
Abstract
Cable bacteria are filamentous members of the Desulfobulbaceae family that oxidize sulfide with oxygen or nitrate by transferring electrons over centimeter distances in sediments. Recent studies show that freshwater sediments can support populations of cable bacteria at densities comparable to those found in marine environments. This is surprising since sulfide availability is presumably low in freshwater sediments due to sulfate limitation of sulfate reduction. Here we show that cable bacteria stimulate sulfate reduction in freshwater sediment through promotion of sulfate availability. Comparing experimental freshwater sediments with and without active cable bacteria, we observed a three- to tenfold increase in sulfate concentrations and a 4.5-fold increase in sulfate reduction rates when cable bacteria were present, while abundance and community composition of sulfate-reducing microorganisms (SRM) were unaffected. Correlation and ANCOVA analysis supported the hypothesis that the stimulation of sulfate reduction activity was due to relieve of the kinetic limitations of the SRM community through the elevated sulfate concentrations in sediments with cable bacteria activity. The elevated sulfate concentration was caused by cable bacteria-driven sulfide oxidation, by sulfate production from an indigenous sulfide pool, likely through cable bacteria-mediated dissolution and oxidation of iron sulfides, and by enhanced retention of sulfate, triggered by an electric field generated by the cable bacteria. Cable bacteria in freshwater sediments may thus be an integral component of a cryptic sulfur cycle and provide a mechanism for recycling of the scarce resource sulfate, stimulating sulfate reduction. It is possible that this stimulation has implication for methanogenesis and greenhouse gas emissions.
Similar content being viewed by others


Introduction
The availability of sulfate in freshwater sediments is low compared with coastal marine systems. Water column concentrations in freshwater systems are generally 2–3 orders of magnitude lower than in marine systems, and while sulfate penetrates meters into marine sediments, the penetration depth of sulfate into freshwater sediments is typically restricted to <10 cm [1]. As a consequence, sulfate reduction typically contributes more to anaerobic mineralization in coastal marine sediments [2] than in freshwater sediments, where methanogenesis dominates the anaerobic carbon turnover at the expense of sulfate reduction [1, 3]. Enhanced supply of sulfate to freshwater sediments, for instance via increased transport from the water column, may however result in stimulated sedimentary sulfate reduction activity and a concurrent decrease in methanogenesis [4].
Experimentally determined sulfate reduction rates (SRR) (i.e., rates determined with the 35SO42− radiotracer method [5]) often exceed the rates that can be derived from reaction transport modeling of sulfate concentrations in the sediment porewater [6,7,8]. For freshwater sediments, this discrepancy can be quite extensive. Bak and Pfennig [9] and Urban et al. [10] thus showed that rates determined from diffusional sulfate fluxes only accounted for 2% of the experimentally derived SRR in Lake Constance, Germany, and Little Rock Lake, Wisconsin, USA, respectively, while in the oligohaline (salinity <2 g kg−1) part of Chesapeake Bay the diffusional flux model could account only for <10% of the sulfate reduction activity [11]. One reason for this discrepancy is that sulfate reduction is accompanied by oxidation of sulfide back to sulfate [12], with the anaerobic sulfide oxidation driven by buried oxidized metals like iron and manganese [7, 13,14,15]. Such coupled sulfate reduction—sulfide oxidation is invisible in the porewater chemistry, and thus represents a cryptic sulfur cycle [6] not accounted for in sulfate gradient analysis of porewater depth profiles. The cryptic sulfur cycle can be an important component of sulfate-limited freshwater environments [16].
Cable bacteria are filamentous bacteria of the Desulfobulbaceae family that perform electrogenic sulfide oxidation (e-SOx) [17, 18] — a process by which the half reaction of sulfide oxidation is coupled to oxygen or nitrate reduction centimeters away [19, 20]. Cable bacteria and e-SOx are present in marine, brackish, freshwater, and aquifer sediments [21,22,23,24]. It has been shown that cable bacteria via e-SOx can enhance the concentration of sulfate in surface layers of marine sediments [25, 26]. Three separate mechanisms can be at play. Firstly, the end product of e-SOx is sulfate [17, 26, 27], which makes cable bacteria potential players in the cryptic sulfur cycle. Secondly, e-SOx promotes dissolution of solid phase sulfides such as iron sulfides through acidification of the porewater [26, 28,29,30]. Subsequent oxidation of the freed sulfide can contribute to further accumulation of sulfate in subsurface layers of the sediment [25, 26, 28]. Finally, e-SOx induces electric fields [21, 26, 31, 32], which initiate ionic drift [33] and thus promote the downward transport of sulfate into the sediment, reducing its loss to the environment [26]. It is very likely that these e-SOx driven mechanisms operate intensively in freshwater sediments colonized by cable bacteria, in particular because electric fields generated through e-SOx, and hence the potential for sulfate retention, are much stronger in freshwater sediments compared with marine sediments [24, 32]. Consequently, the presence of cable bacteria could lead to significant enhancement of sulfate availability in otherwise sulfate-scarce environments, and thereby lift the kinetic limitation [10, 34, 35] of the community of sulfate-reducing microorganisms (SRM), leading to higher SRR in sediments with cable bacteria activity than in sediments without.
In the present study, we test the hypothesis that cable bacteria enhance sulfate availability in freshwater sediments via e-SOx. We further test if such enhancement of sulfate availability is correlated with an increased SRR, and thereby if cable bacteria promote sulfate reduction via e-SOx. In a follow-up experiment, we test if cable bacteria also influence the abundance and community composition of SRM via their alteration of the geochemical environment through e-SOx [29, 30, 36]. This analysis was performed to determine if possibly increased SRRs in cable bacteria-inhabited sediments could be explained by e-SOx-induced shifts in the SRM community structure.
Our study was performed with sediments from two eutrophic lakes: Skanderborg Sø and Vilhelmsborg Sø, both situated in Jutland, Denmark. The presence of e-SOx was addressed through fine-scale measurements of oxygen, sulfide, pH, and electric potential (EP) distributions in microcosms with and without cable bacteria performing e-SOx, prepared with sediments from both lakes. The effect of e-SOx on sulfate availability was addressed through measurement and modeling of the sulfate depth distribution in the microcosms, while the effect of e-SOx on sulfate reduction was addressed through measurements of the SRR using radiotracers. The size and composition of the SRM community was analyzed by qPCR and amplicon sequencing of the gene encoding the beta subunit of the dissimilatory (bi)sulfite reductase (dsrB) [37] in sediments with and without cable bacteria performing e-SOx.
Materials and methods
Sampling
Surface sediment and water were collected from Skanderborg Sø (56°0′45.80″ N, 9°54′58.22″ E) and Vilhelmsborg Sø (56°4′4.14″ N, 10°11′8.89″ E), two shallow, eutrophic, alkaline (pH is 8.3–8.6 according to our data) and dimictic lakes located in Eastern Jutland, Denmark. Skanderborg Sø has an area of 8.7 km2 and an average water depth of 7.8 m (max. 18 m); Vilhelmsborg Sø has an area of 0.019 km2 and an average depth of ~1 m (max. 3 m). Sampling for the biogeochemical experiments was performed in December 2016 and February 2017. Sampling for the follow-up analysis of SRM abundance and community structure was performed in September 2017.
Surface sediments were collected at a water depth of 0.5–1 m. The sediments were collected with a shovel, transferred to a barrel and brought to the laboratory within approximately 1 h after sampling. Surface water was collected in 20 L jars. The concentration of sulfate in the Skanderborg Sø water was 287 ± 16 μM upon sampling and the conductivity was 0.12 S m−1. The sediment was sandy. Concentration of sulfate in the Vilhelmsborg Sø water was 202 ± 3 μM upon sampling and the conductivity was 0.1 S m−1. The sediment was silty.
