
Abstract
Rhizopus microsporus is an early-diverging fungal species with importance in ecology, agriculture, food production, and public health. Pathogenic strains of R. microsporus harbor an intracellular bacterial symbiont, Mycetohabitans (formerly named Burkholderia). This vertically transmitted bacterial symbiont is responsible for the production of toxins crucial to the pathogenicity of Rhizopus and remarkably also for fungal reproduction. Here we show that R. microsporus can live not only in symbiosis with bacteria but also with two viral members of the genus Narnavirus. Our experiments revealed that both viruses replicated similarly in the growth conditions we tested. Viral copies were affected by the developmental stage of the fungus, the substrate, and the presence or absence of Mycetohabitans. Absolute quantification of narnaviruses in isolated asexual sporangiospores and sexual zygospores indicates their vertical transmission. By curing R. microsporus of its viral and bacterial symbionts and reinfecting bacteria to reestablish symbiosis, we demonstrate that these viruses affect fungal biology. Narnaviruses decrease asexual reproduction, but together with Mycetohabitans, are required for sexual reproductive success. This fungal–bacterial-viral system represents an outstanding model to investigate three-way microbial symbioses and their evolution.
Similar content being viewed by others


Introduction
Early-diverging fungal lineages belonging to the phylum Mucoromycota often live in symbioses with bacteria [1, 2]. Fungi are affected by these bacterial symbionts like plants and animals are influenced by their microbiome, suggesting that fungi can also be considered holobionts [3].
One of the best models to study fungal–bacterial symbiosis is the interaction between R. microsporus and Burkholderia rhizoxinica or B. endofungorum [4,5,6]. These endofungal bacteria recently formed the novel genus Mycetohabitans that belongs to Burkholderia sensu lato [7, 8]. Unlike most vertically transmitted endosymbionts, in this system, Koch’s postulates can be fully applied to disentangle functions encoded by either the fungus or the bacterium during their interaction, since both partners can be grown and transformed independently, and the symbiosis can be reestablished through cocultivation [9]. Moreover, recently sequenced genomes of asymbiotic and symbiotic R. microsporus and their endobacteria [10,11,12,13], together with novel “omics” techniques, are helping to decipher how these fungal–bacterial symbioses function and evolve.
The symbioses between R. microsporus and Mycetohabitans spp. have revealed specialized secondary metabolites produced by the bacterial symbionts that are important for Rhizopus’ pathogenicity. The first one elucidated was rhizoxin [9, 14], the potent antimitotic toxin that causes rice seedling blight; the second was rhizonin, one of the first toxins described in Mucorales [15], followed recently by holrhizin A and heptarhizin [16, 17]. Despite high similarity among all bacterial symbionts [5], only rhizoxins and holrhizin A are produced by all Mycetohabitans strains characterized to date, while rhizonin and heptarhizin are distributed in fewer strains. Besides enhancing the metabolic capabilities of their fungal hosts, bacterial endosymbionts control their asexual reproduction [18], and also influence their sexual life cycle, being required for abundant zygospore production [19].
Several mechanisms employed by the bacterial symbionts for interacting with R. microsporus have been investigated in the past 15 years. Bacterial secretion systems play a pivotal role in the functioning of the symbiosis. For instance, a type II secretion system and the production of fungal cell wall-degrading enzymes chitinase and chitosanase enable bacteria to invade its fungal host [20]. A functional type III secretion system, which is assumed to deliver effector proteins to the fungal cytosol, is also required for effective fungal colonization and elicitation of asexual reproduction [21]. Studies focused on the fungal partner revealed that the fungal lipidome undergoes dramatic changes in response to Mycetohabitans. These changes are likely necessary for a mutualistic relationship with bacteria [13].
We recently used RNA sequencing data to further investigate the molecular mechanisms underlying the maintenance of this fungal–bacterial symbiosis [22]. Surprisingly, these analyses led us to the discovery of unexpected microbial partners: the narnaviruses.
What we know today about viruses of the genus Narnavirus (family Narnaviridae) is derived from pioneering studies in Saccharomyces cerevisiae [23,24,25,26]. In yeast, these simplest of known RNA viruses are unencapsidated molecules of positive single-strand RNA of 2.3–2.9 kb [27] that encode a single protein of 80–140 kDa with amino acid sequence motifs characteristic of RNA-dependent RNA polymerases (RdRp) [28]. Genetic and biochemical evidence in yeast showed that these viruses are not encoded in the host genome and localize in the cytoplasm. They form 1:1 ribonucleotide complexes with their RdRp proteins and reach a high copy number (~100,000 copies per cell) under stress conditions, such as growth under nitrogen starvation [29].
Recently, novel narnaviruses had been described in Phytophthora infestans, the oomycete that caused the Irish potato famine [30]; in a strain of the fungus Fusarium poae isolated from wheat [31]; in the protozoan Leptomonas seymouri that parasitizes insects and has been involved in cases of leishmaniasis [32, 33]; in marine invertebrates [34]; in members of the phylum Arthropoda [35], as well as in Caenorhabditis nematodes [36]. These reports show that narnaviruses are not confined to the fungal kingdom and may be capable of horizontal transmission. Narnaviruses do not cause any apparent phenotype in their host. However, in-depth studies have been hampered by the difficulty of generating virus-free strains or by the fact that their eukaryotic hosts harbor a complex and highly diverse viral community [26, 30, 37, 38].
In this study, we demonstrate the presence of two members of the genus Narnavirus in R. microsporus and characterize their incidence and replication during fungal development and reproduction. We also evaluated the influence of symbiotic bacteria and substrate on viral transcription. Finally, we generated combinations of narnaviruses-free, bacteria-free, and bacteria-reinfected strains that allowed us, for the first time, to dissect a tripartite microbial symbiosis between fungi, bacteria and viruses.
Materials and methods
RNA extraction and construction of libraries
Total RNA was extracted, and libraries were constructed as previously described [19]. Briefly, fungal strains 813 and 814 wt (Table 1) and those cured of endobacteria (b−) were grown alone or together for sexual reproduction for 6 days at 30 °C in half-strength PDA. In total, six different conditions were evaluated, each with two biological replicates (12 libraries in total).
RNA-Seq adapter trimming
Quality analyses were done with fastQC ver. 0.11.2, including GC content distribution plots. Adapter removal and low-quality sequences trimming were done with Trimmomatic ver. 0.32. Default parameters were used for all software unless otherwise specified.
