
Abstract
Eukaryote-associated microbiomes vary across host taxa and environments but the key factors underlying their diversity and structure in fungi are still poorly understood. Here we determined the structure of bacterial communities in fungal fruitbodies in relation to the main chemical characteristics in ectomycorrhizal (EcM) and saprotrophic (SAP) mushrooms as well as in the surrounding soil. Our analyses revealed significant differences in the structure of endofungal bacterial communities across fungal phylogenetic groups and to a lesser extent across fungal guilds. These variations could be partly ascribed to differences in fruitbody chemistry, particularly the carbon-to-nitrogen ratio and pH. Fungal fruitbodies appear to represent nutrient-rich islands that derive their microbiome largely from the underlying continuous soil environment, with a larger overlap of operational taxonomic units observed between SAP fruitbodies and the surrounding soil, compared with EcM fungi. In addition, bacterial taxa involved in the decomposition of organic material were relatively more abundant in SAP fruitbodies, whereas those involved in release of minerals were relatively more enriched in EcM fruitbodies. Such contrasts in patterns and underlying processes of the microbiome structure between SAP and EcM fungi provide further evidence that bacteria can support the functional roles of these fungi in terrestrial ecosystems.
Similar content being viewed by others

Introduction
Fungi and bacteria undergo complex interactions ranging from strongly antagonistic and competitive to commensal and mutualistic [1, 2]. Bacteria may promote hyphal growth [3, 4], mycorrhiza formation [5, 6], and production of secondary metabolites [2, 7]. A better understanding of fungal fruitbody microbiome has important implications for fruitbody initiation [8,9,10] and its morphological variation and pigmentation [11, 12]. Fungal fruitbodies, in turn, create a suitable habitat for endofungal bacteria by providing different carbon sources for energy [13,14,15] and protection against environmental stress [7, 16]. Here the term endofungal refers to bacteria occurring within fungal fruitbodies (as opposed to their surface) irregardless of their location in- or outside the host cells.
Previous studies suggest that fruitbody microbiomes are affected by the fungal taxonomic affiliation and genotype, geographic distance and several abiotic factors [17,18,19,20,21]. However, the mechanisms of microbial community formation and the relative importance of each factor remain poorly known. Despite the importance of soil chemistry on bacterial composition [22,23,24], the role of fruitbody chemical characteristics in shaping its microbiome, has not been studied. However, composition of nutrients, vitamins, proteins, mono-, di- and polysaccharides, and several antimicrobial compounds differ considerably among fungal taxa [25,26,27,28].
Fungal species exhibit significant differences in concentration and stoichiometry of C and macronutrients (N, P; [29,30,31]), the content of which varies among the hymenophore, cap and stipe [32, 33], and between fungi with different roles in the environment [30, 31, 34, 35]. Ectomycorrhizal (EcM) fungi, a major symbiotic guild of fungi, supply plants with nutrients and water and receive plant photosynthates in return [36]. By contrast, SAP fungi acquire nutrients from dead organic material, forming one of the main guilds of decomposers [1]. Despite the contrasting nutritional modes of EcM symbionts and saprotrophs (SAP), including most of the macrofungi that form fruitbodies with a stipe and cap (mushrooms), their microbiomes have rarely been compared [37]. Whereas the pH of fruitbodies ranges from 5.5 to 7 [38], its effect on the microbiome in fruitbodies has not been studied. Soil pH and nutrient concentrations are the major determinants of the microbiome structure and functions in soil [39, 40] and fungal fruitbodies [17,18,19]. Growing evidence suggests that soil is the main reservoir for bacterial communities in fungal fruitbodies, where the source community is changed by enrichment or exclusion of specific bacterial groups [19, 37, 41]. However, it remains unclear to what extent and by which mechanisms the soil bacterial community affects the fruitbody microbiome.
This study aims to identify the key determinants of bacterial community composition and diversity across the main functional and phylogenetic groups of mushroom-forming fungi. Fruitbodies of three EcM and three SAP fungal species as well as the nearby soil were sampled to test the relative importance of fungal functional guild and phylogeny, soil, fruitbody chemical characteristics as well as the fruitbody compartment in shaping the structure of the fruitbody microbiome. We hypothesized that fruitbody characteristics have a stronger effect on their microbiome structure than soil parameters.
Material and methods
Study sites and sampling
Altogether 48 mature, not damaged nor decaying fungal fruitbodies from three EcM and three SAP species were collected from two forest sites about 60 km apart in Estonia (Table S1). All samples were collected from the Meenikunno site (∼90,000 m2), except for fruitbodies of Clitocybe geotropa that were sampled at Mälgi; the samples from both areas combined are referred to as main dataset. Mälgi site was included to even the number of SAP species with those of EcM fungi. Meenikunno site was visited several times and Mälgi site once during September–October 2017 and at least five fruitbodies, growing at least 10 m apart, and the underlying soil, were collected from each fungal species. The soil samples included 200–250 g of soil that was taken as 28 subsamples from 10 cm depth within 50 cm radius from each fruitbody (Fig. S1A); in the case of immediate proximity of fruitbodies of two species, one soil sample served for both of these (Fig. S1B). Each soil sample was transferred into a plastic bag and mixed. Altogether 4 g of soil from each sample was weighed into a resealable plastic bag and stored at −20 °C until DNA extraction. A small amount (413–833 mg) of fresh soil from both sampling sites taken at each collecting day, was weighed, dried, and the per-mass water content was calculated. The amount of raw soil needed for DNA extraction (250 mg) was separated from this 4 g of frozen soil and increased accordingly (51–152 mg) to standardize the sample weights based on the calculated water content. The DNA from soil samples was extracted using ZR Soil Microbe DNA Kit (Zymo Research, CA, USA) according to the manufacturer’s instructions.
An additional 61 fruitbodies of 15 species of SAP fungi and 12 species of EcM fungi were collected from eleven forest sites, located all around Estonia to increase the number of host species from both functional guilds and to reduce the host taxonomy effect on the bacterial community structure (Table S2). Unlike in the main dataset, soil was not sampled under these fruitbodies and the subsamples from different parts were pooled and not analyzed separately for obtaining the validation dataset.