Sediment handling and incubation
For the experiments, we adapted the design that has been used in former studies to document e-SOx [20] and to investigate its geochemical implications [26]. This experimental design includes the incubation of macrofauna-free sediments in the presence of an oxic water column (a treatment promoting e-SOx) and in the presence of an oxygen-free water column (a control treatment without e-SOx). The approach presumes that oxygen in the water column, aside from driving e-SOx, does not interfere significantly with the sulfur cycle in the oxygen-free sediment layers below the oxic zone, during the course of the experiment. This presumption is justified as follows: (i) solid-phase oxidized iron and manganese, formed through direct reaction with oxygen in the oxic zone, cannot exert an effect in the anoxic zone without mechanical mixing, e.g., by bioturbation [38], which has been excluded in our setup; (ii) dissolved compounds such as nitrate and nitrite, formed through nitrification in the oxic zone, may extend the impact of oxygen downwards by only a few mm [39,40,41], as nitrification typically accounts for <20% of the oxygen consumption [42]; and (iii) the oxidation of sulfides such as iron sulfides or dissolved hydrogen sulfide to sulfate in the oxygen-affected zone is insignificant in comparison to the oxidation of sulfides in the zone not affected by oxygen [26].
For the biogeochemical experiments the sediment was sieved (mesh size = 0.5 mm) to remove larger fauna and stones; no precautions were taken to prevent reoxidation of reduced iron and manganese present initially in the sediment batch. Five liters of the homogenized sediment was then transferred to each of two 10 L aquaria. After carefully leveling the sediment surface, five liters of in situ water were added gently to minimize sediment resuspension. The resulting sediment and water column height was 10 cm each. The sediment in one aquarium, assigned as ES-free sediment (e-SOx-free sediment), was incubated in the absence, or near absence, of oxygen in the overlying water. Previous studies have shown that absence of oxygen or severe hypoxia (O2 concentrations <10% of air saturation) prevent e-SOx and cable bacteria development [20, 26, 43]. The water column was maintained hypoxic by gently purging N2 gas with 0.04% CO2 (AGA, Sweden) into the aquarium, which was sealed with a lid. The oxygen concentration in the water column was consistently low (0–15 μM) throughout the incubation period of 6 weeks, as measured with a custom-built O2 sensor [44]. To maintain a constant water chemistry, the water column was renewed every 3 days with O2-free water from the field site. The sediment in the other aquarium, assigned as ES-sediment (e-SOx sediment), was incubated in the presence of an air-saturated water column. The overlying water was aerated with a submerged air pump and kept in equilibrium with the atmosphere. As above, the water in the aquaria was renewed every 3 days to maintain a constant water chemistry. Both aquaria were kept at 15 °C for the entire 7-week study period. After 5 weeks of incubation, microprofiles of EP distributions, H2S, pH, and O2 were measured with microsensors. After 6 weeks of incubation, sediment cores were retrieved from both aquaria for measurements of SRR, sulfate concentrations in the sediment porewater, and sediment porosity. The presence of cable bacteria in sediment from the two aquaria was evaluated by fluorescence in situ hybridization (FISH) with probe DSB706 as described previously (Schauer et al. [45], Lucker et al. [46]); see also Fig. S1.)
For the community analysis experiment, the sediment was sieved as described above and then transferred to glass core liners (Height: 10 cm, Inner diameter: 2 cm). For each lake three cores, assigned as ES-cores (e-SOx cores), were placed in an aquarium with oxic lake water, while another three, assigned as ES-free cores (e-SOx-free cores) were placed in an aquarium with oxygen-free lake water. The aquaria were maintained for 6 weeks as described above. After 6 weeks, EP microprofiling was performed to confirm the presence or absence of active cable bacteria in the sediments. At the end of the incubation, the sediment cores were sliced into sections (0–3, 3–21, and 21–40 mm). Subsamples from each of the sections were transferred into sterile microcentrifuge tubes (Eppendorf, Germany) and frozen at −20 °C for later analysis of the SRM community using qPCR and Illumina MiSeq sequencing of the dsrB gene.
Electric potential, oxygen, sulfide, and pH
The depth distribution of the EP was measured with a custom-built EP microelectrode [31] against the Red Rod reference electrode (REF201 Radiometer Analytical, Denmark). Both electrodes were connected to an in-house-made millivoltmeter with a resistance >1014 Ω (Aarhus University, Denmark). The analog signal from the millivoltmeter was digitized for PC-processing using a 16-bit A/D converter (ADC-216; Unisense A/S, Denmark). Depth profiles of H2S, O2, and pH were measured with custom made microsensors [44, 47, 48]. The Red Rod reference electrode was used as reference for the pH measurements. The total sulfide concentration (Σ[H2S] = [H2S] + [HS−] + [S2−]) was calculated from the measured H2S concentration and the pH according to Jeroschewski et al. [47]. All sensors were connected to a four-channel multimeter (Unisense Microsensor Multimeter, Ver 2.01; Unisense A/S, Denmark) with a built-in 16-bit A/D converter. Prior to depth profiling the O2 sensor was calibrated in 0.7 M alkaline ascorbate solution (0 μM O2 at 15 °C) and air-saturated lake water (325 μM O2 at 15 °C). The H2S sensor was calibrated in a darkened calibration chamber containing O2-free HCl. A three point calibration curve in the range 0–50 µM H2S was made by adding fixed amounts of dissolved Na2S from a 10 mM stock solution. Sulfide concentrations in the calibration media were determined on Zn-acetate fixed subsamples as described by Cline [49]. The pH sensor was calibrated in AVS TITRINORM buffers (VWR Chemicals, Denmark) having pH values of 4.0, 7.0, and 9.0, traceable to standard reference material from NIST (www.nist.gov). During profiling, the specific sensor was mounted on a three-dimensional microprofiling system (Unisense A/S, Denmark). The software program SensorTrace PRO (Unisense A/S, Denmark) was used for data acquisition and to control the microprofiling system. The microsensor profiles were measured in grids of 10 × 10 cm. Each of the grid points were separated by two centimeters. In total, 36 profiles were measured with each sensor. For the community analysis experiment, one EP profile was measured per core as described above. No further geochemical analyses were performed.
Sulfate concentrations
Three sediment cores (inner diameter = 5 cm) were sampled from both the ES-and ES-free sediments. The cores were sliced into 3 mm sections down to 30 mm depth, and into 5 mm sections between 30 and 45 mm depth. Each section was transferred to a 50 ml Falcon tube and centrifuged at 3000 × g for 5 min to separate the porewater from the sediment matrix. The extracted porewater was then stored at −20 °C until analysis. Sulfate concentrations were measured by ion chromatography (Dionex IC 3000 system, Dionex, Sunnyvale, California, USA) as described in Roy et al. [50]
Sulfate reduction rates
SRR were determined using the 35SO42− radiotracer method [5, 50]. Previous studies have shown that the co-occurrence of sulfate reduction and sulfide oxidation may lead to underestimation of actual SRR when measured with the 35SO42− radiotracer method since the reduced 35S is converted back into 35SO42− over time [51]. The degree of underestimation increases with the incubation time, as shown by direct measurements and modeling of tracer experiments [51, 52]. Because we expected significant sulfide oxidation in the ES-sediments due to e-SOx, we initially performed time series experiments with these sediments, which were incubated for 1, 5, and 20 min, following the protocol outlined below. Based on the data from these experiments, a standard incubation time of 5 min was chosen for both the ES and ES-free sediments.
