matched to that of MDD recommendations, it is possible that individuals were prescribed the antidepressant for other conditions, e.g. anxiety disorders, obsessive compulsive disorder (OCD), posttraumatic stress disorder (PTSD) and panic disorders, and may have had a misdiagnosis in the first instance. Nevertheless, the high genetic correlation between all antidepressant users and SCID-diagnosed MDD demonstrates that using prescription records with dose and duration exclusions may be a valid method of identifying large samples of proxy MDD cases that are genetically representative of the disorder. It was not possible to account for the use of psychotherapy or ECT (which is advised in patients with severe MDD) and, additionally, no exclusions were applied for the prescription of other psychotropic medications used alongside antidepressants. It is therefore possible that other treatments may have provided more information of response and non-response to antidepressants. We were also only able to obtain prescription records over a 6-year period meaning there are likely to be some individuals who had prescriptions before this period. Despite the 6-week threshold applied here, individuals may also switch antidepressants after 6 weeks continuous treatment, due to side effects such as weight gain [64]. An additional limitation to this method is that it does not account for combination therapies. Although in the UK, combination therapies are often only considered after failure of a second single trial of antidepressant monotherapy [27], therefore by the definition adopted in this current study, they would also be considered treatment resistant. Furthermore, because we integrate prescribing data across a number of different antidepressant drugs and classes, specific associations with treatment resistance within or between classes of prescribed compounds may have been missed. It should also be highlighted that, whilst antidepressant resistance measures were taken over a 6-year period, mood and personality questionnaires were taken at a single time point and may have been impacted by mood or condition (e.g. currently having a depressive episode) of the patient at the time of completing the questionnaire. Lastly, differing covariates were applied in the meta-analyses, e.g. baseline severity in the GENDEP sample but not GS:SFHS.
Whilst the stages of antidepressant resistance measure provide additional information above the binary treatment resistance measure, it differs from models previously defined in the literature. Previous measures (such as the Thase and Rush five-stage model [65], Massachusetts General Hospital staging model [29] and the Maudsley staging model [66]) are significantly more detailed and account for augmentation therapies, ECT, differing antidepressant classes and MDD severity. However, there has also been criticism of these measures as they have arbitrary thresholds and have not been widely validated [67]. In this study, our staging measure was defined solely on antidepressant switching, due to limitations in the available data. This measure is therefore not synonymous with other definitions used in prior studies. Nevertheless, there is no uniformly accepted method of quantifying the degree of antidepressant resistance and this measure will likely capture useful information regarding resistance to antidepressant treatment.
Despite an increased sample size compared to those reported in previous clinical studies of antidepressant response (n = 2897), our numbers are still small for a GWAS and it is likely that we were underpowered. With an increasing availability of electronic records in large biobanks and numerous smaller antidepressant studies, a collaborative effort approach may be required in order to increase sample size for adequate power. To replicate our analysis with adequate power (>0.8) at a MAF of 0.01, it would require a sample size of 7596 cases for antidepressant treatment resistance and 9660 total sample for stages of resistance assuming an OR of 1.6 and beta of 0.3, respectively (Supplemental Table S6; power calculations were completed using QUANTO v1.2). Furthermore, the epigenetic of treatment resistance could be further explored. Recent studies have indicated that methylation levels have been predictive of overall response [68].
With increasing accessibility of electronic health records [69], access to prescription records is becoming possible. In this study, we explored the possibility of utilising this prescription data to examine resistance to antidepressant treatment by inferring drug switching as non-response. We have provided evidence that resistant individuals have a high genetic correlation with neuroticism, psychological distress, schizotypy and mood disorder traits. Furthermore, we demonstrate the need for larger cohorts and collaboration in order to maximise sample size. This study demonstrates the value of this method and, as larger cohort sizes become available, the results of such studies could further inform clinical and research applications.
Disclaimer
This report represents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
References
Ustün T, Ayuso-Mateos J, Chatterji S, Mathers C, Murray C. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–92.
Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800.
Rush A, Trivedi M, Wisniewski S, Nierenberg A, Stewart J, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17.
Uher R, Perroud N, Ng MY, Hauser J, Henigsberg N, Maier W, et al. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am J Psychiatry. 2010;167:555–64.
Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;66:966–75.
Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, et al. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry. 2010;67:133–8.
Tansey KE, Guipponi M, Perroud N, Bondolfi G, Domenici E, Evans D, et al. Genetic predictors of response to serotonergic and noradrenergic antidepressants in major depressive disorder: a genome-wide analysis of individual-level data and a meta-analysis. PLoS Med. 2012;9:e1001326.
Myung W, Kim J, Lim SW, Shim S, Won HH, Kim S, et al. A genome-wide association study of antidepressant response in Koreans. Transl Psychiatry. 2015;5:e672.
Biernacka JM, Sangkuhl K, Jenkins G, Whaley RM, Barman P, Batzler A, et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Transl Psychiatry. 2015;5:e553.
Cocchi E, Fabbri C, Han C, Lee SJ, Patkar AA, Masand PS, et al. Genome-wide association study of antidepressant response: involvement of the inorganic cation transmembrane transporter activity pathway. BMC Psychiatry. 2016;16:106.
Investigators G, Investigators M. Investigators SD. Common genetic variation and antidepressant efficacy in major depressive disorder: a meta-analysis of three genome-wide pharmacogenetic studies. Am J Psychiatry. 2013;170:207–17.
Li QS, Tian C, Seabrook GR, Drevets WC, Narayan VA. Analysis of 23andMe antidepressant efficacy survey data: implication of circadian rhythm and neuroplasticity in bupropion response. Transl Psychiatry. 2016;6:e889.
Fabbri C, Porcelli S, Serretti A. From pharmacogenetics to pharmacogenomics: the way toward the personalization of antidepressant treatment. Can J Psychiatry. 2014;59:62–75.
García-González J, Tansey KE, Hauser J, Henigsberg N, Maier W, Mors O, et al. Pharmacogenetics of antidepressant response: A polygenic approach. Prog Neuropsychopharmacol Biol Psychiatry. 2017;75:128–34.
De Carlo V, Calati R, Serretti A. Socio-demographic and clinical predictors of non-response/non-remission in treatment resistant depressed patients: A systematic review. Psychiatry Res. 2016;240:421–30.
Amital D, Fostick L, Silberman A, Beckman M, Spivak B. Serious life events among resistant and non-resistant MDD patients. J Affect Disord. 2008;110:260–4.
Takahashi M, Shirayama Y, Muneoka K, Suzuki M, Sato K, Hashimoto K. Low openness on the revised NEO personality inventory as a risk factor for treatment-resistant depression. PLoS ONE. 2013;8:e71964.
McGirr A, Van den Eynde F, Chachamovich E, Fleck MP, Berlim MT. Personality dimensions and deep repetitive transcranial magnetic stimulation (DTMS) for treatment-resistant depression: a pilot trial on five-factor prediction of antidepressant response. Neurosci Lett. 2014;563:144–8.
Bacanu SA, Whittaker JC, Nelson MR. How informative is a negative finding in a small pharmacogenetic study? Pharm J. 2012;12:93–95.
Bergen SE, Petryshen TL. Genome-wide association studies of schizophrenia: does bigger lead to better results? Curr Opin Psychiatry. 2012;25:76–82.
Baker LA. Do our “big data” in genetic analysis need to get bigger? Psychophysiology. 2014;51:1321–2.
Wimberley T, Støvring H, Sørensen HJ, Horsdal HT, MacCabe JH, Gasse C. Predictors of treatment resistance in patients with schizophrenia: a population-based cohort study. Lancet Psychiatry. 2016;3:358–66.
O’Dushlaine C, Ripke S, Ruderfer DM, Hamilton SP, Fava M, Iosifescu DV, et al. Rare copy number variation in treatment-resistant major depressive disorder. Biol Psychiatry. 2014;76:536–41.
