
Abstract
Marine sponges often host diverse and species-specific communities of microorganisms that are critical for host health. Previous functional genomic investigations of the sponge microbiome have focused primarily on specific symbiont lineages, which frequently make up only a small fraction of the overall community. Here, we undertook genome-centric analysis of the symbiont community in the model species Ircinia ramosa and analyzed 259 unique, high-quality metagenome-assembled genomes (MAGs) that comprised 74% of the I. ramosa microbiome. Addition of these MAGs to genome trees containing all publicly available microbial sponge symbionts increased phylogenetic diversity by 32% within the archaea and 41% within the bacteria. Metabolic reconstruction of the MAGs showed extensive redundancy across taxa for pathways involved in carbon fixation, B-vitamin synthesis, taurine metabolism, sulfite oxidation, and most steps of nitrogen metabolism. Through the acquisition of all major taxa present within the I. ramosa microbiome, we were able to analyze the functional potential of a sponge-associated microbial community in unprecedented detail. Critical functions, such as carbon fixation, which had previously only been assigned to a restricted set of sponge-associated organisms, were actually spread across diverse symbiont taxa, whereas other essential pathways, such as ammonia oxidation, were confined to specific keystone taxa.
Similar content being viewed by others

Introduction
Marine sponges are a ubiquitous and functionally important component of aquatic ecosystems, providing valuable habitat for a variety of species and coupling the benthic and pelagic zones through the filtration of seawater and recycling of (in)organic nutrients [1,2,3]. Sponges frequently harbor dense, diverse, and stable microbial communities, which can comprise up to 35% of the biomass of high microbial abundance (HMA) sponges [4, 5]. These sponges contain 108–1010 microbial cells g−1 sponge wet weight in contrast to low microbial abundance sponges that contain 105–106 microbes g−1 sponge wet weight [6]. The microorganisms are often host species-specific [7], distinct from those in the surrounding environment [8] and are hypothesized to contribute to many aspects of the sponge’s physiology and ecology [4]. Considering the diversity, abundance, and potential functional importance of their microbiome, sponges are often described as “holobionts”—a complex and interdependent consortium comprising the sponge host and the entire microbiome [4].
Recent studies have suggested that sponge-associated microbial symbionts have the capacity to fix carbon dioxide, metabolize nitrogen, sulfur, and phosphate, and synthesize essential B-vitamins [5]. However, these insights are primarily inferred from 16S rRNA or functional marker gene surveys that either give no information about the functional potential of the symbionts or are limited to analyzing a few discrete pathways that were decided a priori to be of interest. In addition, gene-centric analysis has provided critical functional insights into the sponge microbiome, but could not link function with phylogeny [9]. Only a limited number of symbiont genomes have been retrieved from multiple sponge species [10,11,12,13]. However, the majority of these studies focused on specific lineages that make up a small fraction of the complex sponge microbiome. With sponge microbial richness estimates exceeding 40,000 genetically distinct taxa [7], spread across 50 microbial phyla [5], the majority of sponge symbiont genomes and their functional potential remains undescribed. Here, we used genome-centric metagenomics to characterize 259 high-quality microbial metagenome-assembled genomes (MAGs) retrieved from I. ramosa, an HMA sponge from the Great Barrier Reef (GBR), Australia. The MAGs represent 74% of the microbial community, providing an unprecedented opportunity to characterize the metabolic potential of all major taxa within a sponge microbiome.
Materials and methods
Sample collection, processing, and sequencing
Six Ircinia ramosa (Porifera: Demospongiae, Fig. S13) individuals were collected using SCUBA at 5–7 m water depth at Davies Reef (18°49′ S, 147°38′ E) on the GBR Australia, in October 2016. Samples were held in flow-through aquaria for 2 h during transportation to the Australian Institute of Marine Science Sea Simulator, where the sponges were transferred to a 3600 L mesocosm and kept for 6 months. Directly after transfer to the mesocosm, ~10 cm2 fragments were excised from each specimen (T0) and snap frozen in liquid nitrogen. The six individual sponges were again subsampled after 3 (T3) and 5 (T5) months in the aquaria to determine the composition of the microbiome over time. All excised tissue, comprising both external and internal body tissue, was immediately snap frozen in liquid nitrogen.
Microbial cells were separated from frozen sponge tissue as described previously [14], with the exception that tissue was initially cut into 2 mm2 pieces prior to 1 min homogenization, and cells were filtered twice through 8 µm filters and twice through 5 µm filters. Bacterial pellets were collected after a final centrifugation at 8800 × g for 20 min and cells were frozen at −20 °C. DNA was extracted from cell pellets using the UltraClean Microbial kit (MO BIO, San Diego, USA) according to the manufacturer’s instructions with an additional incubation step of 10 min at 65 °C before bead-beating for 40 s at 5 m s−1 in a FastPrep24 instrument (MP Biomedicals, Santa Ana, CA, USA) to assist lysis. Extracted DNA was further purified using the ZymoResearch (California, USA) DNA Clean and Concentrator kit. DNA was sent to the Australian Centre for Ecogenomics (ACE), Brisbane, libraries were prepared with Illumina Nextera XT, and samples were sequenced on the NextSeq 500 High Output using 2 × 150 bp paired end chemistry. All samples (n = 18) were first sequenced at an average depth of 1.7 Gbp for differential coverage binning and replicates 1, 2, and 4 from T0 were selected for deeper sequencing, with an average depth of 20.6 Gbp, to obtain better coverage of lower abundance members of the microbiome.
Metagenome assembly, taxonomic assignment, and functional annotation
Sequencing adapters were removed using SeqPurge v0.1-852-g5a7f2d2 [15] and all reads were assembled using metaSPades v3.9.0 [16]. Reads were mapped to the resulting assemblies with bamM v1.7.3 (https://github.com/Ecogenomics/BamM), which leverages the Burrows-Wheeler Aligner [17], aligning sequences based on a backward search with the Burrows-Wheeler transformation. Binning was performed with UniteM v0.0.15 (https://github.com/dparks1134/UniteM). In brief, UniteM uses the binning algorithms maxbin v2 v2.2.4, metabat v1 v0.32.4 using all parameter sets (--verysensitive --mb_sensitive --mb_specific --mb_veryspecific --mb_superspecific), metabat v2 v2.12.1, and groopM2 v2 v2.0.0-1, and selects the best MAGs by their CheckM quality scores [18,19,20,21,22].
