
Abstract
The Amazon rainforest is a biodiversity hotspot and large terrestrial carbon sink threatened by agricultural conversion. Rainforest-to-pasture conversion stimulates the release of methane, a potent greenhouse gas. The biotic methane cycle is driven by microorganisms; therefore, this study focused on active methane-cycling microorganisms and their functions across land-use types. We collected intact soil cores from three land use types (primary rainforest, pasture, and secondary rainforest) of two geographically distinct areas of the Brazilian Amazon (Santarém, Pará and Ariquemes, Rondônia) and performed DNA stable-isotope probing coupled with metagenomics to identify the active methanotrophs and methanogens. At both locations, we observed a significant change in the composition of the isotope-labeled methane-cycling microbial community across land use types, specifically an increase in the abundance and diversity of active methanogens in pastures. We conclude that a significant increase in the abundance and activity of methanogens in pasture soils could drive increased soil methane emissions. Furthermore, we found that secondary rainforests had decreased methanogenic activity similar to primary rainforests, and thus a potential to recover as methane sinks, making it conceivable for forest restoration to offset greenhouse gas emissions in the tropics. These findings are critical for informing land management practices and global tropical rainforest conservation.
Similar content being viewed by others
Introduction
Climate change, caused by the anthropogenic release of greenhouse gases [1], is affecting every ecosystem on Earth. Although the majority of greenhouse gases released to the atmosphere are associated with the industrial revolution and fossil fuel combustion, land-use change is a significant contributor. Specifically, tropical deforestation in the last decade has released ~1 Pg C yr−1, an equivalent to 10% of anthropogenic carbon dioxide emissions [1], and 78% of total greenhouse gas emissions in Brazil are caused by land use change [2, 3]. In addition to being biodiversity hotspots of plants and animals, tropical rainforests are large terrestrial carbon sinks. However, rainforest deforestation to create cattle pastures or agricultural fields releases large amounts of stored carbon, converting former terrestrial carbon sinks into major carbon sources [3, 4]. In the Amazon rainforest particularly, over 1 Mha of forest has been lost in 2017 alone [5]. The conversion of primary rainforest (PF) (i.e., mature rainforest older than 150 years) to cattle pasture is a main cause of deforestation in Brazil and not only changes plant diversity but also the microorganisms that drive soil biogeochemical cycling [6].
The methane (CH4) biogeochemical cycle is of interest because of its potency as a greenhouse gas with 86-times the global warming potential of carbon dioxide over a 20-year timescale [1]. Biotic CH4 cycling is controlled by microorganisms, specifically methanogenic archaea that produce CH4, and methanotrophic bacteria that consume CH4 [7, 8]. The balance between these two functional groups determines whether the soil acts as a CH4 source or sink. Under anoxic conditions, soil methanogenic archaea generally metabolize fermentation products such as acetate (acetoclastic methanogenesis) or reduce carbon dioxide with hydrogen (hydrogenotrophic methanogenesis) to produce CH4 [9,10,11]. Methanotrophs are commonly aerobic bacteria from either Gammaproteobacteria, Alphaproteobacteria, or Verrucomicrobia, corresponding to Type I, II, and III methanotrophs, respectively [12, 13]. In addition, anaerobic methane oxidation has been described for wetland soils [14], but for upland soils only the potential for anaerobic oxidation of methane exists [15]. Previous research into the different growth conditions of Type I versus Type II methanotrophs found that Type II methanotrophs generally dominate high CH4, low oxygen environments along with nitrogen- and copper-limiting conditions [16,17,18]. However, Type II methanotrophs have also been found in soils with low CH4 concentrations [19,20,21] likely due to two isoenzymes of the particulate methane monooxygenase that have different affinities for CH4 [22] making them more versatile metabolically. Currently, there is no evidence that organisms related to Verrucomicrobia methanotrophs (Type III) found in soils are methanotrophic, unless these soils are located in geothermal and acidic environments like volcanic mud pots and similarly extreme environments [23].
Researchers have focused on the impact of rainforest-to-pasture conversion on CH4 cycling for decades [24,25,26]. Measurements of infield gas flux generally show soil CH4 consumption across seasons in mature rainforest, while pasture soils emit CH4 [27, 28]. Over the last decade, further research into how tropical land use change influences CH4 cycling microorganisms found varied results. One study observed that the functional biomarkers for methanotrophy (pmoA and mmoX) decreased in cattle pastures with no change to the methanogenesis biomarker (mcrA), while another study observed a decrease in pmoA abundance from Type II methanotrophs and an increase in mcrA in cattle pastures [6, 21]. These previous studies investigated how land use change in the Brazilian Amazon impacts the genomic potential of the soil methane-cycling microbial community, but no study has directly targeted the active community.
Metatranscriptomics, metaproteomics, and stable-isotope probing are increasingly common techniques to target the active microorganisms in an environmental sample [29,30,31,32]. Previous research by our group attempted to use metatranscriptomics and metaproteomics to determine if soil CH4 cycling genes and proteins were differentially expressed between land use types but even with an average of 100 million reads per metatranscriptome were unsuccessful due mostly to low counts of group specific mRNA (unpublished data). Similarly, it is challenging to study complex soil microbial communities due to sparse databases for soil proteomics, especially for tropical soil environments, facing additional issues of protein extraction from soil and obtaining sufficient depth to determine differential protein abundance. Therefore, for this study we used stable-isotope probing to determine the active fraction of the soil microbial community cycling CH4, referred to henceforth as members of the active community. Stable-isotope probing is commonly applied to study CH4 cycling in soil given the specific nature of the substrate and its relevance to climate change [33,34,35]. This technique uses the less abundant isotope of an atom, such as 13C-carbon, to label the microorganisms capable of consuming the 13C and, via their anabolic metabolism, incorporating it into their DNA, which then can be separated by ultracentrifugation from the community DNA. Subsequently, next generation sequencing enables the identification of active community members and provides insight into their functional potential.
The central goal of this study was to determine how the members of the active CH4-cycling microbial community, their functions, and CH4-related metabolic pathways changed across land use types (PF, cattle pasture, and secondary rainforest) and geographically distinct regions of the Brazilian Amazon. We hypothesized that the cause of increased soil methane production in cattle pastures was caused by a decrease in active methanotrophy. To test this hypothesis, we sampled sites at some of the most active deforestation frontiers in northeastern and southwestern Amazonia in the states of Pará (in and around Tapajós National Forest) and Rondônia (Fazenda Nova Vida near Ariquemes), respectively. To determine the community composition and functions of the active methane-cycling microorganisms, we coupled stable-isotope probing (DNA-SIP) with metagenomics, using either 13C-labeled methane (CH4), carbon dioxide (CO2), or sodium acetate (NaAOc) as a substrate. Overall, we observed significant shifts in the active microbial community compositions and their methane-cycling functional genes between land-use types, geographic location, and substrates. Specifically, the abundance and diversity of active methanogens increased with conversion to pasture. Therefore, we conclude that an increased abundance and diversity of active methanogens is causing the overall net positive methane flux in cattle pastures.
