
Abstract
Bacterial infection and inflammation of the airways are the leading causes of morbidity and mortality in persons with cystic fibrosis (CF). The ecology of the bacterial communities inhabiting CF airways is poorly understood, especially with respect to how community structure, dynamics, and microbial metabolic activity relate to clinical outcomes. In this study, the bacterial communities in 818 sputum samples from 109 persons with CF were analyzed by sequencing bacterial 16S rRNA gene amplicons. We identified eight alternative community types (pulmotypes) by using a Dirichlet multinomial mixture model and studied their temporal dynamics in the cohort. Across patients, the pulmotypes displayed chronological patterns in the transition among each other. Furthermore, significant correlations between pulmotypes and patient clinical status were detected by using multinomial mixed effects models, principal components regression, and statistical testing. Constructing pulmotype-specific metabolic activity profiles, we found that pulmotype microbiota drive distinct community functions including mucus degradation or increased acid production. These results indicate that pulmotypes are the result of ordered, underlying drivers such as predominant metabolism, ecological competition, and niche construction and can form the basis for quantitative, predictive models supporting clinical treatment decisions.
Similar content being viewed by others


Introduction
Cystic fibrosis (CF) is characterized by persistent bacterial infection and inflammation of the respiratory tract. Culture-independent studies show that changes in the structure of the rich bacterial communities in CF airways correlate with changes in patients’ clinical states in the short term, and with lung disease progression in the longer term [1,2,3,4,5,6,7,8,9,10,11,12]. Our understanding of the dynamics and ecology of these communities is otherwise limited, especially with respect to community assembly and metabolic activity relative to clinical outcomes. One apparent reason is the individualized acquirement and adaptation of the CF lung microbiome, due to treatment history, patient lifestyle, genotype, and complex immune feedbacks [13, 14]. Therefore, often a strong, patient-associated signature in the data obstructs the view on potential underlying principles. It is unknown, for example, whether or not the CF lung communities comprise distinct types that affect the ecology, function, and pathogenicity of the CF airway microbiome. If causally related, such types could serve as prognostic markers, support treatment decisions, and enable the study of disease patterns across patients [15, 16].
To address this question, we characterized the bacterial communities in 818 sputum samples from 109 CF patients by sequencing the V3–V5 region of the 16S rRNA gene. Airway bacterial communities were partitioned into eight distinct types (“pulmotypes”) by modeling. We assessed the temporal dynamics of the pulmotypes and determined succession patterns along patient trajectories in our data set. We tested whether pulmotypes associated with clinical classifications like clinical state, disease stage, and aggressiveness. A complementary, functional characterization of pulmotypes provided insight on the role of community metabolism in disease development.
Materials and methods
Sputum sample and metadata collection
The sputum samples and DNA sequences included in this study have been reported, in part, in previous studies [11, 14, 17, 18]. During the course of routine medical care, sputum specimens were collected, processed within 4 h for bacterial culture at the Michigan Medicine clinical microbiology laboratory, and then held at 4 °C for up to 5 days until being stored at −80 °C. Storage at 4 °C for this amount of time has been shown to not significantly impact measures of bacterial community composition [19]. Metadata associated with each sputum sample were obtained from review of medical records. Sputum sample collection and medical record review were approved by the University of Michigan Institutional Review Board (HUM00048991). Patient clinical state at the time of sample collection was defined as described previously [11]: baseline; pulmonary exacerbation before antibiotic administration; antibiotic treatment of pulmonary exacerbation; or recovering from pulmonary exacerbation within 21 days after cessation of antibiotics prescribed for exacerbation. Lung disease stage at the time of sample collection was defined based on a measure of the patient’s percent predicted forced expiratory volume in 1 s (ppFEV1): early (ppFEV1 > 70); intermediate (ppFEV1 ≤ 70 ≥ 40); or advanced (ppFEV1 < 40). Lung function was treated categorically due to the imprecise and variable nature of single spirometry measurements, which make them not well suited for use as a continuous variable [20]. Lung disease aggressiveness (mild, moderate, or severe) was defined based on the rate at which ppFEV1 declined relative to patient age, reflecting patient’s overall CF lung disease severity phenotype [21].
DNA extraction, sequencing, and analysis
DNA was prepared from frozen sputum as described previously [14, 18]. Briefly, sputum aliquots were thawed on ice and treated with Sputolysin (EMD Chemicals, San Diego, CA). Samples were mechanically homogenized for 10 s with a tissue homogenizer (Omni International, Kenneshaw, GA) if not dissolved by the Sputolysin. Samples were mechanically disrupted in an UltraClean Fecal DNA bead tube (MO BIO Laboratories, Inc., Carlsbad, CA) by agitation in a Mini-Beadbeater-8 (Biospec Products Inc., Bartlesville, OK) for 1 min on maximum setting. Finally, DNA was purified using a MagNA Pure Compact System (Roche Diagnostics Corp., Indianapolis, IN) automated nucleic acid purification platform according to the manufacturer’s DNA Bacteria v3.1 protocol.
DNA pyrosequencing was performed by the Human Genome Sequencing Center at Baylor College of Medicine. The total 818 samples of this data set were sequenced in 14 batches, i.e., n1 = 64, n2 = 62, n3 = 30, n4 = 27, n5 = 32, n6 = 59, n7 = 61, n8 = 88, n9 = 76, n10 = 39, n11 = 75, n12 = 44, n13 = 85, n14 = 76, using identical methodology. The V3–V5 hypervariable regions of the 16S rRNA gene were amplified by using primer 357F (5’-CCTACGGGAGGCAGCAG-3’) modified with the addition of the 454 FLX-titanium adapter “B” sequence (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-3′) and primer 926R (5’-CCGTCAATTCMTTTRAGT-3’) modified with the addition of unique six- to eight-nucleotide barcode sequences and the 454 FLX-titanium adaptor “A” sequence (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′). Barcode and adaptor sequences can be found at https://www.hmpdacc.org/hmp/doc/HMP_MDG_454_16S_Protocol.pdf. Sequencing was performed via 454 FLX-titanium according to the manufacturer’s specifications.