The fruitbodies were cut lengthwise under the laminar flow using a sterile scalpel. The cut surface was sterilized under UV light for 5 min, followed by a second incision into the exposed inner tissue while avoiding contact with the fruitbody outer surface. For the main dataset, one sample included two 5 mm3 pieces, taken from each fruitbody part—the cap, the middle part, and the lower part of the stipe (Fig. S2A). For the validation dataset, two replicates, both including one piece from each of the three fruitbody parts, were allocated for DNA extraction; the PCR products obtained from the two replicates were pooled before sequencing to comprise one sample per each fruitbody (Fig. S2B). The pieces were crushed using a sterile scalpel.
DNA extraction and PCR
Three DNA extractions from each fruitbody and one from the underlying soil were analyzed separately for the main dataset (Fig. S2A). In the case of the validation dataset two DNA extracts from each fruitbody were allocated for PCR, the products of which were pooled for sequencing (Fig. S2B). The fruitbody material, used for DNA extractions, was comparable in both datasets, because in both cases six fruitbody pieces from opposite parts of three compartments were analyzed in total (Fig. S2). DNA was extracted using the Quick-DNA Fungal/Bacterial Miniprep Kit (Zymo Research, CA, USA) according to the manufacturer’s instructions, except that the samples were centrifuged at the maximum speed (14,680 rpm) and DNA was eluated in two steps (50 μl and 50 μl) to increase the yield.
The 16S rRNA gene variable regions V3–V4 were amplified using bacterial primers 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806RB (5′-GGACTACNVGGGTWTCTAAT-3′). The 25 µl of PCR mix contained 4 µl of sterilized H2O, 5 µl of 5× HOT FIREPol Blend MasterMix (Solis Biodyne, Tartu, Estonia), 0.5 µl of both the forward and reverse primer (200 nM) and 15 µl of the DNA extract. The PCR program included the following steps: 95 °C for 15 min, followed by 27–33 cycles of 95 °C for 30 s, 50 °C for 45 s, and 72 °C for 1 min with a final extension step at 72 °C for 10 min. PCR products were visualized on 1% agarose gel. In total, 27 cycles were used for all the samples, but in case of a weak or no band a higher number of PCR cycles or lower DNA amount were used. All soil samples were amplified using 25 cycles and 2 µl of the DNA extract. The PCR products were purified by using a FavorPrepTM GEL/PCR Purification Kit (Favorgen Biotech Corporation). DNA concentrations were measured with the Qubit dsDNA HS kit (Invitrogen). The samples were sequenced at the Estonian Biocentre (Tartu, Estonia) using the Illumina MiSeq platform in 2 × 300 paired end mode. The sequences obtained from each sample were submitted to Sequence Read Archive and are available under BioProject PRJNA379722 (accession numbers SAMN13377889 to SAMN13378134).
Chemical analysis of soil and fruitbody samples
Total phosphorus (P), nitrogen (N), and carbon (C) contents and pHKCl were measured from both, soil and fruitbody samples. The original amount of each soil sample that was retained after allocating 4 g for the DNA extraction was used for chemical analyses. Similarly, the whole fruitbodies were dried and used for the chemical analyses, except for the pieces that had been taken for the DNA extraction. Total N and C content of oven-dried samples were determined by dry combustion method on a varioMAX CNS elemental analyzer (ELEMENTAR, Germany). Available phosphorus was determined in a Kjeldahl digestion by Fiastar 5000 (AN 5242; Stannous Chloride method, ISO/FDIS 15681). In addition, the content of ammonium nitrogen (NH4–N), extracted with 2 M KCl was determined from soil samples using Flow Injection Analysis (Tecator ASN 65-32/84). All these characteristics were determined at the Plant Laboratory of the Estonian University of Life Sciences. The C:N and N:P ratios were calculated using the mass-based values of C, N, and P. The pH values were unmeasured for some of the fruitbodies because of the insufficient material and the fruitbodies of Cantharellula umbonata were pooled to provide sufficient material for chemical analysis.
Bioinformatic and statistical analysis
Illumina sequencing data were processed using the LotuS pipeline [42], including demultiplexing, quality-filtering, and chimera-checking. Sequences were clustered into OTUs at the 97% similarity level using UPARSE. Taxonomic origin of each OTU was determined based on the Silva database (www.arb-silva.de) using BLAST. USEARCH v10.0.240 [43] was used for generating consensus OTU sequences. All chloroplasts, mitochondria, taxonomically unidentifiable, and eukaryotic OTUs were removed, as well as OTUs containing <5 reads or represented in positive or negative controls. OTUs with read numbers in negative controls constituting <2% of the read numbers in material samples were not removed, but the former share of reads was subtracted from the latter. Eighteen fruitbody samples that contained <1000 reads were excluded from statistical analyses. The fruitbody samples taken separately and in combination with soil samples were rarefied down to 2000 and 7000 reads, respectively. In the validation dataset, samples that contained <1000 reads were excluded from the statistical analyses and samples that contained >5000 reads were rarefied down to 5000.
Data from the three compartments of each fruitbody (cap, the middle, and the lower part of stipe) were treated as separate samples in PERMANOVA, Mantel/Partial Mantel test, and NMDS. Similarly, one sample represents one of the three parts of each fruitbody in the comparison of the relative abundances of different bacterial groups between EcM and SAP fungi as well as in the comparison of Shannon diversity indices among analyzed fungal taxa in the main dataset. For the remaining analyses, reads from the subsamples of individual fruitbody were pooled into one sample in the main dataset. In the validation dataset, one sample exclusively represents the whole fruitbody, regardless of the analysis.
For analyzing the effect of the soil and fruitbody chemical composition on fruitbody bacterial communities, the C, N, P, and NH4N values were log-transformed. The effect of these factors and their ratios on bacterial communities were analyzed using ADONIS function in the vegan package of R (vers.3.6.1, R Development Core Team, 2019). The variables were forward-selected for constructing the best model, according to F-values. The Primer 6 program (Primer-E Ltd, Plymouth, United Kingdom) with the PERMANOVA+ add on package was used to perform the Permanova nested design.