Sediment subsamples were collected from the ES and the ES-free sediments (n = 3 per treatment) in 10 mL plastic syringes modified with a vertical row of holes for tracer injection covered by black gastight tape (Scotch Super 33+ Vinyl Electrical tape; 3M, Minnesota, USA). Twenty-five microliters of carrier-free 35SO42− tracer (1.7 µM) were injected horizontally at 3.5–38.5 mm depths, giving each core an activity of ~400 kBq. This added ~1.5 nM to the concentration of sulfate in the sediment porewater, as in Holmkvist et al. [6]. At the end of the incubation period, each core was sliced into five sections, spanning the depth range 0 to ca. 45 mm. Each section was immediately transferred to a 50 ml Falcon tube containing 5 ml 20% Zn-acetate for inhibition of sulfate reduction, then frozen at −20 °C. For analysis, the samples were thawed at room temperature, and the total reduced inorganic sulfur (TRIS) in the samples was separated from the sulfate by a single-step cold chromium distillation [53]. The radioactivity of 35S contained within the TRIS pool and the SO42− pool was counted in 10 mL Gold Star scintillation cocktail (Meridian Biotechnologies, UK) with a liquid scintillation analyzer (TriCarb 2900TR,Packard Instrument Company, Germany). The SRR was calculated according to Tarpgaard et al. [54].
where φ is the sediment porosity estimated from the density and water content of sediment samples (the porosity was 0.94–0.44 along the depth horizon in Skanderborg Sø sediments, and 0.96–0.64 in the Vilhelmsborg Sø sediment), [SO42−] is the sulfate concentration at a given depth, (here estimated as the mean from three replicated samples), atris is the radioactivity of TRIS pool, atotal is the total radioactivity of the sample including both the radioactivity of the TRIS and SO42− pool (corresponding to 35SO42− initially present in the sample). The factor 1.06 is an isotope fractionation factor proposed by Jorgensen [5].
QPCR and Illumina MiSeq sequencing of the dsrB gene
DNA was extracted from 0.3 g of sediment using the FastDNA Spin Kit for Soil (MP Biomedicals, California, USA) with modifications described in Kamp et al. [55]. Both the dsrB gene and the gene encoding bacterial 16S ribosomal RNA (rRNA) were quantified using SYBR-green based quantitative PCR (qPCR) as described in Jochum et al. [56]. Bacterial 16S rRNA genes were quantified by qPCR using the primers 8Fmod and 338Rabc [57]. DsrB genes were quantified using the primer variant mixtures dsrB-F1a-h and dsrB-4RSI1a-f [58]. PCR amplification and Illumina MiSeq sequencing of an ~350 bp fragment of the dsrB gene were performed using a previously designed primer set (DSR1762Fmix 1–10 and DSR2107Rmix 1–13 [59].
Data analysis
Student t test or Wilcoxon signed rank test were used to test for differences in SRR and sulfate concentrations between ES sediment and ES-free sediment, separately for each sampling site. The latter test was used on data series that did not meet criteria for parametric methods, such as normal distribution. The Shapiro–Wilk test was used to test for normality. Pearson product moment correlation analysis was used to investigate if SRRs were correlated with the sulfate concentration. ANCOVA analysis was used to test if difference in SRR among treatments could be explained by a covariate: the sulfate concentration. All analyses were performed using the R-base package version 3.4.2 [60] as well as the package “lsmeans” [61].
Net production and consumption of sulfate was calculated from inverse modeling of the mean sulfate concentration profiles for each treatment, using the general mass conservation equation [62], which for 1-D systems at steady state has the form:
here dJ(z)/dz is the flux derivative and R(z) the bulk reaction term (R(z) < 0 for net consumption and R(z) > 0 for net production of sulfate).
The flux component J(z) in Eq. 2 was defined from the Nernst–Planck equation modified for porous media [26], as this equation includes both a diffusion component and an ionic drift component and thereby estimates correctly the transports in systems with concentration gradients and electric fields, such as cable bacteria colonized sediments:
here ϕ is the sediment porosity, DS is the diffusion coefficient of sulfate in the sediment, which was estimated from the porosity and the temperature- and salinity-corrected diffusion coefficient of sulfate at infinite dilution (DSO4) using the approximation DS = ϕ2 × DSO4 [63]. DSO4 (0.722 cm−2 d−1) was estimated with the function diffcoeff() from the R-package “marelac” [64]. \(\frac{{dC}}{{dz}}\) denotes the sulfate concentration gradient in the z-direction, n the charge number of the sulfate ion (n = −2), F is the Faraday’s constant (9.65 × 104 coulomb mol−1), R the gas constant (8. 31 J mol−1 K−1), T the temperature (288 K) and \(\frac{{d\psi }}{{dz}}\) is the gradient of the EP. \(\frac{{d\psi }}{{dz}}\) was calculated from polynomial fits of the measured EP distribution in the sediments. A modified version of the software tool PROFILE [65] that implements Eq. 2 with Eq. 3 was used to calculate R(z) in Eq. 2 (Burdorf, Van de Velde and Meysman in prep.)
R(z) estimates the difference between sulfate consumption and gross sulfate production (GSP). With the sulfate consumption being equivalent to sulfate reduction (SRR), GSP at a given depth was calculated as:
The diffusive oxygen uptake (DOU) of the ES sediments was calculated from 20 randomly selected oxygen profiles from each of the lake sediments, using the software tool PROFILE. The diffusion coefficient used in the calculations (DS) was estimated from the porosity and the temperature/salinity corrected diffusion coefficient for oxygen at infinite dilution (DO2) using the approximation DS = ϕ2 × DO2. DO2 (1.621 cm−2 d−1) was estimated with the function diffcoeff() from the R-package “marelac”.
The oxygen penetration depth (OPD) of the ES sediments was defined as the depth where the oxygen concentration was ≤0.1 µM. OPD was estimated for all measured oxygen profiles and reported as the mean ± SE.
For analysis of the SRM community, quality control of raw reads, trimming, chimera removal, and clustering into amplicon sequence variants (ASVs) with DADA2 [66] was carried out as described in detail in Marshall et al. [67], with the exception that the sequencing reads were randomly subsampled down to 4011 reads prior to clustering, to account for differences in sequencing depth between samples. Classification was carried out using an existing dsrAB database and classification scheme [37] updated for this study by Ian Marshall, Aarhus University, to also include cable bacteria dsrAB gene sequences [68]. Cable bacteria ASVs were further identified by aligning them with a database of known cable bacterial dsrB gene sequences using BLASTn. Sequences are available in the GenBank Sequence Read Archive database under accession numbers SAMN10441970—SAMN10442005.
The absolute abundance of a given ASV in a sample was then calculated as the product of its relative abundance and the total number of dsrB genes present in the sample, with the latter being estimated using qPCR. As our comparative analyses focused on the effect of cable bacteria on the dsrB community, all cable bacteria-affiliated dsrB ASVs were removed from the dataset prior to analysis. Richness (here defined as the number of different dsrB ASVs) and the Shannon diversity for ES and ES-free cores were calculated from the dataset using the function estimate richness() from the R-package phyloseq [69]. Student’s t test was used to test for differences in richness, diversity, and general abundance of dsrB ASVs between treatments (ES vs. ES-free cores) for each lake. To address if SRM communities differed between ES and ES-free cores, Bray–Curtis dissimilarity indices were calculated from the dataset separately for the two lakes, using the vegdist() function from the R package vegan [70]. Differences between ES and ES-free cores were evaluated with ANOSIM [71] using the vegan function anosim(). The function exactTest() in the R-package edgeR version 3.20.9 [72] was used to test for significant (FDR-corrected p value <0.05) differences in the abundance of individual dsrB ASVs between ES and ES-free cores. All bioinformatics analyses were performed with nine replicates (data from three cores sliced in three sections) for ES and ES-free cores.
Results
Microelectrode profiles
The OPD of the ES sediments from Vilhelmsborg Sø and Skanderborg Sø, was 3.1 ± 0.1 and 3.7 ± 0.1 mm, respectively (Fig. 1a, c). The DOU of the sediments was 29.0 ± 1.9 and 23.2 ± 1.1 mmol m−2 d−1. Both sediments assigned as ES sediments contained cable bacteria as indicated by FISH (Fig. S1) and displayed features indicating e-SOx: pH extremes were observed with maxima in the oxic zone and minima in deeper oxygen-free layers, and the EP increased with depth. The pH maximum was located in the upper 1 mm of the sediment in both cases and was only ~0.1 U above the value in the water column. The pH minimum (6.2 ± 0.1) was located at 24 mm depth in the Vilhelmsborg Sø sediment. In the Skanderborg Sø sediment the pH minimum (6.4 ± 0.1) was located at 23 mm depth. The EP (measured relative to the water column) increased monotonically in both ES sediments down to 24 mm, to a maximum value of 15.6 ± 0.2 and 12.7 ± 0.6 mV for the Vilhelmsborg Sø and Skanderborg Sø sediment, respectively. From this depth, the EP converged toward an asymptotic value. The electric field (calculated as \(- \frac{{d\psi }}{{dz}}\)) was directed from the sediment toward the water column in both cases. In the ES sediment from Vilhelmsborg Sø, the magnitude of the field increased from approx. 0.6 V m−1 in the oxic zone to a maximum of 1.0 V m−1 in the 7.5–10.5 mm depth interval. Below 10.5 mm, the field strength decreased monotonically to near zero values at ~24 mm depth. The field strength was slightly lower in the Skanderborg Sø sediment. Here, the magnitude of the field increased from ~0.2 V m−1 in the oxic zone to a maximum of 0.9 V m−1 at 9 mm depth. Below this depth, the field strength declined monotonically to near zero values at ~24 mm depth. ∑H2S was below detection limit (2 µM) in sediment from Vilhelmsborg Sø and <7 µM in the sediment from Skanderborg Sø. In the latter, the free ∑H2S was consumed at the border of the oxic zone and below. Net ∑H2S consumption, calculated from the profile, was <5 µmol m−2 d−1.

