
Abstract
Interspecies hydrogen transfer in anoxic ecosystems is essential for the complete microbial breakdown of organic matter to methane. Acetogenic bacteria are key players in anaerobic food webs and have been considered as prime candidates for hydrogen cycling. We have tested this hypothesis by mutational analysis of the hydrogenase in the model acetogen Acetobacterium woodii. Hydrogenase-deletion mutants no longer grew on H2 + CO2 or organic substrates such as fructose, lactate, or ethanol. Heterotrophic growth could be restored by addition of molecular hydrogen to the culture, indicating that hydrogen is an intermediate in heterotrophic growth. Indeed, hydrogen production from fructose was detected in a stirred-tank reactor. The mutant grew well on organic substrates plus caffeate, an alternative electron acceptor that does not require molecular hydrogen but NADH as reductant. These data are consistent with the notion that molecular hydrogen is produced from organic substrates and then used as reductant for CO2 reduction. Surprisingly, hydrogen cycling in A. woodii is different from the known modes of interspecies or intraspecies hydrogen cycling. Our data are consistent with a novel type of hydrogen cycling that connects an oxidative and reductive metabolic module in one bacterial cell, “intracellular syntrophy.”
Similar content being viewed by others
Introduction
Molecular hydrogen is present only in trace concentrations (550 parts per billion) in the Earth’s atmosphere [1], but plays an important part in the global carbon cycle and is a major constituent of microbial metabolism. In anoxic ecosystems it is rapidly produced and consumed by microorganisms resulting in a large turnover [2]. Hydrogen connects different parts of the anaerobic food web and is usually produced by primary fermenters [3]. Fermentations typically yield between 1 and 4 mol of ATP per mol of sugar, and the maximum is only observed if electrons can be blown away into the environment as molecular hydrogen thus allowing the cells to make acetate according to Eq. (1) [4]:
However, hydrogen formation from reduced pyridine nucleotides or flavins is energetically unfavourable and growth according to Eq. (1) requires removal of hydrogen by a syntrophic partner such as a sulfate reducing bacterium, a methanogenic archaeon or an acetogenic bacterium [5,6,7,8]. The latter produces acetate according to Eq. (2):
Since acetogens grow by conversion of H2 + CO2 to acetate, the reaction has to be coupled to net synthesis of ATP [9]. Detailed studies in the acetogenic model organism Acetobacterium woodii estimated the amount of ATP to 0.3 mol per mol of acetate produced [9].
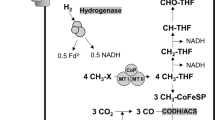
In contrast to methanogenic archaea, acetogenic bacteria do not only grow lithotrophically according to Eq. (2) but also by fermentation [10]. Acetogenesis is a modular metabolism with an oxidative and a reductive branch [11] (Fig. 1). In the oxidative branch, hydrogen (during lithotrophic growth) or an organic carbon and energy source (during heterotrophic growth) are oxidised. Electrons are carried over to the reductive branch (the Wood-Ljungdahl pathway [WLP]) in which 2 mol of CO2 are reduced to acetate according to Eq. (2). Overall, fermentation of fructose to three molecules of acetate by a combination of Eqs. (1) and (2) gives the highest ATP yield in fermenting bacteria of 4.3 mol ATP/mol of sugar [11].

Shown are the oxidation of fructose to acetate in the oxidative branch (left) and the reduction of CO2 to acetate (right) in the reductive branch (WLP). Redox balancing is achieved by a third module, in which the Rnf complex and the electron-bifurcating hydrogenase produce the reductants required for the the WLP. Fd, ferredoxin; Fd2−, reduced ferredoxin; THF, tetrahydrofolate; HDCR, hydrogen-dependent CO2 reductase; CODH/ACS, carbon monoxide dehydrogenase/acetyl-CoA synthetase; Co-FeS-P, corronoid iron-sulfur protein.
