
Abstract
Antibiotic-resistant pathogens pose a significant threat to human health. Several dispersal mechanisms have been described, but transport of both microbes and antibiotic resistance genes (ARGs) via atmospheric particles has received little attention as a pathway for global dissemination. These atmospheric particles can return to the Earth’s surface via rain or snowfall, and thus promote long-distance spread of ARGs. However, the diversity and abundance of ARGs in fresh snow has not been studied and their potential correlation with particulate air pollution is not well explored. Here, we characterized ARGs in 44 samples of fresh snow from major cities in China, three in North America, and one in Europe, spanning a gradient from pristine to heavily anthropogenically influenced ecosystems. High-throughput qPCR analysis of ARGs and mobile genetic elements (MGEs) provided strong indications that dissemination of ARGs in fresh snow could be exacerbated by air pollution, severely increasing the health risks of both air pollution and ARGs. We showed that snowfall did effectively spread ARGs from point sources over the Earth surface. Together our findings urge for better pollution control to reduce the risk of global dissemination of antibiotic resistance genes.
Similar content being viewed by others


Introduction
Antibiotic-resistant pathogens pose a significant threat to human health and have become a serious global challenge in the 21st century. Antibiotic resistance genes (ARGs) are widely disseminated from anthropogenic sources, such as sewage, sludge, hospitals, and animal wastes, into soil [1], freshwater [2, 3], estuarine [4], and marine ecosystems [5, 6]. Terrestrial surface ecosystems have been reported as important reservoirs for ARGs [7, 8]. The continuous discharge of these emerging microbial contaminants could lead to an increase in the background levels of resistance, therefore enhancing the likelihood of global dissemination of ARGs via wind, diffusion, and water vapor loop into air [9, 10].
ARGs could enter the atmosphere adsorbed on biological aerosols from wind-borne dust, wastewater treatment plants, or biomass burning [11,12,13], that are subsequently disseminated globally via jet streams [14,15,16]. These aerosols can then return to the Earth’s surface through deposition in snow and rain, connecting the atmosphere and the Earth’s surface and creating a global ARG cycle [10]. Snowfall is particularly effective in this regard, since it precipitates matter in ice nuclei on a large regional scale. In contrast, rain precipitates particles underneath clouds, which is a much more local process [17,18,19]. Observations from 2008 to 2015 showed that fine particulate matter (PM2.5) was generally detected at twofold higher concentrations in cold seasons than in warmer seasons [20]. Moreover, long-term weather monitoring in China revealed a higher air pollution in colder snow seasons, mainly caused by fossil fuel burning [21, 22]. This may yield more (ARG) deposition onto the land surface. However, the occurrence, fate, and dynamics of ARGs in the atmosphere remain largely unexplored, especially under cold snow conditions.
Antibiotic resistance and ARGs are inherent characteristics of microorganisms [23]. However, the inappropriate use of antibiotics and/or the release of antimicrobial pollutants generates selection pressure on microorganisms, which could lead to a large-scale increase in ARG abundance and drive the generation of new ARGs [4, 8, 10]. ARGs enter atmospheric cycles via evaporation and wind [13, 16, 24], and can be disseminated globally with pollutant transportation [14, 25]. Under the influence of long-term atmospheric circulation, ARGs are completely mixed in the air, and the abundance and profiles of ARGs could reach a relatively stable and homogeneous state. Hence the community composition of ARGs in air may be similar, to a certain extent, at a global scale. However, within the air pollution area, the frequent occurrence of haze under low-pressure airflow conditions in winter, coupled with high-level PM could lead to an increase of the heterogeneity of ARGs in air [11,12,13, 21, 22]. The degree of association between particulate pollution and snow could vary as a function of local pollution. As a result, ARG communities could show different diversities related to local air pollution. Because of the difference in disseminating mode and mechanism, some ARGs will show increase or decrease in abundance with air pollution [4, 9, 11,12,13, 20] and therefore they may be used as an indicators of local air pollution. Against this background, we hypothesize that particulate pollution especially via snowfall could lead to dissemination of ARGs in the atmosphere and that ARGs might serve as biomarkers for anthropogenic atmospheric pollution.
