
Abstract
Long-term symbiotic associations often lead to reciprocal adaptation between the involved entities. One of the main challenges for studies of such symbioses is differentiating adaptation from neutral processes and phylogenetic background. Ambrosia fungi, cultivated by ambrosia beetles as their sole food source, provide an excellent model to study evolutionary adaptation in a comparative framework because they evolved many times, and each origin bears features seemingly convergently adapted to the symbiosis. We tested whether the symbiotic lifestyle of unrelated ambrosia fungi has led to convergence in the key feature of the symbiotic phenotype—nutrition provisioning to the vector beetles. We compared conidia and mycelium content in three phylogenetic pairs of ambrosia fungi and their closely related nonambrosia relatives using an untargeted metabolomic assay. Multivariate analysis of 311 polar metabolites and 14063 lipid features revealed no convergence of nutrient content across ambrosia lineages. Instead, most variation of the metabolome composition was explained by phylogenetic relationships among the fungi. Thus the overall metabolome evolution of each ambrosia fungus is mostly driven by its inherited metabolism rather than the transition toward symbiosis. We identified eight candidate lipid compounds with expression levels different between the swollen ambrosia spores and other tissues, but they were not consistently elevated across ambrosia fungi. We conclude that ambrosia provisions consist either of nonspecific nutrients in elevated amounts, or of metabolites that are specific to each of the ambrosia symbioses.
Similar content being viewed by others
Introduction
Partnering with microbes is a common strategy for insects to deal with nutritional challenges. Complementary biosynthetic pathways of associated microbes increase insect fitness by providing, for example, nutrients lacking in their food [1], symbiont-mediated protections [2], or competitive advantages over conspecifics or other insect species [3]. Some of the most intriguing examples are fungus-farming systems—nutritional symbioses where insect partners cultivate fungal symbionts for nourishment of their progeny. The fungus-farming behavior has evolved multiple times in independent insect lineages including ants, woodwasps, termites, and ambrosia beetles [4, 5]. These fungal symbionts assist the insects by enzymatically extracting nutrients from abundant but recalcitrant plant substrates such as wood and leaves; in turn, the fungi benefit from protection and/or dispersal by the agile insect partners.
Despite the fact that the nutritional roles of these fungal symbionts have long been recognized, the metabolites that underpin the relationship remain entirely unknown. A persistent, obligately nutritional dependence can lead to elaborate evolutionary transitions manifested at the genomic, biochemical, and morphological levels [6]. In many fungus-farming systems, the fungal symbionts have convergently evolved swollen fungal cells that are consumed by the insects, i.e., the gongylidia (swollen hyphal tips) in higher attine ant symbionts, the nodules (swollen spores) in termite symbionts, and the ambrosia cells in ambrosia fungi [7,8,9]. The swollen fungal cells contain large vacuoles for accommodation of more nutrients such as nitrogenous compounds, lipids, carbohydrates [8, 10, 11], and essential enzymes [12]. Many of these fungal-mediated nutrients are advantageous or even indispensable for the normal development of their associated insects [13,14,15]. These enlarged fungal cells are presumably a result of long-term coadaptation with their insect partners. Intriguingly, these fungal symbionts are all derived from free-living fungal lineages, i.e., species that live saprophytically or parasitically in plant tissues, and there is no known reversal from mutualism back to the free-living ecology [16, 17]. The absence of an evolutionary reversal of symbiotic lineages suggests the presence of evolutionary constraints that might enforce the fixation of the symbiotic associations [18]. The inquiry into physiological transitions of these fungal symbionts should, therefore, be considered in a phylogenetic framework to understand the extent to which they are metabolically adapted as nutritional symbionts.
There are few symbiotic systems that manifest a phylogenetic spectrum as diverse as that in ambrosia beetle-fungus symbiosis (hereafter ambrosia symbiosis)—a feature that makes it an excellent model to investigate ecologically adaptive traits in a phylogenetically comparative framework. Ambrosia beetles are an ecological assemblage comprised of over 3400 species [19], with at least 12–16 independent origins which have arisen in two subfamilies of weevils (Curculionidae: Scolytinae and Platypodinae) [20]. Analogously, ambrosia fungi have repeatedly evolved in over ten known distant lineages in Dikarya (Fungi: Ascomycota and Basidiomycota), including the two most studied families that include ambrosia fungi, Ophiostomataceae and Ceratocystidaceae, but also species in Nectriaceae, Bionectriaceae, Saccharomycetaceae, and in two basidiomycete lineages, Peniophoraceae and Meruliaceae [16, 21, 22]. Ambrosia fungi are cultivated by ambrosia beetles as food sources on the walls of their breeding tunnels (galleries) that are usually excavated within declining or recently dead trees. Although ambrosia fungi colonize wood substrates, they do not decompose cellulose (with one known exception, see [23]). Instead, they target more readily available components remaining in freshly dead trees as their carbon sources (including hemicellulose in at least one case) [24].
A recent study has shown that each ambrosia fungus clade possesses a significant catabolic legacy of its own ancestral lineage [25]. This reflects the remarked polyphyletic origins of ambrosia symbiosis as well as the diverse habitats with varied microclimates occupied by the various ambrosia symbioses [22]. This phylogeny-driven catabolism of ambrosia fungi led us to hypothesize that each phylogenetic origin of the ambrosia fungi may synthesize compounds—the beetle provisions—in distinct ways. Consequently, the alternative hypothesis is that of convergent evolution, predicting that the long-term coadaptation between the fungi and the beetles led to convergent metabolic signatures or beetle-preferred compounds among independent ambrosia fungi, just as it led to the morphological convergence of enlarged ambrosia cells.