Electron carriers involved in transferring electrons from the oxidative to the reductive branch in acetogens are NADH, NADPH, or reduced ferredoxin [9]. A third module, the redox balance module, ensures that the different reduced electron carriers from the oxidative module are fed in and are converted to the specific redox carriers required by the WLP [11]. In the model acetogen A. woodii, 2 mol of NADH from glycolysis and 2 mol of reduced ferredoxin from conversion of pyruvate to acetyl-CoA are converted to 1 mol of hydrogen, 2 mol of NADH and 1 mol of reduced ferredoxin by the combined action of the Rnf complex and the electron-bifurcating hydrogenase [11, 12]. NADH is used as reductant for the methylene-tetrahydrofolate (THF) reductase [13] and the methylene-THF dehydrogenase [14], and reduced ferredoxin is used by the CO dehydrogenase/acetyl-CoA synthase as electron donor for CO2 reduction in the carbonyl branch of the WLP [15, 16]. In contrast, the first enzyme used for CO2 reduction in the methyl branch of A. woodii (Fig. 1), the hydrogen-dependent CO2 reductase (HDCR), uses H2 as reductant in vitro [17], indicating the need for electron transfer via hydrogen from the oxidative module (glycolysis) to the reductive module (CO2 reduction). On the other hand, the purified HDCR can also accept electrons from reduced ferredoxin, albeit with 17-fold lower activities [17]. To address a potential hydrogen transfer from the oxidative to the reductive branch of the acetogenic metabolism, we have deleted the only hydrogenase in A. woodii and studied the phenotype of the mutant.
Materials and methods
Growth of A. woodii
A. woodii DSM1030 was cultivated at 30 °C under anoxic conditions in complex medium as previously described [18]. When using the pyrE deletion mutant, 50 mg/l uracil was added to the medium [12]. Unless otherwise stated 20 mM fructose or 20 mM fructose together with H2 + CO2 (80:20 [v/v]) were used as carbon source. Gaseous substrates were used at a pressure of 1.0 × 105 Pa. For growth experiments, concentrations of the used carbon sources were as follows: lactate, 80 mM; ethanol, 50 mM; formate, 100 mM. Minimal medium used for genetic manipulations was prepared as previously described [19] in which yeast extract was omitted and higher amounts of 0.2 g l−1 KH2PO4, 1.35 g l−1 NH4Cl, and 1.5 ml l−1 of selenite/tungstate solution were used and 10 µg ml−1 of D/L-pantothenate was added. For mutant selection, 1.7 µg ml−1 uracil and 1.5 mg ml−1 5-fluoroorotic acid were added.
Genetic modifications
For deletion of the hydBA genes, an uracil auxotroph pyrE deletion mutant was generated using the suicide plasmid pMTL_AW_KO1. The background of the plasmid pMTL_AW_KO1 is pMTL84151 [20] out of which the Gram+ origin of replication was partially deleted by digestion of the vector with XmnI and FspI following a blunt-end ligation. The pyrE deletion cassette was cloned into the multiple cloning site, consisting of a 393 bp upstream flanking region including the first 52 bp of pyrE and a 399 bp downstream flanking region including the last 68 bp of pyrE. Both flanking regions were amplified via PCR, joined by splice-by-overlap-PCR (SOE-PCR) and cloned into the plasmid using BamHI and EcoRI. Transformation and integration of the plasmid into the A. woodii wild type as well as recombination of the plasmid at its homologous regions toward the loss of the pyrE gene has already been described elsewhere [12]. The deletion of 456 bp of the pyrE gene was verified by DNA sequencing analysis [21]. The suicide plasmid pMTL_AW_KO2 for the in-frame deletion of hydBA genes in A. woodii was built in the pMTL_AW_KO1 background. First, the pyrE cassette from Eubacterium limosum KIST612, consisting of the pyrE gene (ELI_0961) and 66 bp of its promoter region, was placed behind the catP resistence marker cassette to be used as a counter selectable marker as described previously [12], generating pMTL_AW_KO1_pyrE_Elim. Second, both fragments of the hydBA deletion cassette, consisting of a 486 bp upstream flanking region including the first 18 bp of hydB and a 462 bp downstream flanking region including the last 15 bp of hydBA, where amplified via PCR, ligated via SOE-PCR and were cloned into pMTL_AW_KO1_pyrE_Elim by using EcoRI and XbaI, which replaced the original pyrE deletion cassette with the hydBA deletion cassette. For plasmid transformation into the pyrE mutant and further integration and recombination of the hydBA deletion cassette, the same protocol as for the pyrE gene deletion, as described above, was followed. The deletion of 3538 bp of the hydBA region was verified by DNA sequencing analysis [21].
Preparation of cell-free extracts
Cells were harvested in the late exponential growth phase and resuspended in lysis buffer, containing 25 mM Tris-HCl buffer, pH 7.8, 420 mM sucrose, 2 mM DTE, 4 µM resazurin, for a 1 h treatment with 2.8 mg ml−1 lysozyme. After washing the protoplasts in analytical buffer, containing 25 mM Tris-HCl, pH 7.6, 20 mM MgSO4, 20 % [v/v] glycerol, 2 mM DTE, 4 µM resazurine, 0.5 mM PMSF, 0.1 mg ml−1 DNAseI, protoplasts were passed twice through a french pressure cell at 110 MPa (Thermo, Needham Hights, MA, USA). The cell-free extract was separated from cell debris by centrifugation at 12,000 × g for 15 min.
