
Abstract
Rapid bacterial growth depends on the speed at which ribosomes can translate mRNA into proteins. mRNAs that encode successive stretches of proline can cause ribosomes to stall, substantially reducing translation speed. Such stalling is especially detrimental for species that must grow and divide rapidly. Here, we focus on di-prolyl motifs (XXPPX) and ask whether their prevalence varies with growth rate. To find out we conducted a broad survey of such motifs in >3000 bacterial genomes across 35 phyla. Indeed, fast-growing species encode fewer motifs than slow-growing species, especially in highly expressed proteins. We also found many di-prolyl motifs within thermophiles, where prolines can help maintain proteome stability. Moreover, bacteria with complex, multicellular lifecycles also encode many di-prolyl motifs. This is especially evident in the slow-growing phylum Myxococcota. Bacteria in this phylum encode many serine-threonine kinases, and many di-prolyl motifs at potential phosphorylation sites within these kinases. Serine-threonine kinases are involved in cell signaling and help regulate developmental processes linked to multicellularity in the Myxococcota. Altogether, our observations suggest that weakened selection on translational rate, whether due to slow or thermophilic growth, may allow di-prolyl motifs to take on new roles in biological processes that are unrelated to translational rate.
Similar content being viewed by others

Introduction
Translation is a fundamental process common to all known forms of life. Cells invest huge amounts of resources into translation. For example, in fast-growing bacterial species like E. coli protein synthesis can account for over 50% of a cell’s total energy budget [1]. What is more, rapid bacterial growth depends on the speed of translation. Specifically, fast-growing bacteria maintain high concentrations of ribosomes, and these ribosomes elongate proteins rapidly during protein synthesis [2]. In addition, bacterial genome characteristics that are correlated with growth rate (rRNA and tRNA gene copy number, as well as codon usage bias) all influence the rate of translation [3,4,5,6].
Multiple factors can negatively impact translation rate and cause ribosomes to pause or “stall” during elongation. These factors include the presence of uncharged tRNAs, rare codons in a translated mRNA, and even specific amino acids encoded by mRNA [7]. Among these amino acids proline stands out. Proline is slow to form peptide bonds due to its structural rigidity and unique status as an N-alkylamino acid [8, 9]. This structural rigidity can contribute to the formation of special secondary structures, like the poly-proline II helix [10, 11], which is associated with the binding domains of signaling proteins [12, 13]. Successive stretches of prolines cause ribosomes to pause translation. The length of this pause—the “strength” of the ribosome stall—depends on the amino acids surrounding the proline stretch [14], the location of the sequence causing the stall within a protein [15], and the translation initiation rate [16].
A special translation factor exists to resolve proline-induced ribosomal stalls. In bacteria this protein is called translation elongation factor P (EFP). EFP is a tRNA mimic that binds to the ribosome between the peptidyl and exit sites [17]. When bound to the ribosome, EFP uses a conserved amino acid residue to interact with the peptidyl-transferase center and accelerate the formation of proline-proline peptide bonds [17]. In many species, this conserved amino acid must be post-translationally modified for EFP to efficiently alleviate stalling [18,19,20]. The importance of EFP and its mitigation of ribosome stalling is underscored by the strong phenotypes caused by its loss. These include diminished growth rate [20,21,22,23,24], loss of motility [25], loss of virulence [20, 22, 24], reduced antibiotic resistance [24, 26], and in some cases, cell death [27].
Although EFP reduces the impact of proline-induced ribosomal stalling, EFP cannot completely eliminate these stalls. Ribosomal profiling shows that E. coli ribosomes still pause at proline residues, albeit much more briefly than in EFP knockout mutants [15]. Indeed, recent work has directly shown that proline motifs lead to ribosomal pausing in wild-type E. coli [28]. Protein evolution may have exploited such unavoidable stalling. For example, in E. coli di-prolyl motifs often occur at the beginning of complex protein domains, and may provide additional time for translational regulation, protein folding, or membrane insertion [29]. Indeed, E. coli appears to prefer rare proline codons for such motifs, effectively lengthening the stall phenotype in these regions [28].
Because EFP cannot fully alleviate proline-induced stalling [15, 28], one would expect that stalling motifs are subject to natural selection, and especially so in fast-growing species under high pressure to maximize their translation rate [2, 4]. Indeed, di-prolyl sequences occur less frequently than expected by chance in the fast-growing E. coli, where highly expressed proteins are especially depleted in these motifs [29]. EFP itself is optimized for high expression in fast-growing bacteria [30], reflecting its importance in maintaining high growth rates.
Slow-growing bacteria are under reduced selection for translational speed [31]. Their genomes have reduced codon usage bias and encode fewer tRNA and rRNA gene copies than their fast-growing counterparts [5, 32]. Therefore, we wondered whether the di-prolyl motifs that can cause ribosome-stalling would be more widespread in slow-growing bacteria. The prevalence of such motifs is unknown outside few well-studied bacterial species, including E. coli [29], S. enterica [33], Bacillus subtilis [23], and several Actinobacteria species [34].
We quantified the occurrence of di-prolyl motifs across more than 3000 bacterial genomes from 35 phyla and found that these motifs are more abundant in genomes with high GC content. This is not surprising, because proline codons are cytosine rich. More importantly, we found that these motifs were more abundant in species with slow predicted growth rates when we controlled for GC content. Di-prolyl motifs are also more abundant in thermophiles, and in species with complex life cycles that involve a multicellular life stage. They are especially abundant in the serine-threonine protein kinases of multicellular species, which are involved in signaling and developmental programs.