Genomic database construction for annotation of RNA-seq reads
A mixed genome reference database was built with eight R. microsporus rRNA gene sequences downloaded from Silva (Table S1), the full genomes of R. microsporus ATCC 52813, ATCC 52814, Mycetohabitans rhizoxinica HKI 454, as well as 38 representative genomes of Burkholderia sensu lato (Table S2). This reference database was built to capture as much R. microsporus and Burkholderia genetic diversity as possible. We also included the genomes of two Candidatus Glomeribacter strains, as they are reported endofungal Betaproteobacteria [39, 40]. RNA-Seq paired-end reads were aligned with bowtie2 ver. 2.2.3 with -local parameter and the resulting BAM alignment files were sorted according to read the name and indexed with samtools 1.3.1. The organism mapping information was obtained by processing alignments with samtools and custom Perl scripts. Organism contribution was summarized, and stacked bar plots were done in R. The GC content and the mapping organism information were obtained for each read pair. GC content information was split into 20 bins with R’s base cut function. Mapping information was summarized as Rhizopus, Burkholderia, or unmapped.
Discovery of narnaviruses in R. microsporus
Unmapped reads were extracted with samtools and assembled with Trinity ver. 2.1.1. The GC content for assembled contigs was calculated in R with the Biostrings letterFrequency function. In order to quantify the expression of assembled contigs, unmapped reads were mapped to the assembled transcripts with bowtie2. Two contigs explained the origin of the high GC content reads. One ORF was found in each of these high GC contigs using Transdecoder ver. 2.1.0. These ORFs were compared with the NCBI nonredundant protein database with BLASTp ver 2.2.3.1. This revealed that these sequences were similar to S. cerevisiae 20S (ScNV-20S) and 23S (ScNV-23S) narnaviruses. Our high GC contigs were then named R. microsporus 20S RNA narnavirus (RmNV-20S) and R. microsporus 23S RNA narnavirus (RmNV-23S), respectively. These sequences are available in GenBank under accession numbers MK204624 and MK204625. Other unmapped assembled transcripts were also analyzed for their open reading frames with Transdecoder ver. 2.1.0. Protein predictions were compared with the NCBI viral protein database with BLASTp 2.2.31+. A total of seven assembled transcripts had a BLASTp hit (E value < 0.1) with a protein annotated as RdRp. Of these proteins, only two had the GDD motif characteristic of viral RdRps: the RmNV-20S and the RmNV-23S.
Organism contribution to RNA-Seq libraries
To measure the contribution of narnaviruses to the RNA-Seq libraries, these two sequences were added to the mixed organism reference genomic database mentioned previously. We included 91 sequences that were similar to RdRps according to blastx comparison with NCBI virus protein sequence database. Paired-end reads were aligned with bowtie2, and mapping information was summarized as Rhizopus, Burkholderia, RmNV-20S, RmNV-23S, RdRp, and unmapped.
Phylogeny construction of RdRp viral proteins
We chose representative amino acid RdRp sequences for narnaviruses, ourmiaviruses, mitoviruses, and members of the Leviviridae. Sequences used for phylogeny are summarized in Table S3. Sequences were aligned with muscle v3.8.31 and the multiple sequence alignment was converted to nexus format with seqmagick. Phylogenies were constructed in MrBayes 3.2.3 (Fig. 1c and Fig. S1) [41]. To sample across fixed amino acid rate matrices, we set (aamodelpr = mixed). Two independent chains were used along with 10 M Monte Carlo Markov chain generations. We also used MEGA7 v7.0.26 to further support our phylogenetic analyses (Fig. S2) [42]. The best fitting evolution models for these sequences were searched for with MEGA and prottest 3.4.2. Amino acid phylogenies were constructed using a maximum likelihood method based on the General Reverse Transcriptase model [43]. A gamma distribution was used to model evolutionary rate differences among sites +G parameter = 1.3618. The rate variation model allowed 1.59% of sites to be evolutionary invariable. All positions containing gaps and missing data were eliminated, resulting in 252 positions in the final dataset. An initial tree for the heuristic search was obtained applying the neighbor-joining algorithm, and 1000 bootstrap replicates were calculated for tree reconstruction. Both phylogenies were visually adjusted with FigTree 1.4.4. for final publication.

a Distribution of RNA-Seq reads in libraries of strain 813 wt and 814 wt by GC content and organism. b Percentage of reads mapping to Burkholderia sensu lato, Rhizopus, RmNV-20S and RmNV-23S narnaviruses, RNA-dependent RNA polymerases (RdRp), and unmapped reads across all libraries, each with two biological replicates. c Phylogenetic tree of selected viral RdRp proteins. The evolutionary history was inferred using MrBayes under a mixed amino acid model. The posterior probability support is shown next to each node. Two independent chains were used along with 10 M Monte Carlo Markov chain generations. The analysis involved 32 amino acid sequences.
Fungal strains and culturing conditions
Wild-type strains used in this study were obtained from the American Type Culture Collection, as described in Table 1. Fungal isolates were grown and maintained in PDA (4 g L−1 potato extract, 10 g L−1 dextrose, and 15 g L−1 agar) at 30 °C. In total, 2 or 3 days old grown mycelia from PDA agar plates were used as inocula for all experiments. Substrates for viral quantification consisted in half-strength PDA (2 g L−1 potato extract, 10 g L−1 dextrose, and 15 g L−1 agar, high nutrient) and for nutrient stress [24, 29], 2 days old grown mycelia from YPAD agar plates (10 g L−1 yeast extract, 20 g L−1 peptone, 40 mg L−1 adenine, 20 g L−1 dextrose, and 20 g L−1 agar) were aseptically transferred to potassium acetate agar (2 g L−1 potassium acetate, 15 g L−1 agar, pH 7.0, low nutrient) and incubated at 30 °C. Biomass was harvested at four developmental stages: lag, exponential, stationary, and decline. Sporangiospores were only collected at the decline phase.
Sexual reproduction was assessed on MEA plates (12.75 g L−1 maltose, 2.75 g L−1 dextrose, 0.78 g L−1 peptone, 2.25 g L−1 glycerol, and 15 g L−1 agar) by co-inoculating disks of mycelia from positive (813 wt) and negative (814 wt) mating type strains at ~5 mm distance for 7 days at 30 °C in darkness.
Spore isolation and quantification
Asexual and sexual spores were detached from agar plates in ~10–15 mL 0.01% Triton X-100 with the aid of a glass spatula. High-quality spores were separated from aggregates and cell debris by filtering the spore suspension through a polyester fiber. For acid nucleic extraction, the spore solution was centrifuged in a 10–80% sucrose gradient at 4500 rcf for 30 min at 4 °C. Sporangiospores and zygospores were harvested from 40% and 50% sucrose fraction, respectively. For all experiments, the sporangiospores were quantified in a Neubauer chamber, and both sporangiospores and zygospores were observed under the light microscope (Leica DM750).