Methods
Site description and sampling
Intact soil cores (5 cm diameter × 10 cm depth) were collected from the Tapajós National Forest and adjacent areas in the State of Pará in June 2016 for DNA-SIP. Another group of soil cores were collected from Fazenda Nova Vida and adjacent areas in the State of Rondônia in April 2017 for DNA-SIP (geographic map and GPS coordinates in Supplementary Methods). For each location, 18 soil cores were collected from each land use type, two primary rainforests (PF1 or PF2), one cattle pasture (P), and one secondary rainforest (SF). Soil cores were collected along a transect ranging from 100 to 200 m with five equidistant sampling points (for additional detail see Supplementary Methods).
Stable-isotope probing
During incubation with stable isotopes, the intact soil cores (~200 g depending on soil density) were stored in gas-tight glass jars in the dark. Soils were incubated at 25 °C for ~7 months due to the low gas exchange at the surface top of the undisturbed soil column (20 cm2) compared to homogenized soil (20–32 × lower rates; unpublished data). Either 25 mL of 13C-carbon dioxide (3% headspace concentration), 1 mL of 13C-sodium acetate (1 mM final concentration, added to the top of each soil core), or 25 mL of 13C-methane (3% headspace concentration) were added every 2 weeks. Equal volumes (1 mL) of sterile water were added to carbon dioxide and methane incubations. Air was added once a week to the methane incubations to ensure an oxic headspace. Pressure was released periodically prior to substrate injection from all jars. The duration of incubation was determined by monitoring the methane gas flux and attempting to ensure 20 mM of substrate was incorporated, following published recommendations to apply 5–500 µM 13C per g of soil [36]. Our target was to incorporate ~100 µM 13C per g of soil, rendering shorter incubation times insufficient. Methane production or consumption was monitored throughout the incubation experiment by gas chromatography (Shimadzu GC-17A, Kyoto, Japan). After incubation, each soil core was sectioned longitudinally into five 2 cm tall segments (numbered 1–5 from top to bottom) and stored frozen at −20 °C until DNA extraction.
For each combination of location (Pará or Rondônia) and transects across each land use type (two PFs, one pasture, one SF), five soil cores were incubated with 13C-substrate and one additional core with 12C-substrate as the control (see Supplementary Methods). For each set of four sampling sites this resulted in a total of six cores for each of the three substrates, or 72 cores total for each location tested.
DNA extraction, quantification, and sample processing
DNA was extracted from 0.25 g of soil from all segments from two of the five 13C soil cores using the DNeasy PowerSoil DNA Extraction kit (Qiagen, Hilden, Germany) to determine the segment with the highest abundance of methanogens or methanotrophs based on the respective functional marker genes using qPCR as described below (see Supplementary Methods). Upon identifying the segment with the highest genomic abundance of methanogens or methanotrophs, DNA was extracted from 4 g of soil from the identified segment of three 13C soil cores and from the 12C-control for each substrate/sample site combination using the DNeasy PowerMax Soil Kit (Qiagen). DNA was quantified fluorometrically using the Qubit dsDNA Broad-Range assay (Invitrogen, Carlsbad, CA). A total of 5 µg of DNA was subjected to ultracentrifugation according to a previously described protocol [36], followed by fractionation of the density gradient into 12 fractions of equal volume. The continuity of the density gradient was confirmed with a refractometer. DNA was precipitated following the published protocol [36] except for the addition of 20 µg linear acrylamide (Invitrogen) instead of glycogen and each fraction was quantified using fluorometry via the Qubit dsDNA High-Sensitivity assay (Invitrogen). To identify the fractions with 13C-labeled DNA, we quantified the abundance of methanogens or methanotrophs in each fraction using qPCR of the respective functional gene marker for a subset of samples compared to their respective 12C-controls (details in Supplementary Methods). We pooled the 12C (~1–5) and 13C (~6–12) fractions, respectively. Since the GC content of microbial DNA can influence DNA density, we sequenced both the light and heavy DNA fractions from our 12C-controls for a total of 12 12C-light DNA, 12 12C-heavy DNA, and 36 13C-heavy DNA samples per location.
Quantitative PCR
The particulate methane monooxygenase alpha subunit gene (pmoA) was amplified using the primer pair A189f/mb661r [37, 38], and the gene for the methyl coenzyme M reductase alpha subunit (mcrA) was amplified using the primer pair mlas/mcra-rev [39]. Standard reaction mixtures and thermocycler conditions are specified in Supplementary Methods.
Sequencing
All DNA library preparation and sequencing were performed at the University of Oregon Genomics and Cell Characterization Core Facility (Eugene, OR) (see Supplementary Methods). Briefly, the three genes of interest (16S rRNA, pmoA, and mcrA) were amplified using custom dual-indexed PCR primers designed by the core facility. For each location, paired-end 300 bp amplicon sequencing of the pooled heavy fractions for three 13C-samples per sample site/substrate combination and the pooled heavy and light fractions for all 12C-controls was completed on an Illumina MiSeq sequencer (Illumina, San Diego, CA). Additionally, 16S rRNA gene sequences from fresh soil in the field were obtained [40] to assess the impact of incubation on the community. For metagenomes, sequencing of the heavy fraction of two 13C-samples per sampling site/substrate combination and all 12C-controls was performed on an Illumina HiSeq4000 across two flow lanes for each location. All sequences were demultiplexed at the core facility.
Soil physical–chemical analysis
Homogenized soil samples stored at 4 °C were processed as described in detail previously [41].
Data and statistical analysis
Amplicon sequences were processed and analyzed using the DADA2 pipeline in QIIME2 [42, 43]. Metagenome sequences were processed and annotated using MG-RAST [44]. GenBank and SEED Subsystem were used for the organismal and functional annotations, respectively. The SEED Subsystem annotation “Methanogenesis strays” is described as “several additional genes and clusters from methanogens.” The influence of homogeneous dispersion within each sample and heterogeneous dispersion between samples was assessed using the Permdisp and Adonis functions, respectively, from the “vegan” package in R on Bray-Curtis dissimilarity matrices made from the annotation tables [45, 46]. To understand the influence of long-term incubation of total community composition, 16S rRNA amplicon sequences from 12C-control samples, light and heavy DNA fractions, and the fresh soil communities from immediately frozen homogenized samples were compared. Briefly, we processed the sequences using the DADA2 pipeline in QIIME2, combined the counts for each ASV in light and heavy DNA fractions to represent the “total” community, then rarefied to 20,000 annotations per sample, and created a Bray-Curtis dissimilarity matrix using the tools described above. To specifically target the active microbial community, the metagenomic annotations were rarefied (vegan:rrarefy) and counts were normalized to the 12C-control for each substrate (see Supplementary Methods). After rarefication, the dissimilarity between land use types within each substrate were analyzed (vegan:Adonis). STAMP v2.1.3 was then used to identify active microorganisms and functions by comparing each individual 13C-sample to their respective 12C-control using Fisher’s exact test [47]. We searched for known methanogens and methanotrophs. All figures were made in R v3.5.1 using ggplot2 [45, 48]. Soil physical–chemical data were analyzed using ANOVA with a Tukey–Kramer post hoc test. Correlation analyses were completed using a Pearson correlation (cor.test) [45].