Phylogenetic distance of the host fungi and the dissimilarity matrix of soil bacterial communities were included into the Mantel and Partial Mantel tests to determine the effect of the host fungus phylogeny and soil microbiome on fungal microbiomes. The phylogenetic tree was constructed using the perl script taxonomy_to_tree.pl following Tedersoo et al. [44], and phylogenetic distance between fungi was calculated using the picante package in R (vers.3.6.1, R Development Core Team, 2019). Soil samples, included in Mantel and Partial Mantel tests, were rarefied to 20,000 sequences. The Bray–Curtis dissimilarity index and Hellinger transformation were used in multivariate analysis.
To visualize the community variation across samples, nonmetric multidimensional scaling (NMDS) plots were generated using the vegan package. Statistical significance of differences in the Shannon diversity indices were determined based on Kruskal–Wallis test, with Wilcoxon test for pairwise comparisons as implemented in ggpubr package of R. Similarly, the relative abundances of the most common bacterial groups at different taxonomic levels were compared between EcM and SAP fungi based on Kruskal–Wallis test results. In addition, multivariate analysis of variance (MANOVA) and analysis of variance (ANOVA) in R dplyr package and vegan package (vers.3.6.1, R Development Core Team, 2019), were used to determine the general as well as factor by factor significance of the variability of chemical parameters between different host taxa, functional guilds as well as between soil and fruitbodies. The t-test was used to determine the significance of the variability of different chemical parameters in pairwise comparisons between fungal species and between fungal species and soil using vegan and ggpubr packages in R. The heatmap, illustrating the Spearman correlations between the relative abundance of 30 most numerous bacterial orders and chemical properties of fruitbodies of EcM and SAP fungi was prepared using ggplot2 package of R. The indicator OTUs associated with each fungal order and functional guild were determined using the Dufrene–Legendre Indicator Species Analysis in labdsv package of R.
Results
Bacterial abundance and community composition in fruitbodies and soil
The main dataset included 2,192,548 reads that were clustered into 9095 OTUs. The 48 fruitbodies from six species yielded in 5418 OTUs based on 880,557 reads (on average 6115 reads per sample), comprising 19 archaeal and 5399 bacterial OTUs. Altogether 7389 OTUs (1,311,991 reads) were recovered from the 41 soil samples, of which 31 and 7358 represented Archaea and Bacteria, respectively. Since the primers used for DNA amplification in the current study perform poorly in amplifying Archaea [45], the few archaeal OTUs had low relative abundance and were excluded from the analyses. The validation dataset of 61 fruitbodies from 27 SAP and EcM species (Table S2) included 4499 OTUs (560,447 reads). The rarefaction curves showed that sequencing depth was adequate for describing the microbial communities in fruitbodies and soil as well as in SAP and EcM fungi in case of both datasets (Fig. S3). All of the following results, unless the use of the validation dataset has been indicated, are based on the main dataset.
The microbial community composition and diversity did not differ significantly between the fruitbody compartments (p > 0.05). Proteobacteria was the most abundant bacterial phylum in the fruitbodies of all analyzed fungal species and soil, followed by Bacteroidetes, Actinobacteria, and Firmicutes in fruitbodies but by Actinobacteria, Acidobacteria, Planctomycetes, and Verrucomicrobia in soil (Fig. S4A). Rhizobium (Rhizobiales, Alphaproteobacteria) and members of the Burkholderia–Caballeronia–Paraburkholderia complex (Burkholderiales, Betaproteobacteria) dominated in fruitbodies (Figs. 1a, b; S4B–D), including in each of the three fruitbody parts, whereas Mycobacterium (Corynebacteriales, Actinobacteria) and Acidipila (Acidobacteriales, Acidobacteriia) dominated in the surrounding soil (Fig. S4). Soil and fruitbodies shared 40.5% of the OTUs, while as much as 68.5% of the fruitbody OTUs were detected also in soil (Fig. S5). However, the structure of bacterial communities was distinct between fruitbody and soil samples, despite the large variation among individual fruitbodies (Figs. 2a; S4), and only 1.1–19% of OTUs overlapped between an individual fruitbody and the underlying soil. The soil bacterial communities had a marginal, although significant effect (Mantel r = 0.116; p = 0.001) on the fruitbody bacterial communities.

Mean relative abundance (%) of most numerous bacterial classes (a) and orders (b) as well as differences of the relative abundance of the most dominant bacterial genera (c–f) inhabiting ectomycorrhizal (EcM) and saprotrophic (SAP) fungal species.

The Bray–Curtis dissimilarity matrix was calculated based on rarified and Hellinger transformed OTU matrix, containing both soil and fruitbody samples, rarefied to ≤7000 reads (a) and fruitbody samples only, rarefied to ≤2000 reads (b). Samples containing less than 1000 reads were removed. Each symbol represents one of the three fruitbody parts or a soil sample. The host names with slashes (a) indicate the soil samples collected under two adjacent fruitbodies. Both host functional guild and phylogenetic group had a significant effect on the bacterial community composition in fruitbodies (PERMANOVA p ≤ 0.001).
Bacterial abundance, diversity, and community composition in fruitbodies of SAP and EcM fungi
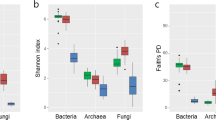
Bacterial diversity differed among all analyzed fungal orders (Fig. 3a) and most of the fungal host species (Fig. S6). The diversity of fruitbody bacteria was significantly higher in SAP compared with EcM fungal species (Figs. 3b; S6). In addition, the proportion of OTUs shared between each fruitbody and its surrounding soil was higher in SAP (mean ± SD: 9.9 ± 4.2%) than in EcM fungal species (4.4 ± 2.7%; Fig. S7). Overall 65.6% of bacterial OTUs in SAP fungi and 50.4% in EcM fungi were represented in the corresponding soil samples (Fig. S8A, B). Altogether 43.9% of the OTUs were shared between SAP and EcM fungi (Fig. S8C).