Generation and characterization of narnaviruses-free, bacteria-free, and bacteria-reinfected R. microsporus strains
The narnaviruses-free strains (nv−) were generated from protoplasts regenerants that grew under chemotherapy using ribavirin or cycloheximide (both from Sigma-Aldrich). In brief, 1 × 104 sporangiospores were inoculated into 250 mL flask with 50 mL PDB up to germination (4–6 h). The germinants were collected by centrifugation at 4500 rcf for 10 min at 4 °C and washed twice in 30 mL 0.6 M KCl. Protoplasts were formed after 4–6 h of incubation in 10 mL digesting buffer (0.6 M KCl, 20 mg mL−1 Lysing enzymes from Trichoderma harzianum, Sigma-Aldrich) at 30 °C with gentle shaking (~60 rpm). Protoplasts were passed through a mesh of polyester fiberfill, and the buffer was removed by centrifugation at 180 rcf for 15 min, and the pellet was carefully suspended in 30 mL 0.6 M KCl. With a cut pipette tip, 100 µL of protoplast suspension was placed in a petri dish and mixed with 20 mL regeneration medium (0.7 M sucrose, 0.5 g L−1 yeast extract, 1.5 g L−1 agar, and 0.6 M KCl) [44]. Regenerants were incubated at room temperature until the appearance of colonies (2–4 days). After that, the colonies were selected and inoculated on plates of half-strength PDA supplemented with 100 µg mL−1 ribavirin or 25 µg mL−1 cycloheximide at room temperature in darkness [45]. Successively, from the periphery of each growing fungal colony, a small piece of mycelia was cut with a scalpel and grown again under chemotherapy. The procedure was repeated until none of the narnaviruses could be detected by RT-PCR (up to 7 and 15 times when using cycloheximide and ribavirin, respectively).
The bacteria-free strains (b−) were obtained by constantly growing mycelia in PDB (4 g L−1 potato extract and 20 g L−1 dextrose) supplemented with 50 µg mL−1 ciprofloxacin until the lack of sporulation, as previously described [9, 18]. Bacteria-reinfected strains (b*) were obtained by coculturing b− strains (colonies of 4–6 days old on half-strength PDA) with Mycetohabitans sp. B7, previously isolated from 814 wt as previously described [9]. The bacterial pre-inoculum was prepared by growing Mycetohabitans sp. B7 in 10 mL TSB-glycerol (17 g L−1 tryptone, 3 g L−1 soytone, 2.5 g L−1 glucose, 5 g L−1 sodium chloride, and 2% glycerol) until OD600 ≥ 0.5. The cells were pelleted at 4500 rcf for 10 min and resuspended in 1 mL TSB-glycerol. The center of the fungal colony was aseptically cut with a sterile scalpel several times, and the bacterial suspension was added. The plates were incubated face up at 30 °C and monitored until sporulation. The genotypes of all strain variants were confirmed by PCR and RT-PCR using specific primers as described below.
All generated strains were characterized on their growth and asexual reproduction on half-strength PDA for six days at 30 °C, while matings with strain 813 wt were assessed in MEA as described above.
Nucleic acid isolation, amplification, and sequencing
Total RNA and gDNA were extracted from fungal mycelia or spores, ground in liquid nitrogen and suspended in 1 mL Trizol reagent (Invitrogen), following the manufacturer’s instructions. Complementary DNA was synthesized using 1 µg of total RNA previously treated with DNase I (Thermo Fisher Scientific) suspended in 9 µL volume with 1 µL of random primer (50 µM) and 1 µL of dNTP mix (10 mM each) for 5 min at 65 °C; later 2 µL of 10X RT Buffer, 4 µL of MgCl2 (25 mM), 2 µL of DTT (0.1 M), and 200 U of Super Script III Reverse Transcriptase (Invitrogen) were added to the reaction tube and incubated for 10 min at 25 °C, followed by 50 min at 50 °C and finally 5 min at 85 °C for inactivation. Narnaviruses were amplified by RT-PCR using two specifically designed pair of primer pairs: Rm_20S_F, Rm_20S_R (5′-TCTCACGTTGTCCGGAAGC-3′, 5′-GCCCAACCGTGAATCAGG-3′) and Rm_23S_F, Rm_23S_R (5′-TTGCCCGCACTAAAGAGGG-3′, 5′-TTTAGGTCACTGGCTGCGG-3′). Both viral amplicons were cloned in vector pJET 1.2 (Thermo Fisher Scientific). Based on the RNA-Seq analyses across all R. microsporus libraries [22], the fungal acetyl-CoA transporter gene (ACoA, protein id 287373), and the bacterial glucose-6-phosphate isomerase gene (GPI, protein id RBRH_01609) were identified as good housekeeping genes. ACoA was amplified using ACoA_F and ACoA_R oligonucleotides (5′-AGAAACCGTTGGCCGAAGAC-3′, 5′-GGCGTCTATGTTGTTGCCGA-3′), while the GPI gene was amplified with GPI_F and GPI_R primers (5′-CAGGAAGTGGTGCGTTTTGG-3′, 5′-TCATCACCGACAATCCGACC-3′). All products and cloned amplicons were confirmed by Sanger Sequencing, conducted at the Genomic Services Laboratory from LANGEBIO—CINVESTAV Irapuato, Mexico.
Quantitative PCR
The absolute and relative quantification of fungal, bacterial, and viral genes was performed in a CFX96 Touch™ Real-Time PCR Detection System (Biorad). A typical PCR reaction comprised 5 µL of 2× Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific), 0.5 µM of each primer, 50 ng of cDNA, and nuclease-free water up to 10 µL volume. The optimized running protocol consisted of denaturation for 2 min at 95 °C; 50 cycles of 95 °C for 30 s, 62 °C for 30 s, and 72 °C for 1 min; and a melting curve from 50.0 to 95 °C, with a 0.5 °C increment each 5 s. For absolute quantification, plasmids of cloned narnaviruses served as standards. The efficiency of fungal and bacterial primers was also estimated by employing serial dilutions of gDNA; no-template control and no-reverse transcriptase reactions were included in every experiment. Output data were examined with CFX Manager™ Software (Biorad).
Statistical analyses
All statistical analyses were performed in R 3.5.1.