Active fraction analysis
In this study, “active” means that the cells were actively growing (anabolically incorporating 13C) and not just metabolically active (catabolic turnover of 13C-substrate independent of growing). The incubations with 13C-labeled substrates determine both actively growing and metabolically active community members, and we used our 12C incubation controls to correct for the metabolically active part. Therefore, an annotation was deemed active, if it was significantly higher (p < 0.05) in the 13C-sample compared to the 12C-control. Samples were normalized to their respective 12C-control with features that had less abundance in the 13C- than 12C-samples being marked as 0 counts. Samples from the same substrate were compared between land use types in STAMP using the multigroup stats function (ANOVA with Tukey–Kramer post hoc test) [49]. All significantly different annotations were checked again to see if they were active in the samples.
Data accessibility
Metagenomes are available publicly on MG-RAST under the following project accession numbers: mgp88468 and mgp86794. All SIP-related raw amplicon sequence files have been deposited on figshare under the following DOI: (https://doi.org/10.6084/m9.figshare.10565552, https://doi.org/10.6084/m9.figshare.10565690, https://doi.org/10.6084/m9.figshare.10565870, https://doi.org/10.6084/m9.figshare.10565897, https://doi.org/10.6084/m9.figshare.10565957, https://doi.org/10.6084/m9.figshare.10565801).
Results and discussion
Active methane-cycling community changes with land use
To understand the active methane-cycling microbial community composition and abundance, we analyzed sequences of both PCR-amplified marker genes (16S rRNA, mcrA, and pmoA) and metagenomes. The amplification-based approach makes our data comparable to many microbial studies that use these biomarkers, but this method comes with the potential issues of primer bias allowing for missed taxonomic groups, lower phylogenetic resolution, and no additional information on ecosystem processes [50, 51]. Therefore, after confirming that enough label was present in the target sample using amplicon-based sequencing, we used metagenomics to gain a deeper understanding of the 13C-labeled methane-cycling community and its supporting members [52,53,54]. The composition of the total soil microbial community, based on 16S rRNA, significantly differed between geographic locations (Rondônia vs. Pará; p = 1e−03, r2 = 0.118), individual land-use types (PF, pasture (P), SF, p = 1e−03, r2 = 0.08), added substrates (CH4, CO2, NaAOc, p = 1e−03, r2 = 0.08), and all interactions of these variables (Fig. 1). When we specifically targeted the active community, we found that only samples incubated with CO2 significantly differed between locations (Pará CO2 p = 3e−03; Rondônia CO2 p = 1.8e−02; Supplementary Figs. 1, 2). It was unsurprising that location is the main driver to differentiate the total microbial community since Rondônia and northwestern Pará are separated by ~1500 km. Also, abiotic factors such as seasonal differences and/or soil physical–chemical properties could be driving these locational differences [55,56,57]. The significant differences in CO2 incubated samples may be due to the similarity of the overall active community across samples, while the methane-consuming or -producing community makes up only a small fraction of that community and the signal is lost when we look at the community composition broadly.

Nonmetric dimensional scaling plot of rarefied 16S rRNA SILVA annotations at genus-level from all samples. The dotted lines outline samples from each substrate incubation (NaAOc = sodium acetate, CO2 = carbon dioxide, and CH4 = methane). The point shapes are based on the substrate incubation with squares = carbon dioxide, circles = methane, and triangles = sodium acetate. The point colors are based on what land use type the sample came from with primary rainforest = green, pasture = orange, and secondary rainforest = blue. The colored lines connect samples from the same geographic location to the centroid (Rondônia = black, Pará = gray). The p values and r2 values for each variable (location, substrate, and land use) and their interactions are derived from the Adonis function in the vegan package.
When we investigated the richness of active methane-cycling communities, we found that pasture samples had the highest active methanogen richness in metagenomes from both locations and regardless of substrate (CO2 or NaAOc); however, it was only significant in Rondônia NaAOc samples (P vs. PF p = 9.6e−03, P vs. SF p = 7.9e−03; Fig. 2). All active methanogens that significantly changed abundance between land-use types were associated with pasture soils in both locations (Table 1). Specifically, Methanosarcina spp. dominated the active methanogens for most samples in both locations regardless of substrate (Fig. 3, Supplementary Tables 1–4). These archaeal species are known to have multiple methanogenesis pathways making them capable of utilizing both 13CO2 and 13NaAOc, likely explaining their dominance in both locations and substrates [58,59,60,61]. In a non-SIP study of these same soils, Methanosarcina spp. contributed significantly to methane flux across land-use types and locations indicating these methanogens are not an artifact of incubation [40].

Significance values (p values) were calculated from an ANOVA with Tukey Honestly Significant Difference Test comparing the richness of active methane-cycling taxa.

All abundances are post-rarefaction and normalized to the respective 12C-control. a The abundance of active methanogens in soils from Pará incubated with 13CO2, (b) the abundance of active methanogens in soils from Rondônia incubated with 13CO2, (c) the abundance of active methanogens in soils from Pará incubated with 13NaAOc, and (d) the abundance of active methanogens in soils from Rondônia incubated with 13NaAOc. Samples on the x-axis are colored by land use type (primary rainforest (PF) = green, pasture (P) = orange, and secondary rainforest (SF) = blue).
We observed a significantly higher abundance of total active methanogens in Rondônia pasture soils compared to both primary and SF samples in 13NaAOc samples (p = 1e−03, p = 3.8e−02, respectively) and compared to SF in 13CO2 samples (p = 9e−03). A similar trend was observed in non-SIP soils from the same locations [40]. There was no significant difference in the abundance of total active methanogens between land-use types for either methanogenic substrate in Pará, but many taxa did significantly change abundance (Table 1). Previous research studies showed mixed findings on methanogen communities’ response to tropical land-use change ranging from no change to increased mcrA gene abundance in pastures [6, 21, 62]. By targeting the active community, we directly show that pasture soils have a higher richness of active methanogens and specific methanogenic taxa significantly increase abundance. This increase in methanogen abundance and richness is likely due to the increased soil carbon cycling occurring in pasture soils [63, 64].
Previous research into methanotrophy across Amazonian land uses found methanotroph abundance to be lower in pasture relative to primary forest soils [6, 21, 62]. Based on these studies, we hypothesized that pasture soils would have the lowest abundance and richness of active methanotrophs. Unlike the active methanogen community, we did not find a consistent association between active methanotroph richness and land-use types across locations. The highest richness was either found in pasture or SF for Pará and Rondônia, respectively, but it was not significant (Fig. 2). The total active methanotroph abundance did not significantly change between land-use types. In both locations and all land-use types, Type II methanotrophs (Alphaproteobacteria) dominated the active methanotroph community (Fig. 4, Supplementary Tables 5, 6). Only one PF sample from Pará was dominated by Type I methanotrophs and only one Type III methanotroph genus was found to be active, Methylacidiphilum, but remained rare (Supplementary Fig. 3; Supplementary Tables 5, 6).

The relative abundance of active methanotrophs was determined by first rarefying metagenome sample abundances, then normalizing taxa counts to the respective 12C-control, and lastly dividing the normalized abundance of methanotrophs annotations of each type (I, II, or III) by the total methanotroph annotations. Type I = gray, Type II = blue, Type III = pink.
