
Abstract
Several Trichonympha protist species in the termite gut have independently acquired Desulfovibrio ectosymbionts in apparently different stages of symbiosis. Here, we obtained the near-complete genome sequence of Desulfovibrio phylotype ZnDsv-02, which attaches to the surface of Trichonympha collaris cells, and compared it with a previously obtained genome sequence of ‘Candidatus Desulfovibrio trichonymphae’ phylotype Rs-N31, which is almost completely embedded in the cytoplasm of Trichonympha agilis. Single-nucleotide polymorphism analysis indicated that although Rs-N31 is almost clonal, the ZnDsv-02 population on a single host cell is heterogeneous. Despite these differences, the genome of ZnDsv-02 has been reduced to 1.6 Mb, which is comparable to that of Rs-N31 (1.4 Mb), but unlike other known ectosymbionts of protists with a genome similar in size to their free-living relatives. Except for the presence of a lactate utilization pathway, cell-adhesion components and anti-phage defense systems in ZnDsv-02, the overall gene-loss pattern between the two genomes is very similar, including the loss of genes responsive to environmental changes. Our study suggests that genome reduction can occur in ectosymbionts, even when they can be transmitted horizontally and obtain genes via lateral transfer, and that the symbiont genome size depends heavily on their role in the symbiotic system.
Similar content being viewed by others


Introduction
Termites efficiently digest lignocellulose in association with gut microbes [1, 2]. The microbial community comprises protists, bacteria, archaea, and viruses, which are unique to termites [3,4,5,6,7]. A prominent feature of this microbiota is a cellular association between protists and prokaryotes, which plays a significant role in the nutrition of phylogenetically basal (so-called ‘lower’) termites [1, 2, 8, 9].
One example of the above phenomenon is the association of several bacterial lineages with the cellulolytic protist genus Trichonympha (phylum Parabasalia). Most Trichonympha species obligatorily harbor ‘Candidatus Endomicrobium trichonymphae’ (phylum Elusimicrobia) [10,11,12] or ‘Candidatus Ancillula trichonymphae’ (phylum Actinobacteria) [13] in the cytoplasm. They are also facultatively associated with ectosymbiotic bacteria, including members of the order Bacteroidales [14] and genus Treponema [14, 15]. In addition to these obligate and facultative symbionts, several Trichonympha species are also associated obligatorily with endosymbiotic ‘Candidatus Adiutrix intracellularis’ [16] and/or ectosymbiotic ‘Candidatus Desulfovibrio trichonymphae’ [13, 16, 17], both of which belong to the class Deltaproteobacteria.
Although none of these symbiotic bacteria have been cultured, previous genomic analyses suggested functions for several of the species. For example, ‘Ca. Endomicrobium trichonymphae’ and ‘Ca. Ancillula trichonymphae’ play a role in upgrading nitrogenous compounds [18,19,20]. ‘Ca. Adiutrix intracellularis’ is capable of reductive acetogenesis using H2 and CO2 and can fix dinitrogen [16]. ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 can oxidize hydrogen via sulfate and/or fumarate respiration and may contribute to the removal of hydrogen that can inhibit cellulose fermentation in the termite gut [21, 22].
Interestingly, different phylotypes of Desulfovibrio ectosymbionts exhibit different manners of association with their respective Trichonympha host species. Whereas phylotype ZnDsv-02 (or ZnvTc-7) (AB444082 and JQ993542) attaches to a shallow groove on the surface of Trichonympha collaris cells in the gut of the termite Zootermopsis nevadensis [16], phylotype ImrTc-15 (JQ993569) is held in a deep invagination on the surface of Trichonympha globulosa in the gut of Incisitermes marginipennis [13]. The localization of phylotype Rs-N31 provides a more unusual example for an ectosymbiont: its cells are almost completely embedded in the surface of Trichonympha agilis in the gut of Reticulitermes speratus, and some Rs-N31 cells can be observed deep within the host cell cytoplasm. Each Rs-N31 cell is connected to the outside of the host cell only by a pore or tube-like structure of ca. 40-nm diameter. The genome is as small as 1.4 Mb, which is only half or less of the genome size of its free-living relatives [21]. Thus, Rs-N31 appears to be an ‘almost intracellular’ symbiont [17, 21]. Rs-N31 cells are coccoid or oval shaped [17, 21], whereas ZnDsv-02 and ImrTc-15 cells are rod shaped [13, 16], more similar to free-living relatives. Phylogenetic analyses of 16S rRNA genes suggested that these three phylotypes were most likely independently acquired by the respective Trichonympha host lineages [13, 21]. Differences in the manner of association and cell morphology between these three phylotypes might be indicative of the depth of the interrelationship with the Trichonympha hosts and the length of time after acquisition as an ectosymbiont.
In this study, we obtained the near-complete sequence of the Desulfovibrio phylotype ZnDsv-02 genome and compared it with known sequences of the genomes of phylotype Rs-N31 and free-living relatives. Our analyses provide evidence of parallel reductive genome evolution of these ectosymbionts and insights into the evolutionary process from free-living to ectosymbiotic and further to an ‘almost intracellular’ lifestyle.
Materials and methods
Termites and protists
Z. nevadensis (family Archotermopsidae) were collected in Hyogo Prefecture, Japan, and kept in the laboratory by feeding pinewood chips [23] (Supplementary methods). The hindgut contents of a worker termite were suspended in sterile Trager’s solution U [24] containing 0.05% bovine serum albumin, and then cells of four Trichonympha species (T. collaris, T. sphaerica, T. campanula, and T. postcylindrica) were collected using an AM6000 inverted micromanipulation microscope system (Leica Microsystems, Wetzlar, Germany). Trichonympha species were identified based on both morphological characteristics and the 18S rRNA gene sequence [25, 26]. Other termite species used for fluorescence in situ hybridization (FISH) or transmission electron microscopy (TEM) were collected as described in the Supplementary methods.
FISH and TEM
FISH analyses were performed as described previously [27, 28]. Probe DSV698 [29] was used in order to detect a broad range of Desulfovibrio bacteria. Probe ZnDsv-02-471, which is specific to Desulfovibrio phylotype ZnDsv-02 [17], was used in this study with newly designed helper probes (Table S1). Co-existing ‘Ca. Endomicrobium trichonymphae’ and ‘Ca. Adiutrix intracellularis’ were detected using specific probes designed in previous studies [16, 25] (Table S1). Cells were observed under a BX51 epifluorescence microscope or FV1000D-IX81 confocal laser scanning microscope (Olympus, Tokyo, Japan). TEM of T. collaris was performed as described previously [28, 30] by collecting 25 T. collaris cells using the micromanipulation system. TEM of T. agilis from R. speratus was performed as described previously [21].