Depth profiles of the ∑H2S concentration (black dots), pH (yellow dots), electric potential (EP) distribution (white dots), and oxygen concentration (red dots) in ES sediment and ES-free sediment from Vilhelmsborg Sø (a, b) and Skanderborg Sø (c, d). Error bars indicate standard error of the mean (n = 36). Only every second point of each data series is plotted for clarity. Two series of pH profiles were measured in the ES-sediments: a series measured with a depth resolution of 100 µm in the upper 6 mm of the sediment and a series measured with a depth resolution of 400 µm in the full domain.
The above-described e-SOx features were not expressed (or only to a much lesser extent) in the sediments assigned as ES-free sediments and no cable bacteria could be detected by FISH. In the sediment from Vilhelmsborg Sø (Fig. 1b), pH decreased monotonically from 8.6 in the water column to an asymptotic value of ~7 at 10 mm depth. An overall but minor decrease in pH with depth (<0.1 U cm−1) was also observed for the Skanderborg Sø sediments (Fig. 1d). The EP measured in the Vilhelmsborg Sø sediment relative to the water column increased monotonically to 2.7 ± 0.9 mV at a depth of 10 mm and thereafter remained constant within the SE. The magnitude of the electric field estimated from the EP signal was 0.4 V m−1 in the top sediment and declined to near zero values at 10 mm depth. While the cause of this field remains unclear, it was likely unrelated to cable bacterial activity as the field remained unaffected by horizontal cuts (data not shown). No changes in EP with depth, and consequently no electric fields, were observed in the Skanderborg Sø sediment. Average ∑H2S was below 6 µM in both sediments, but only detectable in the sediments from Vilhelmsborg Sø (Fig. 1b, d).
Sulfate concentrations
Sulfate concentrations (Fig. 2) were generally higher in the ES sediments than in the ES-free sediments (Wilcoxon signed rank test: p < 2.2 × 10−16, n = 84, for Vilhelmsborg Sø and p = 1.2 × 10−14, n = 84 for Skanderborg Sø). The inventory of sulfate present in the 0–4.25 cm domain of the ES sediment from Vilhelmsborg Sø was more than ten times higher than the inventory present in the same domain of ES-free sediment (27.5 ± 4.5 vs. 2.2 ± 0.2 mmol m−2). For the Skanderborg Sø sediments, the inventory of sulfate present in the 0–3 cm domain of the ES sediment was three times higher than the inventory present in the same domain of ES-free sediment (9.4 ± 0.4 vs. 3.6 ± 1.2 mmol m−2). The difference in sulfate inventories among ES and ES-free sediments were significant for both the sediment from Vilhelmsborg Sø and the sediment from Skanderborg Sø (Student’s t test, p ≤ 0.03, n = 6).

Sulfate concentrations (black dots), R(z) (gray bars) in ES sediment, and ES-free sediment from Vilhelmsborg Sø (a, b) and Skanderborg Sø (c, d). Error bars indicate standard error of the mean (n = 3). Positive values for R(z) signify net production, negative values net consumption of sulfate. The black line represents the modeled sulfate concentration. Dashed line in a and c represents the lower boundary of the oxic zone.
The sulfate profiles measured in ES sediments suggested net production of sulfate in both oxic and anoxic layers. Sulfate profiles measured in the ES-free sediments suggested exclusively consumption. Reaction transport modeling of the mean profiles confirmed this pattern: net sulfate production occurred in the upper 10 mm of the ES sediment from Skanderborg Sø and in the upper 20 mm of the corresponding sediment from Vilhelmsborg Sø. Reaction transport modeling further suggested that the 0–4.5 cm domain of the ES-sediment from Vilhelmsborg Sø was a sulfate source, with an estimated net production of sulfate amounting to 0.98 mmol m−2 d−1. In contrast, the modeling showed that the 0–4.5 cm domain of the ES-free sediment from Vilhelmsborg Sø, as well as both the ES and ES-free sediment from Skanderborg Sø, were sulfate sinks. The net sulfate consumption in these sediments was estimated to 0.77, 0.24, and 0.11 mmol m−2 d−1, respectively.
Sulfate reduction rates
Sulfate reduction was below detection limit in the upper 3.5 mm of the ES sediments, likely because these layers were exposed to oxygen during the incubation (Fig. 3). Below this depth, sulfate reduction was measurable throughout the 4–4.45 cm deep sediment column in both ES and ES-free sediments. Volume-specific SRR in oxygen-free sediment strata were significantly higher in the ES-sediments than in the ES-free sediments (Student’s t test: p = 6.5 × 10−5, n = 27 for the Vilhelmsborg Sø sediment, and p = 0.035, n = 27 for the Skanderborg Sø sediment). The total depth-integrated SRR was 3–4.5-fold higher in these sediments compared to their respective ES-free sediments (13.0 ± 1.0 vs. 2.5 ± 0.9 mmol m−2 d−1 for the Vilhelmsborg sediment and 16.4 ± 4.7 mmol vs. 5.6 ± 0.6 mmol m−2 d−1 for the Skanderborg Sø sediment).

Sulfate reduction rates (gray bars) and sulfate concentrations (black dots) in ES sediments and ES-free sediments from Vilhelmsborg Sø (a, b) and Skanderborg Sø (c, d). Error bars indicate standard error of the mean (n = 3). Dashed line in a and c represents the lower boundary of the oxic zone.
For both lake sediments, the volume-specific SRR was significantly correlated with the sulfate concentration (Pearson product moment correlation, p = 4.98 × 10−6, r = 0.76 for the Vilhemsborg sediments, and p = 6.71 × 10−5 r = 0.69 for the Skanderborg sediments). When this apparent dependency between sulfate and sulfate reduction was taken into account through ANCOVA analysis, no significant difference in SRR between ES-sediments and ES-free sediments were seen (ANCOVA: p = 0.15 for the Vilhelmsborg Sø sediment, and p = 0.31 for the Skanderborg Sø sediment).
Gross sulfate production
GSP, estimated as the sum of SRR and R(z) in Fig. 2, was seen in the entire 0–4 cm domain of both the ES and the ES-free sediments (Fig. 4). For the ES sediment from Vilhelmsborg Sø, the depth-integrated GSP was 13.6 mmol m−2 d−1, and sulfate production in the 3.1 mm deep oxic zone contributed to 2.4% of the overall production. For the ES-free sediment, the depth-integrated rate of GSP was 1.7 mmol m−2 d−1. For the ES sediment from Skanderborg Sø, the depth-integrated rate of GSP was 16.1 mmol m−2 d−1, and sulfate production in the 3.7 mm deep oxic zone contributed to 0.2% of the overall production. For the ES-free sediment, the depth-integrated rate of GSP was 5.5 mmol m−2 d−1.

Gross sulfate production rates estimated from reaction transport modeling and measured SRR for ES and ES-free sediments from Vilhelmsborg Sø (a, b) and Skanderborg Sø (c, d). Dashed line in a and c represents the lower boundary of the oxic zone.
