
Abstract
Vibrio cholerae, the bacterial pathogen responsible for the diarrheal disease cholera, resides in the aquatic environment between outbreaks. For bacteria, genetic variation by lateral gene transfer (LGT) is important for survival and adaptation. In the aquatic environment, V. cholerae is predominantly found in biofilms associated with chitinous organisms or with chitin “rain”. Chitin induces competency in V. cholerae, which can lead to LGT. In the environment, V. cholerae is also subjected to predation pressure by protist. Here we investigated whether protozoal predation affected LGT using the integron as a model. Integrons facilitate the integration of mobile DNA (gene cassettes) into the bacterial chromosome. We report that protozoal predation enhances transformation of a gene cassette by as much as 405-fold. We show that oxidative radicals produced in the protozoal phagosome induces the universal SOS response, which in turn upregulates the integron-integrase, the recombinase that facilitates cassette integration. Additionally, we show that during predation, V. cholerae requires the type VI secretion system to acquire the gene cassette from Escherichia coli. These results show that protozoal predation enhances LGT thus producing genetic variants that may have increased capacity to survive grazing. Additionally, the conditions in the food vacuole may make it a “hot spot” for LGT by accumulating diverse bacteria and inducing the SOS response helping drive genetic diversification and evolution.
Similar content being viewed by others

Introduction
Vibrio cholerae inhabits coastal and estuarine water environments and is the etiological agent of the often-fatal diarrheal disease, cholera [1]. In the environment, V. cholerae is faced with numerous pressures including interactions with free-living heterotrophic protists or protozoa [2]. Protozoa package bacteria into food vacuoles (phagosomes) that become acidified and filled with toxic components that aid digestion, including oxidative radicals. Despite this hostile environment, V. cholerae has evolved mechanisms to resist protozoal predation or manipulate the digestive process in order to create a protective niche [3]. In Acanthamoeba castellanii, V. cholerae can escape digestion and be exocytosed to the environment or shuttled internally to the contractile vacuole where it replicates, remaining during encystment before escaping [4]. In A. castellanii and Tetrahymena pyriformis, food vacuoles containing V. cholerae are expelled to the environment, where they provide a protective habitat and enhance survival and infectious potential [5].
V. cholerae biofilm formation on chitin surfaces provides protection, a nutritive substrate [1] and induces natural competence facilitating lateral gene transfer (LGT) through transformation [6]. Growth on chitin also induces the type VI secretion system (T6SS), resulting in lysis of neighboring cells and subsequent release of DNA [7] that can be taken up for transformation. In addition to DNA uptake, DNA transformation requires integration into a replicon such as the chromosome before it can be expressed and passed onto progeny [8]. V. cholerae pandemic strains have been heavily influenced by LGT [9, 10] as the two main virulence factors, cholera toxin (CTX) that is responsible for diarrhea and an intestinal adhesin and the toxin co-regulated pilus (TCP) are present on mobile genetic elements (MGEs) [11, 12]. MGEs are important in facilitating the integrative step of LGT, particularly where DNA cannot integrate through RecA-mediated homologous recombination. The integron in V. cholerae is a site-specific recombination system that integrates circular MGEs. The integron-integrase is induced by the SOS response [13], a regulatory cascade in bacteria that is induced by the DNA repair protein, RecA, which accumulates around single-stranded DNA (ssDNA) arising from processes such as stalled chromosome replication or LGT and activates its cleavage of the repressor, LexA [14]. Primarily, SOS induces DNA repair genes, however, it also induces site-specific recombinases in MGEs, such as prophages and genomic islands, linking DNA transfer with DNA integration [15, 16].
Here, for the first time, we show that the integron-integrase is expressed in the food vacuoles of protozoa and that this expression is due to oxidative radicals and induction of the SOS response. The induction of the SOS response enhances transformation of a gene cassette by as much as 405-fold by upregulating integron-integrase expression and gene cassette integration. In our assays, the gene cassette was transferred as free DNA or from within an Escherichia coli host, but only when T6SS was functional. Taken together, our results show that protozoal predation enhances genetic diversification that may assist adaptation to grazing pressure and contribute to the evolution of V. cholerae and possibly, to other aquatic bacteria.
Materials and methods
Bacterial strains, plasmids, oligonucleotides, and growth conditions
Bacterial strains, plasmids and primers used in this study are listed in Supplementary Tables 1, 2. Bacterial strains were routinely grown at 37 °C in Luria Bertani (LB) medium. For V. cholerae, kanamycin (Kan), chloramphenicol (Cm) and spectinomycin (Spc) were used at 50, 5, and 100 µg ml−1, respectively. For E. coli WM3064, diaminopimelic acid (DAP) was supplemented to a final concentration of 0.3 mM and Cm and Kan were used at 25 and 50 µg ml−1, respectively. To induce gene expression in strains carrying pSU-pBAD, a final concentration of 0.2% (w/v) L-arabinose was added to the growth medium. Plasmid DNA was extracted using the PureYield Plasmid Miniprep Systems kit (Promega, Wisconsin, USA). PCR fragments were purified using the ISOLATE II PCR and Gel Kit (Bioline, London, England). Oligonucleotides were obtained from Integrated DNA Technologies (Singapore).