Ruderfer DM, Charney AW, Readhead B, Kidd BA, Kähler AK, Kenny PJ, et al. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry. 2016;3:350–7.
Smith B, Campbell H, Blackwood D, Connell J, Connor M, Deary I, et al. Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med Genet. 2006;7:74.
Smith BH, Campbell A, Linksted P, Fitzpatrick B, Jackson C, Kerr SM, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2013;42:689–700.
Joint Formulary Committee. British national formulary. 73 edn. London: BMJ Group and Pharmaceutical Press; 2017.
Souery D, Amsterdam J, de Montigny C, Lecrubier Y, Montgomery S, Lipp O, et al. Treatment resistant depression: methodological overview and operational criteria. Eur Neuropsychopharmacol. 1999;9:83–91.
Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53:649–59.
Conway CR, George MS, Sackeim HA. Toward an evidence-based, operational definition of treatment-resistant depression: when enough is enough. JAMA Psychiatry. 2017;74:9–10.
Ruhé HG, van Rooijen G, Spijker J, Peeters FP, Schene AH. Staging methods for treatment resistant depression. A systematic review. J Affect Disord. 2012;137:35–45.
Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
Iniesta R, Malki K, Maier W, Rietschel M, Mors O, Hauser J, et al. Combining clinical variables to optimize prediction of antidepressant treatment outcomes. J Psychiatr Res. 2016;78:94–102.
Kerr S, Campbell A, Murphy L, Hayward C, Jackson C, Wain L, et al. Pedigree and genotyping quality analyses of over 10,000 DNA samples from the Generation Scotland: Scottish Family Health Study. BMC Med Genet. 2013;14:38.
Nagy R, Boutin TS, Marten J, Huffman JE, Kerr SM, Campbell A, et al. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9:23.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34.
Yang J, Lee S, Goddard M, Visscher P. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S, et al. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet. 2013;9:e1003520.
Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–6.
Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45:730–8.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Hadfield J. MCMC methods for multi-response generalized linear mixed models: The MCMCglmm R package. J Stat Softw. 2010;33:1–22.
International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–83.
Palla L, Dudbridge F. A fast method that uses polygenic scores to estimate the variance explained by genome-wide marker panels and the proportion of variants affecting a trait. Am J Hum Genet. 2015;97:250–9.
Zeng Y, Navarro P, Xia C, Amador C, Fernandez-Pujals AM, Thomson PA et al. Shared Genetics and Couple-Associated Environment Are Major Contributors to the Risk of Both Clinical and Self-Declared Depression. EBioMedicine 2016;14:161–167.
Rozen R, Vockley J, Zhou L, Milos R, Willard J, Fu K, et al. Isolation and expression of a cDNA encoding the precursor for a novel member (ACADSB) of the acyl-CoA dehydrogenase gene family. Genomics. 1994;24:280–7.
Villaseñor A, Ramamoorthy A, Silva dos Santos M, Lorenzo MP, Laje G, Zarate C, et al. A pilot study of plasma metabolomic patterns from patients treated with ketamine for bipolar depression: evidence for a response-related difference in mitochondrial networks. Br J Pharmacol. 2014;171:2230–42.
Wang W, Grimmer JF, Van De Water TR, Lufkin T. Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Dev Cell. 2004;7:439–53.
Papiol S, Arias B, Gastó C, Gutiérrez B, Catalán R, Fañanás L. Genetic variability at HPA axis in major depression and clinical response to antidepressant treatment. J Affect Disord. 2007;104:83–90.
Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31:464–8.
Ling J, Wu X, Fu Z, Tan J, Xu Q. Systematic analysis of gene expression pattern in has-miR-197 over-expressed human uterine leiomyoma cells. Biomed Pharmacother. 2015;75:226–33.
Cohen J. A power primer. Psychol Bull. 1992;112:155–9.