Raw DNA sequences of the total data set were analyzed by using mothur [22, 23] following standard operating procedures (https://www.mothur.org/wiki/454_SOP). Sequences were assigned to operational taxonomic units (OTUs) using an average neighbor algorithm with a 0.03 dissimilarity cutoff (genus level). The consensus taxonomy of each OTU was identified at the genus level using the Bayesian method by aligning the DNA sequences to the Ribosomal Database Project [24] training set containing 9665 bacterial and 384 archaeal 16S rRNA gene sequences [12]. The total number of reads for each community was first rarefied to 1000 to control for differences in sequencing depth before alpha and beta diversity measures were calculated.
Pulmotype classification and statistical models for clinical associations
Bacterial pulmotypes were defined by using the Dirichlet multinomial mixture model (DMM) in mothur. Classification trees for the pulmotypes were generated using JMP software (SAS, Cary, NC).
To determine the relationship between bacterial pulmotypes and clinical outcomes, multinomial mixed models (MMM) of the logit type were used, i.e.,
for k = 1, … ,K – 1 and
where K is the baseline category of dependent variable Y, i is the index of observation and β the regression coefficient. The clinical state and disease stage served as dependent variables Y, pulmotypes as predictors X, and patient identity as random effect. MMMs were fitted using StataSE14 (StataCorp LP, College Station, TX).
PCoA plot was generated based on the Theta-YC distance calculated by mothur. Similarly, PERMANOVA and ANOSIM tests of pulmotypes were conducted in mothur (https://www.mothur.org/wiki/454_SOP).
Random forests analyses were used to identify variables that predict disease aggressiveness for younger patient ages using the R package “randomForest.” The subset of young age samples consisted of 221 samples (mild = 17, moderate = 64, severe = 140) from 49 patients (mild = 7, moderate = 21, severe = 21) age 10–21 years.
Random forest models, cross-validation, and ROC statistics were performed using patient stratification. Random patients were excluded from the procedure and the average of multiple calculations was assessed. This method corrected for potential biases introduced by individual patients. Furthermore, to balance sample numbers between the classes, the larger class was randomly subsampled to achieve sample parity. Unless otherwise indicated, 500 trees and default settings were used for the presented analyses. Cross-validation was performed using rfcv function, averaging over 100 iterations, using ten-fold cross-validation. Variable importance was evaluated as average of 1000 independent random forest models using all features (indicated as full model). Similarly, the ROC curves represent 100 patient-stratified full models and average AUC values were reported. Due to the limited number of samples, a train test split of the data was not performed. Accordingly, the ROC statistics reflect the performance on seen data only.
Significant associations between pulmotypes and disease stage were identified by Fisher exact test. In detail, late stage (C3) was tested against non-late stage (C1 + C2) and early stage (C1) was tested against non-early stage (C2 + C3) for pulmotype 8 and 4 against any other pulmotype.
Principal components regression was performed on the 16S rRNA-derived sample collection in R on rarified, centralized, and scaled data. Y-aware scaling, where FEV1 served as the scaling variable, was used. Principal component analysis was conducted on the FEV1-scaled data and the first two components were used to inform a regression model. The model was validated by assessing the model fit (R2) between the predicted and true FEV1 values. This procedure was performed for all pulmotypes individually, as well as combinations of two or three pulmotypes to identify the best model for FEV1 prediction. For each of the models, all samples in the data collection that were classified to the respective pulmotypes were included for model building. The pipeline was assembled according to http://www.win-vector.com/blog/2016/05/pcr_part2_yaware.
Evaluating pulmotype transitions
To determine the probabilities of transitions between pulmotypes, a sliding window of size 2 was used across all sample time series. Pulmotype transition events were counted between any consecutive samples of every patient; transition categories with less than 3 transition events were excluded from the analysis and counts were normalized against the total of all transition events. To avoid potential patient biases, a leave-one-out strategy was applied. All patients were removed once and the average of all calculations was reported. Implementation was performed in R.
Annotation and dynamics of metabolic profiles
Similar to [25], we assembled a list of cross-feeding metabolites and virulence factors relevant for CF-associated microbiota by combining literature reports with metabolic models in MetaCyc (https://metacyc.org). In particular, we mined reports describing experimental evidence for uptake or export of amino acids, sugars, short-chain fatty acids (SCFA), mucin-derived sugars and O-glycans, siderophores, ammonia, nitrate, nitrite, and CO2, as well as virulence factors produced by the CF microbiome including LPS, ExoS, ExoA, cell toxins, endo toxins, and hemolysin; elastases, proteases, reports on apoptosis and necrosis (“cell death”). Pathways of metabolite production and uptake (i.e., transport, symport, antiport systems) were subsequently confirmed using the MetaCyc cross-organism search for species within the respective genus. The selection of reference metabolic models or genomes was restricted to species relevant for lung disease (search term: “pathogenic,” “oral,” “airways”). The trait profiles of each OTU and literature references are summarized in the Supplementary.
Next, metabolic profiles of pulmotypes were constructed from the trait profiles of all taxa present in the respective pulmotype samples. For each pulmotype, traits were approximated by the mean relative abundance of all taxa hosting the particular trait. The pulmotype trait vectors were correlated using Spearman rank correlation and subsequently transformed to distances. Hierarchical clustering for was performed on the trait distances. All scripts were implemented in R.
To test associations between clinical state and a preceding sequence of pulmotype changes (temporal motifs in the time series), χ2 testing was performed. A contingency matrix was constructed for all available sequences of pulmotypes in three consecutive samples (sliding window extraction). To characterize the clinical state after transitioning through a particular three-pulmotype motif, every individual sequence was labeled with the clinical state (baseline, exacerbation, treatment, recovery) of the third sample in the trajectory.