Growth and maintenance of protozoa
T. pyriformis was routinely cultured in 10 mL of peptone yeast-glucose (PYG) medium in 25 cm2 tissue culture flasks with ventilated caps (Sarstedt Inc., Numbrecht, Germany) and incubated statically at 23 °C. PYG medium consists of 20 g l−1 proteose peptone, 1 g l−1 yeast extract and 0.1 M glucose dissolved in 1 L of 0.1 × M9 Salts Solution (1.28 g L−1 Na2HPO4, 0.3 g L−1 KH2PO4, 0.05 g L−1 NaCl, 0.1 g L−1 NH4Cl). Prior to experiments, 500 µl of T. pyriformis was passaged in 20 mL of 0.35 × Nine Salts Solution (NSS; 6.16 g L−1 NaCl, 0.515 g L−1 Na2SO4, 0.028 g L−1 NaHCO3, 0.0875 g L−1 KCl, 0.014 g L−1 KBr, 0.655 g L−1 MgCl2.6H2O, 0.1435 g L−1 CaCl2.2H2O, 0.0028 g L−1 SrCl2.6H2O and 0.0028 g L−1 H3BO3) [17] and incubated overnight statically at 23 °C to allow the culture to adapt to the nutrient free medium. A. castellanii was routinely cultured and passaged as above with the exception of incubation at 30 °C. Before use in experiments, A. castellanii was passaged for 3 d prior to obtain freshly growing cells, washed with 0.1 × M9 Salts Solution and resuspended in 0.1 × M9 Salts Solution supplemented with 1% glucose. Protozoa were enumerated microscopically using a hemocytometer and numbers were adjusted as described below.
Construction of integron-integrase reporter strain, artificial gene cassette, and mutants
The V. cholerae integron-integrase reporter was created by chitin transformation of p4640 [18], a suicide vector containing an intIA::gfp transcriptional fusion, into V. cholerae A1552. V. cholerae mutants were created by using splicing overlap extension PCR to construct mutated alleles followed by natural transformation [19]. The FRT-cat-FRT cassette interrupting the ΔintIA mutant allele used for selection was excised via FLP-mediated recombination as previously described [20]. The lexA(ind−) mutant was constructed by cloning lexA into pSW4426T, a suicide vector containing an arabinose-inducible ccdB counter-selectable marker and introducing an A91D allele by PCR. Transformations of ccdB constructs were conducted using the CcdB-resistant E. coli ∏3813 strain (Supplementary Table 1). The plasmid containing the mutated lexA(ind−) allele was chitin transformed into A1552 and a recombinant obtained on selective medium. Counter-selection was carried out on non-selective medium containing 0.2% arabinose and colonies screened for the lexA(ind−) allele using amplification refractory mutation system (ARMS) PCR. To investigate gene cassette transfer into the V. cholerae integron, a circular gene cassette that doubles as a plasmid with a conditional origin of replication (oriR6K), pKC01, was constructed. pKC01 was constructed via amplification of a gene cassette (locus tag RS15635 in V. cholerae N16961, NZ_CP028828) with TnFGL3 inserted [21] that was then circularized by ligation. pKC01 contains a kanamycin resistance gene (nptII), promoterless gfpmut3 and lacZ and a single V. cholerae-specific attC site with gfpmut3-lacZ-oriR6K-nptII downstream of the GTTRRRY attC recombination site (see Supplementary Fig. 1 for a schematic of pKC01).
Co-incubation of bacteria with protozoa
Co-incubation assays were conducted in 24-well microtiter plates containing 1 mL of co-culture per well (Nunclon Delta surface, ThermoFisher Scientific, Denmark), as previously described [5, 22]. Briefly, T. pyriformis and A. castellanii were enumerated and adjusted to 103 and 104 cells ml−1, respectively. For A. castellanii, plates were incubated at 23 °C for 1 h to facilitate attachment to the plate surface. Bacterial cells (strains of V. cholerae or E. coli) were added at a final concentration of 108 cells ml−1 for co-incubation with T. pyriformis and 107 cells ml−1 for co-incubation with A. castellanii, and subsequently incubated statically at 23 °C for 4 h.
In LGT co-incubation experiments using pKC01, chitin-competent V. cholerae cells were prepared using commercially available chitin flakes as previously described [19]. Briefly, cells were grown at 30 °C in 10 mL LB to an OD600 of ~0.4 before centrifugation and washing, and then resuspended in 1 mL of 0.7% Sea Salts solution (Sigma Aldrich). To a tube containing 900 μl of sterile 0.7% Sea Salts solution and 50–70 mg chitin flakes, 100 μl of the cell suspension was added and allowed to incubate statically overnight. Subsequently, 500 µl of supernatant was carefully aspirated and discarded and 500 µl of fresh 0.7% Sea Salts solution was added before the tube was rigorously vortexed for 5 min to detach the chitin-competent V. cholerae cells from the chitin flakes. The chitin flakes were left to settle for 2–3 min before the supernatant was carefully aspirated. From multiple tubes, ~5 mL of supernatant was collected, centrifuged and adjusted to an OD600 of 1.0 (equivalent to 109 cells ml−1). pKC01 was provided as both free DNA (5 µg) and as a plasmid in E. coli WM3064 in co-cultures of V. cholerae A1552 and protozoa.
Transformants were extracted from the protozoa for selective plating. For surface-attached A. castellanii, the co-culture was carefully washed three times with sterile 0.1 × M9 containing 1% glucose before an equal volume of the same culture media containing 1% Triton-X was added to lyse the amoeba and release the internalized bacterial cells. In the case of T. pyriformis, co-cultures were aspirated from the plate and gently collected by centrifugation at 400 × g for 10 min before the supernatant was carefully aspirated and discarded. The pellet containing T. pyriformis cells was then gently washed 3 times with sterile 0.35 × NSS and resuspended in an equal volume of 0.35 × NSS containing 1% Triton-X to lyse the protozoa. Appropriate dilutions of lysates were plated on agar plates containing Kan to select for transformants. Transformation frequencies were normalized by dividing the number of Kan resistant CFU by the total number of intracellular CFU [19]. For each transformation, ten random colonies were selected and confirmed to contain the V. cholerae ompW and nptII genes by PCR (Supplementary Table 2).