The boxplots, based on Shannon indices, illustrating the diversity of the microbial communities in three fungal orders (a), in ectomycorrhizal (EcM) and saprotrophic (SAP) fruitbodies and the surrounding soil (b). Significant pairwise differences (p ≤ 0.05) are represented based on Wilcoxon test results. Each symbol represents one of the three fruitbody parts in a but the whole fruitbody in b.
Among the bacterial groups that were common in fruitbodies, the genera Pseudomonas, Sphingomonas, and members of the Burkholderia–Caballeronia–Paraburkholderia complex as well as several families of Alphaproteobacteria were significantly more abundant in SAP than in EcM fruitbodies (Figs. 1c, e, f; S9). By contrast, the relative abundance of the genus Rhizobium (Rhizobiaceae) was significantly higher in EcM than in SAP fungi (Figs. 1d; S9). Several indicator OTUs of Cantharellales and EcM fungi also belonged to Rhizobium but those of SAP fungi and the order Agaricales to the genus Massilia (Burkholderiales; Table S3). Only four indicator OTUs were identified for the order Russulales, the best of which belonged to Burkholderia–Caballeronia–Paraburkholderia group (Table S3).
Determinants of bacterial community structure in fruitbodies
Fungal taxonomy, especially at the host species level (nested in the order-level affiliation; PERMANOVA: Pseudo-F = 7.156; p = 0.001) strongly affected the bacterial community structure in fruitbodies. In addition, the host functional guild significantly affected the microbial community composition in fungal fruitbodies (Table 1). The NMDS plots also distinguished between the bacterial communities of different fungal taxa and to lesser extent between those of EcM and SAP fungi (Figs. 2b; S10A). Analysis of the validation dataset confirmed the significance of the effect of host taxonomy and functional guild on fruitbody bacterial communities (Table 1; Fig. S10B). Mantel tests revealed a strong association between the phylogenetic distance between hosts and the bacterial community composition in fruitbodies (the main dataset: r = 0.496, p = 0.001; Fig. S11; the validation dataset: r = 0.445, p = 0.001).
All measured chemical parameters differed significantly between the fruitbodies of EcM and SAP fungi (MANOVA, approx. F1,36 = 12.26; p < 0.001; Figs. 4; S12) and among studied fungal species (MANOVA, approx. F5,32 = 11.26; p < 0.001; Figs. 4; S12). The fruitbody C:N ratio was the most important factor shaping the endofungal bacterial community composition in fruitbodies (PERMANOVA adj. R2 = 0.107; p < 0.001; Table 1). However, after excluding the taxonomically most distinct fungal taxon (Cantharellus cibarius) with significantly higher C:N ratio than in the other analyzed species (Fig. S12B) from the analysis, the fruitbody pH was the main determinant of the bacterial community structure in fruitbodies (adj. R2 = 0.085; p < 0.001; Table S4). Similarly, the PERMANOVA model based on the validation dataset revealed that the fruitbody pH was the main factor shaping the endofungal bacterial community structure (Table 1). We also detected significant correlations between the relative abundance of several endofungal bacterial groups and the chemical parameters of fruitbodies that often differed between EcM and SAP fungi (Fig. S13). Bacterial diversity (Shannon index) was positively correlated with fruitbody pH as well as fruitbody P and N concentration, but negatively correlated with fruitbody C concentration and C:N ratio based on the Spearman’s correlation test (Fig. S14).

Boxplots illustrating the variation of pH (a), N% (b), P% (c), and C% (d) in fruitbodies of six fungal species and in soil. P values are given for general difference of the variation of these characteristics among soil and fungi (ANOVA) and for pairwise significant differences among five fungal species (t-test); differences between each fungal species and soil were all significant (p < 0.001). The N, P, and C values were log-transformed. Each symbol represents a whole fruitbody or a soil sample. Because of the small size of fruitbodies of Cantharellula umbonata these were pooled for chemical analyses and are presented as one replicate that was not included in these statistical analyses.
Soil parameters had much weaker impact on the bacterial community composition in fruitbodies than host parameters, with only pH (PERMANOVA adj. R2 = 0.002; p = 0.045) and NH4N content (adj. R2 = 0.002; p = 0.043) having significant effect (Table S5). While the content of all measured chemical parameters and their ratios differed significantly between the fruitbodies and the soil (Figs. 4; S12), significant correlations were observed in some cases. For example, the N:P ratio was positively (n = 31; R = 0.67; p < 0.001) and P content negatively (n = 31; R = −0.39; p = 0.029) correlated between EcM fungal fruitbodies and soil. Although the soil parameters did not significantly affect the bacterial diversity in fungal fruitbodies (p > 0.05), they significantly correlated with the diversity of the bacterial communities in soil (Fig. S15), thus affecting the source community of the endofungal bacteria.
Discussion
Our results show that similarly to other organism groups, host intrinsic features have a strong impact on the structure of fungal microbiota that varies in accordance with hosts phylogenetic distance and functional guilds. Previous studies have mostly ascribed the variation among fruitbody bacterial communities to soil characteristics and host identity [17, 19,20,21]. However, fungal functional guild has also been shown to affect the communities of fruitbody inhabiting organisms, such as fungus gnats and bacteria [37, 46]. Our study indicates that the observed effect of host phylogeny and functional guild can be mainly ascribed to the effects of fruitbody chemical properties. In particular, fruitbody pH and C:N were the main determinants of the bacterial community structure, in agreement with patterns described for soil and dead wood [24, 39, 40, 47, 48]. Similarly to previous observations from these two environments [23, 24, 48, 49], we found that fruitbody pH, N%, and P% were positively correlated, while C% as well as C:N ratio were negatively correlated with bacterial diversity in fruitbodies. In addition to C and the macronutrients, differences in carbohydrates and secondary metabolites among distantly related fungal groups [27, 33, 50] may have an additional effect on bacterial community structure in fungi [25, 28, 51,52,53].