Results
Discovery of narnaviruses in symbiotic R. microsporus
In our search for fungal and bacterial genes essential for the maintenance of the symbiosis, we performed and analyzed ribo-depleted RNA-Seq data of two symbiotic R. microsporus strains (813 wt and 814 wt, Table 1) during asexual and sexual reproduction [22]. Most sequence reads mapped to R. microsporus or Burkholderia sensu lato. However, the libraries that involved strain 814 wt showed a clear peak of unmapped reads, with a GC content slightly above the 60.27% expected for M. rhizoxinica (Fig. 1a) [11]. RNA-Seq de novo assembly revealed that two contigs of unknown origin contained the vast majority of unmapped reads. BLASTp analysis revealed that each of these two sequences contained an ORF encoding probable viral RdRp [46]. We further confirmed that these viral sequences comprised 4.0–21.5% of the reads in all libraries in which strain 814 was involved, including those cured of endobacteria (814 wt and b− libraries, Fig. 1b). These two viral sequences represented 6.8 times more RNA-Seq reads than those mapped to Burkholderia in strain 814. Importantly, these sequences were absent in strain 813 (Fig. 1b).
Phylogenetic analyses of these two new viral RdRp proteins showed that they are members of the genus Narnavirus, family Narnaviridae (Fig. 1c, Fig. S1 and S2). One of them is most related to Saccharomyces 20S RNA narnavirus (ScNV-20S), the type strain of the genus (bootstrap 1), while the second is related to Saccharomyces 23S RNA narnavirus. Thus, we named these novel viral species R. microsporus 20S RNA narnavirus (RmNV-20S) and R. microsporus 23S RNA narnavirus (RmNV-23S) following the ICTV taxonomy since sequence similarity is below 50% in both cases (42.9 and 36.7%, respectively) [47]. Finally, we confirmed by RT-PCR and sequencing that strain 814 wt indeed harbors both narnaviruses, but strain 813 does not (Fig. S3).
In-depth mining of all unmapped reads for other putative viral RdRps revealed five additional sequences. However, none of them was abundantly transcribed (together less than 0.01% of reads across all libraries, Fig. 1b) and did not contain the GDD motif characteristic of RdRps. Attempts to amplify viral sequences from the genomic DNA yielded negative results, suggesting that these sequences are not integrated into the fungal genome (Fig. S3).
Viral transcription during fungal development and reproduction
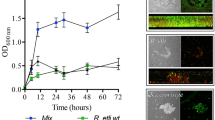
In order to dissect the role of narnaviruses in symbiotic R. microsporus, we evaluated viral replication during fungal development in two different substrates: low and high-nutrient media. The low nutrient medium, based on potassium acetate, has been extensively used in S. cerevisiae to foster ScNV-20S and ScNV-23S replication [25]. The second served as the substrate for our R. microsporus RNA-Seq libraries [19]. As lack of endobacteria impedes asexual reproduction of the fungal host [18], we wondered if the absence of the symbiotic Mycetohabitans would also affect viral transcription. Our qRT-PCR experiments showed that RmNV-20S and RmNV-23S were similarly transcribed in the growth conditions we tested (Fig. 2a, b, F1,349 = 3.7, P = 0.06), but viral copies were affected by the developmental stage (F3,349 = 53.3, P < 0.001), the substrate (F1,349 = 40.7, P < 0.001), and the presence/absence of Mycetohabitans (F1,349 = 49.6, P < 0.001) (Fig. 2a). Comparisons of viral replication for each virus in each substrate revealed that in the low nutrient medium, fungal development (F3,67 = 40.4 and F3,73 = 21.5, P < 0.001), the presence/absence of Mycetohabitans (F1,67 = 48.0 and F1,73 = 19.3, P < 0.001), and the interaction of these two factors (F3,67 = 40.4, P < 0.001; F1,73 = 2.1, P = 0.1) influenced 20S and 23S viral replication. In this substrate, viral copies fluctuated during development and were lowest in the strain devoid of endobacteria at the decline phase (just around 30–150 viruses per ng cDNA, Fig. 2a).

a Absolute quantification of RmNV-20S and RmNV-23S by RT-qPCR in 814 wt and 814 b− strains grown on potassium acetate agar (low nutrient) and half-strength PDA (high nutrient). On the x axis, fungal development from lag (1) to decline (4). Each dot corresponds to a single viral measurement. Bars indicate the arithmetical mean, dotted lines show the tendency, letters and letters with prime symbol above dots represent statistical differences between development stages in RmNV-20S (20S) or RmNV-23S (23S), respectively, by one-way ANOVA followed by Tukey’s post-hoc test, P < 0.05, n = 5 to 31. b Vertical transmission of narnaviruses through asexual and sexual spores. No statistical differences between Narnavirus species were found in sporangiospores or zygospores, but in viral loads in sporangiospores produced in different nutrient conditions by one-way ANOVA, *P < 0.05, n = 3. c Micrographs of reproductive fungal structures. From left to right: sporangiophore, isolated sporangiospore, zygospores formed by mating, and isolated zygospore. All pictures are from 814 wt, except zygospores formed by mating that includes 813 wt strain. Scale bars = 50 µm.
In the high nutrient medium, viral copies were only affected by the developmental stage of the fungus (F3,86 = 82.0 and F3,117 = 72.0, P < 0.001, for 20S and 23S, respectively). In this rich substrate, viruses significantly increased their numbers during development from around 1500 to 4.5 million viral copies per ng cDNA (Fig. 2a).
We further hypothesized that these abundant viruses should be maintained during both asexual and sexual reproduction, since narnaviruses are not known for having an extracellular life phase [26, 47]. To test these hypotheses, we isolated sporangiospores from strain 814 wt and zygospores resulting from the mating between strain 813 wt and 814 wt. Absolute quantification of viral copies in intact sporangiospores and zygospores revealed high numbers of narnaviruses in these reproductive structures (Fig. 2b, c). However, viral titers of RmNV-20S and RmNV-23S were affected by the substrate in which asexual reproduction took place, being significantly higher in the low nutrient medium (one-way ANOVA, *P < 0.05, n = 3). Similar viral copies of the two narnaviruses were also detected in isolated zygospores obtained in MEA plates (Fig. 2b).
Generation and characterization of narnaviruses-free, bacteria-free, and bacteria-reinfected R. microsporus strains
To characterize the role of narnaviruses in symbiotic Rhizopus, we combined the use of an antiviral agent (ribavirin or cycloheximide) and an antibiotic (ciprofloxacin) to generate narnaviruses-free (nv−) and bacteria-free (b−) fungal strains. We additionally reinfected symbiotic bacteria (b*) in all b− strains to investigate if the presence/absence of narnaviruses could influence the re-establishment of the fungal–bacterial symbiosis. As described in Table 1 and exemplified in Fig. 3a, we successfully generated 17 strains derived from R. microsporus 814 wt (nv+b+). Importantly, we generated five independent narnaviruses-free strains to account for the likely effects caused by the employment of ribavirin or cycloheximide (Fig. S4). We also verified the presence/absence of Mycetohabitans by microscopy (Fig. S5). Further, we quantified the abundance of each symbiont under standard growing conditions (day 6th of growth, 30 °C, half-strength PDA) by RT-qPCR (narnaviruses) and qPCR (bacteria and fungi). These results confirmed the lack of narnaviral sequences in narnaviruses-free strains, the absence of bacterial gene copies in all bacteria-free strains, and the presence of only fungal gene copies in the narnaviruses-bacteria-free strains (Fig. 3b). Interestingly, bacterial GPI copies were the only ones that varied in the reinfected strains (ANOVA followed by Tukey’s post-hoc test, P < 0.05, n = 3 to 15), being reduced in the nv− strains when compared with nv+ (Fig. 3b). This suggests that bacterial colonization and/or replication may be reduced in the absence of the narnaviruses. Moreover, relative quantification of bacterial GPI and viral 20S and 23S support the notion that narnaviruses are ~3.9 and 3.3-fold more abundant in the fungal cells than Mycetohabitans (Fig. S6), respectively, consistent with our RNA-seq data.