Microscopic imaging
For confocal laser scanning microscopy (CLSM) of co-cultures containing T. pyriformis and V. cholerae, the samples were collected after 4 h co-incubation: 200 µl of co-culture was stained with the red fluorescent fixable lipophilic dye FM4-64 FX (Invitrogen, Carlsbad CA) according to the manufacturer’s instructions, collected by centrifugation (400 × g, 10 min) and fixed by resuspending in phosphate buffered saline (PBS) containing 2% paraformaldehyde (pH 7.2–7.4) for 10 min at room temperature. Fixed cells were washed three times by centrifugation (400 × g, 10 min) and resuspended in PBS before staining with DAPI (diamidino-2-phenylindole, ThermoFisher Scientific, USA) according to the manufacturer’s instructions. After staining, cells were washed three times as above and resuspended in PBS before mounting on microscope slides; 10 μl of fixed sample was placed on a glass slide and allowed to dry at room temperature for 15 min. After drying, 10 μl of Vectashield HardSet Antifade Mounting Medium (Vector Laboratories, USA) was added and the sample sealed with a glass coverslip (1.5 mm thickness, Neuvitro, USA). Samples were placed in the dark at RT overnight to allow the mounting media to set and imaged the next day or stored at 4 °C for later observation. Mounted samples were imaged using an inverted Nikon A1 confocal laser scanning microscope using 405, 488, and 561 nm excitation to image DAPI, GFP and FM4-64 fluorescence, respectively. Single and Z-stack multi-channel fluorescence images of samples were collected, and corresponding transmitted light (TL) images were obtained using the TL detector. The co-culture containing A. castellanii and V. cholerae were imaged live with wide-field fluorescence microscopy with samples placed in CellCarrier 96—Ultra Microplates (PerkinElmer, UK) after 4 h of co-incubation. All images were analyzed using FIJI [23] and Imaris v9.2 (Bitplane, USA) analysis software.
Quantification of GFP fluorescence
Following co-incubation, T. pyriformis cells were collected by centrifugation (400 × g for 10 min), the supernatant was aspirated, and the pellet gently washed 3 times with sterile 0.35 × NSS. The T. pyriformis was lysed by resuspending in an equal volume of 0.35 × NSS containing 1% Triton-X. For the surface-attached A. castellanii, cells were gently washed three times with sterile 0.1 × M9 and resuspended in an equal volume of 0.1 × M9 containing 1% Triton-X. GFP fluorescence was quantified as previously described [24]. Briefly, 200 µl of sample was transferred into a 96-well microtiter plate (Corning® 96-Well Black Polystyrene Microplate flat bottom clear), GFP fluorescence measured at 488 nm excitation and 530 nm emission wavelengths and optical density at 600 nm were determined using a multimode microplate reader (Spark microplate reader, TECAN, Switzerland). Background fluorescence was determined using cultures containing the wild-type V. cholerae strain. GFP fluorescence was normalized to the cell density of bacterial cells and presented using the following formula:
Quantification and quenching of oxidative radicals
Following a 4 h co-incubation with wild-type V. cholerae, Triton-X (1% final concentration) was added to the protozoan cells and the sample was centrifuged at 20,598 × g for 2 min to collect the supernatant containing the oxidative radicals. Oxidative radicals were measured using the dye 2′,7′-dichlorofluorescin diacetate (DCF-DA) as previously described [25]. Briefly, 10 µM DCF-DA (Sigma, USA) was added and the supernatant incubated at 37 °C for 30 min. The samples (200 µl) were transferred to 96-well microtiter plates (Corning 96-Well Black Polystyrene Microplate flat bottom clear) and fluorescence was measured using a multimode microplate reader (Spark microplate reader, TECAN, Switzerland) with 485 nm excitation and 525 nm emission wavelengths. For quenching experiments, T. pyriformis and A. castellanii cells were treated with 100 mM of the quencher, thiourea (pH 7.0; Sigma, USA) [26], before addition of bacterial cells. Fluorescence data from the reader was divided by 1000 before plotting on a graph.
Quantitative reverse-transcriptase PCR (qRT-PCR)
Total RNA was prepared from co-incubation assays of V. cholerae with T. pyriformis and A. castellanii in 24-well plates. Bacterial cells from within protozoa were collected as described above and RNA was extracted by lysozyme digestion followed by use of the Aurum Total RNA mini kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. The concentration of RNA was measured by spectrophotometry (NanoDrop ND-1000; NanoDrop Technologies). Complementary DNA (cDNA) was prepared from 400 ng RNA from each sample by iScript Reverse Transcription (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions. Quantitative reverse-transcriptase PCR (qRT-PCR) was conducted using SsoAdvanced Universal SYBR Green Master Mix (Bio-Rad, Hercules, CA, USA) and a QuantStudio 6 Flex Real-Time PCR System using V. cholerae integron-integrase specific primers (intIA-1F/intIA-1R) (Supplementary Table 2). Transcription of intIA was determined relative to the transcription of gyrA (gyrA_F/gyrA_R) (Supplementary Table 2) using the comparative Ct (ΔΔCt) method.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 7.01 for Windows, GraphPad Software, La Jolla California, USA (www.graphpad.com). Two-tailed student’s t tests were used to compare means between experimental samples and controls. For experiments with multiple samples, one-way ANOVAs were used with Tukey’s or Dunnett’s Multiple Comparison Test post hoc comparisons of means.