Hydrogenase activity assays
The activity of the electron-bifurcating hydrogenase was determined as described previously [22].
Western blot analysis
For detection of HydB and HydA subunits in cell-free extracs, Western blot analysis was performed as described before [23].
Stirred-tank reactor cultivations
Bioreactor cultivations were conducted in a 2 l working volume Biostat Aplus fermenter (Sartorius, Melsungen, Germany). The vessel was equipped with temperature probe, sparger, baffles, two Rushton-impeller, pH-probe (Hamilton, Bonaduz, Switzerland) and a redox potential probe (Hamilton, Bonaduz, Switzerland). The gas stream into the reactor was maintained at a constant rate by a digital mass-flow controller (Bronkhorst High-Tech, Ruurlo, Netherlands).
Analytical methods
Protein concentrations were determined by the method described by Bradford [24]. Fructose, formate and acetate concentrations were measured enzymatically (Hoffmann-La Roche, Basel, Switzerland). Hydrogen concentrations were determined by gas chromatography (GC) as described previously [17, 25]. Fermentation off-gas analysis was conducted by a Micro-GC (Inficon, Bad Ragaz, Switzerland) equipped with two measurement modules. Module one had argon as carrier gas for determination of hydrogen, oxygen and nitrogen, module two had helium as carrier gas for determination of carbon dioxide and water. Sampling time of the GC was 25 s. The analytical conditions for module one were: injector temperature, 90 °C; column pressure, 2.07 × 105 Pa; column temperature, 80 °C, column, Rt-Molsive 5 Å, 0.25 mm ×10 m with a Rt-Q-Bond, 3 m precolumn and for module two: injector temperature, 90 °C; column pressure, 1.72 × 105 Pa; column temperature, 60 °C, column, Rt-Q-Bond, 0.25 mm × 8 m. Each module was equipped with a thermal conductivity detector and the sampling rate was set to 100 Hz. Analysis time was 90 s for both modules. Off-line samples were taken in 2 ml volume. OD600 was measured in a spectrophotometer, Genesys 10 s UV-Vis (Thermo Scientific, Madison, USA) at a wavelenth of 600 nm. Samples were diluted with 0.9 % NaCl solution, if necessary. Samples were centrifuged at 16,363 × g for 10 min and the supernatant was used for analysis of acetic acid, formic acid and fructose using enzymatic assays as described above.
Results
Deletion of the hydBA genes of the hydrogenase operon (hydCEDBA)
Apart from the hydrogenase module in the HDCR, A. woodii has only one hydrogenase (HydABCD), a soluble, electron-bifurcating enzyme [22]. To delete the genes coding for the two major subunits HydB (Awo_c26980) and HydA (Awo_c26970) of the electron-bifurcating hydrogenase encoded by the hydCEDBA operon (Awo_c27010-Awo_c26970), the suicide plasmid pMTL_AW_KO2 was generated. The plasmid contains homologous flanking regions of ~480 bp upstream and downstream of the hydBA genes each leaving 15 bp plus the start codon of hydB and 12 bp plus the stop codon of hydA intact after deletion. The suicide plasmid was integrated into the chromosome at one of the flanking regions under antibiotic pressure. The following desintegration was forced by the presence of 5-fluoroorotate since the plasmid contains a pyrE gene together with its promoter for production of a functioning orotate phosphoribosyltransferase. However, isolation of mutants using fructose as only carbon source failed. Since this may have been caused by the lack of molecular hydrogen produced by HydABCD, hydrogen was added to the culture. Indeed, colonies were obtained, isolated and characterised. PCR (Fig. 2a) and DNA sequencing analyses confirmed the absence of hydBA in the genome. HydB and HydA could not be detected with antibodies against the two subunits (Fig. 2b) and cell-free extract of these cells did not catalyse hydrogen-dependent NAD+ and ferredoxin reduction. These experiments demonstrate that hydBA were deleted.

Agarose gel showing PCR products of the wild type (WT), pyrE (ΔpyrE) and hydBA mutant (ΔhydBA) using primers which anneal outside the hydBA region (a). Detection of HydB and HydA in crude extract of the wild type and hydBA mutant blotted on a nitrocellulose membrane and by using antibodies against HydB and HydA (b). M, Marker; NC, negative control; WT, A. woodii wild type.