Furthermore, the changes in Pseudomonas relative abundance in these three-pulmotype sequences were monitored and classified as “increase,“ “decrease,“ and “oscillatory” Pseudomonas dynamics. The same three-sample setup was used for testing the association of community stability with treatment (T) or baseline state (B) in the last sample. Here, three consecutive samples with identical pulmotype classification were considered stable, whereas changing pulmotypes were considered unstable. Pulmotype combinations “3-3-3,” “7-7-7,” and “8-8-8” were removed, to avoid potential bias due to highest relative sample numbers collected during treatment in pulmotypes 3, 7, 8. Plots were generated in R with package ggplot and corrplot.
Correlation networks were inferred with SparCC [26] for every pulmotype. Rarified data collection was used and correlation was deemed significant for |r|>0.4 and p value < 0.01 after correction of FDR with Benjamini–Hochberg.
Results
Patient and sputum sample characteristics and DNA sequencing
The characteristics of 109 patients and sputum samples are summarized in Table 1. DNA sequences were binned into 215 bacterial taxa at the genus level. Pseudomonas was the dominant bacterial taxon in the sputum sample set in terms of prevalence (84.7%) and relative abundance (mean 45.4%; range 0.001–99.9%). The other prevalent genera included Streptococcus, Staphylococcus, and Prevotella, each of which was present in at least 50% of the sputum samples. Burkholderia and Achromobacter, although present in lower prevalence, were observed to be the dominant bacterial taxa in some samples (i.e., the taxon with the highest relative abundance).
CF airway bacterial communities stratify into eight pulmotypes
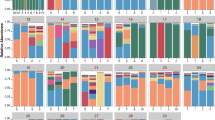
To determine if inherent data structure supports a characterization of the airway bacterial communities into different metacommunities (pulmotypes), we analyzed the 16S rRNA gene sequencing data by using the DMM model [27, 28]. This model partitioned the 818 samples and the bacterial communities therein into eight pulmotypes based on the 50 most abundant bacterial taxa (genus level OTUs), which collectively accounted for 99.6% of the DNA sequence reads in the data set. In Fig. 1A we show the Laplace approximation of the model fit for k = 1–10 Dirichlet components. To exclude potential patient-derived biases, we additionally assessed the Laplace values for 50 DMMs derived from subsampled data by removing two random patients at time. The results confirmed the fundamental patterning in our data (Supplementary Fig. S1). The number of samples within each of these eight pulmotypes ranged from 43 to 152 samples (Fig. 1B) and cluster homogeneity θ distributed as 31.6, 5.6, 15.9, 19.3, 61.4, 32.2, 36.4, 1.5 for pulmotypes 1–8, respectively. Significant differences between pulmotypes with respect to community diversity and richness were observed, with pulmotype 4 being the most diverse and pulmotype 5 the least (Fig. 1C, D). The relative abundances of the top 10 genera across all pulmotypes are shown in Fig. 1E–I, and Supplementary Fig. S2. The average relative abundance of the top 10 genera is shown for all pulmotypes in Supplementary Fig. S2A, and the ratio of typical CF pathogen genera to anaerobic genera is shown in Supplementary Fig. S2B. Typical CF pathogen genera include Pseudomonas, Staphylococcus, Burkholderia, Achromobacter, Stenotrophomonas, and Haemophilus. Anaerobic genera include Streptococcus, Prevotella, Veillonella, Fusobacterium, Gemella, Rothia, Granulicatella, Actinomyces, and Porphyromonas [18]. Pulmotypes 1 and 3 were dominated by Burkholderia and Staphylococcus, showing highest relative abundances respectively. Pseudomonas and Staphylococcus were the dominant taxa in pulmotype 7. Pseudomonas was detected with high relative abundance (≥19.8%) in all sputum samples in pulmotypes 5, 6, and 7. Whereas pulmotype 2 and pulmotype 8 both included bacterial communities dominated by other common CF bacterial taxa such as Achromobacter, Stenotrophomonas, Fusobacterium, and Haemophilus, communities in pulmotype 8 were generally less diverse.

A Partitioning of bacterial communities in 818 sputum samples into eight types based on the Laplace value in the Dirichlet multinomial mixture model. B The distribution of 818 sputum samples among eight pulmotypes. C Community diversity (inverse Simpson) of samples within each pulmotype. D Community richness (observed taxa) of samples within each pulmotype. E–L The relative abundance of the top 10 genera in all samples depicted in each of the eight pulmotypes (short name, color code), i.e., Pseudomonas (Ps, white), Streptococcus (St, red), Prevotella (Pr, green), Staphylococcus (Sa, yellow), Burkholderia (Bu, cyan), Achromobacter (Ac, beige), Veillonella (Ve, olivegreen), Stenotrophomonas (Se, magenta), Fusobacterium (Fu, blue), Haemophilus (Ha, gray). The top and bottom boundaries of each box indicate 1st and 3rd quartile values, respectively, and black lines inside each box represent median values. Ends of the whiskers mark the lowest and highest values within 1.5 times the interquartile range. Outliers, defined as samples with values less than or greater than 1.5 times the interquartile range, are shown as open circles.
In time series of microbial abundance, the strongest signature is often generated by the individual patient [29, 30, 31, 32], which limits the identification of potential clinical patterns that could explain mechanism or provide predictors. Therefore, we tested patient-related factors (disease aggressiveness, CFTR genotype, sex, disease stage, and clinical state), as well as a potential sequencing batch effect for predicting variance in a principal coordinate analysis. The ordination was based on Theta-YC distance between samples that reflect differences in microbial community structure (Fig. 2). The first two ordination components only covered 7.8% of data variability, which is not unusual for data with similar microbiota. We found that both the tested clinical parameters and sequencing batches have low impact on sample beta diversity in this data, i.e., R2 = [0.004, 0.008] and R2 = 0.06, respectively (PERMANOVA, Supplementary Table S1).