16S rRNA amplicon sequencing analysis
To analyse the taxonomic composition of bacteria associated with single Trichonympha cells, the V3–V4 region (ca. 400 bp) of the 16S rRNA gene was amplified by PCR using a Bacteria-specific 341F and 785R primer set (Table S2) and DNA samples prepared by whole-genome amplification (WGA) (see below). The PCR products were subjected to sequencing on an Illumina MiSeq platform as described previously [31]. The sequence reads were trimmed, quality filtered and sorted into amplicon sequence variants (ASVs) using DADA2 v1.12.1 [32, 33] and phylogenetically classified using SINA v1.2.11 [34] with database SILVA SSURef NR99 release 132 [35], as described previously [36]. ASVs with <5 reads were discarded from the analysis.
To examine the abundance and phylogenetic diversity of the genus Desulfovibrio in the gut of various termite and cockroach species, the amplicon sequence datasets (BioProject PRJDB8349) of the 16S rRNA V3–V4 region analyzed in our previous study were used [36]. The sequence reads were sorted into ASVs in the same way described above, except that DADA2 v1.4 was used.
Preparation of DNA from single protist cells and sequencing of the 18S rRNA gene
A single T. collaris cell was dissected into the anterior part that contained Desulfovibrio cells and the posterior part that contained the host nucleus, as described previously [21]. Both parts were collected separately and subjected to WGA using the illustra GenomiPhi V2 Kit (GE Healthcare, Chicago, IL, USA) as described previously [37]. The WGA product from the anterior part was subjected to a second WGA using the illustra GenomiPhi HY Kit (GE Healthcare) as described previously [38]. The resulting product was debranched using RepliPHI Phi29 DNA Polymerase (Epicentre, Madison, WI, USA) and then treated with S1 nuclease (Takara Bio, Shiga, Japan) [39]. To phylogenetically identify the host Trichonympha species, 18S rRNA genes amplified by PCR from WGA samples of the posterior part or whole cells were cloned and sequenced according to the Sanger method as described previously [40] (Supplementary methods).
Genome sequencing, assembly, and binning
Sequencing libraries for the ZnDsv-02 genome were prepared using the TruSeq DNA PCR-Free Library Prep Kit and the Nextera Mate Pair Library Prep Kit (Illumina, Madison, WI, USA). Sequencing was performed using the MiSeq Reagent Kit v3 (600 cycles) on the Illumina MiSeq platform. After adapter removal and quality trimming using cutadapt [41] and prinseq [42], respectively, the resulting reads were assembled into contigs using SPAdes 3.10.1 [43]. The contigs and mate-pair reads were used to generate scaffolds with SCARPA 0.241 [44]. Contigs and scaffolds exhibiting the highest similarity to genome sequences of the genus Desulfovibrio or ‘Ca. Adiutrix intracellularis’ Adiu1 (LQAA00000000) by BLASTn searching against the NCBI non-redundant (nr) nucleotide database were extracted and treated as ‘trusted contigs.’ Sequence reads of 2–5 kb in length, which were obtained on a PacBio RSII platform as described previously [4], were added and re-assembled with the ‘trusted contigs’ using SPAdes 3.10.1. The resulting assemblies were manually inspected and curated as needed. After these processes, the Desulfovibrio genome was still fragmented to 55 contigs. Thus, we incorporated an assembling process as described in the Supplementary methods, and the final contig set was then obtained.
Gene finding, annotation, and comparative genomics
Automatic gene finding and annotation were performed using MiGAP (http://www.migap.org/), RNAmmer 1.2 [45] and tRNAscan-SE 2.0 [46]. By conducting BLASTp searches against the NCBI nr protein database, the gene loci and annotations were manually curated. Pseudogenes were manually identified as described previously [18]. Among 139 single-copy marker genes proposed by Rinke et al. [47], 132 were conserved in Desulfovibrio and used to estimate genome completeness (Dataset S1). Metabolic pathways were predicted using the KEGG automatic annotation server [48]. Clustered regularly interspaced short palindromic repeat (CRISPR) loci were identified using CRISPRCasFinder [49]. Genes associated with secretion systems were detected using TXSScan [50]. Orthologous genes were detected using DomClust [51] with manual corrections. Genes and pseudogenes were classified into non-supervised orthologous groups (NOGs) using EggNOG 4.5.1 [52]. The absence of genes that are functionally important but missing in the ZnDsv-02 genome were re-confirmed by conducting tBLASTn searches against the contigs not included in the ZnDsv-02 assembly, using the amino acid sequences of relatives as queries.
Detection and comparison of single-nucleotide polymorphisms (SNPs) and indels
Quality-filtered MiSeq paired-end reads were mapped to genome sequences using Burrows-Wheeler Aligner 0.7.17 [53]. After removing duplicated reads using SAMtools [54], the average read depth was calculated using DepthOfCoverage implemented in the Genome Analysis Tool Kit (GATK) 3.8.1.0 [55]. The outputs were downsized using DownsampleSam in GATK 4.1.2.0 to adjust the average read depth among samples. Sequence variants were identified using HaplotypeCaller implemented in GATK 4.1.2.0. Reads with mapping quality <30 were eliminated from the analysis. The resulting variants with a read depth of <10 were discarded and then annotated using snpEff 4.3 [56].
Phylogenetic analysis, average nucleotide identity (ANI), and average amino acid identity (AAI)
Nucleotide or amino acid sequences were aligned using MUSCLE [57] with manual corrections, and ambiguously aligned sites were removed using Gblocks [58]. Maximum-likelihood (ML) trees were inferred using MEGA6 [59] or MEGA7 [60]. Amino acid or nucleotide substitution models were selected using model test implemented in MEGA. The robustness of tree topologies was evaluated by 500 bootstrap resamplings. Bayesian trees were also constructed using MrBayes 3.2.1 [61] with four Markov chains running simultaneously for 1,000,000 generations. An ML tree of the 16S rRNA V3–V4 region was inferred, using IQ-TREE 1.6.11 [62] with the ModelFinder option. In this analysis, an ML tree based on near full-length 16S rRNA gene sequences was used as the constraint tree, and the confidence values were calculated using the ultrafast bootstrap approximation and SH-like approximate likelihood ratio test (1000 replicates). ANI and AAI were calculated using the ANI/AAI calculator [63, 64].