A limited number of studies have addressed the nutritional contents of beetle-associated fungi [26, 27], among which sterols [27,28,29] and nitrogenous compounds [10] were inferred as important for the beetles’ development. Yet most studies focused on a single-phylogenetic clade and no metabolite comparison of ambrosia fungi conducted in a phylogenetic framework has been published. Here we utilize the multiple independent evolutionary origins of the ambrosia symbiosis as a natural experiment to explicitly test for metabolomic modifications that transcend phylogenetic relationships. We conducted comparative bioassays of ambrosia fungi and their most closely related nonambrosia species on two types of tissues: ambrosia cells (the swollen asexual spores) and somatic hypha. Four questions were addressed as follows: (1) Have distantly related ambrosia fungi converged on a similar overall metabolome profile? (2) Have ambrosia fungi converged on a similar metabolome profile in the ambrosia cells? (3) What is the relative contribution of phylogeny and lifestyle (ambrosia versus nonambrosia) to the metabolomic profiles of fungi? (4) Are there specific compounds that are consistently increased or decreased in the ambrosia cells across the different origins?
Materials and methods
Fungal isolates
To compare the metabolomic profiles between ambrosia fungi and their nearest available nonambrosia relatives, three phylogenetically independent fungal pairs were selected, listed here with the ambrosia fungus first in each pair: Raffaelea lauricola T.C. Harr., Fraedrich & Aghayeva and Leptographium procerum (W.B. Kendr.) M.J. Wingf. (Ascomycota: Ophiostomataceae); Ambrosiella roeperi T.C. Harr. & McNew and Huntiella moniliformis (Hedgc.) Z.W. de Beer, T.A. Duong & M.J. Wingf. (Ascomycota: Ceratocystidaceae); Flavodon ambrosius D.R. Simmons, You Li, C.C. Bateman & Hulcr and Flavodon flavus (Klotzsch) Ryvarden (Basidiomycota: Meruliaceae). Isolation information for each isolate is listed in Table 1.
Fungal inoculation and sampling
Fungal isolates were inoculated on freshly severed branches of American sweetgum (Liquidambar styraciflua), a tree species that supports regular growth of all our assayed fungi. We emulated the way that ambrosia beetles inoculate fungal isolates on a tree branch by using a sterile toothpick to insert fungal inocula scraped from 2–4-week-old colonies on MEA (malt extract agar, BD Difco) into small holes drilled into autoclave-sterilized sweetgum branches (0.5–1.5 cm in diameter). The inoculated toothpick was tightly wedged into the hole and a flamed metal clipper was used to aseptically remove all of it but the ca. 0.5 cm of the tip inside the inoculation hole. The inoculated branches were put in a sterile jar capped with sterile aluminum foil and were incubated at 25 °C until branches were fully colonized, which took 1–2 months depending on the growth rate of each fungal isolate.
For fungal tissue sampling, the fully colonized branches were placed under an Olympus SZX16 dissecting microscope and fungal tissues were harvested with a sterile microfine tweezer to ensure the separation of fungal spores and somatic hypha. For A. roeperi, ambrosia swollen cells were produced in aggregated form and could be picked up manually to separate from its somatic hypha. For other fungal isolates, inoculated twigs were submerged in sterile deionized water in a 9 cm sterile petri dish. Fungal spores were suspended in the fluid with gentle pipetting and then were collected in 1.5 mL Eppendorf tubes. Somatic hypha was manually collected into 1.5 mL tubes containing 1 mL sterile deionized water using microfine tweezers under a microscope. The suspended spores or suspended hyphal tissues were centrifuged at 14,000 g for 5 min and the supernatant was discarded leaving tissue pellets of fungal spore or hyphal samples. Each tissue sample was redistributed from the tissue pellet and weighed for 30 mg (wet weight) into three technical replicates. All samples were immediately stored in −80 °C for subsequent metabolomic analysis.
Liquid chromatography–tandem mass spectrometry
To obtain the ex vivo metabolomic profiles of assayed fungi, the samples were processed for metabolomic analysis conducted in the Department of Chemistry Mass Spectrometry Research and Education Center at the University of Florida. Samples (10 mg) were resuspended using water and transferred to 2.0 mL homogenization tubes, prefilled with 0.5 mm zirconium beads (TriplePure M-Bio Grade; Midsci; St. Louis, MO). A total volume of 300 µL water was added to the homogenization tubes. Samples were homogenized using a BeadBug microtube homogenizer at five 30-s bursts at 400 rpm. Homogenized fungal material in water was then transferred to a tapered glass centrifuge tube for extraction. A total volume of 1800 μL of 2:1 chloroform:methanol was added to the extraction tube, and thoroughly mixed. The sample was then placed in a freezer (−20 °C) for ~12 h, after which the sample was centrifuged. The bottom layer (lipid layer) was transferred to a fresh 2 dram glass vial. A total volume of 800 μL 2:1 chloroform:methanol was added to the top layer and mixed. The extraction tube was then placed in the freezer (−20 °C) for ~30 min. Then the sample was centrifuged. The top layer (polar metabolite layer) was transferred to a 1.5 mL Eppendorf tube and dried to completeness using a SpeedVac concentrator. The bottom layers were combined in the 2 dram glass vial and dried to completeness under nitrogen. Both layers were reconstituted in 100 µL methanol prior to analysis and transferred to 1.5 mL tapered glass autosampler vials for LCMS analysis.