Solid circles represent pulmotypes 1 (black), 2 (red), 3 (green), 4 (blue), 5 (cyan), 6 (pink), 7 (yellow), and 8 (gray). Separation of bacterial communities in the ordination space is best explained by pulmotypes rather than by patient disease aggressiveness (mild: black; moderate: red; severe: green), CFTR genotype (F508del homozygous: black; F508del heterozygous: red; other genotypes: green; unknown genotypes: blue), sex (male: black; female: red), disease stage (early: black; intermediate: red; advanced: green; unknown: gray), or clinical states (baseline: black; exacerbation: red; treatment: green; recovery: blue; unknown: gray). PERMANOVA, R2 indicated, p value < 0.001 for all tested variables.
We furthermore tested the similarity of the microbiota among pulmotypes and detected that some pulmotypes had distinct bacterial communities (e.g., pulmotypes 1 vs 3, 1 vs 6, and 3 vs 6; analysis of similarity [ANOSIM], R = 1.00, p value < 0.05), others showed indistinguishable community structures (e.g., pulmotypes 2 vs 4; ANOSIM R = 0.08, p value < 0.05) (Supplementary Fig. S3 and Supplementary Table S2).
Recurrent shifts of pulmotypes characterize individual patient trajectories
We asked whether pulmotypes were robust over time and could be used to classify patients into groups with distinct treatment needs and/or clinical states, or whether pulmotypes frequently changed. To address this, we analyzed individual patient trajectories and determined the patient-stratified transition probabilities between pulmotypes of chronologically collected samples. We found that pulmotypes differ in their frequency of appearance and that pulmotypes in consecutive samples shift along recurrent routes. The most frequent route (46% of all transitions) occurred between and within pulmotypes 4, 5, and 6, with pulmotype 6 having the longest uninterrupted stretch (13% of all transitions) across all individual trajectories (Fig. 3A and Supplementary Table S3). The data suggest that compositional changes on this route may organize relative to Pseudomonas in an ongoing process. Also of note, this triangle motif harbors both the pulmotype with highest and lowest community diversity (pulmotype 4 and 5, respectively), indicating that important restructuring can happen in few steps.

A Transition frequencies between pulmotypes (1–8), proportional (%) representation. Transitions were determined using a sliding window approach within patient trajectories. Patient-stratified counting was applied and average values across all calculations were displayed. Node sizes were scaled according to persistence in the same; edge sizes varied according to frequency of transitions between pulmotypes. Edges supported by <3 transitions were removed; transitions that amount to >3% of total were additionally indicated. Node color represents the DMM cluster homogeneity θ of samples associated with the same pulmotype. B Clustering of pulmotypes by their cross-feeding profiles. Cross-feeding potential was approximated by the mean relative abundance of organisms capable of uptake or production for the respective metabolite. Pulmotype trait vectors were hierarchically clustered after transforming Spearman rank correlation to distances. Trait production/uptake are indicated as name/nameup, respectively. AA amino acids, Cell death reports on apoptosis, necrosis, GlcNAc N-acetylglucosamine, GalNAc N-acetylgalactosamine, NOx nitrate, nitrite, Proteases proteases and elastases, Virulence LPS, ExoS, ExoA, cell toxins, endo toxins, hemolysin.
The observed differences in community dynamics and composition led us to hypothesize that pulmotypes were linked to ordered, underlying driver processes such as predominant metabolic activities or ecological competition that caused detectable shifts in airway bacterial community organization and lung function.
To predict the metabolic potential from 16s rRNA amplicon data, reference-based strategies are often used (e.g., [33, 34]). These approaches are limited, as they cannot account for spatial-temporal metabolic variability, strain-specific functional adaptation, or biases in public data repositories with regard to organisms and pathways. To predict the cross-feeding potential of pulmotypes, we combined a database query using MetaCyc [35] with experimental evidence from the literature similar to [25]. This enabled us to focus on a subset of pathways and metabolites relevant for potential interactions in CF lungs: cross-feeding of small molecules for energy metabolism (i.e., carbon source, redox equivalents), mucus-derived glycoproteins and sugars (ecological niche), nitrogen, sulfur and iron assimilation (growth limitation), and metabolites signaling to host cells or for defense (Supplementary Table S4).
We subsequently integrated the function predictions with the prevalence of the genera in the pulmotype classes. We constructed functional profiles for each pulmotype by approximating the relative presence of a trait within the pulmotype by the mean relative abundance of the associated taxon and assessed distance between the profiles on Spearman rank transformed data. We observed that pulmotypes were clearly delineated by their functional trait profiles. Furthermore, a relatively small number of functional traits had a disproportionate impact on pulmotype characterization (Fig. 3B). These included uptake and production of SCFA (propionate, butyrate and acetate), lactate, CO2, NO2– and H2S; uptake of NH3 and NO3–; siderophore shutteling; assimilation of mucus-derived glycoproteins and sugars; triggering of elastase production of host immune cells; apoptosis and necrosis factors (category “cell death” in the figure); and the oxygenation state of the microenvironment. These signature traits suggest an adapted, interacting functional community dwelling at the interface of aerobic and anaerobic conditions in CF airways. Our results support earlier work that highlights the importance of oxygen and other chemical gradients for CF community composition and function [36].
Bacterial pulmotypes correlate with clinical features in CF
We next studied whether bacterial pulmotypes correlate with patient metadata and clinical features and might serve as therapeutic guides. As we have previously reported associations between airway microbiota and patients’ clinical state [18, 37], we sought to determine if bacterial pulmotypes also correlated with clinical states. We implemented a MMM that corrected for patient signature (Supplementary Table S5). The model identified pulmotype 5 as the least likely community type associated with exacerbation. Compared to pulmotype 5, pulmotypes 2, 4, 6, and 8 were 2.6, 2.2, 2.3, and 3.8 times, respectively, more likely to have been collected during exacerbation (p value < 0.05) than during other clinical states. Pulmotype 8 was a marker of antibiotic treatment, insofar as sputum samples of all other pulmotypes had significantly less likelihoods of being collected during antibiotic treatment (p value < 0.05, Supplementary Table S5).