Results
Distribution of Desulfovibrio phylotype ZnDsv-02 in the gut of Z. nevadensis
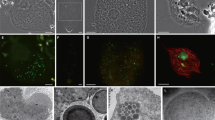
Among the four Trichonympha species present in the gut of Z. nevadensis, Desulfovibrio were detected almost only in T. collaris cell samples by 16S rRNA amplicon sequencing analysis (Fig. S1a) and only in T. collaris cells by FISH analysis using probe DSV698 (data not shown). Almost all of the Desulfovibrio ASVs obtained from the T. collaris cells were identical to (ASV_T4) or differing by one base (ASV_T8) from the ZnDsv-02 sequence (AB444082) (Fig. S1b). ZnDsv-02 cells were specifically detected by FISH using probe ZnDsv-02-471 in the anterior part of T. collaris cells with co-existing ‘Ca. Endomicrobium trichonymphae’ in the posterior part (Fig. 1a, b). ‘Ca. Adiutrix intracellularis’ cells were also detected by FISH in both the anterior and posterior parts of the host cells (Fig. S2). TEM demonstrated that ZnDsv-02 cells were held in hollow-like structures on the host cell surface (Fig. 1c, d). Almost half of the ZnDsv-02 cell surface was exposed to the exterior, unlike the cells of Rs-N31 (Fig. 1e, f) [21]. These results were consistent with the previous study using Z. nevadensis collected in North America [16].

a Phase-contrast image of Trichonympha collaris from the gut of Zootermopsis nevadensis. b Fluorescence in situ hybridization using oligonucleotide probes specific to ZnDsv-02 (6FAM-labeled, green) and ‘Ca. Endomicrobium trichonymphae’ (Texas red-labeled, red) (Table S1). The epifluorescent images were merged. c, d Transmission electron microscopy (TEM) of ZnDsv-02 cells. e, f TEM of Rs-N31 cells. F Host’s flagella. Bars: (a, b), 50 μm; (c–f), 200 nm.
FISH analyses using probe DSV698 also indicated the association of Desulfovibrio bacteria with Trichonympha species in the gut of Reticulitermes yaeyamanus, but not with T. agilis in the gut of Reticulitermes flavipes. No cellular association of Desulfovibrio was observed in the gut protistan community of Coptotermes formosanus and Neotermes koshunensis (data not shown).
Reconstruction and general features of the genome of Desulfovibrio phylotype ZnDsv-02
We reconstructed a draft genome sequence of Desulfovibrio phylotype ZnDsv-02 from a single T. collaris cell in a Z. nevadensis gut (Table 1) and simultaneously determined the complete genome sequence of ‘Ca. Adiutrix intracellularis’ (data not shown). We designated this ‘Ca. Adiutrix intracellularis’ genome sequence as genomovar Adiu2019 to discriminate it from the draft genome obtained in the previous study [16]. In addition, the complete genome sequence of a bacterium belonging to a clade of the order Mycoplasmatales, specific to the termite gut microbiota [65], was reconstructed. Details of these two genomes will be published elsewhere. The ratio of sequence reads mapped to each of the three genome sequences was described in the Supplementary results.
The ZnDsv-02 draft genome consisted of 43 contigs >300 bp (1 611 038 bp in total), and the estimated genome completeness was 99.2%. No contaminating contigs were identified based on CheckM [66] and manual inspection. The estimated genome size (1.6 Mbp) was much smaller than that of free-living relatives, such as Desulfovibrio fairfieldensis (3.7 Mbp) and Desulfovibrio desulfuricans (2.9 Mbp), but larger than that of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 (1.4 Mbp). The largest contig was 232 244 bp, and the N50 value was 90 504 bp. The G + C content (53.6%) was similar to that of Rs-N31 (54.8%), and these were among the lowest G + C contents of known Desulfovibrio species, apart from Desulfovibrio hydrothermalis AM13 (45.1%) and Desulfovibrio salexigens DSM 2638 (47.1%) (Fig. S3). Of 1257 putative protein-coding genes predicted (Dataset S2), 176 returned the highest BLASTp scores to genes of non-Desulfovibrio/Lawsonia/Bilophila bacteria (Table S3). The draft genome contained a single set of the rRNA gene operon, 49 tRNA genes for all amino acids, and 86 pseudogenes. One CRISPR-Cas system was found, with 309 spacer sequences (Dataset S3).
Identification of SNPs and indels
As we were not able to reconstruct the complete genome sequence of ZnDsv-02, we hypothesized that the ZnDsv-02 population on a single host cell was heterogeneous. We therefore estimated the number of SNPs and indels in the ZnDsv-02 and Adiu2019 population genomes as well as those of Rs-N31 [21] and ‘Ca. Endomicrobium trichonymphae’ phylotype Rs-D17 genomovar Ti2015 [19]. When the average read depth was adjusted to the lowest value, 204, the frequencies of the sum of SNPs and indels per kilobase were as follows: 16 (ZnDsv-02), 0.027 (Rs-N31), 0.80 (Adiu2019), and 0.77 (Rs-D17) (Table S4). To evaluate the possibility that only a few distinct genomovars in small amounts contributed to the SNPs in the ZnDsv-02 genome, we inspected each sequence read in two genome regions (length: 100 bp each, read depth: 50 and 41) (Table S5). In both regions, the mapped reads were sorted to eight and six sequence types with different SNP patterns, respectively, and several sequence types were found in similar frequencies (Table S5). Thus, it is likely that the ZnDsv-02 population on a single host cell consisted of several genomovars. The detection of two 16S rRNA ASVs from single T. collaris cells, differing by one base (Fig. S1b), may also be indicative of heterogeneity. Details of the SNP and indel analysis were described in the Supplementary results, Dataset S4 and Fig. S4.
Phylogenomics, ANI, and AAI
A phylogenomic tree was constructed; phylotype ZnDsv-02 clustered with Rs-N31, D. desulfuricans, Desulfovibrio sp. G11, D. fairfieldensis and D. piger (Fig. S5). ZnDsv-02 shared <96% 16S rRNA sequence identity, <79% ANI and <71% AAI with each of these bacteria, including Rs-N31 (Table S6). Among them, D. fairfieldensis CCUG45958 and D. desulfuricans ATCC27774 were selected as representative free-living relatives for further comparisons.
Comparative analysis of the abundance of genes classified by functional category
The genomes of ZnDsv-02, Rs-N31, D. fairfieldensis and D. desulfuricans shared 901 orthologous genes, and 158, 21, 1137, and 475 genes were unique, respectively (Fig. 2, Dataset S5). The number of ZnDsv-02 genes in most NOGs was much fewer than in the free-living relatives and very similar to that of Rs-N31 (Fig. 3). This reduction in the number of genes was conspicuous, particularly in two categories: signal transduction mechanisms [T] and cell motility [N] (Figs. 3 and S6). In both the ZnDsv-02 and Rs-N31 genomes, many genes for response regulator proteins, chemotaxis and flagellar components are missing or pseudogenized; both ectosymbionts are likely nonmotile and have almost lost the ability to respond to environmental changes. Considerable differences were found between the ZnDsv-02 and Rs-N31 genomes in only a few categories: category [L], including DNA methyltransferase and CRISPR-associated proteins; [V], including DNA restriction enzymes; and [U], including the type IV secretion system (Figs. 3 and S6). The ZnDsv-02 genome contains 11 intact restriction-modification (RM) systems, in addition to 1 CRISPR-Cas system, whereas the Rs-N31 genome contains only pseudogenized RM and CRISPR-Cas systems. Among genes involved in DNA repair and recombination, both phylotypes have lost the genes encoding DnaQ, the proofreading 3′–5′ exonuclease subunit of the DNA polymerase III core, and formamidopyrimidine-DNA glycosylase (Fpg), which removes oxidized guanines and prevents the G-C to T-A transversion mutation (Table S7).