Hence, the objective of this study was to investigate the abundance and composition of ARGs in snow so as to identify the potential drivers shaping the biogeography of ARGs and to determine key ARG indicators for air pollution. To achieve these goals, we sampled fresh snow from 2016 to 2019 in 33 cities (29 in China, 3 in North America and 1 in Europe) across the world (13,000-km transect belt) in the Northern Hemisphere. High-throughput quantitative PCR (HT-qPCR) was used to estimate the abundance, number and diversity of ARGs, and the relations between ARG profiles and bacterial communities were examined. Combined with molecular biology results, various statistical analysis methods including partial redundancy analysis, correlation analysis, nonmetric multidimensional scaling analysis (NMDS), fitting curve analysis, and network co-occurrence tests were performed to investigate the biogeography of ARGs in snow and determine key ARG indicators for air pollution.
Methods
Fresh snow sampling
A total of 48 snow sampling sites, 44 fresh snow samples from major cities in China, and three in North America and one in Europe, were collected (across a 13,000-km transect belt) in the Northern Hemisphere (23°05′N-66°05′N). Of these, 22 national-scale sampling sites were taken from China in 2016–2017, and 22 sites were further collected in 2018–2019 to verify the previous results. At the same time, in order to better reveal the occurrence, distribution, and inherent underlying mechanism, we also collected four samples from Munich, Washington, New Jersey, and Edmonton, where the level of local air pollution is lower than that of China. These sample sites have different energy structures, population, socio-economic development levels, and air pollution levels. For each location, five replicate samples were taken at each sampling site (100 × 100 m; all four corners and the central point) and measured separately. Details of sampling sites are presented in the Supplementary Table S1.
Sampling dates were determined from forecasts for snowfall. Fresh snow samples were taken from the beginning until the end of one snow event. When the snow began to land, it was collected on the ground on top of a plastic sheet; the collected samples were stored in a portable freezer and immediately sent to the laboratory. In the laboratory, the snow samples were melted and mixed at 4 °C, and divided into two portions for DNA extraction and chemical analyses. To ensure a sufficient sample size, from each point, we collected at least 30 L of snow (about 4–8 L snow can melt into 1 L of snow water). The extracted DNA was stored at −80 °C until analysis.
Air quality index (AQI)
The AQI is used by Chinese government agency to communicate to the public how polluted the air currently is or how polluted it will, most likely, become. There are six pollutant monitoring items in the AQI: sulfur dioxide (SO2), nitrogen dioxide (NO2), inhalable particles (PM10), PM2.5, carbon monoxide (CO2), and ozone (O3). Public health risks increase with increasing AQI levels, and different countries have their own air quality indices, corresponding to different national air quality standards. The computation of the AQI requires an air pollutant concentration over a specified averaging period; it can be calculated either per hour or per 24 h. An individual score (Individual Air Quality Index) is assigned to each pollutant, and the final AQI is the highest of these six scores (for detailed information, see the Supplementary Table S2).
Backward trajectory analysis
The 2-day (48 h) backward trajectories were calculated every hour at a height of 100 m above the ground level, ending at each snow sampling day, using the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT, NOAA) 4.9 model [26]. The trajectories were then grouped into five clusters using the algorithm of cluster analysis. The clustering of trajectories is based on the total spatial variance method [27], which minimizes the inter-cluster differences among trajectories while maximizing the outer-cluster differences; it has been widely used in previous studies [22, 28].
Sample chemical analysis
Concentrations of NH4+, NO3−, and NO2− were measured using continuous flow analyzers (Germany, SEAL, AA3), while Cl− and SO42− were determined using ion chromatography (USA, Diana, ICS-1000). The Na+, K+, Ca2+, and Mg2+ cations were measured using a full-spectrum direct-reading plasma emission spectrometer (USA, Leeman, Prodigy). Specific-surface area and concentration of particulate matter were measured by a Laser Particle Size Meter (Supplementary Table S3). Socio-economic parameters were collected and calculated from governmental statistical yearbooks, bulletins, and reports (Supplementary Table S1).
DNA extraction and HT-qPCR
The water from the melted snow (0.5–2 L, depending on the pollution level) was filtered through a 0.22-μm-pore-size filter (diameter, 45 mm; Millipore, New Bedford, MA), which was then used for DNA extraction, using the FastDNA Spin Kit for Soil (MP Biomedicals) according to the manufacturer’s protocol. The DNA quality analysis was performed using a NanoDrop2000 UV-visible spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA) and 1% (w/v) agarose gel electrophoresis, and the OD260/OD280 ratio of the DNA was between 1.8 and 2.0. The extracted DNA was quantified using the QuantiFluor dsDNA kit (Promega) in a 96-well microplate reader (SpectraMax M5, Molecular Devices), diluted to 50 ng μL−1 using sterile water, and stored at −80 °C for further analysis.