Although, the total abundance of active methanotrophs did not significantly change between land-use types, the abundance of specific methanotrophs changed in Pará and Rondônia associating with pasture and SF, respectively (Table 1). This was surprising and not what we hypothesized based on previous studies [6, 21, 62]. Several factors should be considered to address this discrepancy. First, our study targeted the microorganisms actively consuming CH4 rather than looking at the total microbial community. Studies of the total microbial community can be influenced by the potential presence of extracellular DNA, which may affect estimates of abundance and diversity [65,66,67]. Additionally, we incubated our samples at CH4 concentrations 16 times greater than those in the atmosphere due to the inability to label the community at low concentrations. Although necessary for the technique, this could influence the composition and activity of the CH4-consuming community. Furthermore, there is a possibility that we incorrectly hypothesized PFs would have the highest methanotroph richness and abundance since these forests are known to be methane sinks [24, 25]. Based on our findings, we hypothesize that active methanotroph abundances do not decrease in pastures. Future research must focus on identifying how environmental variables influence the active methane-cycling community in environmentally-relevant conditions.
Dominant active methanogenesis pathways differed between locations
We next asked which CH4-related metabolic pathways were active across land-use types and how they changed in response to deforestation. We observed active methanotrophy based on the abundance of the genes for particulate methane monooxygenase (pmmo) and soluble methane monooxygenase (smmo) in all 13C-labeled samples and in both locations (Fig. 5; Supplementary Table 7). The pmmo genes were abundant and active in 94% of samples while smmo was active in most SF samples and Rondônia-PF1 (Fig. 5; Supplementary Table 7). The SF and pasture samples at Rondônia significantly increased in pmmo abundance (p = 6e−03; p = 3e−02, respectively) compared to PF (Supplementary Table 7). We found no significant difference in the abundance of any active methanotrophy-related genes across land-use types in Pará (Fig. 5). Overall, Rondônia had a significantly higher relative abundance of pmmo to total methanotrophy annotations compared to Pará (Supplementary Table 7) (p = 1e−04). Soil physical–chemical properties are known to influence the activity of these different methane monooxygenases [68]. Copper is a key component regulating the activity and abundance of these methane monooxygenases having a positive relationship with pmmo abundance [12, 69, 70]. We observed a significantly higher concentration of copper (9×) in Rondônia compared to Pará (p = 7.42E−06) which may explain the increased abundance of active pmmo genes.

The scale is from lowest relative abundance (blue) to highest relative abundance (red) of the genes and is normalized to each respective gene (i.e., column). The metagenome samples are on the y-axis and are colored by land use type (primary rainforest (PF) = green, pasture (P) = orange, and secondary rainforest (SF) = blue) and have the location (Rondônia or Pará) in the label. The metabolic pathways potentially involved in methanotrophy are listed on the x-axis. The dendrogram shows the Euclidean distance between samples.
Regardless of location, the abundance of active methanogenesis genes dominated in pasture compared to other land-use types. Interestingly, in Pará we observed these significant increases in the 13CO2 incubation, while in Rondônia the 13NaAOc incubation accounted for the increased abundance (Fig. 6; Supplementary Table 8). The Pará 13NaAOc incubation presented some significant changes in methanogenesis-related genes including Coenzyme F420 synthesis (p = 8e−05), methanopterin biosynthesis2 (p = 2e−03), and methanogenesis strays (p = 1e−03). The two pasture samples in Pará 13NaAOc incubations performed very differently. Although pastures are considered to be more biotically homogeneous [71], these two samples differed strongly with one sample having about 8.5× more active methanogens (Supplementary Table 3). When the relative abundance of active methanogenesis genes to total annotations was investigated, we identified a significant difference between land-use types in the Pará 13CO2 incubations (PF vs. P p = 1e−03, SF vs. P p = 1.8e−02) and in the Rondônia 13NaAOc incubations (PF vs. P p = 7e−02, SF vs. P p = 1.8e−02) (Supplementary Table 9). In addition to methanogenesis genes changing between land-use types, we observed an increase in carbon cycling activity in Pará pasture soils incubated with 13CO2 (Glycolysis and gluconeogenesis p = 1e−03, Pentose phosphate pathway p = 2e−03, Entner–Doudoroff pathway p = 3e−02). Overall, we found that active methanogenesis was driven by methanogens using the hydrogenotrophic pathway in Pará and the acetoclastic pathway in Rondônia (Supplementary Table 8). This shift in the dominant methanogenesis pathway between locations may be due to differences in the physical–chemical soil parameters or a result of the types of fermentation leading to either more acetate or hydrogen production. Interestingly, the active methanogen community was dominated by Methanosarcina spp. in both locations. Members of the genus Methanosarcina are known to require three different types of hydrogenases for the reduction of CO2 to CH4 with electrons derived from H2 [61]. The significantly increased activity of multiple types of hydrogenases (Energy conserving hydrogenase ferrodixin Ech p = 1.6E−08; membrane bound hydrogenases p = 4.6e−02; Archaeal membrane bound hydrogenases p = 0.048; Coenzyme F420 hydrogenase p = 5e−02) in soils from Pará compared to soils from Rondônia indicates a selection for the hydrogenotrophic pathway. This selection is supported by the increased availability of trace metals (iron) in soils from Pará which are needed by methanogenic hydrogenases [61].

The samples are grouped by location in descending order and include both methanogenic substrates (CO2 and NaAOc). The colors correspond to the land use type (green = primary rainforest, orange = pasture, and blue = secondary rainforest). The shapes of the dots correspond to substrate (circle = CO2, triangle = NaAOc). Active methanogenesis genes includes SEED subsystem annotations as “Methanogenesis,” “Methanogenesis from methylated compounds,” and “Methanogenesis strays,” Methanogenesis strays are “additional genes and clusters from methanogens”. The specific genes associated with “Methanogenesis strays” can be found by searching for the subsystem on the SEED viewer (http://rast.theseed.org/FIG/seedviewer.cgi?page=SubsystemSelect).
Land-use change alters key redox-cycling active taxa
In the context of highly complex soil microbial communities, methanogens need other microorganisms to produce the substrates necessary for this redox reaction to occur. Methanogenesis is one of the least thermodynamically favorable anaerobic reactions; therefore, other redox reactions must transpire prior to methanogenesis [72]. Although we were targeting active methane-cycling microorganisms in this study, the methanogenic substrates used, 13CO2 and 13NaAOc, are not exclusively used by methanogens. Therefore, we investigated which coexisting microorganisms were actively consuming these substrates and thereby interacting with methanogens. Many non-methanogenic but active microbial taxa changed significantly in abundance between land use types in both geographic locations. In the Pará 13CO2 SIP incubations, we observed a significant increase of active Syntrophus aciditrophicus (p = 1E−06) in pasture along with many known sulfate-reducing bacteria (Supplementary Table 9). Syntrophus aciditrophicus is known to promote the growth of Methanospirillum spp., which accounted for 2.76% of the active methanogen community in Pará 13CO2 pasture samples [73]. In the SF, we found a significantly higher abundance of various active nitrifying and sulfur-oxidizing bacteria, such as Nitrobacter and Thioalkalivibrio. Many of these microbial groups are known to utilize CO2 and have thermodynamically preferred redox potentials [74,75,76,77,78,79]. Homoacetogenic taxa, which could potentially compete with methanogens for hydrogen to reduce CO2 belonged to the genus Clostridium and associated with pasture and PF in the 13CO2 incubations (Supplementary Table 9). Rondônia pastures increased in active ammonia-oxidizing microorganisms including Nitrosococcus and Geobacillus species (13CO2 samples; Supplementary Table 9). One potential cause of increased Geobacillus species is the slash and burn process used to create pastures that deposits hydrocarbons in the soil, which these microorganisms are known to use [80,81,82].