Results
The V. cholerae integron-integrase is induced in protozoal phagosomes
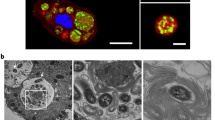
To determine whether the V. cholerae integron-integrase is induced during co-incubation with protozoa, a reporter strain containing a transcriptional fusion of intIA with gfp was co-incubated with the suspension-feeding T. pyriformis or the surface-feeding A. castellanii. Following 4 h of co-incubation, confocal microscopic imaging revealed the presence of GFP-expressing V. cholerae cells in the phagosome of both protozoa (Fig. 1, Supplementary Video 1, Supplementary Fig. 2). In addition, GFP fluorescent cells were observed in cysts (Fig. 1B, pink arrow) of A. castellanii. V. cholerae cells are known to transit to the contractile vacuole from the food vacuole [4] before encystation.

The V. cholerae integron-integrase reporter strain (intIA::gfp) contains a transcriptional fusion of intIA and gfp. The intIA::gfp,ΔrecA strain is the reporter strain with recA deleted. +QT indicates the addition of 100 mM of the quencher, thiourea. A Images show T. pyriformis after feeding on V. cholerae using transmitted light and, composite images from confocal microscopy showing GFP fluorescence (green) and DAPI stain fluorescence (blue) of V. cholerae and the protozoan nucleus, respectively; red (FM4-64FX) for lipophylic membranes and DAPI; and merged fluorescence channels (GFP + FM4-64FX + DAPI) respectively. B Images of amoebae after co-incubation with V. cholerae imaged with transmitted light, wide-field fluorescence microscopy showing GFP fluoresecence and, merged transmission and fluoresecence channels. Pink arrow indicates an ameobal cyst. Lower panels are an inset of the white box indicated in top row panel with the yellow arrow indicating a food vacuole. Scale bar 10 µM.
To quantify the level of integron-integrase induction, the intensity of the GFP fluorescence was measured using a spectrofluorometer after 4 h co-incubation. Compared to the no protozoa control, GFP fluorescence was 53-fold and 15-fold higher when extracted from within T. pyriformis and A. castellanii, respectively (Fig. 2). To confirm intIA induction occurred in the protozoa, qRT-PCR of RNA extracted from purified protozoa was performed and results showed that after 4 h, the relative expression of intIA in the wild type was 29-fold and 4.8-fold higher in T. pyriformis and A. castellanii, respectively, than in the controls (Fig. 3).

GFP fluorescence from V. cholerae strains recovered from within T. pyriformis (+Tp) (A) and A. castellanii (+Ac) (B) and from media controls (−Tp and −Ac). The V. cholerae intIA::gfp reporter strain contains a transcriptional fusion of intIA with gfp. The intIA::gfp,ΔrecA is the reporter strain with recA deleted and the recA-complemented strain (intIA::gfp,ΔrecA/pSU::recA) and vector control strain (intIA::gfp,ΔrecA/pSU). GFP fluorescence of internalized bacterial cells recovered from purified protozoa was measured after 4 h incubation. +QT indicates the addition of 100 mM of the quencher, thiourea. Error bars represent the standard deviations of three independent experiments. Significance was calculated using one-way ANOVA and Tukey’s multiple comparisons test (***p < 0.0001; ****p < 0.00001).

intIA transcript from V. choleraestrains from within T. pyriformis (+Tp) (A) and A. castellanii (+Ac) (B) and from media controls (−Tp and −Ac). intIA transcript was quantified in wild-type (WT), ΔrecA and recA-complemented strain (ΔrecA/pSU::recA) and vector control strain (ΔrecA/pSU). Cells external to the protozoa were removed and total RNA was extracted from purified protozoa. +QT indicates the addition of 100 mM of the quencher, thiourea, to the co-incubation. intIA transcription levels were normalized to gyrA by using the comparative Ct (ΔΔCt) method. Error bars represent the standard deviations of three independent experiments. Significance was calculated using one-way ANOVA and Tukey’s multiple comparisons test (**p < 0.001; ***p < 0.0001; ****p < 0.00001).
Gene cassette transformation is enhanced in intracellular V. cholerae and requires T6SS when the cassette is provided in another bacterial host
Given that the V. cholerae integrase is induced in the phagosomes of T. pyriformis and A. castellanii, an assay was conducted to determine if gene cassette transformation was enhanced. The artificial gene cassette, pKC01 (5 µg), was provided as purified DNA in co-incubation of V. cholerae and protozoa as well as in the no protozoa controls. Only V. cholerae wild-type cells that were pre-grown on chitin produced pKC01 transformants in both the protozoal treatments and controls (Fig. 4A, B). In the presence of T. pyriformis and A. castellanii, transformation efficiency was 405- and 18-fold higher than for chitin transformation in the absence of protists, respectively (Fig. 4A, B). No transformants were obtained using the integron-integrase mutant (ΔintIA) and gene complementation restored the phenotype (Fig. 4A, B). PCR spanning the attI region (using primers IntIAF-2 and VCH3-R) on 10 pKC01 transformants resulted in 4 giving sizes consistent with insertion of pKC01 at attI (~7200-bp), 4 consistent with no insertion at attI (~600-bp) and one amplicon of a larger size (~1500-bp) than the wild type with sequencing determining that an existing V. cholerae gene cassette had relocated into attI (sequence provided in Supplementary Information). Of the four transformants showing no insertion at attI, PCR using primers that bind across the array showed insertion of pKC01 at a site(s) between the 26th and 38th cassettes relative to attI. A suicide plasmid (pCVD442::Δrtx) that can only integrate by homologous recombination did not show enhanced transformation in the presence of protozoa and gave similar efficiencies to pKC01 in the absence of protozoa (Fig. 4A, B) confirming that enhanced pKC01 transformation is due to enhanced integron-integrase expression.