A total of 296 primer sets were selected to investigate genes present in the atmospheric snow DNA. These primer sets targeted resistance genes for all major classes of antibiotics (285 primer sets), transposase genes (8 primer sets), one universal class I integron-integrase gene (intI), and one clinical class 1 integron-integrase gene (cintI), and the 16S rRNA gene. The HT-qPCR was performed using the Wafergen Smart Chip Real-time PCR system at the Key Lab of Urban Environment and Health, Institute of Urban Environment, Chinese Academy of Sciences. For each primer set, a non-template negative control was included. The PCR cycle consisted of 10 min at 95 °C, followed by 40 cycles of denaturation at 95 °C for 30 s and annealing at 60 °C for 30 s. Melting curve analyses were automatically generated by the Wafergen software. All quantitative PCRs were carried out in technical triplicates. Wells with efficiencies beyond the range of 1.7–2.3 or an R2 under 0.99 were discarded. Only data for samples with at least three repeated sampling replicates, which generated amplification products, were regarded as positive and used for further data analysis. The relative copy numbers of ARGs generated by the HT-qPCR were transformed into absolute copy numbers by normalization, using the absolute 16S rRNA gene copy number.
The standard curve method of quantification by the Roche 480 system was used to determine the absolute 16S rRNA copy numbers at the same Key Lab facility. Each qPCR reaction mixture (20 μL) contained 10 μL 2× Light Cycle 480 SYBR Green I Master (Roche Applied Sciences), 1 μg μL–1 bovine serum albumin, 1 μM of each primer, 1 ng μL–1 template DNA, and 6 μL nuclease-free PCR-grade water. The thermal cycle consisted of a 10 min of initial enzyme activation at 95 °C, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s and extension at 72 °C for 15 s. A plasmid control containing a cloned and sequenced 16S rRNA gene fragment (1.39 × 1010 copies per liter) was used to generate eight-point calibration curves from tenfold dilutions for standard calculation. All qPCRs were performed in technical triplicates with negative controls.
Bacterial 16S rRNA gene sequencing
The V4–V5 region of the 16S rRNA gene was amplified, purified, quantified, pooled-, and multiplex-sequenced on an Illumina Miseq platform at Novogene to characterize bacterial communities. Each of the 50-μL PCR reaction mixtures contained 25 μL TaKaRa ExTaq, 0.5 μL bovine serum albumin, 1 μL of each primer, 1 μL DNA as template, and 21.5 μL nuclease-free PCR-grade water. The thermal cycle consisted of 3 min of initial enzyme activation at 94 °C, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 1 min, and extension at 72 °C for 1 min, with a final extension step at 72 °C for 5 min. Raw, paired-end reads were merged to clean sequences after filtering the adapter sequences and removing low-quality reads, ambiguous nucleotides, and barcodes. Raw sequences were demultiplexed and quality-filtered using QIIME [29] and Mothur [30].
Statistical and network analysis
Spearman correlations were performed to test the associations between ARGs and mobile genetic elements (MGEs) in fresh snow samples with bacterial abundances, environmental variables, social-economic factors, and atmosphere climate factors, using the software package SPSS 10.0. One-way ANOVA was used to analyze the differences in ARG abundance under different air pollution levels or elevations, using SPSS 10.0. Heatmaps were generated in Heml. The NMDS analysis was conducted in Canoco 5. Venn plot was generated using VennDiagram package in R 3.6.1. Distance-decay was calculated in the vegan package in R 3.6.1. Partial redundancy analysis was used to delineate the effects of explanatory variables including physiochemical factors (NO3−, NH4+, NO2−, Cl−, SO42−, Na+, K+, Mg2+, Ca2+, specific-surface area, and concentration of particulate matter), social-economic factors (population, GDP, healthcare level (number of hospitals), and sewage treatment (number of sewage treatment plants)), and bacterial community on the ARG profiles of snow samples, using R 3.6.1 with the vegan package.