Taxonomy was assigned to each MAG using GTDB-Tk v0.2.2 (https://github.com/Ecogenomics/GtdbTk), which classifies MAGs based on placement in a reference concatenated gene tree based on a set of 120 bacterial and 122 archaeal markers using a combination of FASTANI and pplacer [23, 24]. GTDB-Tk v0.2.2 annotation is based on the Genome Taxonomy Database (GTDB, http://gtdb.ecogenomic.org) taxonomy R86.3 [25]. MAGs obtained from sub-sampling the same sponge individuals over three time points were de-replicated at 99% identity with Drep v1.0.0 using default parameters [26].
To determine the percentage of the total community captured by the MAGs, the “pipe” and “appraise” functions of SingleM v0.9.0 were used to search for single copy marker genes in both the MAGs and the shallow sequenced raw reads to assess the fraction of marker genes recovered in the MAGs (https://github.com/wwood/singlem). The relative abundance of each MAG from the separate individuals was calculated with coverM v0.2.0 (https://github.com/wwood/CoverM), after de-replicating all 559 MAGs at 95% identity with Drep v2.2.4 [26] to avoid arbitrary mapping between representatives of highly similar genomes. MAGs with a cumulative abundance >2% (sum of MAG abundance across replicates) were included in the heatmap visualized with R v3.5.1 [27]; Fig. S1). To visualize the composition of the I. ramosa microbiome across the three time points, a relative abundance heatmap was created in R v3.5.1 [27], based on 16S rRNA gene sequences recovered from the metagenomics reads using GraftM v0.11.1 [28]. The recovered 16S rRNA gene sequences are representative of all data, including unbinned lineages. Taxonomy was assigned using a GraftM package based on the Silva release NR99 132 [29] after clustering sequences at 97% identity with cd-hit-est v4.8.1 [30, 31]. Relative abundance is calculated as the percentage of reads that match a specific taxonomy string (Fig. S2). GraftM packages can be accessed via https://data.ace.uq.edu.au/public/graftm/7/.
Phylogenetic tree building and calculation of phylogenetic distance and gain
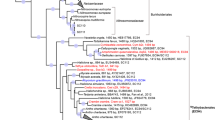
Bacterial and archaeal trees were inferred with GTDB v2.2.2 [25], filtering out MAGs below 40% MAG quality, defined as percentage genome completeness minus five times contamination [25]. Of the 259 de-replicated MAGs produced in this study, 15 were filtered out based on quality and an insufficient amount of amino acids in the multiple sequence alignment (Table S1). A concatenated marker gene tree was inferred using the 244 de-replicated MAGs, together with all publicly available sponge symbiont genomes (MAGs, SAGs, single-cell amplified genomes, and isolate genomes) that were ≥50% complete and possessed ≤10% contamination. For comparison, taxonomy was reassigned to the previously published sponge symbiont genomes using GTDB-Tk v0.2.2 (https://github.com/Ecogenomics/GtdbTk). Phylogenetic distance, defined as the “total branch length spanned by a set of taxa” [32] and phylogenetic gain, defined as the “additional branch length contributed by a set of taxa” [32], were calculated for both the archaeal and bacterial tree with GenomeTreeTk v0.0.41 (https://github.com/dparks1134/GenomeTreeTk) to determine how much additional phylogenetic diversity the MAGs from the current study added to the trees. The tree files were also visualized with Itol v4.2.3 [33] and refined in Inkscape v0.92.3 (https://inkscape.org/) for Fig. 1.

Labels colored in red are MAGs retrieved from this study. Black labels are previously published sponge symbiont genomes (MAGs, SAGs, and isolate genomes). Branch labels display taxonomy at the lowest inferred level. Genomes “GB_GCA_000522425.1” and “RS_GCF_900079095.1” were taxonomically uncharacterized by GTDB-Tk v0.2.2 (https://github.com/Ecogenomics/GtdbTk) and published taxonomy was used (Ca. Entotheonella factor [97] and Ca. Entotheonella palauensis [43], respectively). The bacterial and archaeal tree are rooted to LS-NOB (GB_GCA_001542995.1) and Nitrosopumilus (GCA_001543015.1), respectively.
Metabolic reconstruction of the MAGs
MAGs were annotated with the PFAM and Kyoto Encyclopedia of Genes and Genome (KEGG) Orthologies (KOs) databases [34, 35] using the “annotate” function of EnrichM v0.2.1 (https://github.com/geronimp/enrichM). Genes not annotated by KEGG were considered to encode “hypothetical proteins.” EnrichM’s “classify” function was then used to calculate the completeness of KEGG modules, which are groups of genes organized by steps in a metabolic pathway as defined by KEGG [35]. As we are working with partial genomes, for a function (e.g., carbon fixation) to be assigned to a MAG, the appropriate KEGG module was required to be ≥60% complete and all key enzymes (as defined by the literature) had to be present. A separate module was created for cobalamin because the KEGG module contained only part of the full pathway (Note S1). In addition, ABC transporters were positively assigned if genes for both the substrate specific component and the integral membrane protein were present in the MAG. Bacterial and archaeal trees described in the phylogenetic tree building section (only including MAGs retrieved from this study) were decorated with positively assigned functions from KEGG for Figs. S3–S12. Although we recovered a large proportion of the community (74.2%), the remaining 25.8% of unbinned genomic material may contain functionality we have not described and bioinformatics-based predictions could include incomplete pathways, due to the analysis of incomplete genomes.
GraftM v0.11.1 [28] was additionally used to validate KO annotations in the MAGs and the reads for the key enzymes dimethyl sulfoxide reductase (dmsA), dimethylsulfoniopropionate-dependent demethylase (dmdA), ammonia monooxygenase (amoA), ATP-citrate lyase (acl), Rubisco (rbc), and hydroxylamine dehydrogenase (hao). In brief, GraftM searches for defined key enzymes in MAGs or reads using custom HMMs and inserts them into a reference tree with functional clades [28].