Materials and methods
Analysis of bacterial genomes
We downloaded 3265 bacterial genomes from the Integrated Microbial Genomes (IMG) database [35], selecting only one genome per Average Nucleotide Identity (ANI) cluster to reduce bias towards highly studied species while maximizing the phylogenetic diversity of our dataset. This procedure yielded approximately one representative genome from each species in the database, although we included multiple genomes from a species if the ANI between genomes was below the typical cutoff for species level (less than 96.5, 11 species). We used CheckM [36] to evaluate the quality of these genomes, retaining those which were estimated to be at least 90% complete and contained less than 5% contamination. We also re-assigned taxonomy to the whole dataset using the Genome Taxonomy Database and GTDB-Tool kit (GTDB-Tk) version 0.2.2 [37], and removed any genomes that could not be assigned to a phylum (three genomes). We counted the occurrence of di-prolyl motifs (XXPPX, where X designates any amino acid) in every protein encoded in each genome, using custom python scripts. We count polyproline motifs (XPPPX) as multiple di-prolyl motifs, as such motifs represent independent proline-proline bond formation reactions. The identity of the amino acids surrounding successive prolines (Xs) impacts the severity of the resulting ribosomal stall [14, 29]. We classified each di-prolyl motif according to its predicted stall severity, from weak to medium to strong, using a key derived from a mixture of in vitro and in vivo data [29].
We verified the presence of at least one EFP homolog in nearly every genome using the hmmscan function of HMMER version 3.3.2 [38] to search for the EFP Pfam PF01132. Only the genome of Aquaspirillum serpens did not contain a known EFP homolog. However, because this genome is not fully complete (estimated completeness 98.27% by CheckM), and because it encodes the EFP modification protein earP [20], it likely does encode EFP. Next, we estimated the doubling time associated with each genome using the codon usage bias (CUB) based R package gRodon version 1.8.0 [32]. This package calculates estimated doubling times by comparing the CUB from a set of genes expected to be highly expressed in fast-growing species (ribosomal proteins) to the background codon usage of the genome, with the expectation that fast-growing species use codons corresponding to the most abundant tRNAs to maximize translational rate. This metric provides a good approximation for a species’ doubling time in both whole genomes and metagenomic samples [32].
For a subset of our genomes, we retrieved experimentally measured doubling times from the literature (see Supplementary Dataset S1 for all corresponding citations), with a large proportion of this data coming from a recently compiled database on bacterial phenotypes [39]. We found good agreement between doubling times predicted by gRodon and measured doubling times, especially when only mesophilic species were considered (species with measured doubling times: n = 301, Pearson’s rho = 0.33, p value < 0.0001; mesophiles only: n = 202, Pearson’s rho = 0.44, p value < 0.0001, Fig. S1). In addition, regardless of how accurately CUB reflects measured doubling times, CUB still reflects a species’ investment in optimizing its translation rate.
In order to calculate the median expected expression level of genes, we first used ENCprime [40] to calculate CUB for each individual gene (represented as KEGG KOs) within our genome dataset. Next, we ranked each gene based on its overall CUB, where the highest rank of one corresponds to the gene with the strongest bias and highest predicted expression in each genome. We then took the median of this rank for each gene across all genomes and used these values to approximate its median expression level. Based on this calculation, the five genes with the highest predicted expression level encoded elongation factor Tu, chaperonin GroEL, large subunit ribosomal protein L7/L12, small subunit ribosomal protein S1, and elongation factor Ts. These results are consistent with the expectation that genes related to translation and cell growth should be highly expressed across most genomes. All KEGG annotations were provided by IMG using their annotation pipelines [35].
To identify intrinsically disordered regions (IDRs) within proteins of interest, we used IUPred2A [41] with the “long” option. IUPred computes a “disorder score”, and when this score exceeds a value of 0.5 in a protein region, the region is predicted to be disordered. We calculated an average disorder score for each di-prolyl motif by averaging scores across all five amino acids comprising each motif. We identified serine-threonine kinases by extracting all proteins which fell within KEGG orthology group K08884. Supplementary Datasets S1 and S2 contain additional information on all genomes and all proteins we analyzed, respectively.
Statistical methods
One potentially confounding factor in our analysis is that proline codons are cytosine rich (CCU, CCC, CCA, and CCG), which implies that di-prolyl motifs are inherently more likely to occur in genomes with high GC content. Indeed, the number of coding GC base pairs and the number of di-prolyl motifs are very strongly correlated for genomes in our dataset (Pearson’s rho = 0.94, p value < 0.0001). Because of this correlation, when examining individual proteins and their di-prolyl content, we controlled for the GC content of their encoding gene. We also controlled for protein length. We controlled for both quantities by dividing the total number of nucleotides encoding the di-prolyl motifs (each motif is five amino acids long and thus encoded by 15 base pairs) by the ratio of the total number of base pairs in the gene to the number of GC base pairs in the gene. That is, we preformed all analyses of di-prolyl motifs within genes with the quantity
Another potential confounding factor in our analyses is that GC content and other genomic characteristics are correlated across bacterial phylogenies. In other words, closely related bacteria are more likely to have similar GC content—and thus di-prolyl content—than those that are distantly related. To account for such phylogenetic dependence, we created a phylogenetic tree using 43 concatenated conserved marker genes generated by CheckM [36]. We aligned these sequences using MUSCLE version 3.8.31 [42], and built the phylogenetic tree with FastTree version 2.1.10 [43], using the archaeon Haloquadratum walsbyi as an outgroup (NCBI accession number: GCA_000009185). We used this tree for all subsequent phylogeny-dependent statistical methods.
We calculated Pagel’s λ [44] using the phylosig function from the phytools R package, version 0.6.99 [45]. Pagel’s λ is a measure of the phylogenetic dependence of a trait. A value of λ = 0 indicates that the trait evolved independently of phylogeny, while λ = 1 indicates strong phylogenetic dependency. This calculation confirmed that GC content shows strong phylogenetic dependency (λ = 0.99, p value < 0.0001). Therefore, we controlled for phylogeny in all our genome-based analyses. To this end, we used phylogenetic generalized least squares (PGLS) to measure the contribution of individual genomic characteristics to the prevalence of di-prolyl motifs within our genomes. We also used phylogenetic ANOVA to analyze differences between groups in our dataset. For the PGLS, we used the PGLS function in the R package caper, version 1.0.1 [46] and for the phylogenetic ANOVA we used the phylANOVA function from the R package phytools version 0.7-70 [45]. We performed all statistical analyses and plotting in R version 3.6.2 and created all plots using ggplot2 version 3.3.3 [47]. Quantile-quantile plots (Q-Q plots) of standardized phylogenetic residuals for all plotted PGLS models are presented in Supplementary Materials, demonstrating roughly normal distributions.