Fungal functional guilds affected the correlation between fruitbody and soil chemical parameters, bacterial diversity as well as their community composition. The higher bacterial diversity in SAP fungi may result from higher N and P but lower C content in their fruitbodies compared with EcM fungi, as also observed in previous studies [30, 31, 35]. Differences in fruitbody nutrient content are likely to result from their contrasting carbon sources and the allocation of minerals from EcM fungi to their plant symbionts via mycorrhiza [30, 54]. The N:P ratio and P content in EcM fungi were significantly correlated to these values in soil, as also shown by Kranabetter et al. [31]. In addition, the C:P as well as the N:P ratio of EcM fungi correspond to the values in their host plants and vary more widely over the edaphic gradient than in SAP fungi [31], which may explain the differences between the bacterial communities in EcM and SAP fungi. Although we found that the chemical composition of the host fruitbody differs significantly between EcM and SAP fungi, we could not assess the functional guilds effect in a phylogeny-independent context, because the life strategy and most of the chemical characteristics are phylogenetically conserved [30, 34, 55].
Soil pH and several other chemical properties, previously shown to affect the fruitbody bacterial community structure [17,18,19], had a significant but relatively weak effect compared with that of fruitbody chemistry. Our results suggest that at least part of the “soil effect” in previous studies may have represented the confounding but unmeasured fruitbody chemistry effect instead. Fungi can alter soil conditions and the microbiome near their fruitbodies [18, 56, 57], creating small-scale, nutrient-rich hot spots, where fungal hyphae exude carbohydrates and amino acids [58] with more drastic changes observed after the decomposition of fruitbodies [59]. The higher overlap of bacterial OTUs observed between soil and fruitbodies across the whole sampling site compared with the much lower number of OTUs shared between each fruitbody and its underlying soil, could evidence “hitchhiking” of bacteria along hyphae in soil [60, 61]. However, such pattern is more likely a result from contrasting species richness and turnover between the continuous soil environment and the fungal fruitbodies constituting nutrient-rich islands (with considerably higher pH than soil at our main sampling site).
Variation of chemical parameters may have led to the divergence of the diversity and relative abundance of several bacterial groups in soil and fruitbodies, the extent of which, in turn, was determined by fungal functional guilds. For example, several members of the Proteobacteria, Chlamydiae, Acidobacteria, Planctomycetes, and Verrucomicrobia that clearly prefer soil to fungal fruitbodies [17, 19, 37], were significantly more abundant in SAP than in EcM fungi. These groups may flourish in environments with lower C content, like soil [22, 62, 63] and SAP as compared with EcM fungi. The high C content of EcM fungi may stem from the uptake of plant host photosynthates that are rapidly transferred also to hyphosphere bacteria [64]. Accordingly, Bacteroidetes, preferring high C content [62], was significantly more common in fruitbodies of EcM than SAP fungi. The lower relative abundance of Proteobacteria in EcM may result from the higher C:N ratio and lower pH in their fruitbodies compared with SAP fungi, analogous to observations from soil [40, 48]. At the bacterial genus level, our results confirmed the close association of the Burkholderia–Caballeronia–Paraburkholderia group, Massilia and Pseudomonas with SAP fungi [65, 66]. The high relative abundance of the last two genera in soil have been ascribed to the high N and P contents [57], characteristic also of fruitbodies of SAP fungi. The members of Burkholderiaceae (phylum Proteobacteria), often sensitive to acidic conditions [67], may use SAP fruitbodies to escape from low pH environments [58, 68], like the soil in our main study site. It is possible that other chemical parameters, such as the content of different microelements and/or secondary metabolites in fruitbodies or soil, not measured in this study, also affect the structure of the bacterial community composition in fungal fruitbodies.
The functions of bacterial communities in fungal fruitbodies can currently only be deduced from the role of particular groups documented in other environments. So far, N fixation has been claimed to occur in Tuber magnatum fruitbodies, in cultures of Pleurotus ostreatus mycelium inoculated with a bradyrhizobial strain and in ectomycorrhizae [69,70,71]. Several bacterial groups with N-fixing potential had contrasting relative abundances in fruitbodies of fungi from the two functional guilds. While Rhizobium (Rhizobiaceae) was significantly more abundant in EcM than in SAP fungi, other families of Alphaproteobacteria were more abundant in SAP fungi. For example, the families Beijerinckiaceae and Acetobacteraceae, involving several N-fixing, methanol-degrading members that are often identified in decaying wood [47, 72], were highly abundant in SAP fungi. Our results call for studies to test whether N fixation takes place in fungal fruitbodies and whether its extent and involved bacterial taxa differ with respect to the functional guild of the host fungus.
Endofungal bacteria may support the functionality of the host fungus in a number of ways, such as participating in the release of mineral compounds from soil or in the decomposition processes driven by SAP fungi. We found that the order Micrococcales (Actinobacteria), previously identified to include phosphate and iron releasing mycorrhiza-helper bacteria [73], was significantly more abundant in EcM than in SAP fungi. In addition, several species among Rhizobium, Pseudomonas, and Burkholderia are known as effective phosphate solubilizing and iron mobilizing mycorrhiza-associated bacteria in the mycorrhizosphere [5, 74,75,76]. The high relative abundance of Acidobacteria and members of Burkholderiaceae, primarily characteristic of SAP fungi and decaying material [47, 77, 78], may support the decomposition processes, because these bacterial groups are able to produce a range of enzymes which break down chitin, cellulose, and several other recalcitrant carbon compounds [49, 79, 80]. Bacterial taxa common in fruitbodies of SAP and EcM fungi largely overlap with the most abundant bacterial groups in decaying material and (mycor)rhizosphere, respectively, [47, 77, 81,82,83]. Bacterial communities in fruitbodies may thus mirror the structure and the functions of the microbiome associated with the mycelia in the underlying substrate that give rise to the fruitbodies. Taken together, our results suggest that the structure of the fungal microbiome depends on the joint influence of the fungal nutritional strategy, chemical properties and interactions between fungi and their surrounding soil, which in turn vary depending on fungal functional guilds.