Results and discussion
In total, 18 metagenomes from six Ircinia ramosa biological replicates sampled over three time points were sequenced to produce 559 MAGs (85.97 ± 13.40% completeness, 1.47 ± 1.73% contamination), including 13 archaea and 546 bacteria (Tables S1 and S2). The MAGs contained on average 2974 ± 887 coding sequences, of which 33.45 ± 6.71% were hypothetical proteins. De-replication of the MAGs within each time point reduced the total number to 259, including six archaeal MAGs belonging the phylum Thaumarchaeota (Crenarchaeota as per GTDB) and 253 bacterial MAGS belonging to 14 phyla, including the Acidobacteriota (25 MAGs), Actinobacteriota (43), Bacteroidota (8), Bdellovibrionota (1), Chloroflexota (63), Cyanobacteriota (4), Dadabacteria (6), Gemmatimonadota (21), Latescibacterota (15), Nitrospinota (1), Nitrospirota (5), Patescibacteria (2), Poribacteria (9), and Proteobacteria (50). This community profile is generally consistent with previously described Irciniidae spp. [7, 36, 37] and other HMA sponge microbial surveys [7], though some lineages identified in previous studies have been reclassified in the GTDB taxonomy. For example, Nitrospinota were previously part of the Proteobacteria and the Latescibacterota include the SAUL lineage, also known as PAUC34f (see Table S1 for further taxonomic comparison). However, in contrast to dominant lineages commonly reported in other HMA sponges [38], the microbiome of I. ramosa was dominated by the orders Rhodothermales (Bacteroidota) and Microtrichales (Actinobacteriota; Figs. S1 and S2). Analysis of single copy marker genes present in the reads and MAGs using SingleM showed that the de-replicated MAGs represented 74.2% of the I. ramosa microbial community. Over 60% of the taxa were present in >50% of the replicates across time points (Fig. S2), facilitating determination of the metabolic potential of all dominant taxa.
Ircinia MAGs increase phylogenetic diversity by 32% for archaea and 41% for bacteria
A concatenated genome tree was constructed from 95 previously published sponge symbiont genomes: 27 bacterial isolate genomes, 3 bacterial SAGs, and 65 MAGs (5 archaeal and 60 bacterial; Table S3) and 244 MAGs from this study (6 archaeal and 238 bacterial), that passed the quality criteria (Fig. 1). The MAGs from this study provided a phylogenetic gain (the additional branch length contributed by a set of taxa) of 31.74% for the archaea and 40.51% for the bacteria (Table 1). Although previously published genomes, originating from 24 sponge species, were widely distributed across the phylogenetic tree (Fig. 1), sequencing the microbiome of only one sponge species out of the 9209 presently described sponge species considerably expanded the sponge microbiome phylogenetic tree [39].
Three autotrophic carbon fixation pathways
Sponges obtain nutrients through active filter feeding on organic carbon, but some species have been shown to fix carbon via their microbial photosymbionts [40] or live in symbiosis with methane-oxidizing bacteria [41]. Relatively few sponge symbionts have been identified with autotrophic carbon fixation pathways [42,43,44,45], although carbon exchange between microbes and their host has been confirmed in several species [40, 41, 46, 47]. As microbial carbon acquisition could contribute to sponge metabolism, we searched the I. ramosa MAGs for the six known prokaryotic autotrophic carbon fixation pathways: the Wood-Ljungdahl (WL) pathway, reductive citric acid (rTCA) cycle, 3-hydroxypropionate/4-hydroxybutyrate (HP-HB) cycle, 3-hydroxypropionate (3-HP) bicycle, dicarboxylate/4-hydroxybutyrate (DC-HB) cycle, and the Calvin–Benson–Bassham (CBB) cycle [48]. Metabolic potential for three autotrophic carbon fixation pathways was identified in the I. ramosa microbiome (Figs. 2 and S3, Table S4), with the relative abundance of these MAGs in the six I. ramosa individuals ranging from 0.65 to 5.98%. Consistent with Nitrospirota recovered from other environments, key enzymes of the rTCA cycle, ATP-citrate lyases (aclA and aclB), were identified in all five I. ramosa Nitrospirota MAGs (family UBA8639), with an rTCA module completeness of 90% (Figs. 2 and S3, Table S4). The HP-HB cycle was identified in sponge-associated Nitrosopumilaceae (5 MAGs; Figs. 2 and S3, Table S4), similar to what has been reported for Thaumarchaeota recovered from other sponge species [44, 45]. Furthermore, more than 70% of the genes necessary for carrying out the CBB cycle, including both subunits of Rubisco (rbcS and rbcL) were found in the genus Synechococcus (4 MAGs) and Gammaproteobacteria (3 MAGs), including the family LS-SOB (2 MAGs; Figs. 2 and S3, Table S4). These data show evidence for the presence of multiple carbon fixation pathways within a sponge microbiome and expands our view on the distribution of genes encoding the CBB cycle, which previously had only been identified in sponge-derived MAGs from Cyanobacteria [42] and Entotheonella [43].

HP-HB cycle: 3-hydroxypropionate/4-hydroxybutyrate cycle. Lineages in bold within the taurine metabolism box contain both genes encoding for the taurine transporter (tauACB) and taurine dioxygenase (tauD). Numbers in brackets within the vitamin metabolism stand for the metabolic potential to synthesize one or more of the following B-vitamins: 1: Biotin, 2: Cobalamin, 3: Pantothenate, 4: Pyridoxine, 5: Riboflavin, 6: Thiamine. See Fig. S13 for I. ramosa morphology.
The WL pathway, 3-HP bicycle, and DC-HB cycle were absent in the I. ramosa MAGs (Fig. S3, Table S4). The WL pathway has previously been reported in Poribacteria [49] and the so-called SAUL lineage [50]. However, the contamination of the poribacterial SAG presented in that study was 74.02% as shown in Table S1 from Kamke et al. [51]. Further, key enzymes of the bacterial WL pathway, acetyl-CoA synthase/carbon monoxide dehydrogenase (ACS/CODH) subunits B, C, D, and E, were absent from the SAUL lineage (Note S2). Bacterial and archaeal key enzymes for the WL pathway (acsABCDE and cdhABCDE, respectively) were also absent in our MAGs. Hence evidence for the WL pathway in sponge symbionts is lacking, as is evidence for the DC-HB cycle.
Both radio-labeled and stable isotope-tracer experiments have confirmed that photosymbiont fixed carbon through the CBB pathway can be taken up by the host [40, 46]. However, there is currently no data regarding the fate of carbon from other autotrophic fixation pathways. If symbiont-fixed carbon from each of the pathways gets translocated to the sponge host, the acquired autotrophic carbon could be contributing to the overall high environmental tolerance and plasticity of sponges, particularly to ocean warming, which can cause loss of photosymbionts [52].