The abundance of active Geobacillus, Clostridium, and Sulfolobus spp. increased in Pará 13NaAOc incubated pasture soils (Supplementary Table 10). Some Geobacillus and Clostridium spp. are known to utilize acetate, which may explain their increased abundance in the 13C-labeled community [83, 84]. The denitrifying bacterium Hyphomicrobium denitrificans was active and significantly increased abundance in Pará PF samples along with the genes associated with denitrification (p = 0.02). In the 13NaAOc Rondônia soils, we observed a significant increase in both sulfate-reducing and sulfur-oxidizing microorganisms along with nitrate reducers in SF with many competitors for acetate as a carbon source [77, 85] (Supplementary Table 10). It is well documented that before methanogenesis is able to occur nitrate and sulfate must be depleted as electron acceptors [72]. The increased abundance of active sulfate and nitrate reducers in the Rondônia SF and overall lack of active methanogenesis indicates that these more favorable electron acceptors were still available in the soil during incubation with 13NaAOc inhibiting methanogenesis through substrate competition [86, 87].
Soil physical–chemical parameters increase potential methane production
Land-use change is one of the strongest drivers to alter soil ecosystems. Parallel changes to the soil physical–chemical parameters, physical structure, and aboveground vegetation may provide additional support for increased methanogenesis in pasture soils. Specifically, the compaction caused by cattle grazing creates more anoxic microsites providing more opportunity for methanogenesis to occur [88]. The comparison of soil physical–chemical parameters between the geographic locations presented several significant differences (Supplementary Tables 11, 12). Of note were increased concentrations of sulfur (p = 2.95E−15) and copper (p = 7.42E−06) along with higher pH (p = 1.35E−07) in Rondônia compared to Pará, and total soil acidity (p = 9.29E−11) and total nitrogen (p = 2.31E−06) were significantly higher in Pará soils. For both locations, the soil pH was significantly higher in pasture compared to PFs. Soil bulk density was found to be highest in pasture from both locations (Supplementary Fig. 4). The increased pH in pasture soils likely helps support methanogenesis since optimum process activity is at near neutral pH and quickly decreases as the pH becomes more acidic [89]. Another contributing factor to the increased methanogenesis in pasture soils is due to Urochloa brizantha (formely Brachiaria bizantha) excreting large amounts of carbon as root exudates into the soil [90]. With increased carbon availability in pasture soils, there is overall increased soil microbial activity [91]. All of these changes to the soil in pastures could contribute to the increased methanogenic activity observed in our SIP study. The gas flux trends established for our field sites prior to taking the soil cores are summarized elsewhere [40].
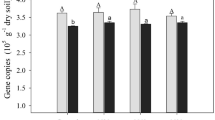
Minimal enrichment of methane-cyclers after incubation
In SIP studies specifically, we target the active microbial groups involved in a biogeochemical process; however, the majority of soil SIP studies use homogenized soil where soil columns get sieved [92,93,94,95,96]. It is clear from the literature that soil structure is an important aspect of microbial activity and carbon cycling; therefore, if possible, microbial activity should be studied under environmentally-relevant conditions. This study shows the feasibility of keeping soil and its assembled microbial communities more similar to the natural environment by incubating soil cores intact. We observed that even after 7 months of incubation, the abundance of functional marker genes (pmoA and mcrA) did not become greatly enriched. Compared to field soils at the time of sampling, there was a small but significant increase of mcrA gene copies in Pará 13CO2 SIP soil (p = 4.6e−03), but no significant difference in soils from Para amended with 13NaAOc (p = 7.2e−01) (Supplementary Fig. 5). Overall, there was no significant difference between the Rondônia SIP and field soils’ mcrA gene abundance. The only significant difference found was between 13CO2-incubated primary forest and pasture samples (p = 4.6e−02). Interestingly, pmoA gene abundance decreased significantly in SIP incubated soils from Pará (p = 1E−05) and Rondônia (p = 4E−07, Supplementary Fig. 6). One possible explanation for the decreased pmoA gene abundance between SIP incubated and field soil is that during the incubation the methanotrophic community was potentially altered. Our comparative analysis of the metagenome data supports this possibility as a 7.7-fold and 4.0-fold increase from Rondônia and Pará, respectively, were observed in obligate methanotroph abundance between 13C vs. 12C-heavy fraction samples. Since primer bias is a common problem, as previously discussed, the change in community could alter the compatibility of the primer to the pmoA sequences of the changed community; thus, potentially presenting a lower pmoA abundance in the SIP than field soils. To further assess any enrichment or change in the microbial community during incubation, we directly compared the original fresh soil that was homogenized and frozen upon collection to the 12C-controls using 16S rRNA amplicon sequences. Overall, we observed that the 7-month substrate incubation did affect the total microbial community (r2 = 0.120, p = 0.001), but less so than location (r2 = 0.143, p = 0.001) and equal to land use (r2 = 0.123, p = 0.001) (Supplementary Table 13; Supplementary Fig. 7). Although we observed a significant effect of substrate incubation on community composition, this difference could also be due to (1) our samples were collected from a single 2 cm section of a 10 cm soil core while original soil samples were immediately homogenized [97, 98] and/or (2) there is large variation among technical replicates from 16S rRNA amplicon sequencing that creates substantial concerns about using a single datapoint (12C-controls) to ascertain dissimilarity of a complex community [99, 100]. Furthermore, the richness observed in our 7-month incubated 12C-controls is similar to that in freshly sampled soils, indicating limited bottleneck effects on the total community composition [40].
Overall, we found that abundances of active methanogen species increased in all pasture samples compared to primary and SF samples, while abundances of active methanotroph species varied by location. Similarly, the abundance of genes representing metabolic pathways associated with methanogenesis increased in all pasture samples compared to other land-use types. Metabolic pathways associated with methanotrophy did not significantly change between land-use types in Pará, and increased in Rondônia in both pasture and SF samples compared to PFs. Although there is potential that these shifts in activity and in abundances of methane-cycling taxa are due to the DNA-SIP incubation process, multiple lines of evidence support that these results are not artifacts of incubation. We determined that the functional biomarker genes mcrA and pmoA did not become greatly enriched during incubation, the methane-cycling taxa associated with field methane fluxes overlap with those taxa we find to be active [40], and the phyla determined dominant in previous studies for these sites changed <10% in our 12C-control samples (original soils ~95% vs. 12C-SIP incubated soils ~85%) [64].