References
de Boer W, Folman LB, Summerbell RC, Boddy L. Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev. 2005;29:795–811.
Kobayashi DY, Crouch JA. Bacterial-fungal interactions: from pathogens to mutualistic endosymbionts. Annu Rev Phytopathol. 2009;47:63–82.
Li Q, Chen CH, Penttinen P, Xiong CH, Zheng L, Huang W. Microbial diversity associated with Tricholoma matsutake fruiting bodies. Microbiology 2016a;85:531–9.
Oh SY, Kim M, Eimes JA, Lim YW. Effect of fruiting body bacteria on the growth of Tricholoma matsutake and its related molds. PLoS One 2018;13:e0190948.
Frey-Klett P, Garbaye J, Tarkka M. The mycorrhiza helper bacteria revisited. N Phytologist. 2007;176:22–36.
Tarkka MT, Drigo B, Deveau A. Mycorrhizal microbiomes. Mycorrhiza. 2018;28:403–9.
Schulz-Bohm K, Tyc O, de Boer W, Peereboom N, Debets F, Zaagman N, et al. Fungus-associated bacteriome in charge of their host behavior. Fungal Genet Biol. 2017;102:38–48.
Cho YS, Kim JS, Crowley DE, Cho BG. Growth promotion of the edible fungus Pleurotus ostreatus by fuorescent pseudomonads. FEMS Microbiol Lett. 2003;218:271–6.
Noble R, Dobrovin-Pennington A, Hobbs PJ, Pederby J, Rodger A. Volatile C8 compounds and pseudomonads influence primordium formation of Agaricus bisporus. Mycologia 2009;101:583–91.
Riedlinger J, Schrey SD, Tarkka MT, Hampp R, Kapur M, Fiedler HP. Auxofuran, a novel metabolite that stimulates the growth of fly agaric, is produced by the mycorrhiza helper Bacterium Streptomyces strain AcH 505. Appl Environ Microbiol. 2006;72:3550–7.
Tauber JP, Gallegos-Monterrosa R, Kovács ÁT, Shelest E, Hoffmeister D. Dissimilar pigment regulation in Serpula lacrymans and Paxillus involutus during inter-kingdom interactions. Microbiology 2018;164:65–77.
Zhou J, Bai X, Zhao R. Microbial communities in the native habitats of Agaricus sinodeliciosus from Xinjiang Province revealed by amplicon sequencing. Sci Rep. 2017;7:15719.
Kalač P. Chemical composition and nutritional value of European species of wild growing mushrooms: a review. Food Chem. 2009;113:9–16.
Rangel-Castro JI, Danell E, Pfeffer PE. A 13 C-NMR study of exudation and storage of carbohydrates and amino acids in the ectomycorrhizal edible mushroom Cantharellus cibarius. Mycologia. 2002;94:190–9.
Warmink JA, Nazir R, van Elsas JD. Universal and species-specific bacterial ‘fungiphiles’ in the mycospheres of different basidiomycetous fungi. Environ Microbiol. 2009;11:300–12.
Guhr A, Borken W, Spohn M, Matzner E. Redistribution of soil water by a saprotrophic fungus enhances carbon mineralization. Proc Natl Acad Sci USA. 2015;112:14647–51.
Li Q, Li X, Chen C, Li S, Huang W, Xiong C, et al. Analysis of bacterial diversity and communities associated with tricholoma matsutake fruiting bodies by barcoded pyrosequencing in Sichuan Province, Southwest China. J Microbiol Biotechnol. 2016b;26:89–98.
Pent M, Hiltunen M, Põldmaa K, Furneaux B, Hildebrand F, Johannesson H, et al. Host genetic variation strongly influences the microbiome structure and function in fungal fruiting-bodies. Environ Microbiol. 2018;20:1641–50.
Pent M, Põldmaa K, Bahram M. Bacterial communities in boreal forest mushrooms are shaped both by soil parameters and host identity. Front Microbiol. 2017;8:836.
Rinta-Kanto JM, Pehkonen K, Sinkko H, Tamminen MV, Timonen S. Archaea are prominent members of the prokaryotic communities colonizing common forest mushrooms. Can J Microbiol. 2018;64:716–26.
Benucci GMN, Bonito GM. The Truffle Microbiome: Species and Geography Effects on Bacteria Associated with Fruiting Bodies of Hypogeous Pezizales. Microb Ecol. 2016;72:4–8.
Eilers KG, Lauber CL, Knight R, Fierer N. Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Biol Biochem. 2010;42:896–903.
Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–31.
Ge Y, Chen C, Xu Z, Eldridge SM, Chan KY, He Y, et al. Carbon/nitrogen ratio as a major factor for predicting the effects of organic wastes on soil bacterial communities assessed by DNA-based molecular techniques. Environ Sci Pollut Res Int. 2010;17:807–15.
Alves M, Ferreira I, Dias J, Teixeira V, Martins A, Pintado M. A review on antimicrobial activity of mushroom (Basidiomycetes) extracts and isolated compounds. Planta Med. 2012;78:1707–18.
Olagbemide PT, Ogunnusi TA. Proximate analysis and chemical composition of Cortinarius species. Eur J Adv Res Biol Life Sci. 2015;3:1–9.
Sanmee R, Dell B, Lumyong P, Izumori K, Lumyong S. Nutritive value of popular wild edible mushrooms from northern Thailand. Food Chem. 2003;82:527–32.
Vieira V, Barros L, Martins A, Ferreira I. Expanding current knowledge on the chemical composition and antioxidant activity of the Genus Lactarius. Molecules. 2014;19:20650–63.
Taylor AFS, Hogbom L, Hogberg M, Lyon AJE, Nasholm T, Hogberg P. Natural 15N abundance in fruit bodies of ectomycorrhizal fungi from boreal forests. N Phytologist. 1997;136:713–20.
Trocha LK, Rudy E, Chen W, Dabert M, Eissenstat DM. Linking the respiration of fungal sporocarps with their nitrogen concentration: variation among species, tissues and guilds. Funct Ecol. 2016;30:1756–68.