First step of nitrification is confined to the Thaumarchaeota
Through their excretion of ammonia, sponges would provide an attractive niche for microorganisms, particularly in ammonium-limited oligotrophic seas [53]. Sponge symbionts are thought to use ammonia as an energy source, thereby detoxifying their host through nitrification [54, 55]. During nitrification, ammonia is consecutively oxidized to hydroxylamine, nitrite, and nitrate [56], catalyzed by ammonia monooxygenases (amoABC), hydroxylamine dehydrogenase (hao), and nitrate oxidoreductases (nxrAB), respectively. The amoA, B, and C subunits were identified in four I. ramosa Thaumarchaeota (family Nitrosopumilaceae) MAGs via KO annotation and placement into a phylogenetic reference tree using an amo-specific GraftM package (Figs. 2 and S4, Table S4). In addition, a search for amoA amongst the raw reads using GraftM showed that amoA was exclusively assigned to archaea. Ammonia oxidizing bacteria and archaea were previously reported to co-occur in a single sponge species [9, 57, 58]. However, as we did not find any evidence for ammonia oxidizing bacteria in I. ramosa, our data suggest that Thaumarchaeota are solely responsible for ammonia oxidation, representing a keystone taxa within the I. ramosa microbiome.
Although graftM identified 13 bacterial MAGs that contained putative haos, these were only distantly related to known haos. As the MAGs containing these hao-homologs did not have the amoA gene necessary for conversion of ammonium to hydroxylamine, we suspect that the hao-homologs do not serve in nitrification, but might rather be involved in the detoxification of hydroxylamine [59] originating from other cellular processes. This finding supports our assertion that the Thaumarchaeota are the only oxidizers of ammonia, as they are able to convert ammonia directly to nitrite themselves [47, 60, 61]. Nitrospirota (3 MAGs), Alphaproteobacteria (9), and Gammaproteobacteria (5) encoded for the genes nxrA and nxrB, that catalyze the conversion of nitrite to nitrate.
Nitrate resulting from nitrification can be recycled through denitrification [62], which is carried out in four steps: nitrate and nitrite reduction (catalyzed by narGHI or napAB and nirK or nirS, respectively), nitric oxide reduction (norBC), and nitrous oxide reduction into nitrogen (nosZ). In the I. ramosa microbiome, narGHI and napAB were identified in the Alphaproteobacteria (10 MAGs), Gammaproteobacteria (6), and Nitrospirota (3; Figs. 2 and S4, Table S4). One Gemmatimonadota (class Gemmatimonadetes) and 18 Gammaproteobacteria MAGs encoded both the norB and norC genes, suggesting that only members of these phyla can convert nitric oxide into nitrous oxide. Absent from all MAGs was nosZ, suggesting that nitrous oxide is not reduced. Full denitrification has so far only been found in sponge-associated Alphaproteobacteria [63] and Entotheonella [43]. Previous studies have shown that only enzymes responsible for the first steps of denitrification (i.e. narGHI, napAB, nirK, and nirS) are enriched in sponge symbionts, while enzymes for the final two steps (i.e., norBC and nosZ) are absent [9, 64, 65], suggesting an incomplete pathway, which would result in the release of nitrous oxide into the surrounding seawater. KOs associated with annamox were absent from all MAGs, as were KOs involved in nitrogen fixation.
Sponge symbionts can translocate nitrogen to the host [46, 66], but the sponge microbiome also contains a diverse array of microbes that assist the host with efficient removal of nitrogenous waste products including ammonium and nitrite [67, 68]. Congruently, the pathways identified in the I. ramosa microbiome were mainly involved in nitrogen removal from the system.
Widespread potential for sulfur metabolism
Taurine is a naturally occurring compound in a wide variety of organisms and taurine-conjugated fatty acids have been identified in Ircinia sp. [69]. The potential of sponge-associated microbes to use taurine via ABC transporters (tauACB) and subsequently convert it to sulfite via taurine dioxygenases (tauD) has been described in a number of lineages, including the Proteobacteria [64, 70] and Ca. Entotheonella [71]. Taurine dioxygenases were identified in I. ramosa MAGs belonging to the Acidobacteriota (21 MAGs), Actinobacteriota (24), Alphaproteobacteria (6), Chloroflexota (10), Gammaproteobacteria (26), Gemmatimonadota (20), Latescibacterota (14), Nitrospinota (1), and Poribacteria (2), of which the tauACB transporter was also identified in the Actinobacteriota (13), Alphaproteobacteria (4), Gammaproteobacteria (4), and Nitrospinota (1), suggesting widespread potential for sponge symbionts to use host derived taurine (Figs. 2 and S5, Table S4). In addition to the cleavage of taurine, sulfite may also originate from the dissimilatory oxidation of sulfide by dissimilatory sulfite reductases, dsrAB and dsrEFH. Interestingly, members of the Gammaproteobacteria (6 MAGs, of which four from the family LS-SOB) are the only taxa capable of dissimilatory sulfide oxidation in the Ircinia holobiont (Figs. 2 and S5, Table S4). Sulfite can in turn be oxidized to sulfate via sulfite oxidase (suox). Genes encoding suox were identified in all phyla except Thaumarchaeota, Cyanobacteriota, Bdellovibrionota, Dadabacteria, and Patescibacteria. Genes involved in dissimilatory sulfate reduction to sulfite (sat and APS reductases aprAB) were identified in Alpha- (3 MAGs) and Gammaproteobacteria (5), and Chloroflexota (5; Figs. 2 and S5, Table S4).
Incomplete oxidation of sulfides can produce thiosulfate, which can be oxidized to sulfate through sulfur-oxidizing proteins (soxAB, soxXYZ), called the SOX complex [72]. Two Alpha- and five Gammaproteobacteria MAGs (similar to the Gammaproteobacteria MAGs capable of sulfide oxidation) were identified with soxAB and soxXYZ, confirming potential to oxidize sulfur through the entire SOX system (Figs. 2 and S5, Table S4). This study demonstrates widespread potential for sulfur metabolism in I. ramosa, which is likely beneficial for both microbes and host. Host derived taurine would provide a source of sulfur for the microbial community while sulfur-oxidizing bacteria could balance sulfide generation from sulfate-reducing bacteria in the sponge [73], providing a detoxifying function and increasing the overall stability of the holobiont.