Capillary-liquid chromatography–mass spectrometry (Cap-LC–MS) was performed on a Bruker Impact II quadrupole time-of-flight mass spectrometer equipped with an Apollo II ion funnel ESI source (Bruker) operated in positive ion mode. The instrument was calibrated using Tune mix purchased from Agilent. The Apollo ESI source was operated at spray voltage of 4.5 kV, capillary temperature of 200 °C, and dry gas at 4.0 L min−1. A full scan was recorded between 150 and 3000 Da at a scan rate of 2 Hz. Data dependent MS/MS spectra were acquired with an inclusion mass of m/z 500–1000 with as many MS/MS acquired in 3 s (dynamic exclusion applied for 3 min). Collision energies were: base m/z 500 CE = 45 eV, base 700 CE base 30 eV, and base m/z 900 CE = 25 eV.
Molecule feature extraction was conducted using MS Convert from Proteowizard 3.0 software followed by analysis by XCMS; binning and alignment tolerances were set to: retention time = ±0.1% + 0.1 min; mass window = ±20 ppm + 5 mDa. Allowed ion species: +H, +Na, +NH4. The output from this processing generated a list of m/z ratios, retention times, and area under the curve values. Polar metabolites and lipid features were identified based on their fragmentation pathways in MS/MS data and against public chemical databases including METLIN [30] and Lipid MAPS [31]. In polar metabolite profiles, two mycelial samples (one H. moniliformis and one F. ambrosius sample) and two spore samples (one L. procerum. and one A. roeperi sample) were discarded following quality control screening, resulting in 32 samples for analysis. In nonpolar profiles (lipid), two mycelial samples (one F. ambrosius and one F. flavus sample) were discarded following quality control screening, resulting in 34 samples for analysis.
Statistical analysis
All analyses were performed using R v.3.5.3. The peak intensities of polar metabolite and lipid profiles of all samples were compiled respectively based on the m/z ratio and retention time of each compound. Compound features that were detected in fewer than half of the sample in the compiled dataset were excluded, resulting in 311 features for polar metabolite profiles and 14,063 features for lipid profiles for analysis. The metabolome of each sample was normalized by calculating the sum of the intensities of all compound features in each sample and then dividing all intensities associated with that sample by its sum. The metabolome of samples was analyzed with a Bray–Curtis dissimilarity matrix using the vegan package for R [32]. The dissimilarity matrix was then projected with NMDS to visualize metabolome similarities among samples.
Convergence of overall metabolite profile test
We hypothesized that ambrosia fungi converged on an overall similar metabolome as a result of having similar ecological strategies (ambrosia lifestyle). To test this hypothesis, we conducted a dissimilarity-based test of homogeneity in multivariate dispersion between ambrosia and nonambrosia isolates [33]. Specifically, this test determines if groups differ in how variable they are among observations within each group. From our hypothesis that ambrosia fungi have converged on a similar metabolome, we predicted that ambrosia fungi would show reduced variability in metabolome when compared with nonambrosia fungi. The test was implemented on a Bray–Curtis dissimilarity matrix using the betadisper function of the vegan package for R [32]. This function identified spatial medians of metabolomic variations of samples between groups (ambrosia and nonambrosia). Mean distances were compared between groups using an analysis of variance.
Convergence of spore metabolite profile test
Given the morphological convergence of swollen spores across all ambrosia fungal lineages, we hypothesized that convergence might also manifest as greater similarity of specific spore metabolomes of the ambrosia fungi, rather than of the overall tissue. To test this hypothesis, we conducted a distance-based, permutational multivariate analysis of variance (PERMANOVA) between ambrosia and nonambrosia isolates. This test measures if groups of observations differ in their composition. From our hypothesis that ambrosia fungi have converged on a similar spore metabolome, we expected to see a significant difference of spore metabolome composition between ambrosia and nonambrosia groups. The test was implemented on a Bray–Curtis dissimilarity matrix using the adonis function of the vegan package for R [32]. We further conducted a homogeneity test on the spore metabolite profiles using betadisper function. Because the betadisper method is less sensitive to the variances among groups, it can help justify any significant result from PERMANOVA that was a result of composition differences of the metabolome.
Metabolome variations attributed to phylogenetic origin versus lifestyle
We conducted a variation partitioning analysis to separate the effects of lifestyle from those of phylogenetic relationships on the metabolome profiles of fungi. Pairwise phylogenetic distances were obtained based on the aligned 28S large subunit rDNA sequences of assayed fungi using the dist.ml function in the phangorn package for R with default settings [34]. The phylogenetic distance matrix was then decomposed to phylogenetic eigenvectors by principal coordinates analysis, using the pcoa function in the ape package for R [35]. Phylogenetic eigenvectors are orthogonal vectors that describe relationships among taxa at various phylogenetic scales [36] and can be used as independent predictor variables in univariate and multivariate modeling [37]. The response variables were the lipid and polar metabolite metabolome values which were averaged for all replicates per tissue (mycelium or spore) and per isolate. The averaged values were Wisconsin double-standardized. To identify significant phylogenetic eigenvectors and to avoid overfitting our final model, we conducted forward multivariate model selection [38] using the forward.sel function of the packfor package for R [39]. We then used variation partitioning to partial out the amount of variation explained by all phylogenetic eigenvectors retained by model selection from the variation explained by lifestyle (ambrosia versus nonambrosia). Variation partitioning was conducted using the varpart function in the vegan package for R [32].