SRM abundance and community composition
EP profiling in ES and ES-free cores (Fig. S2) reproduced the patterns observed in ES and ES-free sediments from biogeochemical experiment, justifying the comparison between the two experiments. Three dsrB ASVs (ASV52, ASV374, and ASV830) were identified as cable bacteria, all ~97% identical to the dsrB of the freshwater species Ca. Electronema palustris [68], which is well within the species threshold for SRM [37]. Only ASV52 was consistently found in all samples, while the other ASVs were rare (<0.05 and <0.02% of all SRM). In Vilhelmsborg Sø, total cable bacteria accounted for 0.7% and 0.2% of the total dsrB pool in the ES and ES-free cores, respectively, which corresponds to 1.6 × 107 ± 3.8×106 and 4.3 × 106 ± 8.9 × 105 gene copies cm−3, respectively. Since no e-SOx was detected in the ES-free cores, the latter dsrB genes likely represent remnant DNA or inactive cells. In Skanderborg Sø, cable bacteria accounted for 0.3% and 0.04% of the total dsrB pool in the ES and ES-free cores, respectively, which corresponds to 3.4 × 105 ± 2.3 × 103 and 3.1 × 104 ± 3.1 × 104 gene copies cm−3, respectively.
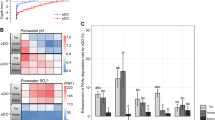
When comparing overall dsrB gene copy numbers to 16S rRNA gene copy numbers for both lakes (Vilhelmsborg Sø: 5.4 × 1010 ± 2.8 × 109 16S rRNA gene copies cm−3 vs. 2.5 × 109 ± 1.9 × 108 dsrB gene copies cm−3; Skanderborg Sø: 1.5 × 109 ± 2.8 × 107 16S rRNA gene copies cm−3 vs. 6.8 × 107 ± 2.0 × 106 dsrB gene copies cm−3), SRM accounted for 4–5% of the total bacterial population, which is a typical SRM fraction for freshwater sediments [73]. In both lakes, dsrB families commonly found in freshwater sediment [73] like Desulfobacteraceae, Desulfobulbaceae, and the Desulfobacca acetoxidans lineage, were among the most abundant SRMs, together with a few unclassified families (Fig. 5). The size, richness, and diversity of the dsrB-carrying communities in both lake sediments was not significantly different between ES and ES-free cores.

Abundance and family-level affiliation of dsrB genes in sediments from Vilhelmsborg Sø (a, b) and Skanderborg Sø (c, d). For each panel, left bars represent ES sediments, and right bars represent ES-free sediments. Stacked bar plots (b, d) show mean relative abundance for the ten most abundant dsrB families detected; oxidative type dsrB families are marked by an asterisk. For unclassified dsrB families, the next higher classified taxonomic rank is given. For details, see Figs. S3 and S4.
In Vilhelmsborg Sø, the abundance of dsrB genes inferred from qPCR was similar in ES and ES-free cores at depths >0.3 mm (Fig. 5a; Student’s t test, p = 0.74). Above this depth, dsrB gene abundance in the ES cores was 66% of that in the ES-free cores. There was no significant difference in ASV richness of the dsrB community between ES and ES-free cores (125 ± 2 vs. 129 ± 2 for the ES and ES-free cores, respectively, Student’s t test, p = 0.11), and the Shannon diversity of the dsrB community was almost identical (3.64 ± 0.04 vs. 3.68 ± 0.02 for the ES and ES-free cores, respectively, Student’s t test, p = 0.386). The same dominant dsrB families were present in both ES and ES-free cores (Fig. 5b), with Desulfobacteraceae, a family from an unclassified lineage, an unclassified family of the Deltaproteobacteria supercluster, and Desulfobulbaceae as the most abundant families (Figs. 5b and S3). ANOSIM analysis of the Bray–Curtis dissimilarity indices suggested a small (R = 0.277) but significant (p = 0.009) dissimilarity between the dsrB communities in the ES and the ES-free cores. However, none of the individual dsrB ASVs showed a significant (FDR-corrected p value <0.05) difference in its abundance between ES and ES-free cores (Table S1).
Likewise, in Skanderborg Sø sediment, dsrB gene abundance was similar in ES and ES-free cores (Fig. 5c; Student’s t test, p = 0.2626), there was no significant difference in ASV richness of the dsrB community between ES and ES-free cores (151 ± 4 vs. 132 ± 12 for the ES and ES-free cores, respectively; Student’s t test, p = 0.14), and the Shannon diversity of the dsrB community was also almost identical (4.40 ± 0.04 vs. 4.34 ± 0.08 for the ES and the ES-free cores, respectively; Student’s t test, p = 0.56). The same dsrB families were most abundant in ES and ES-free cores (Fig. 5d), which were both strongly dominated (>70% of the total community) by unclassified and uncultured lineages, although Desulfobacteraceae, Desulfobulbaceae, and the Desulfobacca acetoxidans lineage were still among the ten most abundant families (Figs. 5d and S4). ANOSIM analysis of the Bray–Curtis dissimilarity indices suggested a significant (p = 0.019) dissimilarity between the dsrB communities in the ES and the ES-free cores, which was however close to the dissimilarity within the dsrB communities in the ES cores and ES-free cores, respectively (R = 0.187); again, none of the individual dsrB ASVs showed a significant (FDR-corrected p value <0.05) difference in its abundance between ES and ES-free cores (Table S1).
Discussion
In the present study, we have analyzed the impacts of cable bacteria and e-SOx on sulfur cycling and SRM communities in freshwater sediments. As our experiments were designed to compare sulfur cycle processes between oxygen-exposed sediments (the ES-sediments) and anoxic sediments (the ES-free sediments), the validity of our conclusions regarding the impact of e-SOx hinges on the assumption that oxygen, aside from driving e-SOx, does not interfere significantly with the sulfur cycle. As our setup excluded bioturbation and thus prevented mechanical transport of metal oxides from the oxic to the anoxic zone, oxygen could only interfere with the sulfur cycle either directly through oxidation of sulfur compounds in the oxic zone or indirectly through the oxic formation and downward diffusion of oxidants like nitrite and nitrate. According to our estimates of GSP (Fig. 5), chemical or biological reoxidation of sulfides to sulfate in the oxic zone of the ES-sediments was insignificant in comparison to the oxidation in the oxygen-free layers: sulfate production in the oxic zone contributed to only 0.2–2.4% of the GSP. Sulfate reduction was undetectable in the oxic zone but readily measured in all ES-sediment layers below 3 mm depth, i.e., those not directly exposed to oxygen (Fig. 3); considering the common redox cascade, where oxygen and nitrate reduction precede sulfate reduction [74], this indicates that the indirect effect of oxygen, e.g., via nitrate, on sulfur cycling was negligible. Therefore, aside from driving e-SOx, oxygen had only a marginal impact on sulfur cycling in the ES-sediments, and the more than threefold higher sulfate inventories and SRR observed in the ES-sediments in comparison to the ES-free sediments thereby suggest that e-SOx enhances sulfate concentrations and increases SRR.
The analysis of SRM communities in ES and ES-free cores suggests on the other hand only very limited influences of cable bacteria-mediated e-SOx on the community structure of SRMs, with no effect on overall abundance, richness, and diversity, and only small changes toward a minor dissimilarity between the SRM communities in ES and ES-free cores (Fig. 5 and Table S1).
Cable bacteria stimulate sulfate accumulation
The 3–10-times larger pools of sulfate in ES sediments as compared to the ES-free sediments were due to the manifestation of three mechanisms associated with e-SOx: (A) enhanced sulfide oxidation; (B) mobilization and oxidation of endogenous sulfur pools; and (C) enhanced downward transport of sulfate.