Transformation assays were assessed in the wild-type (WT), the ΔintIA, ΔrecA, Δhcp1,2, lexA(Ind-) mutants and the intIA-complemented (ΔintIA/pSU::intIA), recA-complemented (ΔrecA/pSU::recA) and hcp1-complemented (Δhcp1,2/pSU-pBAD::hcp1) strains and vector control strains (ΔrecA/pSU, ΔintIA/pSU and Δhcp1,2/pSU). In coincubations with protozoa, V. cholerae must be pre-grown on chitin for successful transfer of DNA. pKC01 and pCVD442::Δrtx (bar data in gray box) were provided as either free DNA (A, B) or carried within E. coli WM3064 (C, D). +QT indicates the addition of 100 mM of the quencher, thiourea, to the co-incubation. Error bars represent the standard deviations of three independent experiments. Significance was calculated using one-way ANOVA and Tukey’s multiple comparisons test (***p < 0.0001; ****p < 0.00001). Where no data is provided for strains and treatments is non-detectable transformation.
To determine whether protozoal predation enhances transformation of pKC01 contained within a neighboring bacterial cell, E. coli WM3064 carrying pKC01 was used as a donor in the gene cassette transformation assay. V. cholerae cells pre-grown on chitin were capable of uptake and integration of pKC01 from E. coli WM3064 at a transformation frequency of 7.73 × 10−5 and 2.06 × 10−5 in T. pyriformis and A. castellanii, respectively (Fig. 4C, D). Transformation of pKC01 was not detected in the no protozoa controls, in the absence of pre-growth on chitin or in the ΔintIA mutant. Complementation of the ΔintIA mutant restored transformation back to wild-type levels (Fig. 4C, D). To determine whether the T6SS is required for transformation of pKC01 from E. coli WM3064, a double hcp mutant incapable of producing T6SS (Δhcp1,2) was tested in transformation assays. No transformants were detected in either the protozoan treatments or in the no protozoa controls when chitin-grown V. cholerae was used (Fig. 4C, D). In contrast, the levels of transformation of the T6SS mutant were comparable to wild-type levels in a standard chitin transformation assay using DNA only (Supplementary Fig. 3). Transformation efficiency of the Δhcp1,2 double mutant was restored to wild-type levels when co-incubated with protozoa (Fig. 4C, D). As with naked DNA, transformation of the suicide plasmid pCVD442::Δrtx provided within E. coli WM3064 was not enhanced in the presence of protists and gave similar efficiencies to pKC01 in the absence of protozoa (Fig. 4C, D).
The V. cholerae SOS response is required for intIA transcription and gene cassette transformation within protozoa
The integron-integrase (intIA) gene is regulated by the SOS response, a global response to DNA damage that involves interaction of the RecA recombinational repair protein with the repressor protein, LexA [13, 15]. To determine whether intIA is induced through the SOS response in protozoal phagosomes, a recA mutation was constructed in the integron-integrase reporter strain. The mutant was co-incubated with T. pyriformis or A. castellanii and analyzed by CLSM and spectrofluorometry. Confocal microscopy revealed no GFP fluorescence in the recA mutant background in T. pyriformis, (Fig. 1A), a finding supported by spectrofluorometry (Fig. 2A). In A. castellanii, wide-field fluorescence microscopy revealed low levels of GFP fluorescence (Fig. 1B) that were substantially reduced compared to the recA+ strain with a tenfold reduction as quantified by spectrofluorometry (Fig. 2B). RT-qPCR of the intIA transcript showed a 27.6-fold reduction in the recA mutant compared to the wild type in T. pyriformis (Fig. 3A). Transcription of intIA was higher in A. castellanii compared to T. pyriformis, although it was 2.6-fold lower than the wild type indicating non-SOS transcription of intIA (Fig. 3B), although this level was insufficient for production of transformants in the gene cassette transformation assay (Fig. 4). The transformation assay revealed non-detectable transformation of pKC01 (either as free DNA or within E. coli WM3064) in the recA mutant strain in both protozoal treatments and no protozoa controls (Fig. 4). Complementation restored the recA mutant to wild type levels in all assays (Figs. 2–4). Transformation of DNA into V. cholerae is single-stranded therefore, pKC01 must be first circularized by RecA before integron-integrase mediation recombination can occur [27]. To confirm that the lack of pKC01 transformants in the recA mutant was due to a dysfunctional SOS-response rather than the requirement for RecA in circularizing pKC01, a lexA(ind-) mutant was engineered that produces a LexA variant resistant to RecA cleavage preventing SOS-induction but maintaining RecA activity [13]. The lexA(ind-) gave non-detectable transformation of pKC01 (either as free DNA or within E. coli WM3064) confirming that the SOS-response is required for transformation in the presence of protozoa.
Oxidative radicals produced by protozoa are required for SOS induction of intIA and LGT in V. cholerae
Protozoa introduce oxidative radicals, such as reactive oxygen species, in phagosomes following internalization of bacteria. We therefore hypothesized that oxidative radicals may be inducing the bacterial SOS response leading to intIA transcription and subsequent pKC01 integration. First, we quantified the level of oxidative radical production in T. pyriformis and A. castellanii during digestion of wild-type V. cholerae with and without treatment with thiourea to confirm that thiourea reduces oxidative radical activity. After 4 h of feeding, protozoal cells were purified and lysed with 1% Triton-X to release intracellular oxidative radicals, which were then stained with the reporter dye, 2′,7′-Dichlorofluorescin Diacetate (DC-FDA). Spectrofluorometry showed that the addition of thiourea reduced intracellular oxidative radicals by ~3-fold in both T. pyriformis and A. castellanii (Fig. 5). Although some DCF-DA fluorescence was observed in the controls, it was 17- and 18-fold higher in the presence of T. pyriformis and A. castellanii, respectively.