Do ambrosia fungi synthesize or lack unique compounds?
Even if ambrosia fungi do not converge on their overall or spore metabolome, it is still possible that they have converged to synthesize specific compounds to meet a particular nutritional requirement of the beetles. Alternatively, ambrosia fungi may have convergently lost the production of specific compounds that are toxic to insects. We tested for the presence of compounds that were significantly higher or lower in amounts in the spores of ambrosia phenotypes compared with their free-living relatives. To obtain the magnitude of the difference in intensity values, the difference between group means (log2 expression values) was calculated for each compound comparing ambrosia and nonambrosia isolates in each phylogenetic clade (i.e., Ophiostomataceae, Ceratocystidaceae, and Meruliaceae). To investigate whether ambrosia fungi yield significantly different amount of each metabolite feature, we conducted Welch’s t-test on the intensity values of each feature between ambrosia and nonambrosia fungi. To account for the issue of recovering false positives resulting from the multiple hypothesis testing, we estimated the false discovery rate using the q-value function of the q-value package for R [40, 41]. Compound features that were significantly higher (p < 0.05 and q < 0.05; significance judged as log2 fold change > 2) or lower (log2 fold change < −2) in amount in ambrosia lineages were cross-compared with examine if they are shared by the three ambrosia fungi. Heatmaps were constructed using pheatmap package for R [42] to visualize the shared metabolite features.
Results
Convergence of the overall metabolome
Testing of homogeneity in multivariate dispersion revealed no convergence among ambrosia fungi, that is, no significant difference in variability among isolates between ambrosia and nonambrosia fungi in lipid profiles, (betadisper, df = 1, F = 0.905, P = 0.348). The polar metabolite profiles showed a marginal significant difference in variability among ambrosia and nonambrosia species (betadisper, df = 1, F = 4.244, P = 0.048), however this appears to be driven by the highly divergent samples of F. flavus mycelia. Both results suggested that ambrosia fungi have not converged on a similar metabolome. Instead, ordinations revealed a strong phylogenetic signal in their metabolome, showing that metabolomes are conserved within lineages throughout the evolutionary transition to the ambrosia lifestyle (Figs. 1 and 2). The technical replicates of each assayed sample were clustered in NMDS ordination, validating our technical methodologies of tissue separation as well as the precision of the mass spectrometry analysis. The ambrosia and nonambrosia fungi within each phylogenetic pair almost fully overlapped in their distribution on the plots (Figs. 1 and 2), with the exception of the mycelial samples of F. flavus in polar metabolite profiles (Fig. 2a). Multivariate analysis also revealed the significant differences of the metabolic compositions between species in each phylogenetic pair (lipids, adonis: df = 2, F = 8.401, r2 = 0.351, P = 0.001; polar metabolites, adonis: df = 2, F = 5.824, r2 = 0.28657, P = 0.001).

a Phylogenetic pairs, not ambrosia fungi, shared similar profiles. Ambrosia: green, nonambrosia: red. b Fungal lineages varied in lipid profiles. Acronyms: Rla (R. lauricola), Lpr (L. procerum), Aro (A. roeperi), Hmo (H. moniliformis), Fam (F. ambrosius), and Ffl (F. flavus). Letters in parentheses after symbol names represent sample types: spore (S) and mycelium (M). Symbols represent technical replicates of each fungal isolate.

a Similar profiles were observed within ambrosia (green) and nonambrosia (red) fungal pairs. b Fungal lineages varied in polar metabolite profiles. Acronyms refer to Fig. 1. Letters in parentheses after symbol names represent sample types: spore (S) and mycelium (M). Symbols represent technical replicates of each fungal isolate.
Convergence of spore metabolites
There was no significant difference in spore metabolome compositions between ambrosia and nonambrosia isolates (lipids, adonis: df = 1, F = 2.017, r2 = 0.112, P = 0.093; polar metabolites, adonis: df = 1, F = 1.644, r2 = 0.105, P = 0.153). An additional homogeneity test indicated no dispersion effect in the spore metabolome (lipids, betadisper: df = 1, F = 0.2068, P = 0.655; polar metabolites, betadisper: df = 1, F = 1.142, P = 0.303), validating that the results in the composition test (adonis) resulted from compositional dissimilarity of the metabolome. These findings show that ambrosia fungi have spore metabolomes that are more similar to their nonambrosia relatives than unrelated ambrosia fungi, refuting the hypothesis of convergence in spore metabolomes among ambrosial lineages. The spore metabolic similarities were also indicated by the clear clustering of samples in the same phylogenetic group on the NMDS plots (Figs. 1b and 2b).
Metabolome variations attributed to phylogenetic origin versus lifestyle
Both lipid and polar metabolite profiles of assayed fungal species were mostly structured by phylogenetic relations among species rather than their lifestyle (ambrosia versus nonambrosia). Model selection retained the first two phylogenetic eigenvectors to explain variation in lipid metabolomes among isolates. These eigenvectors cumulatively explained 15.5% of the variation in lipid profiles among all fungi (Table 2). Of the polar metabolomes, model selection retained only the first phylogenetic eigenvector which explained ~9% of the variation of all fungi (Table 2). A subsequent analysis partitioning the variables of phylogenetic eigenvectors and lifestyle indicated that, in lipid profiles, 16.4% of the metabolome variations were independently explained by phylogenetic eigenvectors, 0% by lifestyle, and 0% by variation shared by both variables. Of the polar metabolome, 10.3% of the metabolite variations were independently explained by phylogenetic eigenvectors, 0% by lifestyle, and 0% by variation shared by both variables. The unexplained residuals were 87.5% and 91.9% in metabolomes of the lipid and polar metabolite, respectively.