Results
Species with large, high GC genomes have many di-prolyl motifs
We quantified the frequency of di-prolyl motifs (XXPPX, where X designates any amino acid) in all proteins within a set of >3000 bacterial genomes from 35 different phyla. Although all di-prolyl motifs (PP) can cause ribosomal stalling, the surrounding amino acids (X) influence the severity of the stall [14]. To assign a stall “strength” to each di-prolyl motif, ranging from strong to medium to weak, we used a published key that has been compiled from in vivo and in vitro proteomic experiments [29]. We found that the number of di-prolyl motifs in each genome varied broadly throughout our dataset. They range from a maximum of 17,841 (1.95 motifs per protein) in Nannocystis exedens (a Myxobacterium with a complex lifecycle) to a minimum of 86 (0.15 motifs per protein) in Mycoplasma cloacale, a poultry-associated pathogen from the family Mycoplasmataceae. The genome of N. exedens also contained the most “strong” di-prolyl motifs at 9665 (1.05 strong motifs per protein), whereas Mesoplasma coleopterae, another pathogen from the Mycoplasmataceae, had the fewest strong motifs at just 17 (0.02 strong motifs per protein).
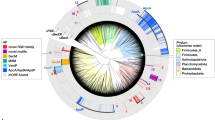
In general, we found that phyla with large, high GC genomes had the highest number of motifs (Actinobacteria, Planctomycetota, and Myxococcota), whereas those with small, low GC genomes had the fewest motifs (Fusobacteria, Campylobacterota, and Thermotogota, Fig. 1). This is not surprising because proline codons are cytosine rich (CCU, CCC, CCA, and CCG), making di-prolyl motifs inherently more likely to occur in large genomes with high GC content. Next, we asked whether any genome-derived characteristics besides GC content and genome size influence the frequency of di-prolyl motifs. When quantifying the influence of these characteristics, we controlled for the shared evolutionary history of our study taxa. Closely related genomes are much more likely to have similar GC content, and therefore similar numbers of di-prolyl motifs, than expected by chance (Pagel’s λ = 0.99, see Methods). In addition, we needed a method that could account for the highly correlated relationship between the frequency of di-prolyl motifs and GC content. To disentangle the contributions of these and other characteristics, while also controlling for phylogeny, we used phylogenetic generalized linear models (PGLS). This statistical method uses a phylogenetic tree to control for phylogenetic relatedness, essentially down-weighting similar observations that originate from closely related species [48], while also accounting for co-correlated variables.

Phyla whose genomes have a low average GC coding content (Fusobacteriota 0.8 Mbp, Campylobacterota 0.7 Mbp, Thermotogota 0.8 Mbp) encode fewer di-prolyl motifs than phyla with high average GC coding content (Actinobacteria 3.2 Mbp, Planctomycetota 3.2 Mbp, Myxococcota 6.3 Mbp). Only phyla represented by at least five genomes in our dataset are shown. Each circle represents one genome. Taxonomy was assigned using GTDB (Genome Database Taxonomy; see Methods). The phylum Proteobacteria was broken down into its corresponding classes and all Firmicutes-adjacent phyla were combined. The top and bottom boundaries of each box represent the 1st and 3rd quartiles, the thick black lines represent the median, and the whiskers indicate values 1.5 times the inter-quartile range.
Thermophiles and microbes with complex life cycles have high levels of di-prolyl motifs
The structural rigidity of proline can also reduce the conformational freedom of polypeptide chains, leading to increased thermo-stabilization [49]. In addition, like slow-growing species, thermophiles are thought to experience weaker selection on growth-associated-traits than mesophiles. This is because high temperatures cause naturally higher rates of catalysis and tRNA diffusion [5], so that thermophiles need to invest less in optimizing growth-associated traits to achieve rapid growth. For these reasons, we hypothesized that di-prolyl motifs may be more abundant in thermophilic bacteria. Testing this hypothesis is complicated by the fact that thermophiles have smaller genomes and shorter proteins than mesophiles [50]. With this in mind, we first performed a phylogenetic ANOVA which confirmed that thermophiles encode more di-prolyl motifs per Mbp of GC coding content than mesophiles (Phylogenetic ANOVA, p value < 0.05, Fig. 2A). Next, we performed a PGLS to verify that thermophiles encoded more di-prolyl motifs when total GC coding sequence size was controlled. We again found that thermophiles encode significantly more di-prolyl motifs than mesophiles (PGLS A, p value < 0.0001, Supplementary Table 1). Psychrophiles did not differ from mesophiles in this respect (PGLS A, p value = 0.53), although this could be due to their comparatively poor representation in our dataset (psychrophiles n = 56).

A Thermophile genomes encode significantly more di-prolyl motifs per GC coding Mbp than mesophiles (Phylogenetic ANOVA, p value < 0.05). Thermophiles are represented by 304 genomes, mesophiles by 2892 genomes: thermophilic mean = 1389 di-prolyl motifs per GC coding Mbp, mesophilic mean = 1150 di-prolyl motifs per GC coding Mbp, thermophilic variance = 202050, mesophilic variance = 153004. B The genomes of species with a complex, multicellular lifecycle encode significantly more di-prolyl motifs per GC coding Mbp than their unicellular counterparts (Phylogenetic ANOVA, p value ≤ 0.001). Multicellular and unicellular species are represented by 61 and 3191 genomes, respectively: multicellular mean = 1902 motifs per GC coding Mbp, unicellular mean = 1157 motifs per GC coding Mbp, multicellular variance = 102622, unicellular variance = 152744.
Anecdotal evidence from our initial data exploration showed that the Myxococcota, which are well-known for their ability to form multicellular fruiting bodies, have the most di-prolyl motifs among all taxonomic groups we examined (Fig. 1), despite not having the highest overall GC content. Myxococcota and other bacteria with complex life cycles rely on cell-cell signaling to orchestrate their developmental programs [51]. Proline-rich regions often occur in the binding domains of signaling proteins, where they mediate protein-protein binding in a highly specific yet reversible manner [13]. Therefore, we wondered whether the genomes of bacteria with complex lifecycles are generally more likely to harbor many di-prolyl motifs.