a Molecular genotyping of strain 814 wt and all its variants. M molecular marker, ACoA fungal housekeeping acetyl-CoA transporter gene (392 pb), GPI bacterial housekeeping glucose-6-phosphate isomerase gene (87 pb), 20S and 23S represent the RmNV-20S (683 pb) and RmNV-23S (537 pb) genomes, respectively. b Absolute quantification of housekeeping fungal and bacterial genes by qPCR (using genomic DNA) and viral load by RT-qPCR (using cDNA). Log10 copies of each symbiont was determined per ng of total nucleic acids extracted. To offset the variation in symbionts proportion per ng of nucleic acids, the fungal gen ACoA was used for normalization of strain-to-strain differences. Nucleic acids were extracted at 6-old day cultures on half-strength PDA at 30 °C. Each symbol corresponds to a single gene measurement and bars indicate the arithmetical mean. Different letters denote statistical differences between strains for each target by one-way ANOVA followed by Tukey’s post-hoc test, P < 0.05, n = 3 to 65 for each strain type.
Effect of narnaviruses and Mycetohabitans on fungal growth and reproduction
Once we succeeded in generating narnaviruses-free and bacteria-free strains and reestablishing symbioses by reinfecting Mycetohabitans (Fig. 3 and Fig. S4), we characterized the impact of narnaviruses and their interaction with symbiotic bacteria, on the fitness of R. microsporus. First, we evaluated the biomass produced under standard conditions finding no significant differences due to narnaviruses across groups of strains (Fig. S7). These results suggest that neither ribavirin/cycloheximide nor ciprofloxacin drastically affected the fungal host.
The absence of RmNV-20S and RmNV-23S during asexual reproduction increased the number of sporangiospores produced by the fungus in comparison to the strains harboring them (Fig. 4a). Since endobacteria are critical for the asexual reproduction of their host, bacteria-free strains do not produce sporangiospores [18]. However, sporulation recovers after reintroduction of the bacterial symbiont, as observed for strain nv+b*. Remarkably, strains nv−b* recovered the same high sporulation phenotype as the nv−b+ strains, suggesting RmNV-20S and RmNV-23S did not directly interfere in the triggering of the asexual reproduction process.

a Sporangiospores produced by 814 wt and its variants on half-strength PDA at 30 °C, after 6 days of culture. b Zygospores produced by the mating of strain 813 wt and 814 wt and its variants on MEA medium at 30 °C. Each symbol represents spores quantified from a single agar plate. Bars indicate the arithmetical mean. Different letters denote statistical differences between strains by one-way ANOVA followed by Tukey’s post-hoc test, P < 0.05, n = 4–6, for each strain as described in Table 1. * most strains nv−b− did not produce zygospores.
Analyses of the sexual reproduction of R. microsporus using 813 wt and all six variant groups of strain 814 revealed that the production of zygospores was nearly abolished when the fungus 814 grew without its viral and bacterial symbionts (Fig. 4b). Only in this case (nv−b−), 17 out of 20 (85%) of the matings rendered 0 zygospores, and the remaining three produced a significantly lower amount of them compared with strain 814 wt (24 vs. 82 zygospores, 70% less). Surprisingly, elimination and reinfection of bacteria in a narnaviruses-positive background increased the number of zygospores produced (Fig. 4b).
Discussion
Our work demonstrates the presence of two novel viral partners of the genus Narnavirus in the symbiosis between R. microsporus and Mycetohabitans species. Strikingly, these viral symbionts were only present in one of the strains analyzed, even though both strains establish symbioses with bacteria. Mycoviruses in filamentous fungi have been studied for decades [48]. Some of them are associated with a reduction of host virulence in plant pathogens [49]. However, in early-diverging fungal lineages, such as the Mucoromycotina, Glomeromycotina, and Mortierellomycotina, the role and evolutionary origins of viruses remain mostly terra incognita [50, 51]. Only the virome of some strains of Rhizophagus and Gigaspora, both arbuscular mycorrhizal fungi (AMF, Glomeromycotina), have been studied. Rhizophagus clarus strain RF1 was found to harbor four dsRNA fragments of different sizes, including a Mitovirus (RcMV1-RF1) and an unclassified mycovirus (GRF1V-M) [52]. Six Mitovirus genomes were associated with strains of R. irregularis and R. diaphanum based on transcriptomic data and in silico analyses [53]. Gigaspora margarita BEG34 harbors endobacteria Candidatus Glomeribacter gigasporarum [54]. It possesses a diverse viral community formed by four mitoviruses, one Ourmia-like virus, one Giardia-like virus, and two sequences related to Fusarium graminearum mycoviruses [55]. These reports highlight the presence of members of the genus Mitovirus in AMF (Glomeromycotina), which only differ from members of the genus Narnavirus in their localization (mitochondria instead of cytoplasm) and codon usage [26]. Recent reports have also shown that members of the Ourmiavirus, which were first identified in plants, are also widespread in fungi [56]. Depending on the RdRp sequences used for the phylogeny, ourmiaviruses form a sister clade to the genus Narnavirus (as shown in Fig. 1c and Figs. S1 and S2), or a sister clade to the genus Mitovirus [56]. These phylogenies strongly suggest that RdRps from ourmiaviruses share a common ancestor with the family Narnaviridae. Since early-diverging fungi, especially AMF (Glomeromycotina), but also Endogone (Mucoromycotina), established the most ancestral symbioses with plants ~480 Ma [57,58,59], the transmission of these positive ssRNA viruses from fungi to plants or vice versa is plausible and should be further investigated [51, 60, 61]. Our finding that symbiotic R. microsporus harbors Mycetohabitans endobacteria and narnaviruses RmNV-20S and RmNV-23S prompts several questions: which symbiosis arose first? Did one symbiosis, viral, or bacterial, facilitate the other? To what extent have each of these symbioses contributed to the diversification of fungi and of Rhizopus in particular?