Metabolic potential for synthesis of six essential B-vitamins
B-vitamins and their cofactors are essential compounds for organismal central metabolism [74, 75]. However, animals cannot synthesize these vitamins themselves and must therefore acquire them through feeding or symbiotic relationships with microorganisms [76, 77]. Previous genomic analyses of sponge microbiomes have highlighted the presence of thiamine (vitamin B1), riboflavin (B2), pyridoxine (B6), biotin (B7), and cobalamin (B12) biosynthesis genes, suggesting a possible role in provisioning vitamins to the sponge host. However, gene-centric analyses could not link these pathways with taxonomy and genomic analyses were focussed on just a few lineages able to metabolize these B-vitamins. Here we show that the potential for biosynthesis of B1, B2, pantothenate (B5), B6, B7, and B12 is widely distributed within the microbiome, with 13 out of the 18 I. ramosa associated microbial phyla containing pathways to synthesize thiamine: Acidobacteriota (17 MAGs), Actinobacteriota (8), Bacteroidota (1), Chloroflexota (21), Dadabacteria (6), Gemmatimonadota (18), Latescibacterota (9), Nitrospinota (1), Nitrospirota (5), Poribacteria (9), and Proteobacteria (22). In addition, the Actinobacteriota (34), Chloroflexota (16), and Proteobacteria (7) encoded for genes necessary for thiamine transport (Figs. 2 and S6, Table S4) and all phyla apart from the Patescibacteria were able to synthesize riboflavin and convert it into the functional coenzymes flavin mononucleotide and flavin adenine dinucleotide (Figs. 2 and S7, Table S4). In addition, the genes necessary to synthesize pantothenate were only absent in Bdellovibrionota and Thaumarchaeota (Figs. 2 and S8, Table S4) and pyridoxine can be synthesized from D-Erythrose 4-phosphate by Acidobacteriota (12 MAGs), Bacteroidota (8), Nitrospirota (5), and Proteobacteria (43; Figs. 2 and S9, Table S4). Further, biotin metabolism was widespread and only absent from MAGs belonging to Bacteroidota, Gemmatimonadota, and Patescibacteria. The biotin transporter was identified in Actinobacteriota (3), Cyanobacteriota (4), Latescibacterota (5), and Proteobacteria (3; Figs. 2 and S10, Table S4). Potential for cobalamin biosynthesis was also identified in four phyla: Chloroflexota (7 MAGs), Cyanobacteriota (4), Proteobacteria (9), and Thaumarchaeota (5; Figs. 2 and S11, Table S4).
The potential for B-vitamin biosynthesis had previously only been reported for single symbiont lineages within any sponge species [14, 38, 78,79,80,81]. Here we reveal extensive taxonomic redundancy in vitamin provisioning within the complex sponge microbiome, including the metabolism of vitamin B5. Although sponges can obtain their B-vitamins through feeding, symbionts likely provide an alternative pathway for vitamin provision, which is supported by the presence of vitamin transporters in the sponge microbiome [45, 65, 82, 83], as well as the presence of vitamin transporters and vitamin catabolism [78] in the sponge host.
Secretion systems and eukaryotic-like proteins in the sponge microbiome
Many symbiotic microorganisms are known to interact with their host via microbial secretion systems, some of which are needle-like protein complexes (Type III, IV, and VI) used to inject effector proteins into an adjacent cell to modulate its metabolism [84]. Eukaryotic-like proteins, such as Ankyrin-repeat (Ank) and WD40 repeat-containing proteins [85, 86], are also thought to be used for bacterial establishment in the host [87, 88]. Surprisingly, only members of the Gammaproteobacteria (3 MAGs) in the I. ramosa microbiome contained genes encoding Type III secretion systems, responsible for the transport of bacterial effector proteins into eukaryotic cells [89], including the order Pseudomonadales (2 MAGs; Fig. S12, Table S4). Secretion Type VI, used by bacterial symbionts to eliminate microbial competitors [90] and to translocate toxic effector proteins into eukaryotic cells [84], was also identified in Pseudomonadales (2 MAGs; Fig. S12, Table S4). KOs associated with Type IV secretion systems, which may play a role in delivering eukaryotic-like proteins into host cells [87], were identified in lineages belonging to the Acidobacteriota and Gemmatimonadota (Fig. S12, Table S4). While Type IV secretion systems have previously been reported to be abundant [9] and enriched [91] in sponge symbionts, overall module completeness was below 60% in I. ramosa MAGs. These data suggest that the majority of the I. ramosa microbiome does not interact directly with the host cells via needle-like secretion systems, but highlights specific lineages that may. Interestingly, the presence of the Type VI secretion system raises the possibility of bacteria–bacteria interactions or potentially parasitism/pathogenesis within the Pseudomonadales.
Ank repeat proteins (ARPs) mediate protein–protein interactions and are one of the most common domains present in eukaryotes, though they are much rarer in bacteria and archaea [89]. ARPs are commonly enriched and actively expressed in sponge symbionts [9, 14, 49, 82] and their presence in microorganisms has been suggested to mediate host–microbe interactions and protect sponge symbionts from host phagocytosis [14, 65, 85]. The average percentage of ARPs in the I. ramosa MAGs was 0.14% of all coding genes (Table S1), ranging from 0 to 0.89% (Acidobacteriota, order Luteitaleales). Of the total MAGs, 27% (including Chloroflexota, Cyanobacteriota, Gemmatimonadota, and Latescibacterota) had an ARP/gene percentage >0.2%, similar to known symbionts (Note S3, [88]) ARPs were absent in multiple lineages, including the Thaumarchaeota, Patescibacteria, Actinobacteriota (family TK06 and order UBA5794), and Chloroflexota (family UBA2991 and order UBA2979), suggesting that these taxa must use alternative mechanisms to evade digestion. Another potential mediator of protein–protein interactions, previously identified as necessary for the infection of rhizobium in root nodules [92], are WD40 repeat-containing proteins. Like ARPs, these proteins are prevalent in sponge symbionts [79, 80]. Of the I. ramosa MAGs, 40% encoded WD40 repeat-containing proteins, with an average WD40 count/gene of 0.08% (Table S1), ranging from 0 to 3.59%. The highest number of WD40 repeat-containing proteins were found in the orders WGA-4E (Poribacteria), Rhodothermales (Bacteroidota), and UBA8231 (Latescibacterota). These findings suggest a role for both ARPs and WD40 repeat-containing proteins in maintaining the stable symbiotic associations evident in I. ramosa. Lineages lacking both ARPs and WD40 repeats must use alternative, yet to be identified, mechanisms for evading host phagocytosis.