Hydrogen restores growth of the hydBA mutant on organic substrates
That growth of the hydBA mutant on fructose is dependent on the addition of hydrogen is in line with the hypothesis that hydrogen is an electron carrier between the oxidative and reductive branch of acetogenesis (Fig. 1). This is also supported by the observation that the mutant did not grow on lactate or ethanol without addition of hydrogen. Formate is an intermediate of the WLP, and is dismutated to CO2 and acetate. As expected, the hydBA mutant did not grow on formate and addition of hydrogen also did not restore growth of the mutant (Table 1). Formate oxidation to CO2 yields hydrogen and obviously, the hydrogenase is essential to reduce NAD and ferredoxin, two electron carriers essential for CO2 reduction, with H2 as reductant.
Hydrogen transfer in A. woodii
Next, we tested for hydrogen consumption and production during growth of the mutant and the wild type in bicarbonate-supplied medium supplemented with 20 mM fructose under an H2 + CO2 (80:20 [v/v]) atmosphere. In theory, 1 mol of hydrogen should be produced by the electron-bifurcating hydrogenase per mol of fructose consumed, which then should be used by the HDCR for CO2 reduction (Fig. 1). The growth rate of wild type and mutant (Fig. 3a) was similar (each ~0.16 h−1) as well as the acetate:fructose ratio of 3:1 in the stationary phase (Fig. 3b), but the mutant cells used only ~20 mM of hydrogen (and 20 mM fructose), in contrast to the wild type which consumed all the hydrogen (Fig. 3a). This result confirms that the HDCR requires hydrogen and that hydrogen is produced by the electron-bifurcating hydrogenase when grown on substrates such as fructose, lactate or ethanol.

Displayed are the optical densities of the wild type (■) and the hydBA mutant (▲) as well as the hydrogen concentration in the headspace of the wild type (×) and the hydBA mutant (+) cultures (a). The fructose consumption of the wild type (◇) and hydBA mutant (▽) together with the concomitant production of acetate in the wild type (◆) and hydBA (▼) mutant was monitored over time (b). (n = 2).
Initial stirred-tank reactor (STR) cultivation of the A. woodii wild type revealed in vivo hydrogen formation
If hydrogen is produced in vivo to connect the oxidative and reductive parts of acetogenesis, then, due to its volatile nature, it should be detectable in the gaseous phase. To determine a possible hydrogen production, a bench-scale anaerobic bioreactor for cell cultivation was set up together with off-gas analysis via GC. Initial cultivations of the A. woodii wild type in bicarbonate-supplied medium with 20 mM fructose as carbon and energy source showed exponentially rising hydrogen flow rates in the off-gas of up to 0.24 µmol min−1 (Fig. 4). Hydrogen levels in the off-gas dropped to 0.1 µmol min−1 after fructose was completely consumed. The cultures reached a maximum OD600 of 2.28 after 24 h of cultivation, the growth rate was 0.19 h−1. From the available 20 mM fructose, 50 mM of acetate was produced after 24 h. Overall 2.4 mol of acetic acid and 8.4 ± 0.2 mmol of hydrogen were produced per mol of fructose consumed. A. woodii requires Na+ [18] and a functional Rnf complex for energy conservation [12]. When grown with fructose in the absence of Na+, the acetate:fructose ratio of the wild type decreased to 2 and 2.1-times more hydrogen was released. Similar results were observed for the rnf deletion mutant. Maximum values for OD600 and growth rate were 1.85 and 0.13 h−1 for the rnf mutant and 1.68 and 0.11 h−1 for the wild type (data not shown). Table 2 summarises the above mentioned fermentation parameters.

The wild type was grown in complex medium with 20 mM fructose as carbon and energy source and the cultivation broth was sparged with 10 ml min−1 20 % CO2 in nitrogen. The upper half shows the average off-line values for OD600 (◆), concentration of fructose (■) and acetic acid (▲) of four cultivations whilst the bottom half shows off-gas flow rates for hydrogen (solid line) and carbon dioxide (dashed line). The grey areas in the bottom half depict the variation of the off-gas values.
Replacing the WLP with other, non-hydrogen consuming reductive pathways restores growth of the hydBA mutant on fructose
H2 + CO2 is converted to formate by the HDCR [17]. Therefore, we speculated that the hydBA mutant should be able to grow on fructose + formate (+ CO2) instead of fructose + CO2, since formate reduction only requires NADH and reduced ferredoxin (Fig. 5). Indeed, the hydBA mutant grew with a doubling time of ~5 h on 20 mM fructose and 100 mM formate, which is slightly slower than the growth of the wild type (~3.5 h–4 h). The final OD600 of the hydBA mutant reached 80–85 % of the final OD600 of the wild type.