We performed a phylogenetic ANOVA and found that bacteria known for multicellular behavior did encode more di-prolyl motifs per GC coding Mbp (Phylogenetic ANOVA, p value ≤ 0.001, Fig. 2B). Next, we modified our PGLS model to include multicellularity as an added variable. We also included growth temperature in the model, because some multicellular bacteria are thermophilic, for example the filamentous thermophile Ardenticatena maritima. We again found that multicellular bacteria have significantly more di-prolyl motifs than unicellular bacteria, independent of total GC coding content and growth temperature (PGLS B, p value < 0.01). This effect was also not driven solely by the Myxococcota—when we removed Myxococcota genomes from our dataset and repeated this analysis, multicellular bacteria still encoded more di-prolyl motifs (see Supplementary Results).
Slow-growing species encode more di-prolyl motifs than fast-growing species
Because di-prolyl motifs can negatively impact translation rate [15, 28], we were curious whether selection for translational speed would influence the number of di-prolyl motifs in a genome. Ideally, we would answer this question using experimentally measured growth rates. Unfortunately, this information is not widely available. As an alternative, we calculated the predicted growth rate of each species in our dataset using a codon usage bias (CUB) centered method, gRodon [32]. CUB refers to the tendency of species to use codons that correspond to the most abundant tRNAs in highly expressed genes. In doing so, these species can increase their translational rate by accelerating tRNA turnover at the ribosome [5]. The degree of CUB in genes encoding ribosomal proteins is well correlated with experimentally measured growth rates in mesophilic species [5, 32, 52] (see Methods, Fig. S1).
Along with predicted growth rates, we also included two other growth-associated traits in this PGLS model: tRNA and rRNA gene copy numbers. One strategy that fast-growing bacteria use to translate proteins rapidly is to ensure that their pool of charged tRNA does not become limiting. Fast-growing species thus often encode multiple copies of the most common tRNA genes [53]. Similarly, fast-growing species tend to encode multiple rRNA gene copies to boost the rate at which rRNA molecules—and consequently ribosomes—are synthesized [54]. When included in our PGLS model, all three growth-associated traits had a significant impact on the number of di-prolyl motifs in a genome, although the significance of rRNA gene copies was weak (PGLS C; predicted doubling time p value < 0.0001, tRNA gene copies p value < 0.005, rRNA gene copies p value < 0.1). Characteristics linked to slow-growth (slower predicted doubling times, fewer tRNA gene copies, and fewer rRNA gene copies) were all associated with more di-proly motifs (Supplementary Table 1).
Using the growth-associated traits of a representative slow (Methylomagnum ishizawai; predicted doubling time = 124 h, tRNA gene copies = 51, rRNA gene copies = 2) and fast growing species (Propionigenium maris; predicted doubling time = 0.11 h, tRNA gene copies = 103, rRNA gene copies = 5) in the equation supplied by the PGLS C model, we found that slow-growth traits resulted in a 14% increase in the number of di-prolyl motifs within a genome, irrespective of GC content (Fig. 3). This can yield a substantial total increase at a high GC content. For example, at a protein-coding GC content of 8 Mbp, slow-growth associated traits yielded an additional 1283 di-prolyl motifs (Fig. 3C).

A Although GC content is the primary determinant of the number of di-prolyl motifs in a genome (PGLS C p value < 0.0001, regression slope b = 1.056), predicted doubling time, tRNA gene copies, and rRNA gene copies also have a significant impact (PGLS C; predicted doubling time p value < 0.0001 b = 0.016, tRNA gene copies p value < 0.005 b = −0.030, rRNA gene copies p value < 0.1, b = −0.003). The two diagonal lines represent the PGLS-predicted relationship between di-prolyl motifs and the GC content of a genome, as calculated using the growth-associated traits of a representative slow and fast-growing species from our dataset. Specifically, the upper blue line represents a prediction based on the growth traits of one of the slowest growing species (Methylomagnum ishizawai; predicted doubling time = 124 h, tRNA gene copies = 51, rRNA gene copies = 2) and the lower red line represents a prediction based on the growth traits of one of the fastest growing species (Propionigenium maris; predicted doubling time = 0.11 h, tRNA gene copies = 103, rRNA gene copies = 5). The slow-growth related traits of Methylomagnum ishizawai result in a 14% predicted increase in di-prolyl motifs. At low GC content (B: enlarged view of the lower left box in (A)), the total impact of growth-associated traits is low (calculated net increase of only 47 di-prolyl motifs per genome at 0.35 Mbp GC content), while at high GC content (C: enlarged view of the upper right box in (A)) the total net increase is substantial (calculated net increase of 1283 di-prolyl motifs at 8 Mbp GC content). The horizontal and vertical axes are plotted on a logarithmic scale.
Although CUB based growth rate metrics predict experimentally measured doubling times well [5, 32] (Fig. S1), we wondered whether the statistical associations we detected would persist if we used experimentally measured doubling times instead. We found such data for 301 (9.2%) species in our dataset (see Supplementary Dataset 1 for details). When we repeated our PGLS analysis on this reduced dataset, we found that species with faster experimentally measured growth rates still encoded fewer di-prolyl motifs than slow-growing species, although rRNA gene copies was no longer a significant predictor (PGLS D, measured doubling times p value < 0.05, tRNA gene copies p value ≤ 0.005, rRNA gene copies p value = 0.440). Removing thermophiles from this analysis significantly improved this relationship (PGLS E, measured doubling times p value ≤ 0.001, tRNA gene copies p value < 0.05, rRNA gene copies p value = 0.315), perhaps because rapid thermophilic growth rates do not necessarily reflect enhanced investment in maximizing translational speed [5]. Using the equation supplied by PGLS E, the growth-associated traits of our representative slow-growing species, Methylomagnum ishizawai (experimentally measured doubling time = 24 h), yielded a 25% increase in di-prolyl motifs over the fast-growing representative Propionigenium maris (experimentally measured doubling time = 0.3 h) (Fig. S2). In sum, both CUB-predicted and experimentally measured growth rates support the notion that fast-growing species encode fewer di-prolyl motifs than slow-growing species.