Our functional studies revealed that RmNV-20S and RmNV-23S replicated similarly in all tested conditions (Fig. 2). These viral sequences are not integrated into the host genome, and RNA-Seq and qRT-PCR confirmed their high titers and changes during fungal development in two substrates (Fig. 2a). Replication of narnaviruses in R. microsporus was also affected by the presence/absence of symbiotic Mycetohabitans, unlike what is known for most mycoviruses in G. margarita BEG53 [55], but only when the fungus was grown on a low nutrient medium. These results suggest that the fungal metabolism is rewired and that endobacteria may provide additional metabolic plasticity to support growth and viral replication under these circumstances. Remarkably, although viral replication diminished in the low nutrient medium, viral inheritance seems higher than in spores produced in rich medium. Do these differences reflect an adaptive trait for the progeny? Single-cell studies on the fungal progeny could shed light on this topic.
We also detected and quantified RmNV-20S and RmNV-23S in the zygospores resulting from the mating of strain 813 wt and 814 wt, suggesting that viable sexual progeny will contain these viruses (Fig. 2b). Multilocus sequence analyses on a vast collection of R. microsporus strains from clinical, food, and environmental sources pointed out that strain 813 and 814 form a well-supported clade within the species R. microsporus, and are indeed the tester strains for sexual reproduction in the species [62]. These facts suggest that RmNV-20S and RmNV-23S could be widespread in natural R. microsporus populations, primarily probably in those located in Eurasia (Table 1).
Horizontal transmission of viruses by cell-to-cell fusion (hyphal anastomosis) has been documented in filamentous fungi [63], even across incompatible vegetative individuals [64]. Anastomosis, however, has not been thoroughly studied in Rhizopus, although is common in other coenocytic fungi such as AMF [65, 66]. Since R. microsporus is often implicated in cases of Mucormycosis in humans [67] and is also a known pathogen of plants, horizontal transmission of narnaviruses RmNV-20S and RmNV-23S to novel hosts should be investigated.
We succeeded in generating the first narnaviruses-free fungal strains by using ribavirin and cycloheximide. Ribavirin has successfully eliminated viruses in filamentous fungi [38] and Phytophthora [68]. Attempts to cure narnaviruses in S. cerevisiae that employed high temperature, the addition of cycloheximide, acridine orange, or guanidine HCl, all failed [47]. Noteworthy, used of ribavirin to cure Narnavirus PiRV-4 in P. infestants also failed [30] but did work for virus PiRV-2 [68], suggesting that susceptibility to different antiviral agents is host and virus dependent. Thanks to this achievement, we could evaluate for the first time the impact of members of the Narnavirus in the biology of their host (Figs. 3 and 4).
By comparing narnaviruses-free, bacteria-free, and bacteria-reinfected fungal strains, we demonstrate that narnaviruses RmNV-20S and RmNV-23S do affect fungal biology. These viruses seem to impose a cost or disadvantage to their host during asexual reproduction by reducing the number of sporangiospores produced (Fig. 4a). This reduction may be due to their very high titers (Fig. 3a). However, our experiments also showed that in strain 814 (M−), they and Mycetohabitans are necessary to ensure sexual reproduction. Remarkably, the production of zygospores was nearly abolished in matings between strains 814 nv−b− and 813 wt (b+). A similar phenotype was observed in bacteria-free matings between strains 813 and 814 [19]. It is still cryptic how symbiotic bacteria and viruses participate in the sexual reproduction of Rhizopus. Based on all these results, we propose that RmNV-20S and RmNV-23S are conditional mutualists of R. microsporus, as they provide benefits to their host only in the presence of the bacterial symbionts. Manipulation of narnaviruses and Mycetohabitans in naturally occurring viruses–bacteria-free Rhizopus strains could help determine if this proposal holds.
In R. clarus RF1, naturally occurring virus-free spores produced significantly more fungal spores in Sorghum than the virus-positive isolates, similar to our observations in R. microsporus. In addition, virus-positive spores of R. clarus reduced plant root and shoot biomass, suggesting a decreased benefit of the plant–fungal symbiosis [52].
RNA silencing represents a critical defense against viral attacks in fungi and other eukaryotes [69]. Since Rhizopus, unlike S. cerevisiae, does encode the Dicer and Argonaute genes needed for a functional RNA silencing system [22], how RmNV-20S and RmNV-23S persist in their host remains an open question.
Finally, it is tempting to speculate that the narnaviral RdRp proteins found here, which seem to be extremely effective for viral replication, could amplify RNA fragments encoded by the fungal or bacterial genomes, leading to molecular changes within the holobiont that could be responsible for some of the phenotypes we observe. This hypothesis warrants further investigation.
In summary, our work represents the first step to understand the evolutionary relationships between Rhizopus, Mycetohabitans, and Narnavirus. Further studies using this fungal–bacterial–viral system will deepen our understanding of the mechanisms and ecological consequences of fungal holobionts.
References
Bonfante P, Desirò A. Who lives in a fungus? The diversity, origins and functions of fungal endobacteria living in Mucoromycota. ISME J. 2017;11:1727–35.
Pawlowska TE, Gaspar ML, Lastovetsky OA, Mondo SJ, Real-Ramirez I, Shakya E, et al. Biology of fungi and their bacterial endosymbionts. Annu Rev Phytopathol. 2018;56:289–309.
Partida-Martínez LP. The fungal holobiont: evidence from early diverging fungi. Environ Microbiol. 2017;19:2919–23.
Partida-Martinez LP, Groth I, Schmitt I, Richter W, Roth M, Hertweck C. Burkholderia rhizoxinica sp. nov. and Burkholderia endofungorum sp. nov., bacterial endosymbionts of the plant-pathogenic fungus Rhizopus microsporus. Int J Syst Evol Microbiol. 2007;57:2583–90.
Lackner G, Möbius N, Scherlach K, Partida-Martinez LP, Winkler R, Schmitt I, et al. Global distribution and evolution of a toxinogenic Burkholderia-Rhizopus symbiosis. Appl Environ Microbiol. 2009;75:2982–6.
Partida-Martinez LP. A model for bacterial-fungal interactions. Saarbrücken: LAP LAMBERT Academic Publishing; 2013.
Estrada-de Los Santos P, Palmer M, Chávez-Ramírez B, Beukes C, Steenkamp ET, Briscoe L, et al. Whole genome analyses suggests that Burkholderia sensu lato contains two additional novel genera (Mycetohabitans gen. nov., and Trinickia gen. nov.): implications for the evolution of diazotrophy and nodulation in the burkholderiaceae. Genes. 2018;9:389.
Oren A, Garrity GM. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2016;66:2463–6.