Disease stage is based on lung function assessment (ppFEV1) and categorized as early, intermediate, or advanced. In a second analysis using MMMs, we tested the association of pulmotypes with different disease stages, while correcting for patient as confounder (Supplementary Table S6). The analysis revealed that pulmotype 4 was most associated with early disease stage. All other pulmotypes were more likely to be observed in intermediate or advanced stages (Supplementary Table S6, upper and middle panel). Notably, pulmotype 4 has the highest diversity of all pulmotypes. Pulmotypes 1 and 5 associated with intermediate disease stage, i.e., they were significantly more likely than pulmotype 4 to be observed in intermediate disease stage compared to other disease stages (MMM, RRR = 9.1, 2.9, respectively, p value < 0.05, Supplementary Table S6). In contrast, pulmotype 8 was associated with advanced disease stage, being 6.6, 9.3, 42.1, and 8.2 times more likely than pulmotypes 2, 3, 4, and 6, respectively (MMM, p value < 0.05, Supplementary Table S6, lower panel).
Pulmotype time dynamics are indicative of clinical state and disease stage
The persistence of a complex lung microbiome requires a sort of equilibrium with host immunity. The discontinuity theory of immunology predicts that host immune response may be triggered more by transient changes in immunogenic patterns than by persistent immune stimuli [38]. Thus, the observed dynamical motifs in patient trajectories (i.e., recurrent pulmotypes shifts) are promising candidates for predicting lung function decline and/or for generating mechanistic hypotheses about lung pathology.
To explore whether temporal motifs of pulmotype change could serve as markers for lung function decline, we tested whether motifs of two or three pulmotypes constituted better predictors of disease stage than individual pulmotypes. To that end, we implemented principal component regression models of all possible pulmotype single, double, and triple combinations and quantified their predictive power (Supplementary Figs. S4 and S5). Pulmotypes 4 and 8 together outperformed all other combinations (model fit R2 = 0.35, F-statistic = 94, p value < 1e–05). Only the triple combination of pulmotype 1, 4 and 8 model performed comparably (R2 = 0.33, F-statistic = 103, p value < 1e–05, Supplementary Fig. S14).
We then tested whether clinical states in patient trajectories could be delineated by querying ecological dynamics of Pseudomonas in three consecutive samples. We hypothesized that the dynamics of Pseudomonas relative to the remaining lung microbiome might coarsely reflect the alveolar microenvironment including oxygenation, pH, and pathogenicity and ultimately relate to disease state. In particular, we looked at increase and decrease of Pseudomonas abundance relative to the rest of the bacterial community. Interestingly, we found that variable relative abundances of Pseudomonas (increasing and decreasing over the three samples) correlated with subsequent baseline states, while a continuous decrease in the relative abundance of Pseudomonas correlated with subsequent exacerbation states consistent with Carmody et al. [11]. An increasing or unchanged relative abundance of Pseudomonas over three time points was associated with subsequent treatment samples (χ2 test, χ2 = 24.2, p value < 0.004).
Finally, we asked more generally whether there existed an association between community stability and immune response. In a similar setup of three consecutive samples, we assessed if opposing clinical states, i.e., baseline or treatment, diagnosed at the time of the last of the three samples correlated with community stability in the previously collected samples. Three samples with the same pulmotype were considered as stable communities, whereas changing pulmotypes were considered unstable. We found a significant correlation of stability with baseline state and changing pulmotypes with antibiotic treatment (χ2 test, χ2 = 4.9, p value = 0.027, Supplementary Fig. S6).
Consistent with previous results, this suggests that ordered ecological processes resulted in dynamic bacterial community structure that associated with clinical states and therefore may have utility in predicting patient clinical trajectories.
Prediction of disease aggressiveness at young age
CF lung disease aggressiveness is defined by a patient’s overall pattern of lung function decline over time, stratifying patients by ppFEV1 relative to their age group. It is a permanent disease characteristic that is currently based on retrospective observation. The ability to predict disease aggressiveness phenotype at early age would presumably have bearing on clinical management. Here we present a proof-of-concept, implementing a random forest model to classify samples for mild, moderate, and severe pathology.
We examined pulmotype communities as potential predictors of disease aggressiveness, as well as patient demographics (Table 1) and individual airway microbiota, only removing disease stage as potential confounder and conducted random forest analysis. We assessed predictors of disease aggressiveness at younger ages using the 0.3 percentile as age cutoff (10–21 years) using balanced models with patient stratification (Fig. 4A–H and Supplementary Fig. S7A–H). Consistently, pulmotypes were top predictors (Fig. 4A and Supplementary Fig. S7A) underlining the importance of the microbiome activity as community. At early age, pulmotypes 4 and 6 were associated with moderate lung disease, whereas pulmotype 2 with severe phenotype (Fig. 4E). At the level of the individual genus, the typical CF pathogen Pseudomonas was most informative (Gini index, accuracy) for classifying moderate from severe disease stages (Fig. 4A, F). Interestingly, the next highest ranks included strict and facultative anaerobic genera like Veillonella, Fusobacterium, and Streptococcus (Fig. 4A, G, H). Fusobacterium has been implicated with mucus degradation [39]; Veillonella and Streptococcus with fermentation. In addition, the F508del homozygous CFTR genotype was associated with typical high risk for severe trajectories (Fig. 4A, D). Furthermore, patients with unknown mutations in our cohort more frequently showed severe disease phenotype.

A Random forest variable importance plot showing predictors ranked by the mean decrease in accuracy and Gini index. The average of 1000 patient-stratified models is shown. B Cross-validation plot depicting average cross-validation error (black) as function of variable numbers. A total of 100 patient-stratified models using 10-fold cross-validation (gray), as well as maximum and minimum errors (red, dashed line) are depicted. C Receiver operator curve of 100 patient-stratified full models, mean AUC = 0.79. D, E Proportion of CFTR genotypes and pulmotypes in mild and moderate disease, corresponding categories color-coded (F508–/– = F508del homozygous; F508+/– = F508del heterozygous; others = no F508del allele; u.k = unknown CFTR genotype). F–H Box plots showing the distribution of Pseudomonas, Streptococcus, and Veillonella relative abundances in moderate and severe disease phenotypes at young age.