Hicks JK, Sangkuhl K, Swen JJ, Ellingrod VL, Müller DJ, Shimoda K, et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin Pharmacol Ther. 2016;102:37–44.
Peters EJ, Reus V, Hamilton SP. The ABCB1 transporter gene and antidepressant response. F1000 Biol Rep. 2009;1:23.
Hodgson K, Tansey K, Dernovsek MZ, Hauser J, Henigsberg N, Maier W, et al. Genetic differences in cytochrome P450 enzymes and antidepressant treatment response. J Psychopharmacol. 2014;28:133–41.
Chen S, Chou WH, Blouin RA, Mao Z, Humphries LL, Meek QC, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther. 1996;60:522–34.
Lobello KW, Preskorn SH, Guico-Pabia CJ, Jiang Q, Paul J, Nichols AI, et al. Cytochrome P450 2D6 phenotype predicts antidepressant efficacy of venlafaxine: a secondary analysis of 4 studies in major depressive disorder. J Clin Psychiatry. 2010;71:1482–7.
Preskorn S, Patroneva A, Silman H, Jiang Q, Isler JA, Burczynski ME, et al. Comparison of the pharmacokinetics of venlafaxine extended release and desvenlafaxine in extensive and poor cytochrome P450 2D6 metabolizers. J Clin Psychopharmacol. 2009;29:39–43.
Kivimäki M, Hamer M, Batty GD, Geddes JR, Tabak AG, Pentti J, et al. Antidepressant medication use, weight gain, and risk of type 2 diabetes: a population-based study. Diabetes Care. 2010;33:2611–6.
Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(Suppl 13):23–29.
Fekadu A, Wooderson S, Donaldson C, Markopoulou K, Masterson B, Poon L, et al. A multidimensional tool to quantify treatment resistance in depression: the Maudsley staging method. J Clin Psychiatry. 2009;70:177–84.
Berlim M, Turecki G. Definition, assessment, and staging of treatment—resistant refractory major depression: a review of current concepts and methods. Can J Psychiatry. 2007;52:46–54.
Menke A, Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialog Clin Neurosci. 2014;16:395–404.
Jensen PB, Jensen LJ, Brunak S. Mining electronic health records: towards better research applications and clinical care. Nat Rev Genet. 2012;13:395–405.
Acknowledgements
This investigation was supported by the Wellcome Trust 104036/Z/14/Z (STRADL, Stratifying Resilience and Depression Longitudinally). Generation Scotland received core funding from the Chief Scientist Office of the Scottish Government Health Directorate CZD/16/6 and the Scottish Funding Council HR03006. We thank all families, practitioners and the Scottish School of Primary Care involved in the recruitment process as well as the entirety of Generation Scotland team; interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, healthcare assistants and nurses. We are grateful to the Sackler Foundation for the generous support of this work. IJD is supported by MRC and BBSRC funding to the University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology (MR/K026992/1).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
AMM has received financial support from Pfizer (formerly Wyeth), Janssen and Lilly and from the Sackler trust. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wigmore, E.M., Hafferty, J.D., Hall, L.S. et al. Genome-wide association study of antidepressant treatment resistance in a population-based cohort using health service prescription data and meta-analysis with GENDEP. Pharmacogenomics J 20, 329–341 (2020). https://doi.org/10.1038/s41397-019-0067-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-019-0067-3
This article is cited by
-
Polygenic risk scores of lithium response and treatment resistance in major depressive disorder
Translational Psychiatry (2023)
-
Exome-wide association study of treatment-resistant depression suggests novel treatment targets
Scientific Reports (2023)
-
Polygenic heterogeneity in antidepressant treatment and placebo response
Translational Psychiatry (2022)
-
Contemporary Genome-Wide Association Studies in Depression: The Critical Role of Phenotyping
Neuroscience and Behavioral Physiology (2022)
-
Understanding genetic risk factors for common side effects of antidepressant medications
Communications Medicine (2021)