The genomes of ZnDsv-02, Rs-N31 and their free-living relatives Desulfovibrio fairfieldensis and Desulfovibrio desulfuricans were compared.

The genomes of Desulfovibrio phylotypes ZnDsv-02 and Rs-N31, and their free-living relatives Desulfovibrio fairfieldensis and Desulfovibrio desulfuricans were compared.
Pseudogenes found in the ZnDsv-02 genome were also classified into NOGs (Fig. S4 and Table S8). In most categories, the pseudogenes were more abundant in Rs-N31 than in ZnDsv-02. The number of pseudogenes in ZnDsv-02 exceeded that in Rs-N31 only in categories [J] and [H] (Fig. S4), including cobalamin biosynthesis proteins. The overall distribution pattern of pseudogenes among NOGs was significantly correlated between ZnDsv-02 and Rs-N31 (two-sided Spearman’s rank correlation test adjusted with Shaffer’s modified Bonferroni procedure, rs = 0.55; p = 0.01).
To identify NOGs contributing to the difference in genome size between the Desulfovibrio ectosymbionts and ectosymbionts belonging to Bacteroidales, the total length of genes assigned to each NOG was compared (Fig. S7). Details were described in the Supplementary Results.
Comparison of predicted metabolic pathways
The metabolic pathways of ZnDsv-02 were predicted and compared with those of Rs-N31 (Fig. 4). Both ZnDsv-02 and Rs-N31 retain pathways to synthesize 20 amino acids and various cofactors (Tables S9 and S10). Although we previously predicted that Rs-N31 cannot synthesize threonine and methionine [21], the gene RSDT_0983, annotated as one of two shikimate kinase genes, likely encodes a ‘missing’ homoserine kinase [67], and we later noted that the biosynthetic pathway from aspartate to methionine is complete in the Rs-N31 genome. Thus, the biosynthetic pathways for methionine and threonine are likely present in Rs-N31 as well as ZnDsv-02. Several genes in the ZnDsv-02 genome involved in cobalamin biosynthesis are either missing or pseudogenized. Although Rs-N31 possesses only a cobalamin-dependent methionine synthase gene (metH), ZnDsv-02 harbors genes for both MetH and cobalamin-independent methionine synthase MetE (Dataset S2).

Pathways found in both phylotypes are shown in black; those found only in Rs-N31 are shown in black with a red ‘X’ mark; those found in Rs-N31, but pseudogenized in ZnDsv-02 are shown in black with a blue cross; those found only in ZnDsv-02 are shown in red; those found in ZnDsv-02, but pseudogenized in Rs-N31 are shown in orange. Flagellar components are absent in both phylotypes, except for fliM in ZnDsv-02.
The genomes of both ZnDsv-02 and Rs-N31 possess pathways for gluconeogenesis and non-oxidative pentose phosphate biosynthesis. The ZnDsv-02 genome retains the glycolytic pathway with the exception of lacking a glucokinase; thus, ZnDsv-02 likely does not utilize sugars, similar to Rs-N31, which has also lost 6-phosphofructokinase and pyruvate kinase [21]. The presence in the ZnDsv-02 genome of a lactate utilization pathway, which is pseudogenized in Rs-N31, suggests that lactate is used as a carbon and energy source. The tricarboxylic acid cycle is incomplete in both phylotypes; each lacks genes for malate dehydrogenase, succinate dehydrogenase and succinyl-CoA synthetase. These ectosymbionts probably cannot use succinate as a carbon source but may use malate and fumarate to produce L-aspartate via an aspartate-ammonia lyase. ZnDsv-02 retains the tripartite ATP-independent periplasmic high affinity transporter DctPQM, which probably imports malate and/or fumarate [68]. DctPQM is missing in Rs-N31, but a putative malate/succinate antiporter, DcuA, is encoded by both genomes. Both ectosymbionts retain the ability to use acetate and CO2 as carbon sources. Genes for the tricarboxylate transporter TctABC found in Rs-N31 are not present in the ZnDsv-02 genome, although the latter instead possesses a Na+-dependent di- or tricarboxylate symporter. Thus, both may use citrate or other carboxylates as carbon sources.
Genes involved in H2 oxidation in Rs-N31, including those encoding dissimilatory sulfite reductase, fumarate reductase (FrdABCD), FoF1-ATPase and several hydrogenases, were also found in the ZnDsv-02 genome, with the exception of the membrane-bound H+-translocating [NiFe] hydrogenase complex EchABCDEF (Table S11). For both ectosymbionts, DcuA and FrdABCD, required for fumarate respiration, exhibited the highest BLASTp scores with those of gammaproteobacteria in the NCBI nr protein database (Fig. S8). Neither phylotype retains the gene for the redox-sensing transcription factor Rex, which functions as a repressor of genes involved in sulfate reduction [69, 70]. Genes encoding catalase, superoxide dismutase and cytochrome bd are absent in the ZnDsv-02 genome, as in the Rs-N31 genome (Table S12). Overall, it is likely that ZnDsv-02 utilizes H2 and lactate as electron donors and sulfate and fumarate as electron acceptors to generate energy via anaerobic respiration.
Genes for nitrogen fixation and transporters for ammonium or urea are absent, suggesting that both ectosymbionts utilize amino acids and oligopeptides as primary nitrogen sources. The gene encoding the aromatic amino acid transporter AroP is absent in the ZnDsv-02 genome, which was found in the genomes of both Rs-N31 and ‘Ca. Endomicrobium trichonymphae’ Rs-D17 [21]. Phylotypes ZnDsv-02 and Rs-N31 possess the biosynthetic pathway for peptidoglycan, but they have lost several genes involved in lipopolysaccharide biosynthesis. Among phylotypes ZnDsv-02 and Rs-N31 and their two free-living relatives, only ZnDsv-02 possesses several components for a type IV secretion system, including VirB5, which may function as an adhesin, although the gene encoding VirB4, which functions as an ATPase and is likely essential for secretion and pilus assembly, is pseudogenized [71] (Table S13, Fig. S9).