(A) In general, depth integrated SRR values were much higher than depth integrated rates of net consumption estimated from reaction transport modeling. For sediments assigned as sulfate sinks, i.e., the ES-free sediment from Vilhelmsborg Sø and the ES and ES-free sediment from Skanderborg Sø, the depth-integrated rate of net sulfate consumption was 33%, 1.5%, and 2% of the experimentally determined SRR, respectively. The discrepancy was even more pronounced for the ES sediment from Vilhelmsborg Sø, as this sediment was a sulfate source, despite high rates of sulfate reduction. This indicates that sulfate reduction in both ES and ES-free sediments was largely driven by sulfate produced within the sediment via sulfide oxidation (the alternative explanation that our 35S-based SRRs were severely overestimated is less likely since experimental studies have shown that the technique can underestimate the indigenous SRR due to reoxidation of the tracer [51]). Our estimates of GSP (Fig. 4) reflect the rates of sulfide oxidation taking place in the sediments. For the Vilhelmsborg Sø sediment, the rate of sulfide oxidation needed to sustain the sulfate production in the oxygen free sediment layers was a factor of 7.8 higher in ES sediment than in the ES-free sediment (13.6 vs. 1.7 mmol m−2 d−1). For the sediment from Skanderborg Sø, the corresponding rate of sulfide oxidation in the ES sediment was ca. 3 times higher than rates estimated for the ES-free sediment (16.1 vs. 5.5 mmol m−2 d−1).
The direct contribution of e-SOx to sulfate production can be estimated from the electron transfer rate of the cable bacteria, which when calculated from the EP profiles as in Damgaard et al. [31] amounted to 44 and 59 mmol electrons m−2 d−1 for the Skanderborg Sø and Vilhelmsborg Sø sediments, respectively. With the transfer of 8 moles of electrons per mole of sulfide being oxidized to sulfate, the rate of sulfate production via e-SOx would amount to 5.4 and 7.4 mmol m−2 d−1 in the Skanderborg Sø and Vilhelmsborg Sø sediments, respectively. According to our EP data (Fig. 1), the cable bacteria activity zone spans the upper 2.4 cm of the sediment [32], and e-SOx would then contribute to 44% of the sulfate production activity in the corresponding oxygen free layers of the Skanderborg Sø sediment (12.3 mmol m−2 d−1) and 100% in the Vilhelmsborg Sø sediment (6.7 mmol m−2 d−1), respectively. It is clear however, that e-SOx cannot fully account for all the sulfate production activity in the ES-sediments inferred from our data, as significant anaerobic sulfate production was present also below the cable bacteria activity zone and in the ES-free sediments (Fig. 5). In these strata, GSP was probably, like for the ES-free sediments, due to sulfide oxidation driven by, e.g., buried oxidized metals like iron and manganese.
(B) Modeling the sulfate profiles measured in the ES-free and ES sediments showed net production of sulfate in oxygen-free sediment layers, and this occurred exclusively in the ES sediments (Fig. 2). Such net production points to the presence of an internal sulfide pool being oxidized, as the reoxidation of sulfide produced from sulfate reduction would lead to no net change in the sulfate concentration profile. Net production of sulfate in the anoxic zone has been observed in marine sediments with e-SOx [26, 28], and in a few sediments from lakes and rivers [75]. In marine sediments with e-SOx, the source of sulfide fueling this sulfate production has been associated to the dissolution of solid phase iron sulfides, as a result of the low pH and low sulfide concentrations induced by e-SOx in the anoxic zone [25, 26]. The mobilized sulfide is then oxidized to sulfate via e-SOx. Though iron chemistry was not addressed in the present study, we expect a similar mechanism operating in the freshwater sediments studied here: pH and sulfide concentrations were similar to those observed in marine sediments, and the color of the suboxic zone in the ES sediments changed from black (diagnostic color for amorphous FeS minerals) to greyish-brown during the course of the incubation.
(C) Modeling the transport of sulfate from the individual components of the Nernst–Planck equation (i.e., the diffusive flux and the drift flux), showed a substantial contribution of the drift flux to the overall transport of sulfate in the ES sediments (Fig. 6) due to the relative strong electric fields present there. Due to the orientation of the electric field, the drift flux was directed downwards and therefore counteracted the upward diffusive flux in the top 10–15 mm of the sediments, leading to a net downward transport of sulfate just below the sediment water interface. Hence, sulfate produced from sulfide oxidation in the oxygen-free zone was retained within the sediment and exposed to further downward transport and metabolic conversions.

Sulfate fluxes in ES-sediments from Vilhelmsborg Sø (a) and Skanderborg Sø (b) Ionic drift (green), diffusional fluxes (red) and net fluxes (black). The diffusive flux was calculated according to: \(J_{\mathrm{diff}} \,=\, - \phi D_{{\mathrm{SO}_{4}}^{2 - }}\frac{{d\left[ {{\mathrm{SO}_{4}}^{2 - }} \right]}}{{dz}}\) and the ionic drift according to: \(J_{\mathrm{drift}} \,=\, \phi D_{{\mathrm{SO}_{4}}^{2 - }}\frac{{nf}}{{RT}}\left[ {{\mathrm{SO}_{4}}^{2 - }} \right]\left( { - \frac{{d\psi }}{{dz}}} \right)\). The net flux was calculated as the sum of the ionic drift and the diffusive flux. Positive fluxes indicate downward transport, negative upward transport. The sulfate concentrations used in the calculations were the modeled concentrations (Fig. 2). All other parameters are specified in the materials and method section.
To summarize the above discussed mechanisms: by lowering the pH and H2S concentration in the sediment, cable bacteria promote mobilization of solid-phase sulfide pools. Their e-SOx metabolism, in combination with conventionally driven sulfide oxidation processes, assures that this sulfide is oxidized to sulfate in the sediment. The electric fields formed through e-SOx assist to retain the produced sulfate in the sediment, and the sulfate is entering a highly efficient reduction-oxidation cycle, which assures regeneration of sulfate almost as fast as it is consumed.
Cable bacteria promote sulfate reduction via enhancement of sulfate concentrations
The 3–4.5-fold increase in the depth-integrated SRR observed in the ES sediments was likely directly linked to the ability of cable bacteria to significantly enhance sulfate availability in an otherwise sulfate-scarce environment, and thereby to soften the kinetic limitation of the SRM community. The correlation analysis performed on data from our biogeochemical experiment showed a significant correlation between SRRs and sulfate concentrations, in line with the hypothesis that sulfate reduction was sulfate limited. Half-saturation constants of the SRM population that support such sulfate limitation of the process may lie in the range of 100–200 µM sulfate. Although half-saturation constants vary considerably among cultured SRM [54], this range has been found for natural SRM populations in Scheldt River freshwater sediments [35]. The ANCOVA analysis, which showed that the difference in SRR between ES and ES-free sediments could be explained by covariation with the sulfate concentration, is in line with the hypothesis that e-SOx-induced sulfate elevation was the main factor driving the enhanced sulfate reduction activity in the ES sediments.
The SRM community analysis performed in our second experiment showed insignificant influence of cable bacteria and e-SOx on the community structure and overall abundance of SRMs. This observation provides evidence against the hypothesis that the 3–4.5-fold higher SRR in ES sediments observed in our first experiment were caused by cable bacteria-induced alternations of the SRM community. Although cable bacteria harbor the dsrB gene and a full sulfate reduction pathway [27], it is unlikely that the stimulated SRR in the ES sediments can be explained with sulfate reduction performed by cable bacteria. Firstly, cable bacteria likely use the sulfate reduction pathway in reverse during e-SOx [27] and typical cable bacteria features indicative of e-SOx (Figs. 1 and S2) would not be present if their major metabolic pathway was sulfate reduction. In addition, the low abundance of cable bacteria-affiliated dsrB ASVs, which were <0.8% of the total pool of ASVs in the ES-cores from both Vilhelmsborg Sø and Skanderborg Sø, implies that sulfate reduction by cable bacteria would contribute very little to the total SRR.
Thus the simplest explanation for the stimulated SRR in the ES sediments, in line with our data, is that cable bacteria promote sulfate reduction via enhancement of sulfate concentrations.