Kranabetter JM, Harman-Denhoed R, Hawkins BJ. Saprotrophic and ectomycorrhizal fungal sporocarp stoichiometry (C: N: P) across temperate rainforests as evidence of shared nutrient constraints among symbionts. N Phytologist. 2019;221:482–92.
Alam N, Amin R, Khan A, Ara I, Shim MJ, Lee MW, et al. Nutritional analysis of cultivated mushrooms in Bangladesh—Pleurotus ostreatus, Pleurotus sajor-caju, Pleurotus florida and Calocybe indica. Mycobiology. 2008;36:228–32.
Rudawska M, Leski T. Macro- and microelement contents in fruiting bodies of wild mushrooms from the Notecka forest in west-central Poland. Food Chem. 2005;92:499–506.
Hobbie EA, Weber NS, Trappe JM. Mycorrhizal vs saprotrophic status of fungi: the isotopic evidence. N Phytologist. 2001;150:601–10.
Vogt KA, Edmonds RL, Grier CC. Biomass and nutrient concentrations of sporocarps produced by mycorrhizal and decomposer fungi in Abies amabilis stands. Oecologia. 1981;50:170–5.
Landeweert R, Hoffland E, Finlay RD, Kuyper TW, van Breemen N. Linking plants to rocks: ectomycorrhizal fungi mobilize nutrients from minerals. TRENDS Ecol Evolution. 2001;16:248–54.
Liu Y, Sun Q, Li J, Lian B. Bacterial diversity among the fruit bodies of ectomycorrhizal and saprophytic fungi and their corresponding hyphosphere soils. Sci Rep. 2018;8:11672.
Konuk M, Afyon A, Yağiz D. Chemical composition of some naturally growing and edible mushrooms. Pak J Bot. 2006;38:799–804.
Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, et al. Structure and function of the global topsoil microbiome. Nature. 2018;560:233–7.
Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010;4:1340–51.
Antony-Babu S, Deveau A, Van Nostrand JD, Zhou J, Le Tacon F, Robin C, et al. Black truffle - associated bacterial communities during the development and maturation of Tuber melanosporum ascocarps and putative functional roles: tuber melanosporum-associated bacterial communities. Environ Microbiol. 2014;16:2831–47.
Hildebrand F, Tadeo R, Voigt A, Bork P, Raes J. LotuS: an efficient and user- friendly OTU processing pipeline. Microbiome. 2014;2:30.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Tedersoo L, Sánchez-Ramírez S, Kõljalg U, Bahram M, Döring M, Schigel D, et al. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Diversity. 2018;90:135–59.
Bahram M, Anslan S, Hildebrand F, Bork P, Tedersoo L. Newly designed 16S rRNA metabarcoding primers amplify diverse and novel archaeal taxa from the environment. Environ Microbiol Rep. 2019;11:487–94.
Põldmaa K, Jürgenstein S, Bahram M, Teder T, Kurina O. Host diversity and trophic status as determinants of species richness and community composition of fungus gnats. Basic Appl Ecol. 2015;16:46–53.
Hoppe B, Krüger D, Kahl T, Arnstadt T, Buscot F, Bauhus J, et al. A pyrosequencing insight into sprawling bacterial diversity and community dynamics in decaying deadwood logs of Fagus sylvatica and Picea abies. Sci Rep. 2015;5:9456.
Zhang X, Xu S, Li C, Zhao L, Feng H, Yue G, et al. The soil carbon/nitrogen ratio and moisture affect microbial community structures in alkaline permafrost-affected soils with different vegetation types on the Tibetan plateau. Res Microbiol. 2014;165:128–39.
Johnston SR, Boddy L, Weightman AJ. Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol Ecol. 2016;92:fiw179.
Ruthes AC, Smiderle FR, Iacomini M. Mushroom heteropolysaccharides: a review on their sources, structure and biological effects. Carbohydr Polym 2016;136:358–75.
Boersma FGH, Warmink JA, Andreote FA, van Elsas JD. Selection of sphingomonadaceae at the base of laccaria proxima and russula exalbicans fruiting bodies. Appl Environ Microbiol. 2009;75:1979–89.
de Carvalho MP, Türck P, Abraham WR. Secondary metabolites control the associated bacterial communities of saprophytic basidiomycotina fungi. Microbes Environ. 2015;30:196–8.
Boersma FGH, Otten R, Warmink JA, Nazir R, van Elsas JD. Selection of Variovorax paradoxus-like bacteria in the mycosphere and the role of fungal-released compounds. Soil Biol Biochem. 2010;42:2137–45.
Taylor AFS, Fransson PM, Högberg P, Högberg MN, Plamboeck AH. Species level patterns in 13C and 15N abundance of ectomycorrhizal and saprotrophic fungal sporocarps. N Phytologist. 2003;159:757–74.
Zanne AE, Abarenkov K, Afkhami ME, Aguilar-Trigueros CA, Bates S, Bhatnagar JM, et al. Fungal functional ecology: bringing a trait-based approach to plant-associated fungi. Biol Rev. 2020;95:409–33.
Danell E, Alström S, Ternström A. Pseudomonas fluorescens in association with fruit bodies of the ectomycorrhizal mushroom Cantharellus cibarius. Mycological Res. 1993;97:1148–52.
Xing R, Yan H, Gao Q, Zhang F, Wang J, Chen S. Microbial communities inhabiting the fairy ring of Floccularia luteovirens and isolation of potential mycorrhiza helper bacteria. J Basic Microbiol. 2018;58:554–63.
Nazir R, Warmink JA, Boersma H, van Elsas JD. Mechanisms that promote bacterial fitness in fungal-affected soil microhabitats. FEMS Microbiol Ecol. 2010b;71:169–85.
Ingelög T, Nohrstedt HÖ. Ammonia formation and soil pH increase caused by decomposing fruitbodies of macrofungi. Oecologia. 1993;93:449–51.