Proteins optimized for translational speed contain few di-prolyl motifs, especially in fast-growing species
Our analysis thus far focused on the incidence of di-prolyl motifs in entire genomes, but this incidence may also vary among proteins within a genome. For example, in E. coli, highly expressed proteins contain fewer di-prolyl motifs than lowly expressed proteins [29]. We wondered whether this link between expression level and di-prolyl motifs exists more generally in the >3000 bacterial genomes we analyzed. To address this question, we used gene annotations from the KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthology database [55], which assigns genes with a common function to a KEGG Orthology (KO) group. This classification provides single-source functional annotations within a hierarchical classification scheme that ranges from coarse-grained, e.g., “genetic information processing”, to fine-grained, e.g., “ribosomal protein-coding”.
Because gene expression data does not exist for the vast majority of our genomes, we used the CUB of individual genes as a proxy for their expression level. Within each genome, we ranked each gene based on its overall CUB, where the highest rank of one corresponds to the gene with the strongest bias and highest predicted expression. We then took the median of this rank for each gene across all genomes (see Methods for details). We found that highly expressed proteins (low median CUB rank) contained significantly fewer di-prolyl motifs, an observation that holds both for all motifs (Spearman’s rho = 0.52, p value < 0.0001, Fig. 4), and for motifs predicted to cause a strong stall (Spearman’s rho = 0.54, p value < 0.0001, Fig. S3). Proteins whose median CUB rank was in the top percentile harbored an order of magnitude fewer di-prolyl motifs per 100 amino acids (AA) than proteins within the bottom CUB percentile (0.002 vs. 0.014 motifs per 100 AA, GC-controlled). These findings place similar observations from E. coli [29] into a broad phylogenetic context.

Proteins expected to be highly expressed (based on CUB, see Methods) contain fewer di-prolyl motifs when protein length and gene GC content are controlled for (Spearman’s rho = 0.52, p value < 0.0001). Each circle represents the average incidence of di-prolyl motifs within a protein (KEGG KO) across all genomes it was identified in. The colors represent the coarse-level function of the KEGG Orthology (KO) group to which each protein belongs. We only included common proteins (present in at least 25% of genomes) in this analysis to reduce bias towards rare proteins. The purple line is a linear regression line, and the shaded area represents the 95% confidence area. The horizontal axis is plotted on a logarithmic scale.
Although highly expressed proteins were generally depleted in di-prolyl motifs, we wondered whether this association would be especially pronounced in fast-growing species. Though we previously showed that fast-growing bacteria encode fewer di-prolyl motifs than slow-growing bacteria in general (Fig. 3), this analysis did not distinguish proteins based on their expected expression level. We hypothesized that fast-growing species would have fewer di-prolyl motifs in proteins expected to be highly expressed, as expression levels generally scale with growth rate [2], and ribosomal stalling is exacerbated by high translation initiation rates [16]. To validate this hypothesis, we identified for each genome those genes expected to be most highly expressed, i.e., genes whose CUB lies within the top percentile. We then calculated the average amount of di-prolyl motifs in the proteins these genes encode. On average, fewer di-prolyl motifs were encoded by genes predicted to be highly expressed in fast-growing species than in slow-growing species, independent of GC content and growth temperature (PGLS F, predicted doubling time p value < 0.0001).
Di-prolyl motifs are enriched in disordered domains of serine-threonine kinases and other signaling proteins
Previous analyses revealed that bacteria capable of multicellular behavior encoded many di-prolyl motifs independent of growth traits and growth temperature (PGLS C, p value ≤ 0.01), with genomes from the Myxococcota containing by far the most di-prolyl motifs in our dataset (Fig. 1). Although we found that di-prolyl motifs located in KEGG-annotated signaling proteins were not responsible for the elevated numbers of these motifs in multicellular bacteria (see Supplementary Results), signaling proteins did contain significantly more di-prolyl motifs in multicellular bacteria (phylogenetic ANOVA, p value ≤ 0.001). A large proportion of these motifs were in protein kinases, and specifically in serine-threonine kinases. Serine-threonine kinases are involved in signal transduction, and work by phosphorylating specific sites on target proteins [56, 57]. Extensive cross-phosphorylation can occur between kinases, modulating their downstream activity in signaling cascades [57]. Multicellular bacteria contain significantly more serine-threonine kinases than unicellular bacteria (Fig. S4, phylogenetic ANOVA p value ≤ 0.001), and these kinases contain many di-prolyl motifs, especially within the Myxococcota. For example, Stigmatella erecta contains 530 di-prolyl motifs spread across the 67 serine-threonine kinases encoded within its genome. Di-prolyl motifs in this species are enriched 45-fold in serine-threonine kinases, such that 3.66% of di-prolyl motifs occur in a set of proteins that make up just 0.08% of its proteome.
The phosphorylation sites of protein kinases are predominately located within intrinsically disordered regions (regions without stable three-dimensional structure—IDRs) [58]. Notably, proline rich sites with high propensity towards forming polyproline II helices (PPII) are evolutionarily conserved at intrinsically disordered phosphorylation sites, where phosphorylation may tune a protein’s propensity to adopt PPII structure [12]. These connections between proline rich PPII sites, phosphorylation, and IDRs led us to ask whether the di-prolyl motifs within serine-threonine kinases were preferentially located within IDRs of these proteins.
We used the disorder prediction software IUPred2A [41] to identify disordered regions in all serine-threonine kinases within our dataset. Although on average only 27% of residues within the 6552 serine-threonine kinases in our dataset were disordered, 72% of di-prolyl motifs were located within these disordered regions, a significant enrichment (χ2 test p value < 0.0001, Fig. S5). In one extreme case, a single serine-threonine kinase from the myxobacterium Stigmatella erecta contained 41 di-prolyl motifs, of which 95% were located within disordered regions (Fig. 5). Though multicellular species contained more serine-threonine kinases than unicellular (Fig. S4), and more di-prolyl motifs within these proteins (phylogenetic ANOVA p value < 0.05), the di-prolyl motifs of both unicellular and multicellular serine-threonine kinases were equally enriched among disordered regions (71% of multicellular motifs occur in IDRs vs 72% of unicellular motifs). Expanding on these findings, we identified four additional common signaling proteins (encoded by at least 25% of genomes in our dataset) whose di-prolyl motifs were significantly enriched within disordered regions (χ2 test Bonferroni corrected p value < 0.001, Fig. S5). These findings indicate that the enrichment of di-prolyl motifs within IDRs may be a general feature of kinases and signaling proteins.