Fd, ferredoxin; Fd2−, reduced ferredoxin.
A. woodi can use caffeate as electron acceptor alternative to CO2 [26, 27]. Reduction of caffeate to hydrocaffeate does not require hydrogen but only NADH as reductant [28] (Fig. 6a). Therefore, the hydBA mutant should be able to grow on, for example, fructose + caffeate. After addition of fructose + caffeate to the wild type and the mutant, growth of the two strains could be detected (Fig. 6b). Doubling times of the mutant and the wild type were similar (~6 h), but growth of the mutant culture stopped much earlier, with a final OD600 of 0.68 compared with 3.2 of the wild type. The mutant also grew on lactate + caffeate (Fig. 6c) or ethanol + caffeate (Fig. 6d), but the growth rate of the hydBA mutant was one fifth and one half of the growth rate of the wild type, respectively. Like for fructose + caffeate, the final OD600 was 0.47 or 0.25 for the mutant and 1.5 or 0.78 for the wild type when grown on lactate + caffeate or ethanol + caffeate, respectively. The lower final OD600 may be due to the inability of the strains to use the HDCR reaction for recapturing CO2 which is produced during the oxidation of the respective substrate.

Fd, ferredoxin; Fd2−, reduced ferredoxin (a). Cells were grown in complex medium supplemented with either 20 mM fructose + 6 mM caffeate (b), 80 mM lactate + 4 mM caffeate (c) or 50 mM ethanol + 4 mM caffeate (d). Growth of the wild type + caffeate (■), hydBA mutant + caffeate (▲), wild type without caffeate (□), hydBA mutant without caffeate (△) were measured over time at 600 nm. Caffeate was added when concentrations dropped below 1 mM and its utilisation by the wild type (×) and hydBA mutant (+) was monitored over time (n = 2).
Discussion
Sulfate reducing bacteria grow by oxidation of lactate or other organic substrates coupled to energy conservation by the reduction of sulfate [29, 30]. They also grow lithotrophically and, indeed, hydrogen is the most effective hydrogen donor for sulfate reduction [4, 29]. This led Odom and Peck to postulate hydrogen cycling as a general mechanism for energy coupling in sulfate reducing bacteria [31] and they postulated this mechanism to be present in methanogens and acetogens as well [5]. According to their model, hydrogen is produced inside the cell and diffuses across the membrane to the periplasm. The authors speculated that the same organism oxidises hydrogen to 2 H+ + 2e− at the periplasmic side of the membrane, thus, producing scalar protons that create a proton motive force (pmf) across the cytoplasmic membrane that drives ATP synthesis. This mechanism requires a soluble, cytoplasmic hydrogenase and a periplasmic, membrane-bound hydrogenase. Biochemical and physiological experiments performed are in line with this hypothesis [32]. More than 35 years later, hydrogen cycling as a mode of pmf generation was directly demonstrated by mutational analyses for the archaeon Methanosarcina barkeri [33]. When grown on methanol, 25 % of the substrate is oxidised to CO2 to generate the electrons needed to reduce the other 75 % to methane. The oxidation of methanol is coupled to the reduction of protons to H2, as catalysed by the F420-reducing hydrogenase. Hydrogen diffuses out of the cell and is harnessed by the membrane-bound Vht hydrogenase that reduces the membrane-integral electron-carrier methanophenazine, the electron donor for the respiratory enzyme, the heterodisulfide reductase [33]. The proton potential established by the heterodisulfide reductase drives the synthesis of ATP in this archaeon [34]. Deletion of the soluble hydrogenase abolished hydrogen formation and deletion of the Vht hydrogenase was lethal, demonstrating nicely that there is hydrogen cycling in one cell, important for energy conservation [33].
The second type of hydrogen transfer in anoxic ecosystems is observed between different species and is called interspecies hydrogen cycling [5]. There, one partner oxidises a substrate linked to the production of hydrogen. Since hydrogen formation is thermodynamically unfavourable, the hydrogen producer can only survive if the hydrogen concentration in the environment is kept low by hydrogen oxidising microorganisms such as methanogenic archaea [6, 7].