Conclusions and perspectives
Cable bacteria enhance sulfate concentrations both directly through their metabolic activity, which includes the oxidation of sulfide to sulfate, and indirectly via both the formation of electric fields as well as mobilization of solid phase sulfide. A sulfate-limited community of SRM then takes advantage of the elevated sulfate concentrations, increasing their cell-specific activities, and driving a general increase in sedimentary SRR. This suggests a mutualistic relationship between cable bacteria and SRMs in sulfate-limited freshwater environments, where cable bacteria support the SRMs with sulfate, which in turn provide sulfide to fuel e-SOx. Via e-SOx, cable bacteria can then be important players in the cryptic sulfur cycle, but as inferred from our data not the major drivers, and more quantitative research on the sulfide oxidation mechanisms are needed to fully constrain this cycle. The ability of cable bacteria to intensify and expand sulfur cycling in freshwater sediments may have implications for the mode of carbon turnover in freshwater sediments, and a future venue of research could be to test if this intensification of sulfur cycling causes cable bacteria to indirectly reduce the rates of methanogenesis, thereby reducing methane emissions from freshwater systems.
References
Holmer M, Storkholm P. Sulphate reduction and sulphur cycling in lake sediments: a review. Freshw Biol. 2001;46:431–51.
Soetaert K, Herman PMJ, Middelburg JJ. A model of early diagenetic processes from the shelf to abyssal depths. Geochim Cosmochim Acta. 1996;60:1019–40.
Capone DG, Kiene RP. Comparison of microbial dynamics in marine and freshwater sediments: contrasts in anaerobic carbon catabolism1. Limnol Oceanogr. 1988;33:725–49.
Canavan RW, Slomp CP, Jourabchi P, Van Cappellen P, Laverman AM, van den Berg GA. Organic matter mineralization in sediment of a coastal freshwater lake and response to salinization. Geochim Cosmochim Acta. 2006;70:2836–55.
Jørgensen BB. Comparison of methods for the quantification of bacterial sulfate reduction in coastal marine-sediments. 1. Measurement with radiotracer techniques. Geomicrobiol J. 1978;1:11–27.
Holmkvist L, Ferdelman TG, Jorgensen BB. A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim Cosmochim Acta. 2011;75:3581–99.
Jørgensen BB, Findlay AJ, Pellerin A. The biogeochemical sulfur cycle of marine sediments. Front Microbiol. 2019;10:1–27.
Jørgensen BB, Parkes RJ. Role of sulfate reduction and methane production by organic carbon degradation in eutrophic fjord sediments (Limfjorden, Denmark). Limnol Oceanogr. 2010;55:1338–52.
Bak F, Pfennig N. Microbial sulfate reduction in littoral sediment of Lake Constance. FEMS Microbiol Lett. 1991;85:31–42.
Urban NR, Brezonik PL, Baker LA, Sherman LA. Sulfate reduction and diffusion in sediments of Little Rock Lake, Wisconsin. Limnol Oceanogr. 1994;39:797–815.
Roden EE, Tuttle JH. Inorganic sulfur turnover in oligohaline estuarine sediments. Biogeochemistry. 1993;22:81–105.
Pellerin A, Bui TH, Rough M, Mucci A, Canfield DE, Wing BA. Mass-dependent sulfur isotope fractionation during reoxidative sulfur cycling: a case study from Mangrove Lake, Bermuda. Geochim Cosmochim Acta. 2015;149:152–64.
Canfield DE. Reactive iron in marine-sediments. Geochim Cosmochim Acta. 1989;53:619–32.
Elsgaard L, Jørgensen BB. Anoxie transformations of radiolabeled hydrogen sulfide in marine and freshwater sediments. Geochim Cosmochim Acta. 1992;56:2425–35.
King GM. Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments. FEMS Microbiol Ecol. 1990;6:131–8.
Pester M, Knorr K-H, Friedrich MW, Wagner M, Loy A. Sulfate-reducing microorganisms in wetlands—fameless actors in carbon cycling and climate change. Front Microbiol. 2012;3:1–19.
Malkin SY, Rao AMF, Seitaj D, Vasquez-Cardenas D, Zetsche E-M, Hidalgo-Martinez S, et al. Natural occurrence of microbial sulphur oxidation by long-range electron transport in the seafloor. ISME J. 2014;8:175–9.
Pfeffer C, Larsen S, Song J, Dong MD, Besenbacher F, Meyer RL, et al. Filamentous bacteria transport electrons over centimetre distances. Nature. 2012;491:218–21.
Marzocchi U, Trojan D, Larsen S, Meyer RL, N.P R, Schramm A, et al. Electric coupling between distant nitrate reduction and sulfide oxidation in marine sediment. ISME J. 2014;8:1682–90.
Nielsen LP, Risgaard-Petersen N, Fossing H, Christensen PB, Sayama M. Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature. 2010;463:1071–4.
Burdorf LDW, Tramper A, Seitaj D, Meire L, Hidalgo-Martinez S, Zetsche EM, et al. Long-distance electron transport occurs globally in marine sediments. Biogeosciences. 2017;14:683–701.
Marzocchi U, Bonaglia S, van de Velde S, Hall POJ, Schramm A, Risgaard-Petersen N, et al. Transient bottom water oxygenation creates a niche for cable bacteria in long-term anoxic sediments of the Eastern Gotland Basin. Environ Microbiol. 2018;20:3031–41.
Muller H, Bosch J, Griebler C, Damgaard LR, Nielsen LP, Lueders T, et al. Long-distance electron transfer by cable bacteria in aquifer sediments. ISME J. 2016:2010–9.
Risgaard-Petersen N, Kristiansen M, Frederiksen RB, Dittmer AL, Bjerg JT, Trojan D, et al. Cable Bacteria in freshwater sediments. Appl Environ Microbiol. 2015;81:6003–11.
Meysman FJR, Risgaard-Petersen N, Malkin SY, Nielsen LP. The geochemical fingerprint of microbial long-distance electron transport in the seafloor. Geochim Cosmochim Acta. 2015;152:122–42.
Risgaard-Petersen N, Revil A, Meister P, Nielsen LP. Sulfur, iron-, and calcium cycling associated with natural electric currents running through marine sediment. Geochim Cosmochim Acta. 2012;92:1–13.
Kjeldsen KU, Schreiber L, Thorup CA, Boesen T, Bjerg JT, Yang T, et al. On the evolution and physiology of cable bacteria. Proc Natl Acad Sci USA. 2019;116:19116–25.
Rao AMF, Malkin SY, Hidalgo-Martinez S, Meysman FJR. The impact of electrogenic sulfide oxidation on elemental cycling and solute fluxes in coastal sediment. Geochim Cosmochim Acta. 2016;172:265–86.
Seitaj D, Schauer R, Sulu-Gambari F, Hidalgo-Martinez S, Malkin SY, Burdorf LDW, et al. Cable bacteria generate a firewall against euxinia in seasonally hypoxic basins. Proc Natl Acad Sci USA. 2015;112:13278–83.
Sulu-Gambari F, Seitaj D, Meysman FJR, Schauer R, Polerecky L, Slomp CP. Cablebacteria control iron–phosphorus dynamics in sediments of a coastal hypoxic basin. Environ Sci Technol. 2016;50:1227–33.
Damgaard LR, Risgaard-Petersen N, Nielsen LP. Electric potential microelectrode for studies of electro-biogeophysics. J Geophys Res. 2014;119:2014JG002665.
Risgaard-Petersen N, Damgaard LR, Revil A, Nielsen LP. Mapping electron sources and sinks in a marine biogeobattery. J Geophys Res (Biogeosci). 2014;119:1475–86.
Bockris JOM, Reddy AKN. Modern electrochemistry—ionics. 2nd ed. New York: Plenum Press; 1998. p. 769.
Ingvorsen K, Jørgensen BB. Kinetics of sulfate uptake by freshwater and marine species of Desulfovibrio. Arch Microbiol. 1984;139:61–6.
Pallud C, Van Cappellen P. Kinetics of microbial sulfate reduction in estuarine sediments. Geochim Cosmochim Acta. 2006;70:1148–62.
van de Velde S, Lesven L, Burdorf LDW, Hidalgo-Martinez S, Geelhoed JS, Van Rijswijk P, et al. The impact of electrogenic sulfur oxidation on the biogeochemistry of coastal sediments: a field study. Geochim Cosmochim Acta. 2016;194:211–32.
Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2014;9:1152.
van de Velde S, Meysman FJR. The influence of bioturbation on iron and sulphur cycling in marine sediments: a model analysis. Aquat Geochem. 2016;22:469–504.
Risgaard-Petersen N. Coupled nitrification-denitrification in autotrophic and heterotrophic estuarine sediments: on the influence of benthic microalgae. Limnol Oceanogr. 2003;48:93–105.
Risgaard-Petersen N, Langezaal AM, Ingvardsen S, Schmid MC, Jetten MSM, Op den Camp HJM, et al. Evidence for complete denitrification in a benthic foraminifer. Nature. 2006;443:93–6.
Risgaard-Petersen N, Meyer RL, Revsbech NP. Denitrification and anaerobic ammonium oxidation in sediments: effects of microphytobenthos and NO3-. Aquat Microb Ecol. 2005;40:67–76.
Jensen K, Sloth NP, Risgaard-Petersen N, Rysgaard S, Revsbech NP. Estimation of nitrification and denitrification from microprofiles of oxygen and nitrate in model sediment systems. Appl Environ Microb. 1994;60:2094–100.
Burdorf LDW, Malkin SY, Bjerg JT, van Rijswijk P, Criens F, Tramper A, et al. The effect of oxygen availability on long-distance electron transport in marine sediments. Limnol Oceanogr. 2018;63:1799–816.
Revsbech NP. An oxygen microsensor with a guard cathode. Limnol Oceanogr. 1989;34:474–8.
Schauer R, Risgaard-Petersen N, Kjeldsen KU, Bjerg JJT, Jørgensen BB, Schramm A, et al. Succession of cable bacteria and electric currents in marine sediment. ISME J. 2014;8:1314–22.
Lücker S, Steger D, Kjeldsen KU, MacGregor BJ, Wagner M, Loy A. Improved 16S rRNA-targeted probe set for analysis of sulfate-reducing bacteria by fluorescence in situ hybridization. J Microbiol Methods. 2007;69:523–8.
Jeroschewski P, Steuckart C, Kuhl M. An amperometric microsensor for the determination of H2S in aquatic environments. Anal Chem. 1996;68:4351–7.
Revsbech NP, Jørgensen BB. Microelectrodes—their use in microbial ecology. Adv Microb Ecol. 1986;9:293–352.
Cline JD. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr. 1969;14:454–8.
Røy H, Weber HS, Tarpgaard IH, Ferdelman TG, Jørgensen BB. Determination of dissimilatory sulfate reduction rates in marine sediment via radioactive 35S tracer. Limnol Oceanogr Methods. 2014;12:196–211.
Moeslund L, Thamdrup B, Barker Jørgensen B. Sulfur and iron cycling in a coastal sediment: Radiotracer studies and seasonal dynamics. Biogeochemistry. 1994;27:129–52.
Xiao K-Q, Beulig F, Røy H, Jørgensen BB, Risgaard-Petersen N. Methylotrophic methanogenesis fuels cryptic methane cycling in marine surface sediment. Limnol Oceanogr. 2018;63:1519–27.
Kallmeyer J, Ferdelman TG, Weber A, Fossing H, Jorgensen BB. A cold chromium distillation procedure for radiolabeled sulfide applied to sulfate reduction measurements. Limnol Oceanogr Methods. 2004;2:171–80.
Tarpgaard IH, Røy H, Jørgensen BB. Concurrent low- and high-affinity sulfate reduction kinetics in marine sediment. Geochim Cosmochim Acta. 2011;75:2997–3010.
Kamp A, Petro C, Roy H, Nielsen S, Carvalho P, Stief P, et al. Intracellular nitrate in sediments of an oxygen-deficient marine basin is linked to pelagic diatoms. FEMS Microbiol Ecol. 2018;94:1–12.
Jochum LM, Chen X, Lever MA, Loy A, Jørgensen BB, Schramm A, et al. Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay. Appl Environ Microb. 2017;83:e01547–17.
Chen X, Andersen TJ, Morono Y, Inagaki F, Jørgensen BB, Lever MA. Bioturbation as a key driver behind the dominance of Bacteria over Archaea in near-surface sediment. Sci Rep. 2017;7:2400.
Lever MA, Rouxel O, Alt JC, Shimizu N, Ono S, Coggon RM, et al. Evidence for microbial carbon and sulfur cycling in deeply buried ridge flank basalt. Science 2013;339:1305–8.
Pelikan C, Herbold CW, Hausmann B, Müller AL, Pester M, Loy A. Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ Microbiol. 2016;18:2994–3009.
R-Core-Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2017. https://www.R-project.org/.
Lenth RV, Least-square means: the R Package lsmeans. 2016;69:332016.
Boudreau BC. Diagenetic models and their implementation. Berlin: Springer; 1997.
Ullman WJ, Aller RC. Diffusion-coefficients in nearshore marine-sediments. Limnol Oceanogr. 1982;27:552–6.
Soetaert K, Petzoldt T, Meysman F. marelac: tools for aquatic sciences. R package version 2.1. 2010. https://CRAN.R-project.org/package=marelac.
Berg P, Risgaard-Petersen N, Rysgaard S. Interpretation of measured concentration profiles in sediment pore water. Limnol Oceanogr. 1998;43:1500–10.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581.
Marshall IPG, Ren G, Jaussi M, Lomstein BA, Jørgensen BB, Røy H, et al. Environmental filtering determines family-level structure of sulfate-reducing microbial communities in subsurface marine sediments. ISME J. 2019;13:1920–32.
Trojan D, Schreiber L, Bjerg JT, Bøggild A, Yang T, Kjeldsen KU, et al. A taxonomic framework for cable bacteria and proposal of the candidate genera Electrothrix and Electronema. Syst Appl Microbiol. 2016;39:297–306.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. Plos ONE. 2013;8:e61217.
Oksanen J, Blanchet FG, Michael Friendly, Kindt R, Legendre P, McGlinn D, et al. Vegan: Community Ecology R package version 25-4. 2019. https://cran.r-project.org.
Clarke KR. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–43.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40.
Wörner S, Pester M. The active sulfate-reducing microbial community in littoral sediment of oligotrophic Lake Constance. Front Microbiol. 2019;10:247.
Froelich PN, Klinkhammer GP, Bender ML, Luedtke NA, Heath GR, Cullen D, et al. Early Oxidation of organic-matter in pelagic sediments of the eastern equatorial atlantic—suboxic diagenesis. Geochim Cosmochim Acta. 1979;43:1075–90.
Jørgensen BB. The sulfur cycle of freshwater sediments: role of thiosulfate. Limnol Oceanogr. 1990;35:1329–42.
Acknowledgements
The authors thank Lars Borregaard Pedersen, Karina Bomholt, Britta Poulsen, Susanne Nielsen, Jeanette Johansen, and Lykke Beinta Bjærge Bamdali for their skillful assistance in the lab. Ian Marshall is thanked for assistance with the bioinformatics. Three anonymous reviewers are acknowledge for their constructive criticism. This study was supported by the Danish National Research Foundation [Agreement nos. DNRF104 and DNRF136] and by a Marie Sklodowska-Curie Individual Fellowship [grant agreement No 656385] awarded to Ugo Marzocchi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sandfeld, T., Marzocchi, U., Petro, C. et al. Electrogenic sulfide oxidation mediated by cable bacteria stimulates sulfate reduction in freshwater sediments. ISME J 14, 1233–1246 (2020). https://doi.org/10.1038/s41396-020-0607-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0607-5
This article is cited by
-
Anwendungen der Kabelbakterien in der Umwelt- und Biotechnologie
BIOspektrum (2024)
-
Cable bacteria with electric connection to oxygen attract flocks of diverse bacteria
Nature Communications (2023)
-
Dissimilatory nitrate reduction by a freshwater cable bacterium
The ISME Journal (2022)
-
Cable bacteria extend the impacts of elevated dissolved oxygen into anoxic sediments
The ISME Journal (2021)
-
Microbial Interactions in Pollution Control Ecosystems
Current Pollution Reports (2021)