Phylogenetic diversity and abundance of bacteria belonging to Desulfovibrio ‘termite cluster I’
As indicated in previous studies [13, 17], the 16S rRNA genes of ZnDsv-02 and Rs-N31 clustered with those obtained exclusively from the termite gut, and the association with Trichonympha species has evolved independently at least three times (Fig. 5a, Table S14 and Supplementary results). To further explore the phylogenetic diversity of this monophyletic cluster (‘termite cluster I’ [17]), we searched our 16S rRNA amplicon datasets obtained from the gut microbiota of 62 termite and 10 cockroach species [36] for sequences assigned to this clade. ASVs from the gut of 22 ‘lower’ termite (non-Termitidae termites, with symbiotic gut protists) species were affiliated with ‘termite cluster I’ (Fig. 5b). No cockroach or ‘higher’ termite (family Termitidae, with no symbiotic gut protists) sequences were assigned to this clade. The 22 ‘lower’ termite species included termite genera that associate and do not associate with Trichonympha. In Z. nevadensis, Hodotermopsis sjoestedti and Reticulitermes species, in which the cellular association of Desulfovibrio symbionts with Trichonympha has been reported [13, 16, 17], the total frequency of ASVs in ‘termite cluster I’ ranged from 2.4 to 16.9% and constituted most of the Desulfovibrio sequences detected in the gut of the respective termite species (Table S15).

The host termites are given in parentheses. Red: the association of Desulfovibrio and Trichonympha was confirmed by FISH; magenta: host termites harbor Trichonympha but the association was not examined by FISH; black: no association of Desulfovibrio and Trichonympha was observed. a A maximum-likelihood (ML) tree of Desulfovibrio (left) and Trichonympha (right) based on near full-length SSU rRNA genes, constructed using MEGA6 and MEGA7 with the K2 + G + I and GTR + G + I nucleotide substitution models, respectively. A total of 1165 and 1471 aligned nucleotides sites were used. Bootstrap confidence values (left; >70%) and posterior probabilities in Bayesian statistics (right; >0.90) are shown for the internal branches. Sequences used as the outgroup and omitted from the tree are listed in Table S14. Black arrows denote the predicted timing when the association of Desulfovibrio with Trichonympha has evolved. b An ML tree of Desulfovibrio based on 16S rRNA amplicon sequence variants, constructed using the TIM3 + F + I + G4 model in IQ-Tree. A total of 418 aligned nucleotide sites were used. SH-like approximate likelihood ratio test values (left; >70%) and ultrafast bootstrap confidence values (right; >70%) are shown. A: Archotermopsis, C: Coptotermes, G: Glyptotermes, H: Hodotermopsis, I: Incisitermes, N: Neotermes, P: Porotermes, R: Reticulitermes, S: Schedorhinotermes, T: Termitogeton, Z: Zootermopsis. *The cellular association of Desulfovibrio with Trichonympha was tested by FISH analysis: *1 in this study; *2 [15]; and *3 [17].
Discussion
Our comparative genomic analysis of Desulfovibrio phylotype ZnDsv-02, ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 and their free-living relatives sheds light on genomic evolution during the course of cellular symbiosis. Although ZnDsv-02 and Rs-N31 were independently acquired by the host Trichonympha lineages (Fig. 5a and Supplementary results), both genomes exhibited very similar reductive genome evolution, as exemplified by the number of genes in each functional category (Fig. 3). It is of particular interest that the number of genes related to signal transduction in ZnDsv-02 has already been reduced to one-tenth or fewer of the number in the free-living relatives, even though ZnDsv-02 is still widely exposed to the exterior environment. A considerable loss of signal transduction-related genes (~40% or fewer of free-living relatives) has also been observed in two ectosymbionts belonging to Bacteroidales, ‘Candidatus Symbiothrix dinenymphae’ [30] and phylotype Barb6XT [72] (Fig. S7). Thus, the loss of regulatory genes responsive to environmental changes can occur in ectosymbionts, suggesting that the protist hosts have provided the ectosymbionts with a stable environment that enables such reductive evolution.
However, the overall degree of genome reduction differs markedly between Desulfovibrio ectosymbionts and those belonging to Bacteroidales. The genome of the former ranges from 1.4 to 1.6 Mb in size, which is approximately half that of their free-living relatives, whereas the latter ranges from 3.6 to 4.8 Mb, which is comparable to their free-living relatives [9, 30, 72]. This difference may be partly attributable to differences in roles necessitated by the symbiotic system. The primary role of the Desulfovibrio ectosymbionts appears to be H2 oxidation [17, 21], the accumulation of which can suppress fermentation in both protist hosts [73] and co-existing Endomicrobium endosymbionts [18, 74]. In contrast, it has been suggested that the Bacteroidales ectosymbionts participate in lignocellulose digestion [9, 30, 72], which involves a large number of genes (Supplementary results and Fig. S7). The difference in cell wall structure and device for attachment to the host cell may also contribute to the genome size difference, although genes of ‘unknown function’ (category [S]) accounted for the largest part of the difference (Fig. S7).
It is noteworthy that the ZnDsv-02 population on the single host protist cell was heterogeneous. The frequency of SNPs and indels in ZnDsv-02 was ~20 times higher than in the endosymbionts ‘Ca. Adiutrix trichonymphae’ Adiu2019 and ‘Ca. Endomicrobium trichonymphae’ Rs-D17 Ti2015 (Table S4). The SNP rate in ZnDsv-02 was also much higher than that in Barb6XT (Table S4) [72]. It is conceivable that genomic variations, probably including rearrangements, led to the failure in reconstructing the complete genome sequence of ZnDsv-02. As the genomes of both ZnDsv-02 and Adiu2019 were reconstructed from the same DNA sample and MiSeq runs, the difference between them would not have been caused by artefacts. It seems unlikely that the sexual reproduction (syngamy) of Trichonympha observed in a Zootermopsis gut [75] was the cause of ZnDsv-02 genome sequence heterogeneity, as the population of co-existing ‘Ca. Adiutrix trichonymphae’ was almost clonal. Thus, we hypothesize that ZnDsv-02 is transmitted horizontally at low frequency among host T. collaris cells. As ZnDsv-02 appears to have lost motility, such horizontal transmission would occur only by chance. We expect that ZnDsv-02 cells are typically transmitted vertically.
The presence of genome sequence variants in the ZnDsv-02 population provides an opportunity to infer the process of the reductive genome evolution. Among 1191 genes that contained SNPs/indels, at least 67 genes likely had pseudogenized variants (Dataset S4), and the rate of pseudogenization/absence of orthologous genes in Rs-N31 was much higher (59%) than in the other 1124 genes (16%) (Supplementary results). Furthermore, the NOG profile of the 67 genes was similar to those of pseudogenes of ZnDsv-02 and Rs-N31 (Fig. S4). These suggest that pseudogenization or loss of genes not essential in the symbiotic system is ongoing in the ZnDsv-02 population, and we hypothesize that horizontal transmission might delay the fixation of such slightly deleterious mutations.