Each circle represents a single di-prolyl motif within the protein, with color indicating the predicted severity of the resulting stall, from strong to medium to weak. A region is predicted to be intrinsically disordered if the IUPred2A disorder score is greater than or equal to 0.5, as indicated by the dashed purple line. The green rectangle at the bottom of the figure indicates the PFAM protein kinase domain (PF00069) which is congruent with the set of ordered residues in this protein, as expected. This protein is encoded by IMG gene 2695004422 in IMG taxon oid 2693429888. The figure design is inspired by default IUPred2A plots [41].
Discussion
Research involving EFP and di-prolyl motifs has largely focused on individual species [24, 25, 27, 29] or proteins [59]. In contrast, we surveyed these motifs in a broad range of genotypically and phenotypically diverse bacteria. Though the exact effect of di-prolyl motifs on translational rate has not been experimentally tested in every species we studied, di-prolyl motifs cause ribosomal stalling in all three domains of life, consistent with the near universal distribution of EFP and EFP homologs [17]. Likewise, every genome in our dataset encoded EFP, with one exception (see Methods). Because proline codons are cytosine rich (CCU, CCC, CCA, and CCG), it is not surprising that the occurrence of di-prolyl motifs is strongly associated with a genome’s GC content (Fig. 1). We thus focused on patterns of di-prolyl motif occurrence that cannot be explained by GC content alone. We found such patterns in three groups of bacteria: slow-growing, thermophilic, and multicellular species.
We were especially interested in potential associations between di-prolyl content and growth rates. Indeed, we found that fast-growing species encode significantly fewer di-prolyl motifs than slow-growing species (PGLS C, Fig. 3). This relationship holds whether we estimate growth rate indirectly from CUB (PGLS C, Fig. 3), or directly from experimental measurements (PGLS E, Fig. S2). In addition, while proteins expected to be highly expressed generally encode fewer di-prolyl motifs (Fig. 4 and S3), this trend is exacerbated in fast-growing species (PGLS F). These findings are based on analyses of variance that correct for phylogenetic relatedness and allow the impact of co-correlated traits to be quantified independently (PGLS, Supplementary Table 1).
Several highly expressed proteins critical to cell function contain di-prolyl motifs that cannot be removed without a resulting loss of function. For example, the valine tRNA synthetase ValS contains a poly-proline motif that is highly conserved, critical to charge tRNAVal efficiently, and important to prevent its mischarging with threonine [59]. This implies that some di-prolyl motifs are maintained in the face of negative selection pressure because the benefit of their specific biochemical activity outweighs any impact they may have on translational rate. If the genomes of some species can encode more di-prolyl motifs because of weaker selection on translational rate, these motifs may also acquire new and useful roles unrelated to their effect on protein translation.
One candidate for such a role is to stabilize proteins in the high temperature environments experienced by thermophilic bacteria. Proline residues can increase the thermostability of proteins by at least two mechanisms. First, their rigid structure reduces the degrees of freedom available to a protein [49]. Second, increasing the proportion of prolines and other hydrophobic residues enhances thermostability by reducing accessibility to the protein core [49]. Thermophiles have smaller genomes and shorter proteins than mesophiles [50]. However, when we controlled for the proportion of their genome that encodes proteins, we found that thermophiles encoded more di-prolyl motifs than mesophiles (PGLS A, Fig. 2A). Thermophiles are thought to experience relaxed selection on translation rate because catalysis naturally proceeds more rapidly at higher temperatures [5]. Relaxed selection on translational speed in thermophiles may allow prolines to exist where they would otherwise be detrimental for translational rate, and thus help stabilize a thermophilic proteome.
Another potential role for di-prolyl motifs exists in bacteria with complex, multicellular lifecycles, most notably the Myxococcota (Fig. 1). We found that multicellular bacteria contained significantly more di-prolyl motifs than unicellular bacteria (PGLS B, Fig. 2B). These organisms rely on signaling proteins to orchestrate their complex lifecycles [60, 61], which often contain proline-rich regions with their binding domains [12, 13]. Though we found signaling proteins were not exclusively responsible for this effect (Supplementary Results), multicellular species do encode more of the di-prolyl rich signaling proteins serine-threonine kinases compared to unicellular bacteria (Fig. S4). These kinases contain an outsized proportion of di-prolyl motifs, which can be enriched up to 45-fold in some Myxococcota compared to their background occurrence within the proteome. Serine-threonine kinases play central roles in signaling between cells by modulating the activity of their target proteins through phosphorylation [57] and have been linked to cellular complexity in multicellular bacteria [56].
Bacterial kinases are known to cross and auto-phosphorylate, creating extensive signaling networks [57]. The phosphorylation sites of kinases are enriched in intrinsically disordered regions (IDRs), where phosphorylation can trigger changes in 3D-structure that alter downstream activity [58]. Interestingly, proline rich regions with high propensity towards forming polyproline II helices (PPII) also occur primarily within IDRs [62], and are evolutionarily conserved at phosphorylation sites [12]. Following these connections, we found that di-prolyl motifs within serine-threonine kinases are enriched among IDRs, as illustrated by a specific example in Fig. 5. Furthermore, we found that the di-prolyl motifs of four other common signaling proteins (present in >25% of our genomes) were significantly enriched among IDRs (Fig. S5). Three of these signaling proteins are known to be phosphorylated (response regulator RegA [63], sensor kinase CheA [64], and an OmpR family sensor kinase [65]). Phosphorylation can have a dramatic effect on local bias towards PPII structures, in effect tuning a protein’s propensity towards adopting a PPII structure [12, 66]. As PPII structures commonly form the binding domains of signaling proteins [12, 13], a connection between phosphorylation, PPII formation (or collapse), and the modulation of signaling protein activity is appealing. However, verifying these connections and determining their biological significance remains a task for future work.