Partida-Martinez LP, Hertweck C. Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature. 2005;437:884–8.
Lackner G, Moebius N, Partida-Martinez L, Hertweck C. Complete genome sequence of Burkholderia rhizoxinica, an endosymbiont of Rhizopus microsporus. J Bacteriol. 2011;193:783–4.
Lackner G, Moebius N, Partida-Martinez LP, Boland S, Hertweck C. Evolution of an endofungal lifestyle: deductions from the Burkholderia rhizoxinica genome. BMC Genom. 2011;12:210.
Horn F, Üzüm Z, Möbius N, Guthke R, Linde J, Hertweck C. Draft genome sequences of symbiotic and nonsymbiotic Rhizopus microsporus strains CBS 344.29 and ATCC 62417. Genome Announc. 2015;3:e01370-14.
Lastovetsky OA, Gaspar ML, Mondo SJ, LaButti KM, Sandor L, Grigoriev IV, et al. Lipid metabolic changes in an early divergent fungus govern the establishment of a mutualistic symbiosis with endobacteria. Proc Natl Acad Sci USA. 2016;113:15102–7.
Partida-Martinez LP, Hertweck C. A gene cluster encoding rhizoxin biosynthesis in “Burkholderia rhizoxina”, the bacterial endosymbiont of the fungus Rhizopus microsporus. ChemBioChem. 2007;8:41–5.
Partida-Martinez LP, Looß CF, de, Ishida K, Ishida M, Roth M, Buder K, et al. Rhizonin, the first mycotoxin isolated from the zygomycota, is not a fungal metabolite but is produced by bacterial endosymbionts. Appl Environ Microbiol. 2007;73:793–7.
Niehs SP, Scherlach K, Hertweck C. Genomics-driven discovery of a linear lipopeptide promoting host colonization by endofungal bacteria. Org Biomol Chem. 2018;16:8345–52.
Niehs SP, Dose B, Scherlach K, Roth M, Hertweck C. Genomics-driven discovery of a symbiont-specific cyclopeptide from bacteria residing in the rice seedling blight fungus. Chembiochem Eur J Chem Biol. 2018;19:2167–72.
Partida-Martinez LP, Monajembashi S, Greulich K-O, Hertweck C. Endosymbiont-dependent host reproduction maintains bacterial–fungal mutualism. Curr Biol. 2007;17:773–7.
Mondo SJ, Lastovetsky OA, Gaspar ML, Schwardt NH, Barber CC, Riley R, et al. Bacterial endosymbionts influence host sexuality and reveal reproductive genes of early divergent fungi. Nat Commun. 2017;8:1843.
Moebius N, Üzüm Z, Dijksterhuis J, Lackner G, Hertweck C. Active invasion of bacteria into living fungal cells. eLife. 2014;3:e03007.
Lackner G, Moebius N, Hertweck C. Endofungal bacterium controls its host by an hrp type III secretion system. ISME J. 2011;5:252–61.
Bermúdez-Barrientos, J. Roberto. Exploring the molecular mechanisms maintaining the Rhizopus microsporus—Burkholderia rhizoxinica symbiosis. Irapuato: CINVESTAV-Irapuato; 2016.
Rodriguez-Cousiño N, Esteban LM, Esteban R. Molecular cloning and characterization of W double-stranded RNA, a linear molecule present in Saccharomyces cerevisiae. Identification of its single-stranded RNA form as 20S RNA. J Biol Chem. 1991;266:12772–8.
Esteban LM, Rodriguez-Cousiño N, Esteban R. T double-stranded RNA (dsRNA) sequence reveals that T and W dsRNAs form a new RNA family in Saccharomyces cerevisiae. Identification of 23S RNA as the single-stranded form of T dsRNA. J Biol Chem. 1992;267:10874–81.
Wejksnora PJ, Haber JE. Ribonucleoprotein particle appearing during sporulation in yeast. J Bacteriol. 1978;134:246–60.
Hillman BI, Cai G. The family narnaviridae: simplest of RNA viruses. In: Ghabrial SA, editor. Advances in virus research, Vol 86: Mycoviruses. San Diego: Elsevier Academic Press Inc.; 2013. pp 149–76.
Rodríguez-Cousiño N, Solórzano A, Fujimura T, Esteban R. Yeast positive-stranded virus-like RNA replicons. 20 S and 23 S RNA terminal nucleotide sequences and 3’ end secondary structures resemble those of RNA coliphages. J Biol Chem. 1998;273:20363–71.
García-Cuéllar MP, Esteban R, Fujimura T. RNA-dependent RNA polymerase activity associated with the yeast viral p91/20S RNA ribonucleoprotein complex. RNA. 1997;3:27–36.
Matsumoto Y, Fishel R, Wickner RB. Circular single-stranded RNA replicon in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1990;87:7628–32.
Cai G, Myers K, Fry WE, Hillman BI. A member of the virus family narnaviridae from the plant pathogenic oomycete Phytophthora infestans. Arch Virol. 2012;157:165–9.
Osaki H, Sasaki A, Nomiyama K, Tomioka K. Multiple virus infection in a single strain of Fusarium poae shown by deep sequencing. Virus Genes. 2016;52:835–47.
Lye L-F, Akopyants NS, Dobson DE, Beverley SM. A narnavirus-like element from the trypanosomatid protozoan parasite Leptomonas seymouri. Microbiol Resour Announc. 2016;4:e00713–16.
Grybchuk D, Akopyants NS, Kostygov AY, Konovalovas A, Lye L-F, Dobson DE, et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc Natl Acad Sci USA. 2018;115:E506–15.
Shi M, Lin X-D, Tian J-H, Chen L-J, Chen X, Li C-X, et al. Redefining the invertebrate RNA virosphere. Nature. 2016;540:539–43.
Fauver JR, Akter S, Morales AIO, Black WC, Rodriguez AD, Stenglein MD, et al. A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery. Virology. 2019;528:181–97.
Richaud A, Frézal L, Tahan S, Jiang H, Blatter JA, Zhao G, et al. Vertical transmission in Caenorhabditis nematodes of RNA molecules encoding a viral RNA-dependent RNA polymerase. Proc Natl Acad Sci USA. 2019;116:24738–47.
Zoll J, Verweij PE, Melchers WJG. Discovery and characterization of novel Aspergillus fumigatus mycoviruses. PLoS ONE. 2018;13:e0200511.
Niu Y, Yuan Y, Mao J, Yang Z, Cao Q, Zhang T, et al. Characterization of two novel mycoviruses from Penicillium digitatum and the related fungicide resistance analysis. Sci Rep. 2018;8:5513.