Following co-incubation with wild-type (WT) V. cholerae A1552, the level of internal oxidative radicals in T. pyriformis (A) and A. castellanii (B) were measured using the fluorescent dye DCF-DA and fluorescence spectrometry. +QT indicates the addition of 100 mM of the quencher, thiourea, to the co-incubation. Error bars represent the standard deviations of three independent experiments. Significance was calculated using one-way ANOVA and Tukey’s multiple comparisons test (**p < 0.001; ***p < 0.0001).
Using CLSM we showed that the addition of thiourea resulted in a substantial reduction in GFP-fluorescing cells in both protozoa (Fig. 1). Spectrofluorometry showed that the addition of thiourea resulted in a 7.4- and 2.3-fold reduction of GFP fluorescence in T. pyriformis and A. castellanii, respectively, compared to the no thiourea controls (Fig. 2). RT-qPCR showed thiourea treatment resulted in a 1.8- and 1.6-fold decrease in intIA transcription in the wild type compared to the no thiourea T. pyriformis and A. castellanii controls respectively (Fig. 3). Additionally, thiourea decreased pKC01 transformation in the chitin-grown wild type by 535-fold and 47-fold in T. pyriformis and A. castellanii, respectively (Fig. 4). Interestingly, although transformation of free pKC01 was detected in the no protozoa controls (Fig. 4), thiourea had no impact on transformation levels indicating that increased levels of pKC01 transformation in protozoa is due solely to the production of oxidative radicals. These oxidative radicals induced the SOS response above levels occurring during LGT alone. When pKC01 was provided by E. coli WM3064, thiourea was found to reduce transformation frequency in the wild type by 389- and 45-fold in T. pyriformis and A. castellanii, respectively (Fig. 4). Complemented mutants incubated with protozoa in the presence of thiourea showed transformation efficiency similar to the wild type (Fig. 4).
Discussion
V. cholerae is a highly infective human pathogen but most of its lifecycle is spent in the aquatic environment as biofilms on chitinous surfaces [28, 29] where it becomes naturally competent and therefore, capable of LGT-mediated genetic diversification [6]. In the environment, V. cholerae is present in mixed communities in the water column and in biofilms [2] and is under constant grazing presure from protozoa [30]. Although protozoa may have feeding preferences, they will graze upon different bacterial groups [31,32,33] accumulating and concentrating diverse bacteria in food vacuoles [3], providing an opportunity for genetic exchange [3, 34]. In this study, we demonstrate the uptake of V. cholerae and E. coli by two different predators, the filter feeding T. pyriformis and the raptorial feeder, A castellanii. We demonstrate that gene cassette transformation is enhanced in V. cholerae when co-incubated with protozoa, and that this is due to increased expression of the integron-integrase and gene cassette integration in the phagosome. While other studies have demonstrated that food vacuoles are micro-niches for conjugation and transduction [34,35,36,37], we identify that the conditions of the food vacuole enhance transformation by upregulating the integrative step of LGT. We show that V. cholerae was transformed with a gene cassette from E. coli when they were inside the food vacuoles of both T. pyriformis and A. castellanii, and that this required a functional T6SS indicating that it was utilized to lyse neighboring cells to access DNA. That we detected no gene cassette transformants in the controls lacking protozoa indicates that protozoa are important in facilitating the close association of donor and recipient cells required for T6SS-mediated LGT (see Fig. 6 for an illustrative demonstration). Additionally, we observed that transformation rates differed between our two protozoal models which is likely related to feeding rates being higher in the T. pyriformis ciliate versus the surface-feeding A. castellanii [38] which affects the number of bacteria exposed to the phagosome environment.

V. cholerae forms mixed biofilms on chitin substates in the water column becoming naturally competent. Protozoal feeding results in mixed bacteria in the food vacuole where oxidative radicals induce the V. cholerae SOS response increasing expression of the integron-integrase and enhancing integration of transferred gene cassettes. V. cholerae utilizes type VI secretion to acquire DNA from other bacteria in the food vacuole.
Another important aspect of this study was demonstrating that the integron-integrase is induced through the SOS response via the action of protozoan-produced oxidative radicals in the phagosomes. Laterally transferred DNA activates the SOS reponse which induces the integron-integrase thus linking DNA transfer with DNA integration [13, 18]. However, in this study, it is the presence of oxidative radicals that are shown to enhance integron-integrase mediated gene cassette transformation in V. cholerae. Conversely, RecA-mediated homologous recombination was not enhanced in the presence of protists indicating that at least in this context, there is a limit to recA induction and confirming that increased transformation of pKC01 is due to upregulated integron-integrase and not increased DNA uptake. Since thiourea reduced transformation efficiency in co-incubation with protozoa but not when V. cholerae was grown on chitin, it can be concluded that SOS is activated above the level induced by the presence of ssDNA from LGT and is due to oxidative radicals. Additionally, low levels of non-SOS regulated integron-integrase induction was observed in A. castellanii although this was insufficient to produce transformants. Nevertheless, it shows that SOS-independent integron-integrase induction occurs in the phagosome with catabolite control a likely pathway since it has previously been observed to control the V. cholerae intIA [39]. As the SOS response is conserved in all bacteria and is important in generating genetic diversity through mutation, recombination and LGT [14], protozoal predation is likely an important driver of bacterial adaptation. Importantly, the class 1 integron-integrase which is important in the lateral transfer of antibiotic resistance genes is also regulated by the SOS response [13] and so the protozoal phagosome environment may be an important niche for transfer of antibiotic resistance genes in other bacteria. Additionally, prior studies have reported that the SOS response enhances the transfer of multiple MGEs in bacteria including the lysogenic CTXϕ in V. cholerae [40] and others involved in antibiotic resistance [13, 18, 41,42,43,44,45,46] so, lateral transfer of DNA in this context likely goes beyond gene cassettes.