Combined with molecular biology results, environmental factors, socio-economic factors, and natural factors were analyzed in parallel to identify potential drivers shaping the biogeography of ARGs in snow. Partial redundancy analysis, correlation analysis, fitting curve analysis, and network co-occurrence analysis were performed to determine key ARG indicators for air pollution and biogeochemical cycles. Specifically, Spearman’s rank correlations were calculated in the R environment using psych package and the correlation between two items was considered statistically robust with r > 0.8 and a p value < 0.01 according to Li et al. [31]. Network visualization was conducted on the interactive platform of Gephi with Fruchterman Reingold placement algorithm [32]. The network parameters such as high average degree, high closeness centrality, and low betweenness centrality were used to identify the key ARG subtypes in each module [33]. Spearman’s correlation coefficients were calculated between the ARG subtypes and the total abundance or AQI, where ARGs with Spearman’s correlation coefficients > 0.7 and a p value < 0.05. The linear regressions between ARG indicators and total ARGs or AQI were analyzed using Origin 9.0. The p values were adjusted with a multiple testing correction using the Benjamini–Hochberg method to reduce the chances of obtaining false-positive results in R environment [34]. The broken line plot was used to fit the correlation between the abundance of ARG indicators and total ARGs. All bar charts, scatter diagrams, pie charts, and broken line graphs were generated in Origin 9.0.
Results
ARGs and MGEs in snow
A total of 205 different ARGs and 10 MGEs were detected from the subset of urban fresh snow samples (Fig. 1a), respectively ranging from 1.55 × 104 to 3.83 × 107 and from 2.07 × 103 to 7.38 × 106 copies L−1 snow water. The detected ARGs and MGEs showed a high temporal and spatial heterogeneity (one-way ANOVA, p = 0.041 and p = 0.046, respectively). The abundance of ARGs and MGEs were significantly correlated with AQI and fine particulate matter (PM2.5) (all <p = 0.05, Supplementary Table S4). Analysis of the detected ARGs showed that the most common genes were most likely involved in conferring resistance to aminoglycosides, β-lactams, or were multidrug resistance determinants (Fig. 1b). Antibiotic transformation/deactivation was the dominant resistance mechanism encoded by the ARGs, followed by efflux pumps, and cellular protection (Fig. 1c and Supplementary Fig. S1). The relative abundance, the number of ARGs detected and bacterial abundance all increased significantly with increasing air pollution (all <p = 0.05) (Fig. 1b, d, and Supplementary Table S4).

a Sampling sites and years of atmospheric snow. A total of 48 snow sampling sites, 44 fresh snow samples from major cities in China and three in North America and one in Europe, were collected across the world, covering different climatic and geological zones. The sites were affected by different climatic, socio-economic, and physiochemical factors, at various degrees. The size of the solid circles represents the AQI value. b Absolute abundance, numbers of different ARGs and MGEs, and bacterial abundance at sampling sites, binned by different AQI values. Sampling sites are divided into different pollution levels based on AQI intervals of <50, 50–100, and >100, representing good, moderate, and polluted air, respectively. The unit of abundance was copies per liter of snow water. Error bars represent standard error (SE) of the replicates at each site (n = 3). c The pie charts show the proportions of four resistance mechanisms (antibiotic deactivation, efflux pumps, cellular protection, and others) under different pollution levels. d Heatmap of detected ARGs, classified by resistance mechanisms, from snow collected under different AQI values. Only ARGs of which the total abundance was accounted over 60% of each resistance mechanism were shown. The values of the heatmap was the log of ARG absolute abundance.
Partial redundancy analysis showed that a suite of environmental factors (NO3−, NH4+, NO2−, Cl−, SO42−, Na+, K+, Mg2+, and Ca2+ levels, specific-surface area, and concentration of particulate matter) explained over 86% of the observed variations in ARGs (Supplementary Fig. S2 and Table S5). Increasing levels of air pollution significantly and positively influenced ARG abundance (Supplementary Fig. S3). Temperature, solar radiation, and socio-economic factors such as population, GDP, medical level, and sewage treatment conditions were not significantly correlated with any of the ARGs (Supplementary Table S6). In summary, particulate pollution of the atmosphere was strongly correlated with the occurrence and dissemination of ARGs in air, and subsequent snowfall resulted in pollution of the land surface with both particulate matter and ARGs.