In contrast to ZnDsv-02, the frequency of SNPs and indels in the Rs-N31 genome was ~30 times lower than in the two endosymbionts (Table S4). This suggests that those endosymbionts can also undergo chance horizontal transmission, even though the rate would be much lower than that expected for ZnDsv-02. Rs-D17 retains a functional CRISPR-Cas system and several intact RM systems [19, 76], but all CRISPR-Cas and RM systems are pseudogenized in Rs-N31 [21]. This suggests that Rs-D17 can still come into contact with phages that may be phagocytosed with wood particles and other luminal contents by host Trichonympha cells, whereas Rs-N31 may not, possibly due to the difference in their specific localization. We therefore hypothesize that cytoplasmic endosymbionts such as Rs-D17 can also be transmitted horizontally among host protist cells via phagocytosis as a very rare occurrence.
An apparently intact CRISPR-Cas system and several RM systems are also present in the ZnDsv-02 genome, suggesting that ZnDsv-02 is exposed to phages and other extracellular DNA. This provides the opportunity for lateral gene transfer (LGT), and indeed, ZnDsv-02 possesses many genes not found in other desulfovibrios (Table S3). Such LGT and phage infection would also contribute to the genomic variations among ZnDsv-02 cells. This situation of ZnDsv-02 with chance horizontal transmission and acquisition of foreign genes is somewhat similar to that of ‘recently host–restricted symbionts’ of insects [77] such as Serratia symbiotica, of which genome size is approximately half that of its free-living relatives [78]. However, in ectosymbiosis with protists in the termite gut, host restriction appears to be not enough for massive genome reduction. We suggest that massive genome reduction occurs when bacteria become restricted to a specific function in addition to host restriction, and that genome reduction is initiated even when bacteria can still be transmitted horizontally by chance and acquire genes via LGT. The direct cause of pseudogenization and relatively low G + C content (Fig. S3) in ZnDsv-02 and Rs-N31 could be loss of the dnaQ and fpg genes, which are required for DNA replication proofreading and repair of oxidized guanine, respectively (Table S7). The loss of dnaQ is occasionally observed in intracellular bacteria such as Mycobacterium leprae [79] and mycoplasmas [80] and suggested to be the cause of their genome reduction [79, 80]. The loss of dnaQ might be advantageous for rapid adaptation to their novel niches.
Interestingly, our 16S rRNA amplicon sequencing analysis detected many ASVs that clustered with the Desulfovibrio ectosymbionts of Trichonympha in ‘termite cluster 1’ from diverse ‘lower’ termite species that do not harbor Trichonympha (Fig. 5b). Our FISH analyses indicated that at least several of those Desulfovibrio phylotypes are not associated with any protist cells; they are probably free-swimming bacteria. In any case, it seems likely that the common ancestor of ‘termite cluster 1’ acquired the potential and preference to become an ectosymbiont of protists. A likely key factor would be acquisition of DcuA, which is essential for fumarate respiration [81], via LGT (Fig. S8). DcuA enables the bacteria to proliferate under the low sulfate concentrations in the termite gut [17], if the bacteria can obtain enough malate from the protist host. In turn, Desulfovibrio ectosymbionts remove H2 in close vicinity to the hydrogenosomes [21].
Although the pattern of gene loss was very similar between ZnDsv-02 and Rs-N31 (Figs. 3 and S6), differences that likely reflect the respective symbiotic relationships were found, in addition to the defense mechanisms. For example, ZnDsv-02 retains most subunits of the type IV secretion system (Fig. S9), which is absent in Rs-N31. As the genome also contains several genes that function in the conjugal transfer system, the type IV secretion system might have played a role in exchanging DNA entities such as plasmids in the ancestor of ZnDsv-02. Because gene components essential for secretion (virB4, B7) and DNA transfer (traI) [71] are psedogenized or absent (Dataset S2 and Table S13), we hypothesize that ZnDsv-02 retains the remaining components only for attachment to the host cell surface, whereas Rs-N31 has completely lost these components. The ability to use lactate is another prominent difference. Utilization of lactate as a carbon and energy source is common in Desulfovibrio [82, 83], but the related genes are pseudogenized in Rs-N31. This may indicate that lactate is difficult to take up in the ‘almost intracellular’ lifestyle, and/or that using H2 as the sole energy source more benefits the Trichonympha host.
The results of our study thus provide new insights into the evolutionary process of cellular symbiosis. Although ZnDsv-02 cells remain widely exposed to the exterior environment as ectosymbionts and can undergo horizontal transmission and LGT events, the genome reduction has already begun. We emphasize the importance of the functional reduction associated with becoming a symbiont, and genome analyses of more diverse mutualistic ectosymbionts may lead to further understanding of mechanisms of genome reduction, which have been investigated mostly in intracellular symbionts and parasitic ectosymbionts [84,85,86].
Based on differences in 16S rRNA sequence, ANI and AAI [63, 64], as well as its independent acquisition by the host protist lineage, we believe that phylotype ZnDsv-02 should be considered distinct from ‘Candidatus Desulfovibrio trichonymphae.’ We therefore propose the novel species name ‘Candidatus Desulfovibrio kirbyi’ for this bacterium.
Description of ‘Candidatus Desulfovibrio kirbyi’ sp. nov
Desulfovibrio kirbyi (kir’byi. N.L. gen. n. Kirbyi, of Kirby; named after Harold Kirby), a microbiologist who reported the presence of this bacterium as ‘peripheral granules’ in 1932 [87]. The bacteria are short, rod-shaped cells with dimensions of 1.0–2.3 μm (mean ± SD, 1.5 ± 0.3; n = 40) by 0.4–0.8 μm (0.6 ± 0.1), based on FISH analysis. The bacteria specifically attach to the cell surface of T. collaris in the gut of Z. nevadensis. The assignment is based on the 16S rRNA gene sequence (AB444082) and specific hybridization with probe ZnDsv-02-471 (Table S1). This species corresponds to phylotype ZnDsv-02 and the draft genome sequence was deposited in the DDBJ under BioSample SAMD00200503.
Data availability
The sequence data obtained in this study were deposited in the DDBJ under BioProject PRJDB9170 for the genome sequences and the 18S rRNA gene sequences of four Trichonympha species. BioProject PRJDB9250 will appear for the 16S rRNA gene sequence datasets of bacteria associated with four Trichonympha species. The sequence data for the 16S rRNA ASVs of Desulfovibrio obtained from various termite and cockroach species will be available at https://doi.org/10.6084/m9.figshare.12137268.
References
Hongoh Y. Toward the functional analysis of uncultivable, symbiotic microorganisms in the termite gut. Cell Mol Life Sci. 2011;68:1311–25.
Brune A. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol. 2014;12:168–80.
Hongoh Y. Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci Biotechnol Biochem. 2010;74:1145–51.
Pramono A, Kuwahara H, Itoh T, Toyoda A, Yamada A, Hongoh Y. Discovery and complete genome sequence of a bacteriophage from an obligate intracellular symbiont of a cellulolytic protist in the termite gut. Microbes Environ. 2017;32:112–7.