Whether or not hydrogen cycling occurs in the ecophysiologically relevant group of acetogenic bacteria remained to be established. In this report, we show clear evidence that A. woodii releases hydrogen into the atmosphere but only ~8.4 mmol hydrogen per mol of fructose was liberated, which is less than 10 % of the acetic acid generated, but similar to the result of Braun et al. [35]. This result indicates that the HDCR efficiently captures the hydrogen produced by the electron-bifurcating hydrogenase. If the electron-bifurcating hydrogenase is missing, cells growing on fructose, ethanol or lactate are unable to produce hydrogen needed for CO2 reduction by the HDRC and therefore are unable to grow, except when they grow mixotrophically on fructose + hydrogen or fructose + formate. This is also clear evidence that hydrogen evolution takes place inside the cell and hydrogen is directly used within the cell before it can diffuse through the cell membrane. Since A. woodii lacks a membrane-bound hydrogenase, hydrogen oxidation is not linked to energy conservation and, therefore, does not fall into the category “intraspecies hydrogen cycling linked to energy conservation”, as postulated by Odom and Peck [5].
In contrast, A. woodii combines the metabolic features of two syntrophic partners in one bacterial cell. Depending on the environmental conditions A. woodii can play the part of the fermenting partner as in coculture with a methanogen [36] or the hydrogen consuming partner in syntrophic interactions. When grown together with methanogens on H2 + CO2, acetogens usually would be outcompeted by methanogens since methanogenesis from H2 + CO2 delivers much more energy than acetogenesis [37]. However, under certain conditions acetogens dominate as H2 + CO2 -consuming partner in syntrophic interactions. For example, hydrogen-utilising acetogens compete successfully with hydrogen-utilising methanogens in wood-feeding termites [38]. Further studies showed that acetogens can outcompete methanogens for hydrogen at a pH of 6.2 and also at more acidic pH values [39] and at low temperature, for example at an in situ temperature of 4 °C in sediments of Lake Constance [40]. The partial pressure of hydrogen measured in pore water is too low to allow growth of pure cultures and it is speculated that the in situ partial pressure of hydrogen might be higher for acetogens living in close proximity to the hydrogen-producing organism [38]. Here, we demonstrate that, in addition, A. woodii can also play both parts—the fermenting and the hydrogen consuming part—in one cell. This is the closest proximity one can get. We propose to call this novel type of hydrogen cycling that connects an oxidative and reductive metabolic module in one bacterial cell “intracellular syntrophy.”
Acetogenic microorganisms are phylogenetically very divers [41]. Acetogenesis has been found in different phylogenetic clades of the Bacteria and has been studied there for the last hundred years, but rather recently acetogenesis was also found in different phyla of the Archaea. This is not only based on genomic but also on physiological analyses [42,43,44]. These archaea are supposed to grow autotrophically on H2 + CO2 to produce acetate but also on organic substrates. It is postulated that they ferment the organic substrates such as short chain fatty acids to acetate, alcohols and molecular hydrogen [43]. At the same time the WLP can act as electron sink to make the fermentation energetically possible, which makes fermentation independent from a syntrophic partner [44]. Our mutational analyses in A. woodii fully support this model.
The coupling of hydrogen-dependent CO2 reduction to hydrogen-producing fermentations allows acetogens to grow, for example, in the deep biosphere on substrates that are otherwise inaccessible for energetic reasons. Hydrogen production from fermentation is common in aquatic sediments and hydrogen production and consumption is essential for anaerobic food webs [45]. Hydrogen production from organic substrates also puts a new perspective on the origin of the eukaryotic cell [43]. Many acetogens have just one soluble hydrogenase like A. woodii [46], others such as Moorella thermoacetica [47], Thermoanaerobacter kivui [48] or Heimdallarchaeota and Odinarchaeota [43] have membrane-bound, ion-translocating hydrogenase activities as hydrogen consuming respiratory enzymes. Their role in hydrogen transfer is still an open question.
References
Novelli PC, Lang PM, Masarie KA, Hurst DF, Myers R, Elkins JW. Molecular hydrogen in the troposphere: Global distribution and budget. J Geophys Res. 1999;104:30427–44.
Piché-Choquette S, Constant P. Molecular hydrogen, a neglected key driver of soil biogeochemical processes. Appl Environ Microb. 2019;85:02418–18.
Schink B. Synergistic interactions in the microbial world. Antonie Van Leeuwenhoek. 2002;81:257–61.
Thauer RK, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bact Rev. 1977;41:100–80.
Odom JM, Peck HD Jr. Hydrogenase, electron-transfer proteins, and energy coupling in the sulfate-reducing bacteria Desulfovibrio. Annu Rev Microbiol. 1984;38:551–92.