The community composition and diversity of atmospheric ARGs in urban fresh snow was further investigated. The NMDS analysis of ARG profiles showed that there was no significant clustering under different air pollution levels or under the same pollution level (Supplementary Fig. S4), which implied that the atmospheric ARGs shared a similar dominant community. In seventy percent of the total samples, the three most abundant ARGs at the resistance class were similar, namely, multidrug, aminoglycoside, and β-lactamase resistance genes. The ARG diversity measures showed that the numbers of unique ARG subtypes in polluted sites were significantly higher than that in clean sites (Supplementary Fig. S5). For example, aadA1-01 was only detected in polluted sites, but was absent from clean sites. Moreover, the Chao1 index of ARGs increased significantly with the increase of air pollution (r = 0.412, p < 0.05, Supplementary Fig. S6). These results indicated that the composition of dominant resistance classes of ARGs was independent of the air pollution levels, while air pollution could increase the richness of ARGs.
Co-occurrence of atmospheric ARGs, MGEs, and bacteria
The elevated interaction among ARGs and MGEs with increasing air pollution was also confirmed by Spearman’s rank correlation-based network analysis (Fig. 2, Supplementary Tables S7 and S8). ARG abundances were correlated at a high significance level with the abundance of transposase genes across all AQI levels (all <p = 0.05), and significantly correlated with integron-integrase genes at polluted sites (Supplementary Fig. S7 and Table S9). Specifically, the most frequently detected tnpA genes (tnpA-02, tnpA-07, and Tp614 genes), were closely connected with ARGs in networks from the different environmental samples (Supplementary Table S10). These genes mediate gene transfer within chromosomes or plasmids [35,36,37], suggesting a significant contribution of horizontal gene transfer to ARG dissemination. Network co-occurrence analysis of ARGs and the 16S rRNA genes of co-occurring bacteria had a strong positive correlation at the family level, while at the genus level, the diversity of bacteria and their connections with ARGs increased with higher pollution levels (Fig. 2). For example, the vancomycin resistance gene vanC-03 exhibited a stronger connection with a wider range of bacteria in polluted sites (Supplementary Table S11). Pseudomonas only connected with aadA5-01 in clean sites while they showed significant connections with both ARGs (aac(6′)-Ib(aka aacA4)-01, aadA5-01, aadA5-02, tetR-02) and MGEs (intI-1(clinic), tnpA-01) in polluted sites (Supplementary Table S11).This suggests that air pollution may promote co-occurrence of atmospheric ARGs, MGEs, and bacteria.

In the ARG and MGE networks, nodes and edges are colored according to the homology of ARGs and MGEs. The nodes with a high degree of significance are labeled, being classified into the aadA family, vancomycin resistance, the class A and C β-lactamase families, and transposase, respectively. Node size was proportional to node degree; edges represent interactions between nodes. In the bacterial and ARG and MGE networks, nodes were colored for the types of ARGs and genera of bacteria. Edges were colored for the interactions with ARGs and bacteria, respectively (see Supplementary Tables S7 and S8 for detailed network parameters).
Air pollution and dissemination of ARGs
Since ARGs can be recovered in samples deposited from the atmosphere, air movement potentially promotes the dissemination of resistance genes. In this study, ARG abundance and diversity were similar (one-way ANOVA, p > 0.05) among sampling sites below and above an elevation of 500 m (regions below altitude 500 m are more significantly affected by human activities). The correlation between ARGs, cations, and anions also showed no significant differences above and below 500 m (Supplementary Fig. S8). No distance-decay pattern was observed for ARG profiles in snow along the entire 13,000-km transect (Fig. 3b, all >p = 0.05). Taken together, these findings suggest that atmospheric ARGs were fully mixed via vertical and horizontal air movement. Meanwhile, there were no significant relationships between microbial community similarity and geographical distance in the ranges of 4000 km and 13,000 km (Supplementary Fig. S9). This suggests that ARGs are present as intracellular DNA, but also as extracellular DNA in contaminated particles (in accordance with previous results in sediments [38], wastewater [39] and sludge [40]), and are thus more susceptible to atmospheric dissemination.