Conclusions
Land-use change from rainforest to pasture stimulates the soil methanogenic community in the Brazilian Amazon. Using undisturbed soil columns for SIP incubations, we were able to ascertain that methanogen abundance and activity is significantly higher in pastures compared to both primary and SFs which could drive methane emissions from the soil of Brazilian cattle pastures. Future studies should focus on identifying what specific environmental factors are responsible for increased methanogenesis in pasture soils (i.e., pH, vegetation, compaction, nutrient inputs from livestock, carbon or trace element availability, etc.), so that land management can better mitigate CH4 emissions. Another important finding was that SFs in both locations exhibited active methanotrophy and decreased methanogenesis similar to levels observed in primary forests, suggesting they have recovered as methane sinks. Through large forest restoration efforts occurring in the tropics, there is potential to see these forests recover with enough time to overcome excess CH4 production. It is currently unknown how long SFs take to recover as a CH4 sink, and how widespread this recovery is geographically. Adoption of best management practices in pastures can compensate for a small fraction of the impact of deforestation on net emission of greenhouse gases and the loss of carbon from Amazonia. With the currently accelerating expansion of land-use change in Amazonia understanding which players might assist mitigation of concomitant greenhouse gas production is increasingly important for all agricultural management [101].
References
IPCC. Climate change 2013: the Physical Science Basis. Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change. In: Stocker TF, Qin D, Plattner G-K, Tignor M, Allen SK, Boschung J, et al. editors. Cambridge, United Kingdom and New York, USA: Cambridge University Press; 2013. p. 1535.
De Azevedo TR, Junior CC, Junior AB, dos Santos Cremer M, Piatto M, Tsai DS, et al. SEEG initiative estimates of Brazilian greenhouse gas emissions from 1970 to 2015. Sci Data. 2018;5:180045.
West TAP, Börner J, Fearnside PM. Climatic benefits from the 2006-2017 avoided deforestation in Amazonian Brazil. Front For Glob Change. 2019;2:52.
Malhi Y, Roberts JT, Betts RA, Killeen TJ, Li W, Nobre CA. Climate change, deforestation, and the fate of the Amazon. Science. 2008;319:169–73.
Curtis PG, Slay CM, Harris NL, Tyukavina A, Hansen MC. Classifying drivers of global forest loss. Science. 2018;361:1108–11.
Paula FS, Rodrigues JLM, Zhou J, Wu L, Mueller RC, Mirza BS, et al. Land use change alters functional gene diversity, composition and abundance in Amazon forest soil microbial communities. Mol Ecol. 2014. https://doi.org/10.1111/mec.12786.
Evans PN, Boyd JA, Leu AO, Woodcroft BJ, Parks DH, Hugenholtz P, et al. An evolving view of methane metabolism in the Archaea. Nat Rev Microbiol. 2019;17:219–32.
Serrano-Silva N, Sarria-Guzmán Y, Dendooven L, Luna-Guido M. Methanogenesis and methanotrophy in soil: a review. Pedosphere. 2014;24:291–307.
Zinder SH. Physiological ecology of methanogens. In: Ferry JG, editor. Methanogenesis: ecology, physiology, biochemistry and genetics. New York, NY, USA: Chapman & Hall; 1993. p. 128–206.
Schink B, Stams AJM. Syntrophism among prokaryotes. In: Rosenberg E, DeLong F, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes: prokaryotic communities and ecophysiology. Berlin: Springer; 2013. p. 471–93.
Costa KC, Leigh JA. Metabolic versatility in methanogens. Curr Opin Biotechnol. 2014;29:70–5.
Semrau JD, DiSpirito AA, Yoon S. Methanotrophs and copper. FEMS Microbiol Rev. 2010;34:496–531.
van Teeseling MCF, Pol A, Harhangi HR, van der Zwart S, Jetten MSM, Op den Camp HJM, et al. Expanding the verrucomicrobial methanotrophic world: description of three novel species of Methylacidimicrobium gen. nov. Appl Environ Microbiol. 2014;80:6782–92.
Zheng Y, Hou L, Chen F, Zhou J, Liu M, Yin G, et al. Denitrifying anaerobic methane oxidation in intertidal marsh soils: Occurrence and environmental significance. Geoderma. 2020;357:113943.
Zheng Y, Wang H, Liu Y, Zhu B, Li J, Yang Y, et al. Methane-dependent mineral reduction by aerobic methanotrophs under hypoxia. Environ Sci Technol Lett. 2020;7:606–12.
Murrell JC, Dalton H. Nitrogen fixation in obligate methanotrophs. J Gen Microbiol. 1983;129:3481–6.
Graham DW, Chaudhary JA, Hanson RS, Arnold RG. Factors affecting competition between type I and type II methanotrophs in two-organism, continuous-flow reactors. Microb Ecol. 1993;25:1–17.
Amaral JA, Knowles R. Growth of methanotrophs in methane and oxygen counter gradients. FEMS Microbiol Lett. 1995;126:215–20.
Dunfield PF, Liesack W, Henckel T, Knowles R, Conrad R. High-affinity methane oxidation by a soil enrichment culture containing a type II methanotroph. Appl Environ Microbiol. 1999;65:1009–14.
Bull ID, Parekh NR, Hall GH, Ineson P, Evershed RP. Detection and classification of atmospheric methane oxidizing bacteria in soil. Nature. 2000;405:175–8.
Meyer KM, Klein AM, Rodrigues JLM, Nüsslein K, Tringe SG, Mirza BS, et al. Conversion of Amazon rainforest to agriculture alters community traits of methane-cycling organisms. Mol Ecol. 2017;26:1547–56.
Baani M, Liesack W. Two isoenzymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc Natl Acad Sci USA. 2008;105:10203–8.
Sharp CE, Smirnova AV, Graham JM, Stott MB, Khadka R, Moore TR, et al. Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ Microbiol. 2014;16:1867–78.
Keller M, Goreau TJ, Wofsy SC, Kaplan WA, McElroy MB. Production of nitrous oxide and consumption of methane by forest soils. Geophys Res Lett. 1986;10:1156–9.
Steudler PA, Melillo JM, Feigl BJ, Neill C, Piccolo MC, Cerri CC. Consequence of forest-to-pasture conversion on CH4 fluxes in the Brazilian Amazon Basin. J Geophys Res Atmos. 1996;101:18547–54.
Fearnside PM. Greenhouse gases from deforestation in Brazilian Amazonia: net committed emissions. Climatic Change. 1997;35:321–60.
Fernandes SAP, Bernoux M, Cerri CC, Feigl BJ, Piccolo MC. Seasonal variation of soil chemical properties and CO2 and CH4 fluxes in unfertilized and P-fertilized pastures in an Ultisol of the Brazilian Amazon. Geoderma. 2002;107:227–41.
Wick B, Veldkamp E, de Mello WZ, Keller M, Crill P. Nitrous oxide fluxes and nitrogen cycling along a pasture chronosequence in Central Amazonia, Brazil. Biogeosciences. 2005;2:175–87.
Tveit AT, Urich T, Svenning MM. Metatranscriptomic analysis of active peat soil microbiome. Appl Environ Microbiol. 2014;80:5761–72.
Singer E, Wagner M, Woyke T. Capturing the genetic makeup of the active microbiome in situ. ISME J. 2017;11:1949–63.