While the presented classifiers are limited as diagnostic tool due to the number of early age samples (n = 221) and patients (n = 49), our results suggest that pulmotypes and other medicable features have potential for both early markers of disease aggressiveness and targets of intervention strategies.
Discussion
Steady progress has been made toward understanding the airway microbiome in persons with CF. Studies have shown that airway bacterial communities are much more complex than previously appreciated, variable between CF patients, relatively resistant to perturbation, and dependent on the individual’s clinical state and lung disease stage, e.g., Stressmann et al. [13]. The dynamics of these communities, particularly with respect to their assembly and functional attributes, are less well described. This study characterized the bacterial communities in 818 sputum samples collected from 109 CF patients and partitioned these communities into eight pulmotypes based on community structure (relative abundance of community members). Analysis of predicted metabolic activity of these pulmotypes and recurrent transitions among them suggested associations between community activity and lung disease progression.
Our analyses show that communities dominated by Pseudomonas, the genus most often associated with CF lung disease, could be divided into at least four distinct pulmotypes based on the composition of the remaining species (Fig. 1 and Supplementary Fig. S2). These pulmotypes, each with distinct inferred metabolic attributes, were associated with different clinical features of CF lung disease. Although pulmotype 5 is characterized by the greatest relative abundance of Pseudomonas, it is strongly associated with stable (baseline) clinical state and intermediate lung disease stage. This is an interesting finding, as communities dominated by canonical pathogens are commonly thought of as dysbiotic, disease-driving microbiota. Pulmotype 5 displays a tight association among Veillonella, Prevotella, Fusobacterium, and Pseudomonas (Supplementary Fig. S8) and at a metabolic level by shuttling of the SCFAs propionate, acetate, butyrate, as well as lactate and amino acids (Fig. 3 and Supplementary Fig. S9). Interestingly, pulmotype 5 also exhibited the highest cluster homogeneity among pulmotypes suggesting robustness of these communal patterns.
Moreover, anaerobes like Fusobacterium, Prevotella, Veillonella, and Streptococcus effectively utilize mucin as a carbon source in the lung by foraging mucin glycans [39, 40]. Mucins exhibit a diverse O-glycan architecture that can be utilized as a carbon source once hydrolyzed from the mucin backbone [41, 42]. While Pseudomonas lacks this capability, it benefits significantly from the assimilation of available mucin building blocks like N-acetylglucosamine, fucose, and galactose, as well as secondary anaerobic fermentation products from the microbial community. Interestingly, the presence of mucin glycans has also been reported to attenuate Pseudomonas virulence in the lung [43]. In summary, these properties of pulmotype 5 suggest a link between the ecological niche of available mucin carbon, a microbial community facilitating Pseudomonas and clinically stable periods.
In contrast, pulmotype 8, was more likely to be observed in samples collected during antibiotic treatment for exacerbation and at advanced disease stage despite a lower relative abundance of Pseudomonas compared to pulmotype 5 (Supplementary Fig. S2). Of note, pulmotype 8 also displayed the lowest cluster homogeneity θ, indicating a relative dissimilarity of underlying samples as well as noisy cluster association and noisy transition dynamics (Fig. 3A). Yet, the observed levels of Pseudomonas are consistent with previous observations of a decline in both the relative and absolute abundance of Pseudomonas around the time of exacerbation of respiratory symptoms in CF [11]. Together, these data suggest that community dominance by Pseudomonas, typically believed to be the major driver of lung disease, is in and of itself not the major determinant of clinical status. The independence of Pseudomonas relative abundance on the clinical state in the pulmotypes rather indicates that temporal ecological dynamics and metabolites derived from the entire microbial community impact the virulence of Pseudomonas [40, 44]. The prevalent shifts among pulmotypes 4, 5, and 6 further suggested an ordered ecological process. Besides Pseudomonas, preponderant taxa in these communities included the non-classical CF pathogens Streptococcus, Prevotella, and Veillonella that have been associated with decreased airway inflammation in CF [45]. Similarly, our results likely imply that a diverse, less immunogenic community (early stage, high diversity, pulmotype 4) adapted through community rearrangement to the complex environment of the CF airway (pulmotypes 5, 6). Moreover, robust co-occurrence patterns among Prevotella, Veillonella, and Streptococcus (Supplementary Fig. S8) and an overlapping functional context (Supplementary Fig. S9) led us to speculate that some metabolic cross-talk could exist. Targeted experimental work will be required to decipher the functional role of non-classical organisms in CF.
To characterize differences in metabolic cross-feeding between pulmotypes, we predicted export and assimilation pathways from 16S rRNA amplicon data combining database and literature query [25]. This reference-based strategy could not account for short-term or intermediate-term metabolic variability and organism biases, a limitation that we share with other amplicon studies. It provides, however, an intuition about the prevalence of possible ecological interactions.
As sputum passes through the oropharynx during expectoration, there is potential for bacterial species residing in this space to “contaminate” the sample. We recently have shown, however, that species present in saliva have a minimal impact on measures of bacterial community structures in expectorated CF sputum [46]. As such, we do not believe that saliva contamination of sputum accounts for the pulmotypes described in our analyses.