Nazir R, Tazetdinova DI, van Elsas JD. Burkholderia terrae BS001 migrates proficiently with diverse fungal hosts through soil and provides protection from antifungal agents. Front Microbiol. 2014;5:598.
Warmink JA, Nazir R, Corten B, van Elsas JD. Hitchhikers on the fungal highway: The helper effect for bacterial migration via fungal hyphae. Soil Biol Biochem. 2011;43:760–5.
Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–64.
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 2011;43:1450–5.
Gorka S, Dietrich M, Mayerhofer W, Gabriel R, Wiesenbauer J, Martin V, et al. Rapid transfer of plant photosynthates to soil bacteria via ectomycorrhizal hyphae and its interaction with nitrogen availability. Front Microbiol. 2019;10:168.
Ghodsalavi B, Svenningsen NB, Hao X, Olsson S, Nicolaisen MH, Al-Soud WA, et al. A novel baiting microcosm approach used to identify the bacterial community associated with Penicillium bilaii hyphae in soil. PLoS One. 2017;12:e0187116.
Haq IU, Calixto RO, da R, Yang P, dos Santos GMP, Barreto-Bergter E, et al. Chemotaxis and adherence to fungal surfaces are key components of the behavioral response of Burkholderia terrae BS001 to two selected soil fungi. FEMS Microbiol Ecol. 2016;92:fiw164.
Shin D, Lee Y, Park J, Moon HS, Hyun SP. Soil microbial community responses to acid exposure and neutralization treatment. J Environ Manag. 2017;204:383–93.
Nazir R, Boersma FGH, Warmink JA, van Elsas JD. Lyophyllum sp. strain Karsten alleviates pH pressure in acid soil and enhances the survival of Variovorax paradoxus HB44 and other bacteria in the mycosphere. Soil Biol Biochem. 2010a;42:2146–52.
Izumi H, Anderson IC, Alexander IJ, Killham K, Moore ERB. Diversity and expression of nitrogenase genes (nifH) from ectomycorrhizas of Corsican pine (Pinus nigra). Environ Microbiol. 2006;8:2224–30.
Jayasinghearachchi HS, Seneviratne G. Can mushrooms fix atmospheric nitrogen? J Biosci. 2004;29:293–6.
Barbieri E, Ceccaroli P, Saltarelli R, Guidi C, Potenza L, Basaglia M, et al. New evidence for nitrogen fixation within the Italian white truffle Tuber magnatum. Fungal Biology. 2010;114:936–42.
Hoppe B, Kahl T, Karasch P, Wubet T, Bauhaus J, Buscot F, et al. Network analysis reveals ecological links between N-fixing bacteria and wood-decaying fungi. PLos One. 2014;9:e88141.
Pavić A, Stanković S, Saljnikov E, Krüger D, Buscot F, Tarkka M, et al. Actinobacteria may influence white truffle (Tuber magnatum Pico) nutrition, ascocarp degradation and interactions with other soil fungi. Fungal Ecol. 2013;6:527–38.
Calvaruso C, Turpault MP, Leclerc E, Frey-Klett P. Impact of ectomycorrhizosphere on the functional diversity of soil bacterial and fungal communities from a forest stand in relation to nutrient mobilization processes. Microb Ecol. 2007;54:567–77.
Fontaine L, Thiffault N, Paré D, Fortin JA, Piché Y. Phosphate-solubilizing bacteria isolated from ectomycorrhizal mycelium of Picea glauca are highly efficient at fluorapatite weathering. Botany. 2016;94:1183–93.
Rodríguez H, Fraga R. Phosphate solubilizing bacteria and their role in plant growth promotion. Biotechnol Adv. 1999;17:319–39.
Hervé V, Le Roux X, Uroz S, Gelhaye E, Frey-Klett P. Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium during wood decay. Environ Microbiol. 2014;16:2238–52.
Sun H, Terhonen E, Kasanen R, Asiegbu FO. Diversity and community structure of primary wood-inhabiting bacteria in boreal forest. Geomicrobiol J. 2014;31:315–24.
López-Mondéjar R, Brabcová V, Štursová M, Davidová A, Jansa J, Cajthaml T, et al. Decomposer food web in a deciduous forest shows high share of generalist microorganisms and importance of microbial biomass recycling. ISME J. 2018;12:1768–78.
Stopnisek N, Zühlke D, Carlier A, Barberán A, Fierer N, Becher D, et al. Molecular mechanisms underlying the close association between soil Burkholderia and fungi. ISME J. 2016;10:253–64.
Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–38.
Timonen S, Hurek T. Characterization of culturable bacterial populations associating with Pinus sylvestris —Suillus bovinus mycorrhizospheres. Can J Microbiol. 2006;52:769–78.
Uroz S, Oger P, Morin E, Frey-Klett P. Distinct ectomycorrhizospheres share similar bacterial communities as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol. 2012;78:3020–4.
Acknowledgements
We thank Leho Tedersoo and three anonymous reviewers for constructive comments on the paper. We also thank Rasmus Puusepp for laboratory assistance. Funding was provided by Estonian Research Council grants (PUT1317 and IUT20-30), Swedish Research Council (Vetenskapsrådet, grant no: 2017‐05019) and the European Union through the European Regional Development Fund (the Center of Excellence EcolChange).
Author information
Authors and Affiliations
Contributions
MP, MB, and KP contributed to study design and writing; MP performed molecular and data analysis.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pent, M., Bahram, M. & Põldmaa, K. Fruitbody chemistry underlies the structure of endofungal bacterial communities across fungal guilds and phylogenetic groups. ISME J 14, 2131–2141 (2020). https://doi.org/10.1038/s41396-020-0674-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0674-7
This article is cited by
-
Fusarium fruiting body microbiome member Pantoea agglomerans inhibits fungal pathogenesis by targeting lipid rafts
Nature Microbiology (2022)
-
Bacterial communities associated with mushrooms in the Qinghai-Tibet Plateau are shaped by soil parameters
International Microbiology (2022)
-
Widespread bacterial diversity within the bacteriome of fungi
Communications Biology (2021)