Keiblinger KM, Wilhartitz IC, Schneider T, Roschitzki B, Schmid E, Eberl L, et al. Soil metaproteomics—comparative evaluation of protein extraction protocols. Soil Biol Biochem. 2012;54:14–24.
Kroeger ME, Nüsslein K. Stable isotope probing—detection of active microbes in soil. In: Elsas JDV, editor. Modern soil microbiology, 3rd ed. Boca Raton: CRC Press; 2019.
Esson K, Lin X, Kumaresan D, Chanton JP, Murrell JC, Kostka JE. Alpha- and gammaproteobacterial methanotrophs codominate the active methane-oxidizing communities in an acidic boreal peat bog. Appl Environ Microbiol. 2016;82:2363–71.
Dumont MG, Radajewski SM, Miguez CB, McDonald IR, Murrell JC. Identification of a complete methane monooxygenase operon from soil by combining stable isotope probing and metagenomic analysis. Environ Microbiol. 2006;8:1240–50.
Lu Y, Leuders T, Friedrich MW, Conrad R. Detecting active methanogenic populations on rice roots using stable isotope probing. Environ Microbiol. 2005;7:326–36.
Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW, et al. DNA stable-isotope probing. Nat Protoc. 2007;2:860–6.
Holmes AJ, Costello A, Lidstrom ME, Murrell JC. Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol Lett. 1995;132:203–8.
Costello A, Lidstrom ME. Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl Environ Microbiol 1999;65:5066–74.
Steinberg LM, Regan JM. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl Environ Microbiol. 2008;74:6663–71.
Meyer KM, Morris AH, Webster K, Klein AM, Kroeger ME, Meredith LK, et al. Belowground changes to community structure alter methane-cycling dynamics in Amazonia. 2020. https://www.biorxiv.org/content/10.1101/2020.03.10.984807v1. in review.
Navarrete AA, Tsai SM, Mendes LW, Faust K, de Hollander M, Cassman NA, et al. Soil microbiome responses to the short-term effects of Amazonian deforestation. Mol Ecol. 2015;24:2433–48.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
Keegan KP, Glass EM, Meyer F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol Biol. 2016;1399:207–33.
R Core Team. R: a language and environment for statistical computing. Vienna Austria: R Foundation for Statistical Computing; 2018. https://www.r-project.org/.
Oksanen JF, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: community ecology package. R package version. 2018;2:4–6. https://CRAN.R-project.org/package=vegan.
Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–4.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Spring-Verlag; 2016.
Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–4.
Pinto AJ, Raskin L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE. 2012;7:e43093.
Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–98.
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to sequencing and analysis. Nat Biotechnol. 2017;35:833–44.
Coyotzi S, Pratscher J, Murrell JC, Neufeld JD. Targeted metagenomics of active microbial populations with stable-isotope probing. Curr Opin Biotechnol. 2016;41:1–8.
Eyice Ö, Namura M, Chen Y, Mead A, Samavedam S, Schäfer H. SIP metagenomics identifies uncultivated Methylophilaceae as dimethylsulphide degrading bacteria in soil and lake sediment. Int Soc Microb Ecol J. 2015;9:2336.
Cardoso D, Sarkinen T, Alexander S, Amorim AM, Bittrich V, Celis M, et al. Amazon plant diversity revealed by a taxonomically verified species list. Proc Natl Acad Sci USA. 2017;114:10695–700.
Sgouridis F, Ullah S. Soil greenhouse gas fluxes, environmental controls, and the partitioning of N2O sources in UK natural and seminatural land use types. J Geophys Res Biogeosciences. 2017. https://doi.org/10.1002/2017JG003783.
Wanyama I, Pelster DE, Butterbach-Bahl K, Verchot LV, Martius C, Rufino MC. Soil carbon dioxide and methane fluxes from forests and other land use types in an African tropical montane region. Biogeochemistry. 2019;143:171–90.
Lackner N, Hintersonnleitner A, Wagner AO, Illmer P. Hydrogenotrophic methanogenesis and autotrophic growth of Methanosarcina thermophila. Archaea. 2018;2018:4712608.
Ferry JG. Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Curr Opin Biotechnol. 2011;22:351–7.
Welander PV, Metcalf WW. Loss of the mtr operon in Methanosarcina blocks growth on methanol, but not methanogenesis, and reveals an unknown methanogenic pathway. Proc Natl Acad Sci USA. 2005;102:10664–9.
Mand TD, Metcalf WW. Energy conservation and hydrogenase function in methanogenic archaea, in particular the genus Methanosarcina. Microbiol Mol Biol Rev. 2019;83:e00020–19.
Lammel DR, Feigl BJ, Cerri CC, Nusslein K. Specific microbial gene abundances and soil parameters contribute to C, N, and greenhouse gas process rates after land use change in Southern Amazonian Soils. Front Microbiol. 2015;6:1057.
Kroeger ME, Delmont TO, Meyer KM, Guo J, Khan K, Rodrigues JLM, et al. New biological insights into how deforestation in Amazonia affects soil microbial communities using metagenomics and metagenome-assembled genomes. Front Microbiol. 2018;9:1635.
Pedrinho A, Mendes LW, Merloti LF, de Cassia da Fonseca M, de Souza Cannavan F, Tsai SM. Forest-to-pasture conversion and recovery based on assessment of microbial communities in Eastern Amazon rainforest. FEMS Microbiol Ecol. 2019;95:fiy236.
Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland MS, Fierer N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol. 2017;2:16242.
Lennon JT, Muscarella ME, Placella SA, Lehmkuhl BK. How, when, and where relic DNA affects microbial diversity. MBio. 2018;9:e00637–18.
Sirois SH, Buckley DH. Factors governing extracellular DNA degradation dynamics in soil. Environ Microbiol Rep. 2019;11:173–84.
Mancinelli RL. The regulation of methane oxidation in soil. Annu Rev Microbiol. 1995;49:581–605.
Guggenheim C, Brand A, Bürgmann H, Sigg L, Wehrli B. Aerobic methane oxidation under copper scarcity in a stratified lake. Sci Rep. 2019;9:4817.
Knapp CW, Fowle DA, Kulczycki E, Roberts JA, Graham DW. Methane monooxygenase gene expression mediated by methanobactin in the presence of mineral copper sources. Proc Natl Acad Sci USA. 2007;104:12040–5.
Rodrigues JLM, Pellizari VH, Mueller R, Baek K, da C Jesus E, Paula FS, et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc Natl Acad Sci USA. 2013;110:988–93.
Hoehler T, Gunsalus RP, McInerney MJ. Environmental Constraints that Limit Methanogenesis. In Timmis KN, editor. Handbook of Hydrocarbon and Lipid Microbiology. Berlin, Heidelberg: Springer; 2010. p. 635–54.
Elshahed MS, Bhupathiraju VK, Wofford NQ, Nanny MA, McInerney MJ. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl Environ Microbiol. 2001;67:1728–38.
Zehnder AJB, Stumm W. Geochemistry and biogeochemistry of anaerobic habitats. In Zehnder AJB editor. Biology of Anaerobic Microorganisms. New York, NY, USA: John Wiley & Sons; 1988. p. 1–38.
Wolfe RS. My kind of biology. Annu Rev Microbiol. 1991;45:1–35.