Analysis of the functional potential of pulmotypes demonstrated a limited suite of metabolites that distinguish pulmotypes and that likely derive from microbial metabolisms or the immune environment. For example, products of microbial nitrogen metabolism, such as ammonia or nitrite, and assimilation of nitrate, likely derived from immune cell activity (e.g., [47]), were found to be distinguishing features of pulmotypes. Nitrogen metabolism is a critical feature of CF airway community activity with denitrification and nitrogen cycling playing critical roles in sustaining mixed bacterial populations in vivo [48]. Also, SCFAs including formate, acetate, butyrate, and propionate were significant features. Theses derive from microbial degradation of complex polysaccharides, such as respiratory mucins, into simple sugars followed by fermentation. SCFAs contribute to an acidic pH in airways, are reported to trigger IL-8-dependent recruitment of neutrophils, mediate host–microbe interactions, and likely play crucial roles in microbial community function [49, 50]. Carbon dioxide and its soluble form bicarbonate are centrally engaged in the pathogenesis of CF lung disease and are found to contribute to pulmotype classification. Interestingly, bicarbonate has also been linked to increased growth of Pseudomonas [51]. Finally, hydrogen sulfide, which is a strong readout for a highly reduced environment without available oxygen, is also relevant for pulmotypes. The presence of hydrogen sulfide in CF airways strongly suggests the existence of variable redox states that enable the growth and survival of a spectrum of aerobic and anaerobic bacteria in the lower respiratory tract [52,53,54].
The lung microbiomes in persons with CF display individual, as well as regional biases [55]. The models used in this work accurately reflect the characteristics of the studied patient cohort and are regionally predictive. This also includes the correct identification of intervals with acute microbial treatment (e.g., pulmotype 8). The presented concepts, however, are globally transferable and we propose that classifiers built on regional data collections should be developed to support clinical decisions. In the case of DMM classifiers to be designed for the stratification of clinical samples, we recommend examining the vector of sample allocation probabilities [27] as a confidence measure and/or predictor variable. Due to its continuous and multidimensional nature, this may be superior to factorial cluster labels for machine learning and can support distinguishing between minor and major microbiome changes in patient trajectories.
While the absolute number of Dirichlet community clusters may differ with quality, scale, and region of input data, they provide the rational basis for comparing different patient trajectories in the face of strong patient-derived microbiome signature.
In summary, we show that recurrent community types with distinctive community metabolism and pathogenicity characterize the disease trajectory of our CF cohort. These pulmotypes are promising models to unravel mechanistic patterns at the microbiome–host interface in CF.
Data availability
The sequencing data are available in the NCBI Bioproject database (accession numbers: PRJNA423040, PRJNA756039). Detailed SRA accession numbers are additionally listed as Supplementary information.
References
Filkins LM, Hampton TH, Gifford AH, Gross MJ, Hogan DA, Sogin ML, et al. Prevalence of Streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol. 2012;194:4709–17.
Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, et al. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One. 2012;7:e45001.
Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML, et al. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci USA. 2012;109:13769–74.
Guss AM, Roeselers G, Newton IL, Young CR, Klepac-Ceraj V, Lory S, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5:20–9.
Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, et al. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci USA. 2007;104:20529–33.
Brown PS, Pope CE, Marsh RL, Qin X, McNamara S, Gibson R, et al. Directly sampling the lung of a young child with cystic fibrosis reveals diverse microbiota. Ann Am Thorac Soc. 2014;11:1049–55.
Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe. 2015;18:307–19.
Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci USA. 2008;105:15070–5.
van der Gast CJ, Walker AW, Stressmann FA, Rogers GB, Scott P, Daniels TW, et al. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–91.
Zhao J, Carmody LA, Kalikin LM, Li J, Petrosino JF, Schloss PD, et al. Impact of enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS One. 2012;7:e33127.
Carmody LA, Zhao J, Schloss PD, Petrosino JF, Murray S, Young VB, et al. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann Am Thorac Soc. 2013;10:179–87.
Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One. 2010;5:e11044.
Stressmann FA, Rogers GB, van der Gast CJ, Marsh P, Vermeer LS, Carroll MP, et al. Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax. 2012;67:867–73.
Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–14.
Rogers GB, Bruce KD, Hoffman LR. How can the cystic fibrosis respiratory microbiome influence our clinical decision-making? Curr Opin Pulm Med. 2017;23:536–43.
Widder S, Knapp S. Microbial metabolites in cystic fibrosis: a target for future therapy? Am J Respir Cell Mol Biol. 2019;61:132–3.
Mahboubi MA, Carmody LA, Foster BK, Kalikin LM, VanDevanter DR, LiPuma JJ. Culture-based and culture-independent bacteriologic analysis of cystic fibrosis respiratory specimens. J Clin Microbiol. 2016;54:613–9.
Carmody LA, Caverly LJ, Foster BK, Rogers MAM, Kalikin LM, Simon RH, et al. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS One. 2018;13:e0194060.
Zhao J, Li J, Schloss PD, Kalikin LM, Raymond TA, Petrosino JF, et al. Effect of sample storage conditions on cultureindependent bacterial community measures in cystic fibrosis sputum specimens. J Clin Microbiol 2011;49:3717–8.
Hnizdo E, Yu L, Freyder L, Attfield M, Lefante J & Glindmeyer HW. The precision of longitudinal lung function measurements: Monitoring and interpretation. Occup Environ Med 2005;62:695–701.
Konstan MW, Wagener JS, VanDevanter DR. Characterizing aggressiveness and predicting future progression of CF lung disease. J Cyst Fibros. 2009;8:S15–19.
Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16s rRNA-based studies. PLoS One. 2011;6:e27310.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145.
Sung J, Kim S, Cabatbat JJT, Jang S, Jin YS, Jung GY, et al. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat Commun 2017;8:15393.
Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687.
Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7:e30126.
Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509:357–60.
Price KE, Hampton TH, Gifford AH, Dolben EL, Hogan DA, Morrison HG, et al. Unique microbial communities persist in individual cystic fibrosis patients throughout a clinical exacerbation. Microbiome 2013;1:27.
Carmody LA, Zhao J, Kalikin LM, LeBar W, Simon RH, Venkataraman A, et al. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 2015;3:12.
de Dios Caballero J, Vida R, Cobo M, Maiz L, Suarez L, Galeano J, et al. Individual patterns of complexity in cystic fibrosis lung microbiota, including predator bacteria, over a 1-year period. mBio 2017;8::e00959–17.