Our observations are consistent with previously drawn connections between cellular complexity and polyproline motifs, with more complex organisms containing higher numbers of such motifs [10]. The multicellular bacteria in our dataset are generally slow-growing, with an average predicted doubling time of 8.4 hours. As a result, selection on translational rate is weaker in these species, which may allow prolines to accumulate where they would otherwise be discouraged. Indeed, it could be informative to interrogate possible regulatory functions of EFP in multicellular species, as EFP likely influences the expression of signaling proteins highly enriched in di-prolyl motifs.
In conclusion, our observations suggest active selection against di-prolyl motifs in a broad range of fast-growing species, and in highly expressed proteins of such species. Such selection is likely driven by high pressure on optimizing translational rate. Wherever such selection is relaxed, di-prolyl motifs may be free to proliferate and take up new roles. One of these roles may be to ensure proteome stability in thermophiles. Another may be to help cells in simple multicellular prokaryotes communicate. However, the causal role of di-prolyl motifs in any of these processes is unclear. For example, did multicellular bacteria emerge from slow-growing unicellular bacteria, where a high incidence of di-prolyl motifs facilitated kinase-based cell signaling and helped establish multicellularity? Or did multi-cellular bacteria emerge from fast-growing unicellular bacteria, such that their reduced growth rate, kinase-based signaling, and the importance of di-prolyl motifs emerged only secondarily? These and other questions about the biological functions and evolutionary origins of di-prolyl motifs provide exciting directions for future work.
Data availability
All genomes used in this study are publicly available from JGI’s IMG database [35]. Taxon IDs corresponding to every genome are listed in Supplementary Dataset 1, along with the genomic characteristics calculated for this study. Results from analyses of individual proteins are presented in Supplementary Dataset 2 and results of all PGLS models are listed in Supplementary Table 1. R scripts and all files needed to reproduce these analyses are available at https://github.com/tessbrewer/proline_project.
References
Russell JB, Cook GM. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol Rev. 1995;59:48–62.
Klumpp S, Scott M, Pedersen S, Hwa T. Molecular crowding limits translation and cell growth. PNAS. 2013;110:16754–9.
Pedersen S. Escherichia coli ribosomes translate in vivo with variable rate. EMBO J. 1984;3:2895–8.
Ran W, Higgs PG. Contributions of speed and accuracy to translational selection in bacteria. PLoS One. 2012;7:e51652.
Vieira-Silva S, Rocha E. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2009;6:1–15.
Roller BRK, Stoddard SF, Schmidt TM. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat Microbiol. 2016;1:1–7.
Buskirk AR, Green R. Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos Trans R Soc B. 2017;372:20160183–11.
Wohlgemuth I, Brenner S, Beringer M, Rodnina MV. Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem. 2008;283:32229–35.
Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC. Slow peptide bond formation by proline and other N-alkylamino acids in translation. PNAS. 2009;106:50–54.
Mandal A, Mandal S, Park MH. Genome-wide analyses and functional classification of proline repeat-rich proteins: potential role of eIF5A in eukaryotic evolution. PLoS One. 2014;9:e111800–13.
Adzhubei AA, Sternberg MJE, Makarov AA. Polyproline-II helix in proteins: structure and function. J Mol Biol. 2013;425:2100–32.
Elam WA, Schrank TP, Campagnolo AJ, Hilser VJ. Evolutionary conservation of the polyproline II conformation surrounding intrinsically disordered phosphorylation sites. Protein Sci. 2013;22:405–17.
Ball LJ, Kühne R, Schneider-Mergener J, Oschkinat H. Recognition of proline-rich motifs by protein-protein-interaction domains. Angew Chem Int Ed Engl. 2005;44:2852–69.
Starosta AL, Lassak J, Peil L, Atkinson GC, Virumäe K, Tenson T, et al. Translational stalling at polyproline stretches is modulated by the sequence context upstream of the stall site. Nucleic Acids Res. 2014;42:10711–9.
Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015;11:13–21.
Hersch SJ, Elgamal S, Katz A, Ibba M, Navarre WW. Translation initiation rate determines the impact of ribosome stalling on bacterial protein synthesis. J Biol Chem. 2014;289:28160–71.
Lassak J, Wilson DN, Jung K. Stall no more at polyproline stretches with the translation elongation factors EF‐P and IF‐5A. Mol Microbiol. 2016;99:219–35.
Yanagisawa T, Sumida T, Ishii R, Takemoto C, Yokoyama S. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nature. 2010;17:1136–43.
Park J-H, Johansson HE, Aoki H, Huang BX, Kim H-Y, Ganoza MC, et al. Post-translational modification by beta-lysylation is required for activity of Escherichia coli elongation factor P (EF-P). J Biol Chem. 2012;287:2579–90.
Lassak J, Keilhauer E, Fürst M, Wuichet K, Gödeke J, Starosta AL, et al. Arginine-rhamnosylation as new strategy to activate translation elongation factor P. Nat Chem Biol. 2015;11:266–70.
Tollerson R, Witzky A, Ibba M. Elongation factor P is required to maintain proteome homeostasis at high growth rate. PNAS. 2018;115:1–6.
Peng WT, Banta LM, Charles TC, Nester EW. The chvH locus of Agrobacterium encodes a homologue of an elongation factor involved in protein synthesis. J Bacteriol. 2001;183:36–45.
Rajkovic A, Hummels KR, Witzky A, Erickson S, Gafken PR, Whitelegge JP, et al. Translation control of swarming proficiency in Bacillus subtilis by 5-amino-pentanolylated elongation factor P. J Biol Chem. 2016;291:10976–85.
Navarre WW, Zou SB, Roy H, Xie JL, Savchenko A, Singer A, et al. PoxA, YjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol Cell. 2010;39:209–21.
Hummels KR, Kearns DB. Suppressor mutations in ribosomal proteins and FliY restore Bacillus subtilis swarming motility in the absence of EF-P. PLoS Genet. 2019;15:e1008179–27.
Rajkovic A, Erickson S, Witzky A, Branson OE, Seo J, Gafken PR, et al. Cyclic rhamnosylated elongation factor P establishes antibiotic resistance in Pseudomonas aeruginosa. MBio. 2015;6:1–9.
Yanagisawa T, Takahashi H, Suzuki T, Masuda A, Dohmae N, Yokoyama S. Neisseria meningitidis translation elongation factor P and its active-site arginine residue are essential for cell viability. PLoS One. 2016;11:e0147907–27.