McInerney MJ, Sieber JR, Gunsalus RP. Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol. 2009;20:623–32.
Stams AJ, Plugge CM. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol. 2009;7:568–77.
Müller N, Griffin BM, Stingl U, Schink B. Dominant sugar utilizers in sediment of Lake Constance depend on syntrophic cooperation with methanogenic partner organisms. Environ Microbiol. 2008;10:1501–11.
Schuchmann K, Müller V. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol. 2014;12:809–21.
Drake HL, Gößner AS, Daniel SL. Old acetogens, new light. Ann N. Y Acad Sci. 2008;1125:100–28.
Schuchmann K, Müller V. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microbiol. 2016;82:4056–69.
Westphal L, Wiechmann A, Baker J, Minton NP, Müller V. The Rnf complex is an energy coupled transhydrogenase essential to reversibly link cellular NADH and ferredoxin pools in the acetogen Acetobacterium woodii. J Bacteriol. 2018;200:e00357–18.
Bertsch J, Öppinger C, Hess V, Langer JD, Müller V. A heterotrimeric NADH-oxidizing methylenetetrahydrofolate reductase from the acetogenic bacterium Acetobacterium woodii. J Bacteriol. 2015;197:1681–9.
Ragsdale SW, Ljungdahl LG. Purification and properties of NAD-dependent 5,10-methylenetetrahydrofolate dehydrogenase from Acetobacterium woodii. J Biol Chem. 1984;259:3499–503.
Ragsdale SW, Wood HG. Acetate biosynthesis by acetogenic bacteria. Evidence that carbon monoxide dehydrogenase is the condensing enzyme that catalyzes the final steps in the synthesis. J Biol Chem. 1985;260:3970–7.
Shanmugasundaram T, Ragsdale SW, Wood HG. Role of carbon monoxide dehydrogenase in acetate synthesis by the acetogenic bacterium, Acetobacterium woodii. BioFactors. 1988;1:147–52.
Schuchmann K, Müller V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science. 2013;342:1382–5.
Heise R, Müller V, Gottschalk G. Sodium dependence of acetate formation by the acetogenic bacterium Acetobacterium woodii. J Bacteriol. 1989;171:5473–8.
Schölmerich MC, Katsyv A, Sung W, Mijic V, Wiechmann A, Kottenhahn P, et al. Regulation of lactate metabolism in the acetogenic bacterium Acetobacterium woodii. Environ Microbiol. 2018;20:4587–95.
Heap JT, Pennington OJ, Cartman ST, Minton NP. A modular system for Clostridium shuttle plasmids. J Microbiol Methods. 2009;78:79–85.
Sanger FS, Nickelen F, Coulson AR. DNA-sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–7.
Schuchmann K, Müller V. A bacterial electron-bifurcating hydrogenase. J Biol Chem. 2012;287:31165–71.
Hess V, Vitt S, Müller V. A caffeyl-coenzyme A synthetase initiates caffeate activation prior to caffeate reduction in the acetogenic bacterium Acetobacterium woodii. J Bacteriol. 2011;193:971–8.
Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of proteine-dye-binding. Anal Biochem. 1976;72:248–54.
Basen M, Geiger I, Henke L, Müller V. A genetic system for the thermophilic acetogenic bacterium Thermoanaerobacter kivui. Appl Environ Microbiol. 2018;84:e02210–17.
Tschech A, Pfennig N. Growth yield increase linked to caffeate reduction in Acetobacterium woodii. Arch Microbiol. 1984;137:163–7.
Dilling S, Imkamp F, Schmidt S, Müller V. Regulation of caffeate respiration in the acetogenic bacterium Acetobacterium woodii. Appl Environ Microbiol. 2007;73:3630–6.
Hess V, Gonzalez JM, Parthasarathy A, Buckel W, Müller V. Caffeate respiration in the acetogenic bacterium Acetobacterium woodii: a coenzyme A loop saves energy for caffeate activation. Appl Environ Microbiol. 2013;79:1942–7.
Postgate JR. Recent advances in the study of the sulfate-reducing bacteria. Bacteriol Rev. 1965;29:425–41.
Traore AS, Hatchikian CE, Belaich JP, Le Gall J. Microcalorimetric studies of the growth of sulfate-reducing bacteria: energetics of Desulfovibrio vulgaris growth. J Bacteriol. 1981;145:191–9.
Odom JM, Peck HD Jr. Hydrogen cycling as a general mechanism for energy coupling in the sulfate-reducing bacteria. FEMS Microbiol Lett. 1981;12:47–50.