Do ambrosia fungi synthesize or lack unique compounds?
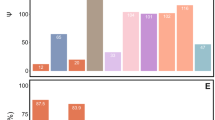
We detected compounds that were significantly higher or lower in abundance in spores of ambrosia fungi when compared with nonambrosial lineages. There were eight compound features in lipid profiles shown to be significantly different between ambrosia and nonambrosia fungi across phylogenetic pairs (Table 3 and Fig. 3). In contrast, we found no evidence of shared, significantly different compound features in polar metabolite profiles. Within individual ambrosia fungus species, we recovered 207, 1122, and 187 lipid compounds that were significantly higher in amount in ambrosia fungi in R. lauricola, A. roeperi, and F. ambrosius, respectively. Conversely, 560, 813, and 205 lipid compounds were significantly lower in amount in the same respective ambrosia lineages. Of the polar metabolomes, we recovered 3, 8, and 16 metabolites which were significantly higher in amount in ambrosia lineages in R. lauricola, A. roeperi, and F. ambrosius. Only one ambrosia fungus, A. roeperi, displayed any significantly lower in amount of polar metabolites than in its nonambrosial counterpart (nine features); there was no analogous reduction of polar metabolite detected in the ambrosial Ophiostomataceae and Meruliaceae.

Columns represented technical replicates of fungal isolates. Categories and main classes of identified lipids referred to LIPID MAPS online database. SP02 represents ceramides, GP12 represents glycerophosphoglycerophosphoglycerols, GP01 represents glycerophosphocholines. Acronyms on columns represent phylogenetic groups of samples: Oph (Ophiostomataceae), Cer (Ceratocystidaceae), and Mer (Meruliaceae).
By matching the mass and fragmentation pathways of compound features in MS/MS data, we putatively identified the function classes of the eight shared, significantly different features in the ambrosia lipid metabolome. They included two glycerophospholipids and a sphingolipid. However, the expression levels were not consistent across phylogenetic lineages (Table 3 and Fig. 3). A glycerophospholipid [CL(54:6), GP12] and a sphingolipid [Cer(t44:0(OH)), SP02] were detected higher in ambrosia species in the ambrosial Ophiostomataceae and Ceratocystidaceae, but lower in Meruliaceae. The glycerophospholipids [PC(30:4), GP01] were reduced in ambrosia lineages of Ophiostomataceae and Meruliaceae, but expressed higher in Ceratocystidaceae. We were unable to identify the other five shared lipid compounds as they were not detected in the MS/MS identification step and not present in public databases. These five compound features also showed no consistent pattern of expression pattern across the three phylogenetic pairs (Fig. 3).
We further examined features putatively identified as ergosterols and their derivatives as their importance in beetles’ development was implied in Norris et al. [26] and Bentz and Six [29]. However, no shared ergosterols and derivatives were found to be consistently highly expressed in all ambrosial lineages (Fig. 4).

Chemical common names and compositions referred to MS/MS data retrieved from LIPID MAPS online database. Acronyms on columns refer to Fig. 3. Chemical composition: episteryl palmitoleate = C44H74O2 + H, ergosteryl palmitoleate = C44H72O2 + Na, episteryl oleate1 = C46H78O2 + H, ergosteryl oleate = C46H76O2 + NH4, episteryl oleate2 = C46H78O2 + NH4, 16:3-Glc-Campesterol = C50H82O7 + NH4, 18:3-Glc-Campesterol = C52H86O7 + NH4, 18:2-Glc-Campesterol = C52H88O7 + NH4, and 22:2-Glc-Campesterol = C56H96O7 + NH4.
Discussion
Ambrosia fungi displayed no convergence of the overall metabolome. Even the adaptative apparatus, ambrosia cells, displayed greater similarity to metabolomes of tissues (spores and mycelia) of phylogenetically related free-living fungi than to other ambrosia fungi. Therefore, in terms of the overall metabolome, ambrosia symbiosis does not seem to require substantial changes to the fungal cell contents on the evolutionary pathway to the symbiotic lifestyle. Each origin of ambrosia fungus retains a considerable metabolic legacy from its ancestor and synthesizes metabolites in its distinct way. Thus, ambrosia symbioses are an ecologically diverse suite of many functionally distinct, mutualistic associations.
Several nonpolar compounds (lipids) were repeatedly different in abundance in ambrosia species compared with their nonambrosia relatives across phylogenetic ranges. Although these shared compounds were elevated in only two of the three assayed ambrosia lineages, they warrant further research as candidate compounds for the maintenance of the mutualism.
Conserved metabolome of each ambrosia fungus
Based on lipid and polar metabolomes, we found that phylogenetic relatedness of assayed fungi was a strong predictor of the metabolic variation. The ambrosia lifestyle does not indicate common metabolome profiles between phylogenetic lineages. If there are any features of the metabolome modulated by the symbiotic adaptation, they do not seem to be detectable in the metabolome profiles of our assayed fungi, even inside the adaptative ambrosia cells. Although surprising, these results were in line with the recent findings based on variation in carbon source utilization which showed that each origin of the ambrosia symbiosis has followed a distinct evolutionary trajectory [25]. From the perspective of ambrosia ecology, most ambrosia beetles are the early colonizers of freshly dead trees, where many readily accessible nutrients are available and pose a relatively less competitive environment to their fungal symbionts. The longevity of each individual gallery is relatively short, typically several months, making the proliferation of symbionts in such a short while the priority to ensure entering the transportation organ (mycangium) of the new beetle generation [43]. It appears that, rather than a metabolism convergent on nutrient provisioning, the ambrosia fungi may have metabolism optimized for their ecological niches.