Strittmatter AW, Liesegang H, Rabus R, Decker I, Amann J, Andres S, et al. Genome sequence of Desulfobacterium autotrophicum HRM2, a marine sulfate reducer oxidizing organic carbon completely to carbon dioxide. Environ Microbiol. 2009;11:1038–55.
Badziong W, Ditter B, Thauer RK. Acetate and carbon dioxide assimilation by Desulfovibrio vulgaris (Marburg), growing on hydrogen and sulfate as sole energy source. Arch Microbiol. 1979;123:301–5.
Watson SW, Waterbury JB. Characteristics of two marine nitrite oxidizing bacteria, Nitrospina gracilis nov. gen. nov. sp. and Nitrococcus mobilis nov. gen. nov. sp. Arch Microbiol. 1971;77:203–30.
Sorokin DY, Tourova TP, Lysenko AM, Mityushina LL, Kuenen JG. Thioalkalivibrio thiocyanoxidans sp. nov. and Thioalkalivibrio paradoxus sp. nov., novel alkaliphilic, obligately autotrophic, sulfur-oxidizing bacteria capable growth on thiocyanate, from soda lakes. IJSEM. 2002;52:657–64.
Kukla J, Whitfeld T, Cajthaml T, Baldrian P, Veselá-Šimáčková H, Novotný V, et al. The effect of traditional slash-and-burn agriculture on soil organic matter, nutrient content, and microbiota in tropical ecosystems of Papua New Guinea. Land Degrad Dev. 2018;30:166–77.
Banat IM, Marchant R. Geobacillus activities in soil and oil contamination remediation. In: Logan NA, De Vos P, editors. Endospore-forming soil bacteria, soil biology 27, Berlin Heidelberg: Springer-Verlag; 2011. p. 259–70.
Durrer A, Margenot AJ, Silva LCR, Bohannan BJM, Nüsslein K, van Haren J, et al. Beyond total carbon: long-term effect of deforestation on Amazonian soils. Biogeochemistry BIOG-D-19-00139. in review.
Nazina TN, Tourova TP, Poltaraus AB, Novikova EV, Grigoryan AA, Ivanova AE, et al. Taxonomic study of aerobic thermophilic bacilli: descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G.th. IJSEM. 2001;51:433–46.
Schnürer A, Schink B, Svensson BH. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. IJSEM. 1996;46:1145–52.
Myhr S, Torsvik T. Denitrovibrio acetiphilus, a novel genus and species of dissimilatory nitrate-reducing bacterium isolated from an oil reservoir model column. IJSEM. 2000;50:1611–9.
Yao H, Conrad R, Wassmann R, Neue HU. Effect of soil characteristics on sequential reduction and methane production in sixteen rice paddy soils from Chine, the Philippines, and Italy. Biogeochemistry. 1999;47:269–95.
Achtnich C, Bak F, Conrad R. Competition for electron donors among nitrate reducers, ferric iron reducers, sulfate reducers, and methanogens in anoxic paddy soil. Biol Fertil Soils. 1995;19:65–72.
Verchot LV, Davidson EA, Cattânio JH, Ackerman IL. Land-use change and biogeochemical controls of methane fluxes in soils of Eastern Amazonia. Ecosystems. 2000;3:41–56.
Wagner R, Zona D, Oechel W, Lipson D. Microbial community structure and soil pH correspond to methane production in Arctic Alaska soils. Environ Microbiol. 2017;19:3398–410.
Feigl BJ, Melillo J, Cerri CC. Changes in the origin and quality of soil organic matter after pasture introduction in Rôndonia (Brazil). Plant Soil. 1995;175:21–9.
Salimon CI, Davidson EA, Victoria RL, Melo AWF. CO2 flux from soil in pastures and forests in southwestern Amazonia. Glob Change Biol. 2004;10:833–43.
Wilhelm RC, Singh R, Eltis LD, Mohn WW. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 2019;13:413–8.
Pepe-Ranney C, Campbell AN, Koechli CN, Berthrong S, Buckley DH. Unearthing the ecology of soil microorganisms using a high-resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front Microbiol. 2016;7:703.
Murase J, Hordijk K, Tayasu I, Bodelier PLE. Strain-specific incorporation of methanotrophic biomass into eukaryotic grazers in a rice field soil revealed by PLFA-SIP. FEMS Microbiol Ecol. 2011;75:284–90.
Radajewski S, Ineson P, Parekh NR, Murrell JC. Stable-isotope probing as a tool in microbial ecology. Nature. 2000;403:646–9.
Cébron A, Bodrossy L, Chen Y, Singer AC, Thompson IP, Prosser JI, et al. Identity of active methanotrophs in landfill cover soil as revealed by DNA-stable isotope probing. FEMS Microbiol Ecol. 2007;62:12–23.
Mathew RP, Feng Y, Githinji L, Ankumah R, Balkcom KS. Impact of no-tillage and conventional tillage systems on soil microbial communities. Appl Environ Soil Sci. 2012;2012:548620.
Eilers KG, Debenport S, Anderson S, Fierer N. Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem. 2012;50:58–65.
Zhou J, Wu L, Deng Y, Zhi X, Jiang Y-H, Tu Q, et al. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011;5:1303–13.
Wen C, Wu L, Qin Y, Van Nostrand JD, Ning D, Sun B, et al. Evaluation of the reproducibility of amplicon sequencing with Illumina MiSeq platform. PLoS ONE. 2017. https://doi.org/10.1371/journal.pone.0176716.
Berenguer E, Gardner TA, Ferreira J, Aragao LE, Camargo PB, Cerri CE, et al. Developing cost-effective field assessments of carbon stocks in human-modified tropical forests. PLoS ONE. 2015;10:e0133139.
Acknowledgements
The authors thank the owners and staff of Agropecuaria Nova Vida for logistical support and permission to work on their property. We also thank all collaborating private landowners of Santarem for their support and access to their land. We would like to thank the Large-Scale Biosphere-Atmosphere Program (LBA), coordinated by the National Institute for Amazon Researchers (INPA), for the use and availability of data for logistical support and infrastructure during field activities. We are grateful to Liana Chesini, Joice Ferreira, and Rede Amazônia Sustentável for logistical support during field activities. Additionally, we are grateful to Alexandre Pedrinho and Wagner Piccinini for assistance with fieldwork and Kiran Khan, Alex Thompson, Luke Gibney, and Rachel Feldman for assistance processing samples.
Funding
This project was supported by the National Science Foundation—Dimensions of Biodiversity (DEB 1442183), NSF-FAPESP 446 (2014/50320-4), by the Agriculture and Food Research Initiative Competitive Grant 2009-447 35319-05186 from the US Department of Agriculture National Institute of Food and Agriculture, and by the U.S. Department of Energy Joint Genome Institute through the Office of Science of the U.S. Department of Energy under Contract DE-AC02-442 05CH11231. Assistance fieldwork in Pará was in part supported by NERC NE/K016431/1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kroeger, M.E., Meredith, L.K., Meyer, K.M. et al. Rainforest-to-pasture conversion stimulates soil methanogenesis across the Brazilian Amazon. ISME J 15, 658–672 (2021). https://doi.org/10.1038/s41396-020-00804-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00804-x