Whelan, FJ, Heirali AA, Rossi L, Rabin HR, Parkins MD, & Surette MG. Longitudinal sampling of the lung microbiota in individuals with cystic fibrosis. PLoS One 2017:12:e0172811.
Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, et al. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems 2016;1.
Douglas, GM, Maffei, VJ, Zaneveld, JR, Yurgel, SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 2020;38:685–8.
Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res 2018;46:D633–D639.
Quinn RA, Comstock W, Zhang T, Morton JT, da Silva R, Tran A, et al. Niche partitioning of a pathogenic microbiome driven by chemical gradients. Sci Adv 2018;4:eaau1908.
Quinn RA, Whiteson K, Lim YW, Zhao J, Conrad D, LiPuma JJ, et al. Ecological networking of cystic fibrosis lung infections. NPJ Biofilms Microbiomes. 2016;2:4.
Pradeu T, Vivier E. The discontinuity theory of immunity. Sci Immunol. 2016;1:AAG0479.
Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathog. 2016;12:e1005846.
Adamowicz EM, Flynn J, Hunter RC, Harcombe WR. Cross-feeding modulates antibiotic tolerance in bacterial communities. ISME J. 2018;12:2723–35.
Rose MC & Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev 2006;86:245–78.
Tailford LE, Crost EH, Kavanaugh D. & Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet 2015;6:81.
Wheeler KM, Carcamo-Oyarce G, Turner BS, Dellos-Nolan S, Co JY, Lehoux S, et al. Mucin glycans attenuate the virulence of Pseudomonas aeruginosa in infection. Nat Microbiol 2019;4:2146–54.
Twomey KB, O'Connell OJ, McCarthy Y, Dow JM, O'Toole GA, Plant BJ, et al. Bacterial cis-2-unsaturated fatty acids found in the cystic fibrosis airway modulate virulence and persistence of Pseudomonas aeruginosa. ISME J 2012;6:939–50.
Zemanick ET, Wagner BD, Robertson CE, Ahrens RC, Chmiel JF, Clancy JP, et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J 2017;50:1700832.
Lu J, Carmody LA, Opron K, Simon RH, Kalikin LM, Caverly LJ, et al. Parallel analysis of cystic fibrosis sputum and saliva'reveals overlapping communities and an opportunity for sample decontamination. mSystems 2020;5.
Jones KL, Hegab AH, Hillman BC, Simpson KL, Jinkins PA, Grisham MB, et al. Elevation of nitrotyrosine and nitrate concentrations in cystic fibrosis sputum. Pediatr Pulmonol 2000;30:79–85.
Quinn RA, Lim YW, Maughan H, Conrad D, Rohwer F, Whiteson KL. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. mBio. 2014;5:e00956–00913.
Mirkovic B, Murray MA, Lavelle GM, Molloy K, Azim AA, Gunaratnam C, et al. The role of short-chain fatty acids, produced by anaerobic bacteria, in the cystic fibrosis airway. Am J Respir Crit Care Med. 2015;192:1314–24.
Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against flu by shaping Ly6c(-) patrolling monocyte hematopoiesis and CD8(+) t cell metabolism. Immunity. 2018;48:992–1005.e1008.
Flynn JM, Phan C, Hunter RC. Genome-wide survey of Pseudomonas aeruginosa PA14 reveals a role for the glyoxylate pathway and extracellular proteases in the utilization of mucin. Infect Immun. 2017;85:e00182–17.
Jorth P, Ehsan Z, Rezayat A, Caldwell E, Pope C, Brewington JJ, et al. Direct lung sampling indicates that established pathogens dominate early infections in children with cystic fibrosis. Cell Rep. 2019;27:1190–204.e1193.
Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37:783–92.
Cowley ES, Kopf SH, LaRiviere A, Ziebis W, Newman DK. Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic, and extremely reduced due to hydrogen sulfide formation. mBio. 2015;6:e00767.
Cuthbertson L, Walker AW, Oliver AE, Rogers GB, Rivett DW, Hampton TH, et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020;8:45.
Acknowledgements
We gratefully acknowledge the many individuals who provided the sputum samples used in this study. We thank the Michigan Medicine clinical microbiology laboratory for the generous assistance in obtaining sputum samples for this study. We thank Prof. Barbara Bailey for fruitful discussions about random forests. This study was funded by NHLBI grants 1RC1HL100809 and 1R01HL136647 to JJL. Additional support was provided by NIH CTSA grant UL1RR024986 and the Charles Woodson Pediatric Research Fund. SW was supported by the Austrian Science Fund (FWF) Elise Richter V585-B31.
Author information
Authors and Affiliations
Contributions
SW provided conceptualization, investigation, methodology, formal analysis, visualization, writing—original draft, and writing—review and editing. JZ conducted methodology, formal analysis, visualization, writing—original draft, and writing—review and editing. LAC contributed investigation, data curation, writing—original draft, and writing—review and editing. QZ and PDS provided software and supported data curation. LMK contributed to data curation, project administration, and writing—review and editing. JJL contributed to conceptualization, funding acquisition, resources, supervision, writing—original draft, and writing—review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Widder, S., Zhao, J., Carmody, L.A. et al. Association of bacterial community types, functional microbial processes and lung disease in cystic fibrosis airways. ISME J 16, 905–914 (2022). https://doi.org/10.1038/s41396-021-01129-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01129-z
This article is cited by
-
Limited effects of azithromycin on the oropharyngeal microbiome in children with CF and early pseudomonas infection
BMC Microbiology (2023)
-
CDSS for Early Recognition of Respiratory Diseases based on AI Techniques: A Systematic Review
Wireless Personal Communications (2023)
-
CF-Seq, an accessible web application for rapid re-analysis of cystic fibrosis pathogen RNA sequencing studies
Scientific Data (2022)