If fungal provisioning to the beetle does not occur via any specific metabolite(s), it may occur via an overall increased volume of many generic nutritional compounds, or compounds not assayed in this study. Termitomyces, the fungus cultivated by termites, exceeds free-living mushrooms primarily in the overall content of protein which were not assayed here [44]. In mushrooms eaten by humans, the most nutritious species are also characterized by overall high-protein content and essential amino acid content, rather than by individual compounds [45]. Therefore, we recommend testing for protein content, amino acid content, and overall-nutrition content in ambrosia fungi in future studies.
The phylogeny-dependent metabolomes of ambrosia fungi may also highlight the diverse ecological contexts of ambrosia fungi. The three phylogenetic pairs utilized here represent diverse ambrosia ecologies. Flavodon ambrosius is the only ambrosia fungus known to degrade the structural components of wood [23]. Raffaelea lauricola is specialized on freshly dead Lauraceae and is rarely sampled from any other plant family [46]. Ambrosiella roeperi specializes on small-diameter branches with reduced water content [22]. These three distinct ecological contexts are likely to drive the metabolism of the fungi in very different directions, which may make it difficult to detect subtle changes by our exploratory metabolomic investigation.
Although the paired sampling design is one of the main strengths of our study, the number of phylogenetic replicates is limited. There are at least eight other origins of the ambrosia phenotype in fungi, and some of them inhabit very different ecological contexts [22]. Metabolomes of such diverse fungi might be deeply affected by the phylogenetic origins and ecological requirements, which can be seen in the portion of unexplained residuals of metabolome variations. To reach a greater statistical strength, future studies should cover the ambrosia diversity more comprehensively, with a deeper sampling within each phylogenetic clade, and with focus on clades that also occupy the same niche.
Specific compounds in ambrosia tissues
Although a clear pattern of expression levels of compounds was not present in our assayed fungi, we have recovered eight lipids (nonpolar compounds), consistently diverging in the three ambrosia and nonambrosia phylogenetic pairs. The putatively identified three shared lipids belong to two lipid classes, sphingolipid and glycerophospholipid, both of which play key roles in cell membrane formation as well as other biological characters [47, 48]. Any potential biological relevance of these compounds to the ambrosia beetles requires further investigation. The other five unidentified, shared lipids have highlighted the difficulty of metabolome annotation, which represents one of the major obstacles in untargeted metabolomics research [49].
The shared eight lipid compounds may indicate the importance of carbon and energy-rich compounds to the ambrosia symbiosis. The few available studies of the metabolism of the ambrosia symbiosis inferred that ergosterols and nitrogenous compounds might be the ‘metabolic currency’ of this symbiosis [10, 26, 28, 29]. Our results suggested that the effect of these ‘currencies’ on ambrosia symbiosis might exist, but perhaps not in the form of being a trait that is unique to fungi involved in the symbiosis. Several classes of sterols were detected in the lipid metabolome of all our assayed fungi, including ergosterols and their derivatives. None of these sterols were preferentially up- or down-expressed across ambrosia lineages (Fig. 4). Ergosterol is a ubiquitous, vital component of fungal cell membrane; it exists in almost all fungi but is absent in animals and plants [50]. The acquisition and utilization of ergosterol by insects, especially for most ambrosia beetles living in the nutrient-poor xylem, is indispensable for their normal growth [51]. Concentration of ergosterol in fungi generally ranges from 2.6 to 42 μg/ml [52], which should readily meet the nutritional need of ergosterol for most scolytine beetles as suggested in [27] and [29].
One previous nutritional study of fungal isolates of Xyleborus suggested that the tested fungi could sustain the growth of their beetle partners [53]. The study design is questionable because of the unclear symbiont identity (the genus Xyleborus is not known to culture Fusarium species). However, even a potential symbiont mismatch would indicate that beetles may be sustained by compounds found in many fungal lineages recovered from the surrounding environment, even in vitro. In addition, it has been demonstrated that ergosterol contents were not significantly different between symbiotic and nonsymbiotic fungi of Dendroctonus [29]. These findings, coupled with the absence of any shared, significantly increased sterols across the ambrosia fungi in our sample, imply that a strong selection of the overexpressed phenotype of ergosterol and/or a specific type of ergosterol might not be a prerequisite for fungi to give rise to the ambrosia symbiosis.
Conclusion
Disentangling phenotypic adaptations from neutral processes of evolution remains a challenge for most symbiosis studies. The ambrosia symbiosis, one of the most ecologically and phylogenetically diverse symbiotic systems, is an exceptional natural model to empirically test for phenotypic modifications that transcend phylogenetic relationships. Our results highlighted an emerging notion that each ambrosia symbiosis represents a functionally distinct symbiosis. We believe that each of the ambrosia symbioses will be best understood within its specific ecological context and phylogenetic origin.
Very few invasive ambrosia beetles and fungi have had noticeable negative impacts, but those few pestiferous species have been notably destructive, even catastrophic [54, 55]. It is difficult to make biosecurity decisions when the impactful species are not distinguished from the harmless ones. With the increasing accessibility of ‘omics’ technologies, more information is becoming available about the relationships between the vector beetles and their fungal associates including the pathogens. A greater understanding of these life forms is critical for effective, evidence-based decisions of agencies.