Conclusions
Genomic recovery of most members of the I. ramosa microbiome has provided unprecedented insights into the metabolic potential of a complex sponge microbiome. Previous investigations of the sponge microbiome [12] have suggested that microbes can be partitioned into distinct groups based on broad-scale analysis of all metabolic pathways (Clusters of Orthologous Groups) present in the MAGs. One of these groups was shown to encode higher numbers of genes for carnitine metabolism while a second was enriched for genes for sulfated polysaccharide degradation. These findings suggest a degree of metabolic specialization between microbial groups but do not preclude the existence of other pathways that might be shared between lineages. Here, we reveal considerable redundancy in pathways for autotrophic carbon fixation, nitrogen and sulfur metabolism, and vitamin synthesis. Sponges are sessile organisms, residing in a dynamic environment. Changes in the pumping activity of the sponge can rapidly modify the environmental conditions of the microbiome—for instance, shifting the mesohyl region from oxic to anoxic and vice versa [73, 93]. In such an environment, a variety of metabolic traits from the taxonomically-diverse microbiome increases the physiological plasticity of the holobiont under changing environmental conditions. The identification of three autotrophic carbon fixation pathways (rTCA cycle, HP-HB cycle, and CBB cycle) raises questions about the extent to which the sponge host relies on microbial symbionts for carbon acquisition. Importantly, functional redundancy in carbon fixation could increase the stability of the sponge under changing environmental conditions [94]. Supporting this hypothesis are observations of sponge bleaching, which, in comparison to coral bleaching, is rarely lethal [95, 96]. In addition, elevated inorganic carbon availability appears to reduce the degree of stress in sponges exposed to ocean warming [95]. Future functional physiological studies should test the relative contribution of each carbon fixation pathway to host nutrition under different environmental conditions.
This study also identified potential keystone taxa within the I. ramosa microbiome, with Thaumarchaeota (family Nitrosopumilaceae) as the sole taxa able to perform the ammonia oxidation step of nitrification and Gammaproteobacteria (predominantly the family LS-SOB) as the only sulfide oxidizers. Assigning potential functions to specific taxa through metagenomic analysis will allow these taxa to be targeted in functional physiological studies. Loss of keystone taxa, due to environmental stress or other perturbations, might have detrimental effects on the sponge host and its direct environment. For example, in the absence of ammonia oxidizers, toxic ammonia would not be converted into nitrate and cycled through the sponge holobiont.
Current genomic resources for reef invertebrate microbiomes are limited. Using an integrative metagenomic approach we reveal extensive metabolic potential and taxonomic and functional redundancy in the sponge microbiome. Establishment of a comprehensive genomic baseline for a sponge microbiome provides a valuable platform for future functional studies aimed at validating and quantifying the contribution of each of the putative symbiotic functions to host health. Studies employing (meta)transcriptomics, proteomics, immunohistochemical and stable isotope-tracer experiments should be undertaken to confirm expression and track the fate of specific compounds within the holobiont. Future studies aimed at understanding the sponge as a holobiont should also move towards the retrieval of host genomes so that the metabolic potential of the sponge and its symbionts can be assessed for metabolic complementarity.
Data availability
All data for this study can be found under the Bioproject ID PRJNA555144. The metagenomic reads for I. ramosa are available at NCBI under the accession numbers SRR9841427-SRR9841447 and the MAGs can be found under the accession numbers VXLI00000000-VYGU00000000, which are also shown in Table S1.
Change history
05 February 2020
The supplementary information files were initially linked incorrectly but have since been corrected. All files were available at publication.
References
Bell JJ. The functional roles of marine sponges. Estuar Coast Shelf Sci. 2008;79:341–53.
De Goeij JM, Van Oevelen D, Vermeij MJ, Osinga R, Middelburg JJ, de Goeij AF, et al. Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science. 2013;342:108–10.
Beazley LI, Kenchington EL, Murillo FJ, Sacau MdM. Deep-sea sponge grounds enhance diversity and abundance of epibenthic megafauna in the Northwest Atlantic. ICES J Mar Sci. 2013;70:1471–90.
Webster NS, Thomas T. The sponge hologenome. MBio. 2016;7:e00135–16.
Pita L, Rix L, Slaby BM, Franke A, Hentschel U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome. 2018;6:46.
Hentschel U, Fieseler L, Wehrl M, Gernert C, Steinert M, Hacker J, et al. Microbial diversity of marine sponges. In: Mueller W, editor, Marine molecular biotechnology. Berlin; Springer: 2003. p. 59–88.
Thomas T, Moitinho-Silva L, Lurgi M, Björk JR, Easson C, Astudillo-García C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870.
Taylor MW, Tsai P, Simister RL, Deines P, Botte E, Ericson G, et al. ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 2013;7:438.
Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, et al. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci USA. 2012;109:E1878–87.
Karimi E, Keller-Costa T, Slaby BM, Cox CJ, da Rocha UN, Hentschel U, et al. Genomic blueprints of sponge-prokaryote symbiosis are shared by low abundant and cultivatable Alphaproteobacteria. Sci Rep. 2019;9:1999.
Podell S, Blanton JM, Neu A, Agarwal V, Biggs JS, Moore BS, et al. Pangenomic comparison of globally distributed Poribacteria associated with sponge hosts and marine particles. ISME J. 2019;13:468–81.
Slaby BM, Hackl T, Horn H, Bayer K, Hentschel U. Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 2017;11:2465–78.
Diez-Vives C, Esteves AIS, Costa R, Nielsen S, Thomas T. Detecting signatures of a sponge-associated lifestyle in bacterial genomes. EMR. 2018;10:433–43.
Thomas T, Rusch D, DeMaere MZ, Yung PY, Lewis M, Halpern A, et al. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 2010;4:1557–67.
Sturm M, Schroeder C, Bauer P. SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinforma. 2016;17:208.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Graham ED, Heidelberg JF, Tully BJ. BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ. 2017;5:e3035.
Imelfort M, Parks D, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ. 2014;2:e603.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2015;32:605–7.
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114.
Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinforma. 2010;11:538.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004.
Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8.
R Core Team R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2013). http://www.R-project.org/.