As a defense against grazing, generating genetic diversity is important for producing strain(s) that may have an advantage. Apart from the acquisition of new gene cassettes, induction of the integron-integrase causes variation in the cassette array such as rearrangements (observed in this study with relocation of a cassette into attI in one of our transformants) or deletions of which the latter is known to cause changes in surface properties and biofilm formation in at least one Vibrio species [47, 48]. Additionally, the SOS response can cause other genetic changes such as elevated mutation, recombination or movement of MGEs [14]. Due to the overlap of protozoal survival factors with virulence factors, it is hypothesized that protozoal predators may contribute to the emergence of pathogens [49] and our study demonstrating a link between generation of genetic diversity and protozoal grazing selection supports this idea. As V. cholerae is released from protozoa into the environment as expelled food vacuoles (EFVs) and these packaged cells are more resistant to environmental stress and more infectious in vivo [5], the cycling of V. cholerae through protozoa in EFVs provides opportunity for creation of genetic variants that survive long enough to be “tested” in human disease. Taken together, this study shows that the food vacuoles of protozoa may be an important niche where V. cholerae generates genetic diversity through SOS-induced LGT.
References
Conner JG, Teschler JK, Jones CJ, Yildiz FH. Staying alive: Vibrio cholerae's cycle of environmental survival, transmission, and dissemination. Microbiol Spectr. 2016;4. https://doi.org/10.1128/microbiolspec.VMBF-0015-2015.
Sakib SN, Reddi G, Almagro-Moreno S. Environmental role of pathogenic traits in Vibrio cholerae. J Bacteriol. 2018;200:e00795–17.
Espinoza-Vergara G, Hoque MM, McDougald D, Noorian P. The impact of protozoan predation on the pathogenicity of Vibrio cholerae. Front Microbiol. 2020;11:17.
Van der Henst C, Scrignari T, Maclachlan C, Blokesch M. An intracellular replication niche for Vibrio cholerae in the amoeba Acanthamoeba castellanii. ISME J. 2016;10:897–910.
Espinoza-Vergara G, Noorian P, Silva-Valenzuela CA, Raymond BBA, Allen C, Hoque MM, et al. Vibrio cholerae residing in food vacuoles expelled by protozoa are more infectious in vivo. Nat Microbiol. 2019;4:2466–74.
Meibom KL, Blokesch M, Dolganov NA, Wu C-Y, Schoolnik GK. Chitin induces natural competence in Vibrio cholerae. Science. 2005;310:1824–7.
Borgeaud S, Metzger LC, Scrignari T, Blokesch M. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science. 2015;347:63–7.
Le Roux F, Blokesch M. Eco-evolutionary dynamics linked to horizontal gene transfer in Vibrios. Annu Rev Microbiol. 2018;72:89–110.
Cho YJ, Yi H, Lee JH, Kim DW, Chun J. Genomic evolution of Vibrio cholerae. Curr Opin Microbiol. 2010;13:646–51.
Chun J, Grim CJ, Hasan NA, Lee JH, Choi SY, Haley BJ, et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci USA. 2009;106:15442–7.
Karaolis DKR, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci USA. 1998;95:3134–9.
Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–4.
Guerin É, Cambray G, Sanchez-Alberola N, Campoy S, Erill I, Da Re S, et al. The SOS response controls integron recombination. Science. 2009;324:1034.
Baharoglu Z, Mazel D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev. 2014;38:1126–45.
Cambray G, Sanchez-Alberola N, Campoy S, Guerin É, Da Re S, González-Zorn B, et al. Prevalence of SOS-mediated control of integron integrase expression as an adaptive trait of chromosomal and mobile integrons. Mob DNA. 2011;2:6.
Fornelos N, Browning DF, Butala M. The use and abuse of LexA by mobile genetic elements. Trends Microbiol. 2016;24:391–401.
Mårdén P, Tunlid A, Malmcrona-Friberg K, Odham G, Kjelleberg S. Physiological and morphological changes during short term starvation of marine bacterial islates. Arch Microbiol. 1985;142:326–32.
Baharoglu Z, Bikard D, Mazel D. Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet. 2010;6:e1001165.
Marvig RL, Blokesch M. Natural transformation of Vibrio cholerae as a tool—optimizing the procedure. BMC Microbiol. 2010;10:155.
Blokesch M. TransFLP—a method to genetically modify Vibrio cholerae based on natural transformation and FLP-recombination. JoVE. 2012;68:e3761.
Cameron DE, Urbach JM, Mekalanos JJ. A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proc Natl Acad Sci USA. 2008;105:8736–41.
Noorian P, Hu J, Chen Z, Kjelleberg S, Wilkins MR, Sun S, et al. Pyomelanin produced by Vibrio cholerae confers resistance to predation by Acanthamoeba castellanii. FEMS Microbiol Ecol. 2017;93:fix147.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Kicka S, Trofimov V, Harrison C, Ouertatani-Sakouhi H, McKinney J, Scapozza L, et al. Establishment and validation of whole-cell based fluorescence assays to identify anti-mycobacterial compounds using the Acanthamoeba castellanii - Mycobacterium marinum host-pathogen system. PLoS ONE. 2014;9:e87834.