References
Douglas AE. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol. 1998;43:17–37.
Brownlie JC, Johnson KN. Symbiont-mediated protection in insect hosts. Trends Microbiol. 2009;17:348–54.
Six DL. Ecological and evolutionary determinants of bark beetle—fungus symbioses. Insects. 2012;3:339–66.
Mueller UG, Gerardo NM, Aanen DK, Six DL, Schultz TR. The evolution of agriculture in insects. Annu Rev Ecol Evol S. 2005;36:563–95.
Wermelinger B, Thomsen IM. The woodwasp Sirex noctilio and its associated fungus Amylostereum areolatum in Europe. In: Slippers B, de Groot P, Wingfield MJ editors. The sirex woodwasp and its fungal symbiont: research and management of a worldwide invasive pest. Dordrecht: Springer; 2012. pp. 65–80.
Moran NA. Symbiosis as an adaptive process and source of phenotypic complexity. PNAS. 2007;104:8627–33.
Batra LR, Michie MD. Pleomorphism in some ambrosia and related fungi. Trans Kans Acad Sci. 1963;66:470–81.
Quinlan RJ, Cherrett JM. The role of fungus in the diet of the leaf-cutting ant Atta cephalotes (L.). Ecol Entomol. 1979;4:151–60.
Leuthold RH, Badertscher S, Imboden H. The inoculation of newly formed fungus comb with Termitomyces in Macrotermes colonies (Isoptera, Macrotermitinae). Ins Soc. 1989;36:328–38.
Ayres MP, Wilkens RT, Ruel JJ, Lombardero MJ, Vallery E. Nitrogen budgets of phloem-feeding bark beetles with and without symbiotic fungi. Ecology. 2000;81:2198–210.
Hyodo F, Tayasu I, Inoue T, Azuma J-I, Kudo T, Abe T. Differential role of symbiotic fungi in lignin degradation and food provision for fungus-growing termites (Macrotermitinae: Isoptera). Funct Ecol. 2003;17:186–93.
Schiøtt M, Rogowska-Wrzesinska A, Roepstorff P, Boomsma JJ. Leaf-cutting ant fungi produce cell wall degrading pectinase complexes reminiscent of phytopathogenic fungi. BMC Biol. 2010;8:156.
Nobre T, Rouland-Lefèvre C, Aanen DK. Comparative biology of fungus cultivation in termites and ants. In: Bignell DE, Roisin Y, Lo N editors. Biology of termites: a modern synthesis. Dordrecht: Springer; 2011. pp. 193–210.
Suen G, Teiling C, Li L, Holt C, Abouheif E, Bornberg-Bauer E, et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011;7:e1002007.
Bracewell RR, Six DL. Experimental evidence of bark beetle adaptation to a fungal symbiont. Ecol Evol. 2015;5:5109–19.
Massoumi Alamouti S, Tsui CK, Breuil C. Multigene phylogeny of filamentous ambrosia fungi associated with ambrosia and bark beetles. Mycol Res. 2009;113:822–35.
Jordal BH, Cognato AI. Molecular phylogeny of bark and ambrosia beetles reveals multiple origins of fungus farming during periods of global warming. Bmc Evol Biol. 2012;12:133.
Nygaard S, Hu H, Li C, Schiøtt M, Chen Z, Yang Z, et al. Reciprocal genomic evolution in the ant–fungus agricultural symbiosis. Nat Commun. 2016;7:ncomms12233.
Wood SL. Bark and ambrosia beetles of South America (Coleoptera, Scolytidae). In: Monte L editor. Provo, Utah: Bean Life Science Museum; 2007.
Johnson AJ, McKenna DD, Jordal BH, Cognato AI, Smith SM, Lemmon AR, et al. Phylogenomics clarifies repeated evolutionary origins of inbreeding and fungus farming in bark beetles (Curculionidae, Scolytinae). Mol Phylogenet Evol. 2018;127:229–38.
Li Y, Simmons DR, Bateman CC, Short DPG, Kasson MT, Rabaglia RJ, et al. New fungus-insect symbiosis: culturing, molecular, and histological methods determine saprophytic polyporales mutualists of Ambrosiodmus ambrosia beetles. PLoS ONE. 2015;10:e0137689.
Hulcr J, Stelinski LL. The ambrosia symbiosis: from evolutionary ecology to practical management. Annu Rev Entomol. 2017;62:285–303.
Kasson MT, Wickert KL, Stauder CM, Macias AM, Berger MC, Simmons DR, et al. Mutualism with aggressive wood-degrading Flavodon ambrosius (Polyporales) facilitates niche expansion and communal social structure in Ambrosiophilus ambrosia beetles. Fungal Ecol. 2016;23:86–96.
De Fine Licht HH, Biedermann PHW. Patterns of functional enzyme activity in fungus farming ambrosia beetles. Front Zool. 2012;9:13.
Huang Y-T, Skelton J, Hulcr J. Multiple evolutionary origins lead to diversity in the metabolic profiles of ambrosia fungi. Fungal Ecol. 2019;38:80–8.
Norris DM, Baker JM, Chu HM. Symbiontic interrelationships between microbes and ambrosia beetles. III. Ergosterol as the source of sterol to the insect. Ann Entomol Soc Am. 1969;62:413–4.
Kok LT, Norris DM, Chu HM. Sterol metabolism as a basis for a mutualistic symbiosis. Nature. 1970;225:661–2.