Boyd JA, Woodcroft BJ, Tyson GW. GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res. 2018;46:e59-e.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2.
Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–42.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2015;44:D279–85.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015;44:D457–62.
Erwin PM, Lopez-Legentil S, Gonzalez-Pech R, Turon X. A specific mix of generalists: bacterial symbionts in Mediterranean Ircinia spp. FEMS Microbiol Ecol. 2012;79:619–37.
Esteves AI, Hardoim CC, Xavier JR, Goncalves JM, Costa R. Molecular richness and biotechnological potential of bacteria cultured from Irciniidae sponges in the north-east Atlantic. FEMS Microbiol Ecol. 2013;85:519–36.
Bayer K, Jahn MT, Slaby BM, Moitinho-Silva L, Hentschel U. Marine sponges as chloroflexi hot spots: genomic insights and high-resolution visualization of an abundant and diverse symbiotic clade. MSystems. 2018;3:e00150–18.
Van Soest RWM, Boury-Esnault N, Hooper JNA, Rützler K, de Voogd NJ, Alvarez B, et al. World Porifera database. (2018) http://www.marinespecies.org/porifera.
Wilkinson CR. Net primary productivity in coral reef sponges. Science. 1983;219:410–2.
Vacelet J, Fiala-Médioni A, Fisher C, Boury-Esnault N. Symbiosis between methane-oxidizing bacteria and a deep-sea carnivorous cladorhizid sponge. Mar Ecol Prog Ser. 1996;145:77–85.
Gao Z-M, Wang Y, Tian R-M, Wong YH, Batang ZB, Al-Suwailem AM, et al. Symbiotic adaptation drives genome streamlining of the cyanobacterial sponge symbiont “Candidatus Synechococcus spongiarum”. MBio. 2014;5:e00079–14.
Liu F, Li J, Feng G, Li Z. New genomic insights into “Entotheonella” symbionts in Theonella swinhoei: Mixotrophy, anaerobic adaptation, resilience, and interaction. Front Microbiol. 2016;7:1333.
Moitinho-Silva L, Díez-Vives C, Batani G, Esteves AI, Jahn MT, Thomas T. Integrated metabolism in sponge–microbe symbiosis revealed by genome-centered metatranscriptomics. ISME J. 2017;11:1651–66.
Tian RM, Sun J, Cai L, Zhang WP, Zhou GW, Qiu JW, et al. The deep-sea glass sponge Lophophysema eversa harbours potential symbionts responsible for the nutrient conversions of carbon, nitrogen and sulfur. Environ Microbiol. 2016;18:2481–94.
Achlatis M, Pernice M, Green K, Guagliardo P, Kilburn MR, Hoegh-Guldberg O, et al. Single-cell measurement of ammonium and bicarbonate uptake within a photosymbiotic bioeroding sponge. ISME J. 2018;12:1308.
Moeller FU, Webster NS, Herbold CW, Behnam F, Domman D, Albertsen M, et al. Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella basta. Environ Microbiol. 2019;21:527234.
Hügler M, Sievert SM. Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Annu Rev Mar Sci. 2011;3:261–89.
Siegl A, Kamke J, Hochmuth T, Piel J, Richter M, Liang C, et al. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 2011;5:61–70.
Astudillo‐García C, Slaby BM, Waite DW, Bayer K, Hentschel U, Taylor MW. Phylogeny and genomics of SAUL, an enigmatic bacterial lineage frequently associated with marine sponges. Environ Microbiol. 2018;20:561–76.
Kamke J, Sczyrba A, Ivanova N, Schwientek P, Rinke C, Mavromatis K, et al. Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 2013;7:2287–300.
Bell JJ, Bennett HM, Rovellini A, Webster NS. Sponges to be winners under near-future climate scenarios. Biosci. 2018;68:955–68.
Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nat Rev Microbiol. 2012;10:641–54.
Bayer K, Schmitt S, Hentschel U. Physiology, phylogeny and in situ evidence for bacterial and archaeal nitrifiers in the marine sponge Aplysina aerophoba. Environ Microbiol. 2008;10:2942–55.
Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev. 2007;71:295–347.
Ward BB. Nitrification in marine systems. In: Capone DG, Bronk DA, Mulholland MR, Carpenter EJ, editors. Nitrogen in the marine environment. 2nd ed. Amsterdam: Elsevier Press; 2008. p. 199–262.
Han M, Li Z, Zhang F. The ammonia oxidizing and denitrifying prokaryotes associated with sponges from different sea areas. Micro Ecol. 2013;66:427–36.
Radax R, Hoffmann F, Rapp HT, Leininger S, Schleper C. Ammonia-oxidizing archaea as main drivers of nitrification in cold-water sponges. Environ Microbiol. 2012;14:909–23.
Mohammadi SS, Pol A, van Alen T, Jetten MS, Op den Camp HJ. Ammonia oxidation and nitrite reduction in the verrucomicrobial methanotroph Methylacidiphilum fumariolicum SolV. Front Microbiol. 2017;8:1901.
De la Torre JR, Walker CB, Ingalls AE, Könneke M, Stahl DA. Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ Microbiol. 2008;10:810–8.
Könneke M, Bernhard AE, José R, Walker CB, Waterbury JB, Stahl DA. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature. 2005;437:543–6.
Maldonado M, Ribes M, van Duyl FC. Nutrient fluxes through sponges: biology, budgets, and ecological implications. Adv Mar Biol. 2012;62:113–82.
Bondarev V, Richter M, Romano S, Piel J, Schwedt A, Schulz-Vogt HN. The genus pseudovibrio contains metabolically versatile bacteria adapted for symbiosis. Environ Microbiol. 2013;15:2095–113.
Karimi E, Slaby BM, Soares AR, Blom J, Hentschel U, Costa R. Metagenomic binning reveals versatile nutrient cycling and distinct adaptive features in alphaproteobacterial symbionts of marine sponges. FEMS Microbiol Ecol. 2018;94:74.
Liu M, Fan L, Zhong L, Kjelleberg S, Thomas T. Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 2012;6:1515–25.
Fiore CL, Baker DM, Lesser MP. Nitrogen biogeochemistry in the Caribbean sponge, Xestospongia muta: a source or sink of dissolved inorganic nitrogen? PLoS ONE. 2013;8:e72961.
Hoffmann F, Radax R, Woebken D, Holtappels M, Lavik G, Rapp HT, et al. Complex nitrogen cycling in the sponge Geodia barretti. Environ Microbiol. 2009;11:2228–43.