Krafczyk R, Qi F, Sieber A, Mehler J, Jung K, Frishman D, et al. Proline codon pair selection determines ribosome pausing strength and translation efficiency in bacteria. Commun Biol. 2021;4:1–11.
Qi F, Motz M, Jung K, Lassak J, Frishman D. Evolutionary analysis of polyproline motifs in Escherichia coli reveals their regulatory role in translation. PLoS Comput Biol. 2018;14:e1005987–19.
Karlin S, Mrázek J, Campbell A, Kaiser D. Characterizations of highly expressed genes of four fast-growing bacteria. J Bacteriol. 2001;183:5025–40.
Dethlefsen L, Schmidt TM. Performance of the translational apparatus varies with the ecological strategies of bacteria. J Bacteriol. 2007;189:3237–45.
Weissman JL, Hou S, Fuhrman JA. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. PNAS. 2021;118:1–10.
Hersch SJ, Wang M, Zou SB, Moon K-M, Foster LJ, Ibba M, et al. Divergent protein motifs direct elongation factor P-mediated translational regulation in Salmonella enterica and Escherichia coli. MBio. 2013;4:1–10.
Pinheiro B, Scheidler CM, Kielkowski P, Schmid M, Forné I, Ye S, et al. Structure and function of an elongation factor P subfamily in Actinobacteria. Cell Rep. 2020;30:4332–42. e5.
Chen I-MA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45:D507–D516.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925–7.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195–16.
Madin JS, Nielsen DA, Brbic M, Corkrey R, Danko D, Edwards K, et al. A synthesis of bacterial and archaeal phenotypic trait data. Sci Data. 2020;7:1–8.
Novembre JA. Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol. 2002;19:1390–4.
Erdos G, Dosztányi Z. Analyzing protein disorder with IUPred2A. Curr Protoc Bioinforma. 2020;70:1–15.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50.
Pagel M. Inferring the historical patterns of biological evolution. Nature. 1999;401:877–84.
Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2011;3:217–23.
Orme D, Freckleton R, Thomas G, Petzoldt T, Fritz S, Isaac N, et al. caper: comparative analyses of phylogenetics and evolution in R. 2018; https://CRAN.R-project.org/package=caper.
Wickham H. ggplot2: elegant graphics for data analysis. 2016. Springer-Verlag New York.
Symonds MRE, Blomberg SP. A primer on phylogenetic generalized least squares. In: Garamszegi L (eds). Modern phylogenetic comparative methods and their application in evolutionary biology. (Springer, Berlin, Heidelberg, 2014) pp. 105–30.
Watanabe K, Suzuki Y. Protein thermostabilization by proline substitutions. J Mol Catal B Enzym. 1998;4:167–80.
Sabath N, Ferrada E, Barve A, Wagner A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol Evol. 2013;5:966–77.
Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, et al. Evolution of sensory complexity recorded in a myxobacterial genome. PNAS. 2006;103:15200–5.
Long AM, Hou S, Ignacio-Espinoza JC, Fuhrman JA. Benchmarking microbial growth rate predictions from metagenomes. ISME J. 2020;15:1–13.
Rocha EPC. Codon usage bias from tRNA’s point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–86.
Klappenbach JA, Dunbar JM, Schmidt TM. rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol. 2000;66:1328–33.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114.
Perez J, Castaneda-García A, Jenke-Kodama H, Muller R, Munoz-Dorado J. Eukaryotic-like protein kinases in the prokaryotes and the myxobacterial kinome. PNAS. 2008;105:15950–5.
Shi L, Pigeonneau N, Ravikumar V, Dobrinic P, Macek B, Franjevic D, et al. Cross-phosphorylation of bacterial serine/threonine and tyrosine protein kinases on key regulatory residues. Front Microbiol. 2014;5:1–13.
Jakob U, Kriwacki R, Uversky VN. Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev. 2014;114:6779–805.
Starosta AL, Lassak J, Peil L, Atkinson GC, Woolstenhulme CJ, Virumäe K, et al. A conserved proline triplet in Val-tRNA synthetase and the origin of elongation factor P. Cell Rep. 2014;9:476–83.
Nariya H, Inouye S. A protein Ser/Thr kinase cascade negatively regulates the DNA-binding activity of MrpC, a smaller form of which may be necessary for the Myxococcus xanthus development. Mol Microbiol. 2006;60:1205–17.
Stein EA, Cho K, Higgs PI, Zusman DR. Two Ser/Thr protein kinases essential for efficient aggregation and spore morphogenesis in Myxococcus xanthus. Mol Microbiol. 2006;60:1414–31.
Iakoucheva LM, Radivojac P, Brown CJ, OConnor TR, Sikes JG, Obradovic Z, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49.
Elsen S, Swem LR, Swem DL, Bauer CE. RegB/RegA, a highly conserved redox-responding global two-component regulatory system. Microbiol Mol Biol R. 2004;68:263–79.
Tawa P, Stewart RC. Kinetics of CheA autophosphorylation and dephosphorylation reactions. Biochemistry. 1994;33:7917–24.
Yoshida T, jian CaiS, Inouye M. Interaction of EnvZ, a sensory histidine kinase, with phosphorylated OmpR, the cognate response regulator. Mol Microbiol. 2002;46:1283–94.
Cho M-H, Wrabl JO, Taylor J, Hilser VJ. Hidden dynamic signatures drive substrate selectivity in the disordered phosphoproteome. PNAS. 2020;117:1–11.
Acknowledgements
We acknowledge funding from the European Research Council under Grant Agreement No. 739874, as well as from Swiss National Science Foundation grant 31003A_172887, and from the University Priority Research Program in Evolutionary Biology at the University of Zurich. We thank members of the Wagner lab for helpful discussions, particularly Andrei Papkou and Pouria Dasmeh, and Michael Engel for figure design input.
Author information
Authors and Affiliations
Contributions
TEB and AW conceived and designed the project. TEB performed all computational analyses. TEB and AW wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Brewer, T.E., Wagner, A. Translation stalling proline motifs are enriched in slow-growing, thermophilic, and multicellular bacteria. ISME J 16, 1065–1073 (2022). https://doi.org/10.1038/s41396-021-01154-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01154-y