Morales-Ramos JA, Rojas MG, Sittertz-Bhatkar H, Saldaña G. Symbiotic relationship between Hypothenemus hampei (Coleoptera: Scolytidae) and Fusarium solani (Moniliales: Tuberculariaceae). Ann Entomol Soc Am. 2000;93:541–7.
Bentz BJ, Six DL. Ergosterol content of fungi associated with Dendroctonus ponderosae and Dendroctonus rufipennis (Coleoptera: Curculionidae, Scolytinae). Ann Entomol Soc Am. 2006;99:189–94.
Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, et al. METLIN: a technology platform for identifying knowns and unknowns. Anal Chem. 2018;90:3156–64.
Fahy E, Sud M, Cotter D, Subramaniam S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007;35:W606–12.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, et al. vegan: Community ecology package, R package version 2. 2013.
Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–53.
Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90.
Diniz-Filho JAF, Bini LM, Rangel TF, Morales-Castilla I, Olalla-Tárraga MÁ, Rodríguez MÁ, et al. On the selection of phylogenetic eigenvectors for ecological analyses. Ecography. 2012;35:239–49.
Tedersoo L, Mett M, Ishida TA, Bahram M. Phylogenetic relationships among host plants explain differences in fungal species richness and community composition in ectomycorrhizal symbiosis. N Phytol. 2013;199:822–31.
Blanchet FG, Legendre P, Borcard D. Forward selection of explanatory variables. Ecology. 2008;89:2623–32.
Dray S, Legendre P, Blanchet G. packfor: Forward Selection with permutation (Canoco p. 46). R package version 00-7/r58. 2009.
Storey JD, Tibshirani R. Statistical significance for genomewide studies. PNAS. 2003;100:9440–5.
Dabney A, Storey JD, Warnes GR. qvalue: Q-value estimation for false discovery rate control. R package version 2. 2010.
Kolde R. Pheatmap: pretty heatmaps. R package version. 2012;61:617.
Li Y, Ruan Y-Y, Stanley EL, Skelton J, Hulcr J. Plasticity of mycangia in Xylosandrus ambrosia beetles. Insect Sci. 2019;26:732–42.
Botha WJ, Eicker A. Nutritional value of Termitomyces mycelial protein and growth of mycelium on natural substrates. Mycol Res. 1992;96:350–4.
Boa E. Wild Edible Fungi: A Global Overview of Their Use and Importance to People. Daya Publishing House; 2004.
Saucedo-Carabez JR, Ploetz RC, Konkol JL, Carrillo D, Gazis R. Partnerships Between Ambrosia Beetles and Fungi: Lineage-Specific Promiscuity Among Vectors of the Laurel Wilt Pathogen, Raffaelea lauricola. Microb Ecol. 2018;76:925–40.
Bittman R. Glycerolipids: chemistry. In: Roberts GCK, editor. Encyclopedia of biophysics. Springer: Heidelberg, Berlin; 2013. pp. 907–14.
Singh A, Del Poeta M. Sphingolipidomics: An Important Mechanistic Tool for Studying Fungal Pathogens. Front Microbiol. 2016;7:501.
Chaleckis R, Meister I, Zhang P, Wheelock CE. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr Opin Biotech. 2019;55:44–50.
Schwadorf K, Müller HM. Determination of ergosterol in cereals, mixed feed components, and mixed feeds by liquid chromatography. J Assoc Anal Chem. 1989;72:457–62.
Clayton RB. The utilization of sterols by insects. J Lipid Res. 1964;5:3–19.
Pasanen A-L, Yli-Pietilä K, Pasanen P, Kalliokoski P, Tarhanen J. Ergosterol content in various fungal species and biocontaminated building materials. Appl Environ Microbiol. 1999;65:138–42.
Baker JM, Norris DM. A complex of fungi mutualistically involved in the nutrition of the ambrosia beetle Xyleborus ferrugineus. J Invertebr Pathol. 1968;11:246–50.
Fraedrich SW, Harrington TC, Rabaglia RJ, Ulyshen MD, Mayfield AE, Hanula JL, et al. A fungal symbiont of the redbay ambrosia beetle causes a lethal wilt in redbay and other Lauraceae in the southeastern United States. Plant Dis. 2008;92:215–24.
Mendel Z, Protasov A, Sharon M, Zveibil A, Yehuda SB, O’Donnell K, et al. An Asian ambrosia beetle Euwallacea fornicatus and its novel symbiotic fungus Fusarium sp. pose a serious threat to the Israeli avocado industry. Phytoparasitica. 2012;40:235–8.
Acknowledgements
The authors thank Adam Wong for suggestions regarding the metabolomic analysis. This project was partially funded by the U.S. Forest Service, U.S. Department of Agriculture—Animal and Plant Health Inspection Service (USDA APHIS) and the National Science Foundation. The UF Mass Spectrometry Research and Education Center was funded by NIH grant S10 OD021758-01A1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, YT., Skelton, J. & Hulcr, J. Lipids and small metabolites provisioned by ambrosia fungi to symbiotic beetles are phylogeny-dependent, not convergent. ISME J 14, 1089–1099 (2020). https://doi.org/10.1038/s41396-020-0593-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0593-7
This article is cited by
-
Fungal mutualisms and pathosystems: life and death in the ambrosia beetle mycangia
Applied Microbiology and Biotechnology (2021)
-
Bark beetle mycobiome: collaboratively defined research priorities on a widespread insect-fungus symbiosis
Symbiosis (2020)