Odom JM, Peck HD Jr. Localization of dehydrogenases, reductases, and electron transfer components in the sulfate-reducing bacterium Desulfovibrio gigas. J Bacteriol. 1981;147:161–9.
Kulkarni G, Mand TD, Metcalf WW. Energy conservation via hydrogen cycling in the methanogenic archaeon Methanosarcina barkeri. mBio. 2018;9:e01256–18.
Deppenmeier U, Müller V. Life close to the thermodynamic limit: how methanogenic archaea conserve energy. Results Probl Cell Differ. 2008;45:123–52.
Braun K, Gottschalk G. Effect of molecular hydrogen and carbon dioxide on chemo-organotrophic growth of Acetobacterium woodii and Clostridium aceticum. Arch Microbiol. 1981;128:294–8.
Winter JU, Wolfe RS. Methane formation from fructose by syntrophic associations of Acetobacterium woodii and different strains of methanogens. Arch Microbiol. 1980;124:73–9.
Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–91.
Breznak JA, Brune A. Role of microorganisms in the digestion of lignocellulose by termites. Annu Rev Entomol. 1994;39:453–87.
Phelps TJ, Zeikus JG. Influence of pH on terminal carbon metabolism in anoxic sediments from a mildly acidic lake. Appl Environ Microbiol. 1984;48:1088–95.
Conrad R, Bak F, Seitz HJ, Thebrath B, Mayer HP, Schütz H. Hydrogen turnover by psychotrophic homoacetogenic and mesophilic methanogenic bacteria in anoxic paddy soil and lake sediment. FEMS Microbiol Lett. 1989;62:285–94.
Drake HL, Küsel K, Matthies C. Ecological consequences of the phylogenetic and physiological diversities of acetogens. Antonie Van Leeuwenhoek. 2002;81:203–13.
He Y, Li M, Perumal V, Feng X, Fang J, Xie J, et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol. 2016;1:16035.
Spang A, Stairs CW, Dombrowski N, Eme L, Lombard J, Caceres EF, et al. Proposal of the reverse flow model for the origin of the eukaryotic cell based on comparative analyses of Asgard archaeal metabolism. Nat Microbiol. 2019;4:1138–48.
Orsi WD, Vuillemin A, Rodriguez P, Coskun ÖK, Gomez-Saez GV, Lavik G, et al. Metabolic activity analyses demonstrate that Lokiarchaeon exhibits homoacetogenesis in sulfidic marine sediments. Nat Microbiol. 2019;5:248–55.
Conrad R, Schink B, Phelps TJ. Thermodynamics of H2-consuming and H2-producing metabolic reactions in diverse methanogenic environments under in situ conditions. FEMS Microbiol Ecol. 1986;38:353–60.
Poehlein A, Schmidt S, Kaster A-K, Goenrich M, Vollmers J, Thürmer A, et al. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS ONE. 2012;7:e33439.
Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, et al. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ Microbiol. 2008;10:2550–73.
Schölmerich MC, Müller V. Energy conservation by a hydrogenase-dependent chemiosmotic mechanism in an ancient metabolic pathway. Proc Natl Acad Sci USA. 2019;116:6329–34.
Acknowledgements
This work was funded by a grant from the Bundesministeriums für Bildung und Forschung (BMBF) for the OBAC-project (ERA-IB-16–018) and from the European Research Council (ERC) for the ACETOGENS project (741791). Open access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AW and VM conceived the study. AW, SC, FO, and VNS conducted the experiments. AW, FO and VM wrote the manuscript with input from all authors. All documents were edited and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wiechmann, A., Ciurus, S., Oswald, F. et al. It does not always take two to tango: “Syntrophy” via hydrogen cycling in one bacterial cell. ISME J 14, 1561–1570 (2020). https://doi.org/10.1038/s41396-020-0627-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0627-1
This article is cited by
-
Developing a genetic engineering method for Acetobacterium wieringae to expand one-carbon valorization pathways
Biotechnology for Biofuels and Bioproducts (2023)
-
Reprogramming the metabolism of an acetogenic bacterium to homoformatogenesis
The ISME Journal (2023)
-
Lactate formation from fructose or C1 compounds in the acetogen Acetobacterium woodii by metabolic engineering
Applied Microbiology and Biotechnology (2023)
-
Formate-driven H2 production by whole cells of Thermoanaerobacter kivui
Biotechnology for Biofuels and Bioproducts (2022)
-
Microbial Interactions in Pollution Control Ecosystems
Current Pollution Reports (2021)