Ghignone S, Salvioli A, Anca I, Lumini E, Ortu G, Petiti L, et al. The genome of the obligate endobacterium of an AM fungus reveals an interphylum network of nutritional interactions. ISME J. 2012;6:136–45.
Uehling J, Gryganskyi A, Hameed K, Tschaplinski T, Misztal PK, Wu S, et al. Comparative genomics of Mortierella elongata and its bacterial endosymbiont Mycoavidus cysteinexigens. Environ Microbiol. 2017;19:2964–83.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Dimmic MW, Rest JS, Mindell DP, Goldstein RA. rtREV: an amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J Mol Evol. 2002;55:65–73.
Zhang L, Fu Y, Xie J, Jiang D, Li G, Yi X. A novel virus that infecting hypovirulent strain XG36-1 of plant fungal pathogen Sclerotinia sclerotiorum. Virol J. 2009;6:96.
Niu Y, Zhang T, Zhu Y, Yuan Y, Wang S, Liu J, et al. Isolation and characterization of a novel mycovirus from Penicillium digitatum. Virology. 2016;494:15–22.
Routhier E, Bruenn JA. Functions of conserved motifs in the RNA-dependent RNA polymerase of a yeast double-stranded RNA virus. J Virol. 1998;72:4427–9.
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2012.
Ghabrial SA, Castón JR, Jiang D, Nibert ML, Suzuki N. 50-plus years of fungal viruses. Virology. 2015;479–80:356–68.
Hillman BI, Suzuki N. Viruses of the chestnut blight fungus, Cryphonectria parasitica. Adv Virus Res. 2004;63:423–72.
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia. 2016;108:1028–46.
Dolja VV, Koonin EV. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018;244:36–52.
Ikeda Y, Shimura H, Kitahara R, Masuta C, Ezawa T. A novel virus-like double-stranded RNA in an obligate biotroph arbuscular mycorrhizal fungus: a hidden player in mycorrhizal symbiosis. Mol Plant-Microbe Interact. 2012;25:1005–12.
Neupane A, Feng C, Feng J, Kafle A, Buecking H, Lee Marzano S-Y. Metatranscriptomic analysis and in silico approach identified mycoviruses in the arbuscular mycorrhizal fungus Rhizophagus spp. Viruses-Basel. 2018;10:707.
Bianciotto V, Lumini E, Bonfante P, Vandamme P. ‘Candidatus glomeribacter gigasporarum’ gen. nov., sp. nov., an endosymbiont of arbuscular mycorrhizal fungi. Int J Syst Evol Microbiol. 2003;53:121–4.
Turina M, Ghignone S, Astolfi N, Silvestri A, Bonfante P, Lanfranco L. The virome of the arbuscular mycorrhizal fungus Gigaspora margarita reveals the first report of DNA fragments corresponding to replicating non-retroviral RNA viruses in fungi. Environ Microbiol. 2018;20:2012–25.
Li CX, Zhu JZ, Gao BD, Zhu HJ, Zhou Q, Zhong J. Characterization of a novel ourmia-like mycovirus infecting Magnaporthe oryzae and implications for viral diversity and evolution. Viruses. 2019;11:223.
Bidartondo MI, Read DJ, Trappe JM, Merckx V, Ligrone R, Duckett JG. The dawn of symbiosis between plants and fungi. Biol Lett. 2011;7:574–7.
Desirò A, Faccio A, Kaech A, Bidartondo MI, Bonfante P. Endogone, one of the oldest plant-associated fungi, host unique Mollicutes-related endobacteria. New Phytol. 2015;205:1464–72.
Lutzoni F, Nowak MD, Alfaro ME, Reeb V, Miadlikowska J, Krug M, et al. Contemporaneous radiations of fungi and plants linked to symbiosis. Nat Commun. 2018;9:5451.
Roossinck MJ. Evolutionary and ecological links between plant and fungal viruses. New Phytol. 2019;221:86–92.
Andika IB, Wei S, Cao C, Salaipeth L, Kondo H, Sun L. Phytopathogenic fungus hosts a plant virus: a naturally occurring cross-kingdom viral infection. Proc Natl Acad Sci USA. 2017;114:12267–72.
Dolatabadi S, Walther G, Ende van den AHGG, Hoog de GS. Diversity and delimitation of Rhizopus microsporus. Fungal Divers. 2013;64:145–63.
Ghabrial SA. Origin, adaptation and evolutionary pathways of fungal viruses. Virus Genes. 1998;16:119–31.
Hamid MR, Xie J, Wu S, Maria SK, Zheng D, Assane Hamidou A, et al. A novel deltaflexivirus that infects the plant fungal pathogen, Sclerotinia sclerotiorum, can be transmitted among host vegetative incompatible strains. Viruses. 2018;10:295.
Chagnon P-L. Ecological and evolutionary implications of hyphal anastomosis in arbuscular mycorrhizal fungi. FEMS Microbiol Ecol. 2014;88:437–44.
de Novais CB, Pepe A, Siqueira JO, Giovannetti M, Sbrana C, de Novais CB, et al. Compatibility and incompatibility in hyphal anastomosis of arbuscular mycorrhizal fungi. Sci Agric. 2017;74:411–6.
Itabangi H, Sephton-Clark PCS, Zhou X, Insua I, Probert M, Correia J, et al. A bacterial endosymbiont enables fungal immune evasion during fatal mucormycete infection. 2019:584607.
Cai G, Fry WE, Hillman B. PiRV-2 stimulates sporulation in Phytophthora infestans. Virus Res. 2019;271:197674.
Nuss DL. Mycoviruses, RNA silencing, and viral RNA recombination. Adv Virus Res. 2011;80:25–48.
Acknowledgements
Authors acknowledge María Nélida Vázquez Sánchez for technical support; Luis Delaye for advice on the phylogenetic analyses, as well as Karin Grothe, Robert Winkler, and the three anonymous referees for their useful comments. LPPM is thankful to Consejo Nacional de Ciencia y Tecnología (CONACyT) in Mexico, which financed most of this research with grant FOINS-2015-01-006.
Author information
Authors and Affiliations
Contributions
LPPM, ANEV, JRBB, and CAG: designed and planned research; ANEV, JRBB, JFCR, GCL, FMC, AMV, DACP, LPPM: performed experiments and analyzed the data; SJM and TEP generated RNA-Seq libraries and provided R. microsporus wild-type strains; LPPM secured funding; LPPM and ANEV wrote the paper; all authors read, improved, and approved the final version of this document.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Espino-Vázquez, A.N., Bermúdez-Barrientos, J.R., Cabrera-Rangel, J.F. et al. Narnaviruses: novel players in fungal–bacterial symbioses. ISME J 14, 1743–1754 (2020). https://doi.org/10.1038/s41396-020-0638-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0638-y