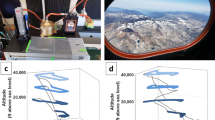
Plane graphs of airflow trajectory under different air qualities with a-1 AQI < 50 (good), a-2 50 < AQI < 100 (moderate), and a-3 AQI > 100 (polluted), and a-4 stereogram of the pressure variation of air trajectory under different pollution levels. The green bar indicates clean sampling sites where the airflow trajectory showed both horizontal and vertical dimensions; the orange bar indicates light pollution, and red bars indicate heavy pollution, where the airflow descent trajectory decreased. b Distance-decay analysis of ARG similarity revealed a significant spatial pattern of ARG distribution. National scale, from 0 to 4000 km, Pearson’s r = −0.28, p = 0.002. Global scale, from 0 to 13,000 km, Pearson’s r = −0.006, p > 0.05. c Correlation between AQI value and the abundance of typical ARG families (aadA family, vancomycin resistance, the class A and C β-lactamase families) (expressed as copies per L snow water, plotted on a log scale). The left column shows that parts of the ARG subtypes of the families were significantly positively related to AQI. The upper two graphs on the right column show that parts of the subtypes of the vancomycin resistance and aadA family showed no obvious difference as pollution increased, while the latter graph shows that some in the class A β-lactamase families decreased with increasing pollution levels.
Since we have data on the abundance of ARGs; the settling velocity and mass of snowflakes; and the number of snowflakes per cubic meter, we were able to estimate the rate of ARG deposition via snowfall. ARGs flux via snowfall was calculated to be between 7 ± 3 and 1007 ± 474 copies per m2 per second (Supplementary Fig. S10 and Table S12). Detailed explanations of these calculations are shown in the Supplementary data. The estimated ARG flux showed significant correlation with AQI and PM2.5 (all <p = 0.05) with strong heterogeneity in the air (Supplementary Table S13), further emphasizing the dissemination of antibiotic resistance via air pollution.
Each sampling site has different prevailing wind directions that change with climatic conditions and seasons (Fig. 3a and Supplementary Fig. S11). This means that weather patterns act to diffuse ARGs at a regional scale. Distance-decay analysis showed a significant negative correlation between the similarity of ARG profiles and the geographical distance within 4000 km (r = −0.28, p < 0.001, Fig. 3b). Other than horizontal directions in all sampling sites, airflow trajectory in pristine sampling sites showed both up and down directions, while in polluted sites, it shifted to downward movements due to the low pressure (700–1000 hPa atmospheric pressure, Supplementary Fig. S12). This resulted in the accumulation of resistance genes, which is consistent with the static nature of haze without the external force of atmospheric movement, such as winds [20]. This was also consistent with the results that there was a background value of resistance genes in the atmosphere, and air pollution led to the dissemination of some specific genes.
Curve fitting and co-occurrence network reveal resistance genes as markers of atmospheric pollution
The network co-occurrence analysis showed that the vancomycin resistance genes, the aadA family, and the class A and C β-lactamase gene families were the key connecting ARG families under different pollution conditions (Fig. 2 and Supplementary Table S14). Through component analysis, we found that these three ARG families and their subtype genes possessed distinct distribution characteristics (Fig. 3c). The abundance of the vancomycin resistance genes and the aadA family showed an increasing or unchanged trend with increased pollution, while the class A and C β-lactamase families showed an increase or decreasing trend. This made it possible to identify potential indicators for total abundance of ARGs, and the severity of air pollution.
The co-occurrence patterns among ARG subtypes revealed that the genes blaCTX-M-04 and blaCMY2-02 in the class A and C β-lactamase families, aadA1 and aadA2-02 in the aadA family, and vanC-03 belong to the vancomycin resistance were the hub of their respective modules (Fig. 4a and Supplementary Table S15). Correlation and linear fitting analysis also showed that the abundance distributions of blaCTX-M-04, blaCMY2-02, aadA1, aadA2-02, and vanC-03 were in agreement with total ARG abundance, while the blaSFO abundance showed an opposite trend (Fig. 4a). Moreover, they showed different co-efficient with increasing air pollution levels. ARGs blaCMY2-02, aadA1, vanC-03 showed an increasing trend, aadA2-02 and blaCTX-M-04 showed a stable trend, and blaSFO gene showed a decreasing trend (Fig. 4b and Supplementary Table S16). Therefore, we propose that blaCTX-M-04, blaCMY2-02, aadA1, aadA2-02, vanC-03, and blaSFO genes are most suitable indicators, or proxies, to estimate the abundance of other co-occurring ARG subtypes, total ARGs, and the severity of air pollution.