Ye Q, Zhang C, Wang Z, Feng Y, Zhou A, Xie S, et al. Induction of oxidative stress, apoptosis and DNA damage by koumine in Tetrahymena thermophila. PLoS ONE. 2019;14:e0212231.
Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen species. Science. 2013;339:1210–3.
Vit C, Richard E, Fournes F, Whiteway C, Eyer X, Lapaillerie D, et al. Cassette recruitment in the chromosomal Integron of Vibrio cholerae. Nucleic Acids Res. 2021;49:5654–70
Tamplin ML, Gauzens AL, Huq A, Sack DA, Colwell RR. Attachment of Vibrio cholerae serogroup O1 to zooplankton and phytoplankton of Bangladesh waters. Appl Environ Microbiol. 1990;56:1977–80.
Rawlings Tonya K, Ruiz Gregory M, Colwell Rita R. Association of Vibrio cholerae O1 El Tor and O139 Bengal with the copepods Acartia tonsa and Eurytemora affinis. Appl Environ Microbiol. 2007;73:7926–33.
Matz C, Kjelleberg S. Off the hook—how bacteria survive protozoan grazing. Trends Microbiol. 2005;13:302–7.
Gonzalez JM, Sherr EB, Sherr BF. Size-selective grazing on bacteria by natural assemblages of estuarine flagellates and ciliates. Appl Environ Microbiol. 1990;56:583–9.
González Juan M, Iriberri J, Egea L, Barcina I. Differential rates of digestion of bacteria by freshwater and marine phagotrophic protozoa. Appl Environ Microbiol. 1990;56:1851–7.
Wey Jennifer K, Jürgens K, Weitere M. Seasonal and successional influences on bacterial community composition exceed that of protozoan grazing in river biofilms. Appl Environ Microbiol. 2012;78:2013–24.
Aijaz I, Koudelka GB. Tetrahymena phagocytic vesicles as ecological micro-niches of phage transfer. FEMS Microbiol Ecol. 2017;93:fix030.
Schlimme W, Marchiani M, Hanselmann K, Jenni B. Gene transfer between bacteria within digestive vacuoles of protozoa. FEMS Microbiol Ecol. 1997;23:239–47.
McCuddin ZP, Carlson SA, Rasmussen MA, Franklin SK. Klebsiella to Salmonella gene transfer within rumen protozoa: Implications for antibiotic resistance and rumen defaunation. Vet Microbiol. 2006;114:275–84.
Matsuo J, Oguri S, Nakamura S, Hanawa T, Fukumoto T, Hayashi Y, et al. Ciliates rapidly enhance the frequency of conjugation between Escherichia coli strains through bacterial accumulation in vesicles. Res Microbiol. 2010;161:711–9.
Parry JD. Protozoan grazing of freshwater biofilms. Adv Appl Microbiol. 2004;54:167–96.
Baharoglu Z, Krin E, Mazel D. Connecting environment and genome plasticity in the characterization of transformation-induced SOS regulation and carbon catabolite control of the Vibrio cholerae Integron Integrase. J Bacteriol. 2012;194:1659–67.
Quinones M, Kimsey HH, Waldor MK. LexA cleavage Is required for CTX prophage induction. Mol Cell. 2005;17:291–300.
Lindsay JA, Ruzin A, Ross HF, Kurepina N, Novick RP. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol Microbiol. 1998;29:527–43.
Maiques E, Úbeda C, Campoy S, Salvador N, Lasa Í, Novick Richard P, et al. β-Lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J Bacteriol. 2006;188:2726–9.
Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427:72–4.
Úbeda C, Maiques E, Knecht E, Lasa Í, Novick RP, Penadés JR. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol Microbiol. 2005;56:836–44.
Chittò M, Berger M, Klotz L, Dobrindt U. Sub-inhibitory concentrations of SOS-response inducing antibiotics stimulate integrase expression and excision of pathogenicity islands in uropathogenic Escherichia coli strain 536. Int J Med Microbiol. 2020;310:151361.
Baharoglu Z, Mazel D. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob Agents Chemother. 2011;55:2438–41.
Rapa RA, Shimmon R, Djordjevic SP, Stokes HW, Labbate M. Deletion of integron-associated gene cassettes impact on the surface properties of Vibrio rotiferianus DAT722. PLoS ONE. 2013;8:e58430.
Rapa R, Labbate M The function of integron-associated gene cassettes in Vibrio species: the tip of the iceberg. Front Microbiol. 2013;4:385.
Adiba S, Nizak C, van Baalen M, Denamur E, Depaulis F. From grazing resistance to pathogenesis: the coincidental evolution of virulence factors. PLoS ONE. 2010;5:e11882.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
MHR, KRM, DM, and ML designed the study, analysed and interpreted the data. MHR performed the experiments. LC assisted with microscopy, GEV, AR, MMH, and PN assisted in the design and running of other experiments. MHR and ML wrote the paper. ML and DM provided funding. All authors reviewed and provided critical feedback of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rahman, M.H., Mahbub, K.R., Espinoza-Vergara, G. et al. Protozoal food vacuoles enhance transformation in Vibrio cholerae through SOS-regulated DNA integration. ISME J 16, 1993–2001 (2022). https://doi.org/10.1038/s41396-022-01249-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-022-01249-0
This article is cited by
-
Protistan predation selects for antibiotic resistance in soil bacterial communities
The ISME Journal (2023)
-
Phagotrophic protists preserve antibiotic-resistant opportunistic human pathogens in the vegetable phyllosphere
ISME Communications (2023)