Schläppy M-L, Schöttner SI, Lavik G, Kuypers MM, de Beer D, Hoffmann F. Evidence of nitrification and denitrification in high and low microbial abundance sponges. Mar Biol. 2010;157:593–602.
Emura C, Higuchi R, Miyamoto T. Irciniasulfonic acid B, a novel taurine conjugated fatty acid derivative from a Japanese marine sponge, Ircinia sp. Tetrahedron. 2006;62:5682–5.
Gauthier M-EA, Watson JR, Degnan SM. Draft genomes shed light on the dual bacterial symbiosis that dominates the microbiome of the coral reef sponge Amphimedon queenslandica. Front Mar Sci. 2016;3:196.
Lackner G, Peters EE, Helfrich EJ, Piel J. Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc Natl Acad Sci USA. 2017;114:E347–56.
Tian RM, Wang Y, Bougouffa S, Gao ZM, Cai L, Bajic V, et al. Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur‐oxidizing bacterium in sponge. Environ Microbiol. 2014;16:3548–61.
Hoffmann F, Larsen O, Thiel V, Rapp HT, Pape T, Michaelis W, et al. An anaerobic world in sponges. Geomicrobiol J. 2005;22:1–10.
Combs GF Jr, McClung JP. The vitamins: fundamental aspects in nutrition and health. 5th ed. San Diego: Academic Press Inc; 2016.
Tomiki T, Saitou N. Phylogenetic analysis of proteins associated in the four major energy metabolism systems: photosynthesis, aerobic respiration, denitrification, and sulfur respiration. J Mol Evol. 2004;59:158–76.
Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. Algae acquire vitamin B 12 through a symbiotic relationship with bacteria. Nature. 2005;438:90–3.
Moriyama M, Nikoh N, Hosokawa T, Fukatsu T. Riboflavin provisioning underlies Wolbachia’s fitness contribution to its insect host. MBio. 2015;6:e01732–15.
Fiore CL, Labrie M, Jarett JK, Lesser MP. Transcriptional activity of the giant barrel sponge, Xestospongia muta Holobiont: molecular evidence for metabolic interchange. Front Microbiol. 2015;6:364.
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y-i, Sugahara J, et al. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci USA. 2006;103:18296–301.
Kamke J, Rinke C, Schwientek P, Mavromatis K, Ivanova N, Sczyrba A, et al. The candidate phylum Poribacteria by single-cell genomics: new insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS ONE. 2014;9:e87353.
Liu MY, Kjelleberg S, Thomas T. Functional genomic analysis of an uncultured δ-proteobacterium in the sponge Cymbastela concentrica. ISME J. 2011;5:427–35.
Díez-Vives C, Moitinho-Silva L, Nielsen S, Reynolds D, Thomas T. Expression of eukaryotic‐like protein in the microbiome of sponges. Mol Ecol. 2017;26:1432–51.
Moitinho-Silva L, Seridi L, Ryu T, Voolstra CR, Ravasi T, Hentschel U. Revealing microbial functional activities in the Red Sea sponge Stylissa carteri by metatranscriptomics. Environ Microbiol. 2014;16:3683–98.
Costa TR, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, et al. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol. 2015;13:343–59.
Nguyen MT, Liu M, Thomas T. Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol Ecol. 2014;23:1635–45.
Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci. 1999;24:181–5.
Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–4.
Jernigan KK, Bordenstein SR. Ankyrin domains across the tree of life. PeerJ. 2014;2:e264.
Al-Khodor S, Price CT, Kalia A, Kwaik YA. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 2010;18:132–9.
Speare L, Cecere AG, Guckes KR, Smith S, Wollenberg MS, Mandel MJ, et al. Bacterial symbionts use a type VI secretion system to eliminate competitors in their natural host. Proc Natl Acad Sci USA. 2018;115:E8528–37.
Karimi E, Ramos M, Gonçalves J, Xavier JR, Reis MP, Costa R. Comparative metagenomics reveals the distinctive adaptive features of the Spongia officinalis endosymbiotic consortium. Front Microbiol. 2017;8:2499.
Yano K, Shibata S, Chen WL, Sato S, Kaneko T, Jurkiewicz A, et al. CERBERUS, a novel U-box protein containing WD-40 repeats, is required for formation of the infection thread and nodule development in the legume–Rhizobium symbiosis. Plant J. 2009;60:168–80.
Schläppy M-L, Hoffmann F, Røy H, Wijffels RH, Mendola D, Sidri M, et al. Oxygen dynamics and flow patterns of Dysidea avara (Porifera: Demospongiae). J Mar Biol Assoc UK. 2007;87:1677–82.
McCann KS. The diversity–stability debate. Nature. 2000;405:228–33.
Bennett HM, Altenrath C, Woods L, Davy SK, Webster NS, Bell JJ. Interactive effects of temperature and pCO 2 on sponges: from the cradle to the grave. Glob Chang Biol. 2017;23:2031–46.
Cowart J, Henkel T, McMurray S, Pawlik JR. Sponge orange band (SOB): a pathogenic-like condition of the giant barrel sponge, Xestospongia muta. Coral Reefs. 2006;25:513.
Wilson MC, Mori T, Rückert C, Uria AR, Helf MJ, Takada K, et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature. 2014;506:58–62.
Acknowledgements
We acknowledge Andrea Severati and other staff at the Australian Institute of Marine Science SeaSim facility who assisted with sample collection and sponge husbandry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Engelberts, J.P., Robbins, S.J., de Goeij, J.M. et al. Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J 14, 1100–1110 (2020). https://doi.org/10.1038/s41396-020-0591-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0591-9
This article is cited by
-
The road forward to incorporate seawater microbes in predictive reef monitoring
Environmental Microbiome (2024)
-
Unveiling microbial guilds and symbiotic relationships in Antarctic sponge microbiomes
Scientific Reports (2024)
-
Prokaryotic communities of the French Polynesian sponge Dactylospongia metachromia display a site-specific and stable diversity during an aquaculture trial
Antonie van Leeuwenhoek (2024)
-
A role for encrusting, endolithic sponges in the feeding of the parrotfish Scarus rubroviolaceus? Evidence of further trophic diversification in Indo-Pacific Scarini
Coral Reefs (2024)
-
Trait biases in microbial reference genomes
Scientific Data (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}