a Co-occurrence network analysis of ARG biomarkers and their correlation with total ARGs. The network shows the potential interaction among the ARG biomarkers (different colors). The line plots show the abundance of each biomarker (different colors) along with the total ARG abundance (gray) in all sampling sites. Biomarkers were colored differently for both the nodes in the network and the lines in the curves, with vanC-03 in blue, blaCMY2-02 in magenta, aadA1 in light cyan, blaCTX-M-04 in dark cyan, aadA2-02 in yellow, and blaSFO in light gray. The linear regression curves in the upper left of each line plot show the correlation between abundance of potential biomarkers and the total ARGs. b Correlation between potential biomarkers and AQI. Data for 2016–2017 and 2018–2019 are shown in the black and the blue plot, respectively. The blue line with light cyan represents the linear regression of biomarker and AQI in total snow samples from 2016 to 2019. The gray line with light gray represents the linear regression of biomarker and AQI in the snow samples from 2016 to 2017.
Further verification of temporal and spatial scales
We collected further fresh snow samples in 2018–2019 to verify the results of the initial study. A total of 245 ARGs and 12 MGEs were detected from the 22 urban fresh snow samples (Supplementary Fig. S13), with absolute abundances ranging from 2.64 × 104 to 2.29 × 106 copies L−1. Both ARG and MGE abundances were significantly correlated with air pollution (all <p = 0.05, Supplementary Table S4). Resistance genes against β-lactams, aminoglycosides, and multidrug resistance determinants were again the most prevalent genes, in good agreement with the initial results. Both transposase and integron genes showed strong relationships with ARGs (all <p = 0.05, Supplementary Table S17), indicating that horizontal transfer promotes the potential spread of ARGs. In particular, the integron-integrase gene was significantly correlated with almost all resistance genes in polluted sites (p < 0.05), but not in clean sites (Supplementary Table S17), again consistent with previous results.
Component analysis showed that the proposed ARG indicator genes showed similar trends with air pollution and total ARG abundance, although some ARG subtypes of the aadA family, vancomycin resistance, and the class A and C β-lactamase families showed different trends with increasing pollution levels. The blaSFO gene in the class A β-lactamase family decreased in the follow-up study with increasing air pollution. The aadA1 in the aadA family, the blaCMY2-02 gene in the class C β-lactamase family, and vanC-03 were strongly and positively related with AQI (all <p = 0.05), while aadA2-02 in the aadA family and blaCTX-M-04 in the class A β-lactamase family showed no significant differences under distinctive contamination conditions (one-way ANOVA, p > 0.05). Here, we showed again that all six indicator genes could predict the trends in total ARGs and air pollution conditions.
Discussion
In this study we report ARG pollution in fresh snow on a planetary scale. ARGs are widely distributed among environmental bacteria, which exist in air mainly in the form of biological aerosols adsorbed onto particle surfaces [41, 42]. Particulate matter with sizes less than 10 μm can be transported over long distances in the atmosphere and have a relatively long residence time [14, 25, 43, 44]. Diverse and abundant air-borne ARGs can then return to the land surface via precipitation, especially snowfall. This indicates that ARGs from major point sources (sewage [3, 45], sludge [46], hospitals [45], and animal manure [1]) can potentially contaminate the entire surface of the Earth. This generates an “environmental ARG loop” (Fig. 5), connecting human activity to the whole earth’s surface via deposition of particulate pollutants, including those that carry resistant bacteria or resistance genes.

The ARGs generated from hospitals, livestock farms, and sewage treatment plants (among others) could emit from terrestrial surface soil and water ecosystems into the atmosphere through wind action, water evaporation, and dust transport, and return to ground again via wet or dry precipitation. The increase of particulate matter (PM) in the air will significantly increase ARGs’ habitats. Particularly in winter, these ARGs mainly come back to the surface via snowfall deposition, making it more accessible again to the human, animals, and other pathogenic microorganisms. The ARGs cycle among the atmosphere, terrestrial and aquatic ecosystems will result in a continuity of hazardous exposure to ARGs. The red, yellow, and blue points represent various ARGs, and the gray irregular circle represents particulate matter.