Tikhe CV, Husseneder C. Metavirome sequencing of the termite gut reveals the presence of an unexplored bacteriophage community. Front Microbiol. 2018;8:2548.
Brune A, Dietrich C. The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu Rev Microbiol. 2015;69:145–66.
Bourguignon T, Lo N, Dietrich C, Šobotník J, Sidek S, Roisin Y, et al. Rampant host switching shaped the termite gut microbiome. Curr Biol. 2018;28:649–54.
Ohkuma M, Noda S, Hattori S, Iida T, Yuki M, Starns D, et al. Acetogenesis from H2 plus CO2 and nitrogen fixation by an endosymbiotic spirochete of a termite-gut cellulolytic protist. Proc Natl Acad Sci USA. 2015;112:10224–30.
Treitli SC, Kolisko M, Husník F, Keeling PJ, Hampl V. Revealing the metabolic capacity of Streblomastix strix and its bacterial symbionts using single-cell metagenomics. Proc Natl Acad Sci USA. 2019;116:19675–84.
Stingl U, Radek R, Yang H, Brune A. “Endomicrobia”: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl Environ Microbiol. 2005;71:1473–9.
Ikeda-Ohtsubo W, Brune A. Cospeciation of termite gut flagellates and their bacterial endosymbionts: Trichonympha species and ‘Candidatus Endomicrobium trichonymphae’. Mol Ecol. 2009;18:332–42.
Ohkuma M, Sato T, Noda S, Ui S, Kudo T, Hongoh Y. The candidate phylum ‘Termite Group 1’ of bacteria: phylogenetic diversity, distribution, and endosymbiont members of various gut flagellated protists. FEMS Microbiol Ecol. 2007;60:467–76.
Strassert JF, Köhler T, Wienemann TH, Ikeda-Ohtsubo W, Faivre N, Franckenberg S, et al. ‘Candidatus Ancillula trichonymphae’, a novel lineage of endosymbiotic Actinobacteria in termite gut flagellates of the genus Trichonympha. Environ Microbiol. 2012;14:3259–70.
Hongoh Y, Sato T, Noda S, Ui S, Kudo T, Ohkuma M. Candidatus Symbiothrix dinenymphae: bristle-like Bacteroidales ectosymbionts of termite gut protists. Environ Microbiol. 2007;9:2631–5.
Izawa K, Kuwahara H, Sugaya K, Lo N, Ohkuma M, Hongoh Y. Discovery of ectosymbiotic Endomicrobium lineages associated with protists in the gut of stolotermitid termites. Environ Microbiol Rep. 2017;9:411–8.
Ikeda-Ohtsubo W, Strassert JF, Köhler T, Mikaelyan A, Gregor I, McHardy AC, et al. ‘Candidatus Adiutrix intracellularis’, an endosymbiont of termite gut flagellates, is the first representative of a deep-branching clade of Deltaproteobacteria and a putative homoacetogen. Environ Microbiol. 2016;18:2548–64.
Sato T, Hongoh Y, Noda S, Hattori S, Ui S, Ohkuma M. Candidatus Desulfovibrio trichonymphae, a novel intracellular symbiont of the flagellate Trichonympha agilis in termite gut. Environ Microbiol. 2009;11:1007–15.
Hongoh Y, Sharma VK, Prakash T, Noda S, Taylor TD, Kudo T, et al. Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Natl Acad Sci USA. 2008;105:5555–60.
Izawa K, Kuwahara H, Kihara K, Yuki M, Lo N, Itoh T, et al. Comparison of intracellular “Ca. Endomicrobium trichonymphae” genomovars illuminates the requirement and decay of defense systems against foreign DNA. Genome Biol Evol. 2016;8:3099–107.
Strassert JF, Mikaelyan A, Woyke T, Brune A. Genome analysis of ‘Candidatus Ancillula trichonymphae’, first representative of a deep-branching clade of Bifidobacteriales, strengthens evidence for convergent evolution in flagellate endosymbionts. Environ Microbiol Rep. 2016;8:865–73.
Kuwahara H, Yuki M, Izawa K, Ohkuma M, Hongoh Y. Genome of ‘Ca. Desulfovibrio trichonymphae’, an H2-oxidizing bacterium in a tripartite symbiotic system within a protist cell in the termite gut. ISME J. 2017;11:766–76.
Hongoh Y, Ohkuma M. Termite gut flagellates and their methanogenic and eubacterial symbionts. In: Hackstein JHP, editor. Microbiology monographs: (Endo) symbiotic methanogenic archaea. Heidelberg: Springer-Nature; 2018.
Yashiro T, Mitaka Y, Nozaki T, Matsuura K. Chemical and molecular identification of the invasive termite Zootermopsis nevadensis (Isoptera: Archotermopsidae) in Japan. Appl Entomol Zool. 2018;53:215–21.
Trager W. The cultivation of a cellulose-digesting flagellate, Trichomonas termopsidis, and of certain other termite protozoa. Biol Bull. 1934;66:182–90.
Ikeda-Ohtsubo W. Endomicrobia in termite guts: symbionts within a symbiont. Phylogeny, cospeciation with host flagellates, and preliminary genome analysis (Dissertation). Germany: Philipps-Universität Marburg; 2007.
Tai V, James ER, Perlman SJ, Keeling PJ. Single-cell DNA barcoding using sequences from the small subunit rRNA and internal transcribed spacer region identifies new species of Trichonympha and Trichomitopsis from the hindgut of the termite Zootermopsis angusticollis. PLoS One. 2013;8:e58728.
Noda S, Ohkuma M, Yamada A, Hongoh Y, Kudo T. Phylogenetic position and in situ identification of ectosymbiotic spirochetes on protists in the termite gut. Appl Environ Microbiol. 2003;69:625–33.
Murakami T, Onouchi S, Igai K, Ohkuma M, Hongoh Y. Ectosymbiotic bacterial microbiota densely colonize the surface of thelastomatid nematodes in the gut of the wood-feeding cockroach Panesthia angustipennis. FEMS Microbiol Ecol. 2019;95:fiy238.
Manz W, Eisenbrecher M, Neu TR, Szewzyk U. Abundance and spatial organization of Gram-negative sulfate-reducing bacteria in activated sludge investigated by in situ probing with specific 16S rRNA targeted oligonucleotides. FEMS Microbiol Ecol. 1998;25:43–61.
Yuki M, Kuwahara H, Shintani M, Izawa K, Sato T, Starns D, et al. Dominant ectosymbiotic bacteria of cellulolytic protists in the termite gut also have the potential to digest lignocellulose. Environ Microbiol. 2015;17:4942–53.
Murakami T, Segawa T, Dial R, Takeuchi N, Kohshima S, Hongoh Y. Bacterial microbiota associated with the glacier ice worm is dominated by both worm-specific and glacier-derived facultative lineages. Microbes Environ. 2017;32:32–39.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43.
Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
Utami YD, Kuwahara H, Igai K, Murakami T, Sugaya K, Morikawa T, et al. Genome analyses of uncultured TG2/ZB3 bacteria in ‘Margulisbacteria’ specifically attached to ectosymbiotic spirochetes of protists in the termite gut. ISME J. 2019;13:455–67.
Sato T, Kuwahara H, Fujita K, Noda S, Kihara K, Yamada A, et al. Intranuclear verrucomicrobial symbionts and evidence of lateral gene transfer to the host protist in the termite gut. ISME J. 2014;8:1008–19.
Hongoh Y, Sharma VK, Prakash T, Noda S, Toh H, Taylor TD, et al. Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science. 2008;322:1108–9.
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, et al. Sequencing genomes from single cells by polymerase cloning. Nat Biotechnol. 2006;24:680–6.
Murakami T, Segawa T, Bodington D, Dial R, Takeuchi N, Kohshima S, et al. Census of bacterial microbiota associated with the glacier ice worm Mesenchytraeus solifugus. FEMS Microbiol Ecol. 2015;91:fiv003.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Donmez N, Brudno M. SCARPA: scaffolding reads with practical algorithms. Bioinformatics. 2013;29:428–34.
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. bioRxiv. 2019. https://doi.org/10.1101/614032.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185.
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–W251.
Abby SS, Cury J, Guglielmini J, Néron B, Touchon M, Rocha EPC. Identification of protein secretion systems in bacterial genomes. Sci Rep. 2016;6:23080.
Uchiyama I. MBGD: microbial genome database for comparative analysis. Nucleic Acids Res. 2003;31:58–62.
Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42:D231–D239.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118.; iso-2; iso-3. Fly. 2012;6:80–92.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91.
Rodriguez-R LM, Konstantinidis KT. Bypassing cultivation to identify bacterial species. Microbe. 2014;9:111–8.
Hongoh Y, Ohkuma M, Kudo T. Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiol Ecol. 2003;44:231–42.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Price MN, Zane GM, Kuehl JV, Melnyk RA, Wall JD, Deutschbauer AM, et al. Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics. PLoS Genet. 2018;14:e1007147.
Valentini M, Storelli N, Lapouge K. Identification of C4-dicarboxylate transport systems in Pseudomonas aeruginosa PAO1. J Bacteriol. 2011;193:4307–16.
Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, et al. Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol. 2012;194:1145–57.
Christensen GA, Zane GM, Kazakov AE, Li X, Rodionov DA, Novichkov PS, et al. Rex (encoded by DVU_0916) in Desulfovibrio vulgaris Hildenborough is a repressor of sulfate adenylyl transferase and is regulated by NADH. J Bacteriol. 2015;197:29–39.
Fronzes R, Christie PJ, Waksman G. The structural biology of type IV secretion systems. Nat Rev Microbiol. 2009;7:703–14.
Tai V, Carpenter KJ, Weber PK, Nalepa CA, Perlman SJ, Keeling PJ. Genome evolution and nitrogen fixation in bacterial ectosymbionts of a protist inhabiting wood-feeding cockroaches. Appl Environ Microbiol. 2016;82:4682–95.
Inoue JI, Saita K, Kudo T, Ui S, Ohkuma M. Hydrogen production by termite gut protists: characterization of iron hydrogenases of parabasalian symbionts of the termite Coptotermes formosanus. Eukaryot Cell. 2007;6:1925–32.
Zheng H, Dietrich C, Brune A. Genome analysis of Endomicrobium proavitum suggests loss and gain of relevant functions during the evolution of intracellular symbionts. Appl Environ Microbiol. 2017;83:e00656–17.
Messer AC, Lee MJ. Effect of chemical treatments on methane emission by the hindgut microbiota in the termite Zootermopsis angusticollis. Microb Ecol. 1989;18:275–84.
Zheng H, Dietrich C, Hongoh Y, Brune A. Restriction-modification systems as mobile genetic elements in the evolution of an intracellular symbiont. Mol Biol Evol. 2015;33:721–5.
McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2011;10:13–26.
Burke GR, Moran NA. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 2011;3:195–208.
Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–11.
Naito M, Morton JB, Pawlowska TE. Minimal genomes of mycoplasma-related endobacteria are plastic and contain host-derived genes for sustained life within Glomeromycota. Proc Natl Acad Sci USA. 2015;112:7791–6.
Ullmann R, Gross R, Simon J, Unden G, Kröger A. Transport of C4-dicarboxylates in Wolinella succinogenes. J Bacteriol. 2000;182:5757–64.
Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, et al. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol. 2004;22:554–9.
Keller KL, Wall JD. Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front Microbiol. 2011;2:135.
George EE, Husnik F, Tashyreva D, Prokopchuk G, Horák A, Kwong WK, et al. Highly reduced genomes of protist endosymbionts show evolutionary convergence. Curr Biol. 2020;30:925–33.
Nicks T, Rahn-lee L. Inside out: archaeal ectosymbionts suggest a second model of reduced-genome evolution. Front Microbiol. 2017;8:384.
Moran NA, Bennett GM. The tiniest tiny genomes. Annu Rev Microbiol. 2014;68:195–215.
Kirby H. Flagellates of the genus Trichonympha in termites. Univ Calif (Berkeley) Publ Zool. 1932;37:349–476.
Acknowledgements
We are grateful to Kenji Matsuura for kindly providing Z. nevadensis colonies with pinewood chips for rearing and to Wakako Ikeda-Ohtsubo for helpful advice regarding morphological identification of Trichonympha species. We also thank Miho Kiyooka, Masahiro Yuki, Kazuki Izawa, Takuya Yoshioka, and Katsura Igai for assisting with experiments. Sanger sequencing was done in the RIKEN BSI. TEM was assisted by Keiko Ikeda in the Biomaterial Analysis Center in the Tokyo Institute of Technology. This study was financially supported by IFO to HK (G-2018-1-039) and KAKENHI grants from JSPS to YH (16H04840, 17H01510, 17K19423, 20H02897, and 20H05584) and to MO (17H01447 and 19H05689).
Author information
Authors and Affiliations
Contributions
YH, HK, and MO conceived research and provided equipment and reagents; HK, MT, and YH designed research; MT and HK performed experiments; MT, YH, and HK analyzed genomic data; TM and MT analyzed amplicon data; MT performed phylogenetic analysis; KT performed additional FISH on Rf, Ry, Cf and Nk specimens; RK and TI conducted additional genome assembling; AT conducted PacBio sequencing; MT and YH wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takeuchi, M., Kuwahara, H., Murakami, T. et al. Parallel reductive genome evolution in Desulfovibrio ectosymbionts independently acquired by Trichonympha protists in the termite gut. ISME J 14, 2288–2301 (2020). https://doi.org/10.1038/s41396-020-0688-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0688-1