Furthermore, our results provide direct evidence that precipitation of ubiquitous ARGs in fresh snow was exacerbated by air pollution. We found a significant impact of air pollution on both the diversity and abundance of ARGs and MGEs. Integrons are widely known for their role in the transfer of ARGs [47]. There was a strong significant positive correlation between integrons and ARGs and poor air quality, while there was hardly any correlation with good air quality, suggesting that particulate pollution may accelerate both ARG dissemination and horizontal transfer of ARGs, which is consistent with previous results that the cities with different air pollution levels have different levels of ARGs [11, 48]. The possible microbial mechanism is that bacteria, such as the Pseudomonas detected in this study, could be metabolically active in the snow water and multiply with nutrients derived from the air pollution, especially in highly polluted air [18, 49]. This is also potentially driven by selection of resistance determinants by co-polluting selective agents. An increasing number of studies record metals [50], antibiotics [12, 51], pathogens [52, 53], and microplastics [54,55,56] as being associated with air-borne particulates, and this effect is worse during haze episodes [57, 58]. These selective agents are known to interact, and could drive co-selection, because resistance genes for these various agents are often clustered in genomic islands and plasmids. Therefore, particulate matter could be a hotspot for the development of new resistance combinations due to co-contamination of particles with toxic chemicals. We believe that air pollution can translate local pollution of ARGs into global impact, which gives a further urgent need for a strong reduction of air pollution.
Our finding that ARG transport by air could be exacerbated by anthropogenic air pollution is an additional strong indication for the impact of the Anthropocene to changes in geological, ecological, sociological, and anthropological influences in the recent Earth history [59]. Increased population and human activities have dramatically changed atmospheric biogeochemical cycles and leave strong imprints [60, 61]. Geochemical isotopic markers (such as 15N, 13C, and 18O) can be used to trace human activities, but are currently limited for environmental samples >100 years old without disturbance, with a higher accuracy at the millennium scale [50, 62,63,64,65]. It is still unknown how to retrieve the human footprint at the centennial scale under obvious allochthonous disturbance. Compared with stable and passive geochemical markers, community structure and genetic information of microorganisms are significantly shaped by the external environments, thereby quickly reflecting these imprints. In this study, we estimated the flux of ARGs (7 ± 3 to 1007 ± 474 copies per m2 per second) in snowfall carried down from the air. Such large volume of ARGs will leave a distinct mark in sediment and ice thus potentially form clear stratigraphic signatures. The time point of commercial development and large-scale application for each antibiotic is different. In this study we found that a subset of six unique resistance genes can be used as biomarkers to trace human activities. Further environmental ARG bioindicator research may focus on centennial-scale dating in sediments and ice cores to estimate the lower boundary of the Anthropocene, to complement chemical and mineralogical dating methods. In summary, particulate matter pollution can accelerate ARG dissemination, and snowfall monitoring can potentially be used as a mean to surveil global dissemination of ARGs. In addition to the geochemistry and atmospheric physics, we provide a novel biological method to study the influences of human activities on the atmospheric cycle across a 100-year scale. Future environmental ARG biomarker research should focus on other centennial-scale dating studies, such as sediment core, soil core, and ice core analysis to estimate the human footprint.
Data availability
Sequence data of the bacterial 16S rRNA genes generated by the Illumina platform were deposited in the NCBI Sequence Read Archive (SRA) database (SRA accession no. SRR6660990-6661015). The authors declare that the other metadata supporting our findings are available within this article and the Supplementary documents. Other data could be requested from the corresponding author.
References
Tang XJ, Lou CL, Wang SX, Lu YH, Liu M, Hashmi MZ, et al. Effects of long-term manure applications on the occurrence of antibiotics and antibiotic resistance genes (ARGs) in paddy soils: evidence from four field experiments in south of China. Soil Biol Biochem. 2015;90:179–87.
Baquero F, Martínez JL, Cantón R. Antibiotics and antibiotic resistance in water environments. Curr Opin Biotech. 2008;19:260–5.
Czekalski N, Diez EG, Buergmann H. Wastewater as a point source of antibiotic-resistance genes in the sediment of a freshwater lake. ISME J. 2014;8:1381–90.
Zhu YG, Zhao Y, Li B, Huang CL, Zhang SY, Yu S, et al. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat Microbiol. 2017;2:3435–40.
Marti E, Variatza E, Luis, Balcazar J. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 2014;2:36–41.
Niu ZG, Zhang K, Zhang Y. Occurrence and distribution of antibiotic resistance genes in the coastal area of the Bohai Bay, China. Mar Pollut Bull. 2016;107:245–50.
Huang YJ, Cheng MM, Li WH, Wu LH, Chen YS, Luo YM, et al. Simultaneous extraction of four classes of antibiotics in soil, manure and sewage sludge and analysis by liquid chromatography-tandem mass spectrometry with the isotope-labelled internal standard method. Anal Methods UK. 2013;5:3721–31.
Zhang SH, Yang GL, Hou SG, Zhang TJ, Li ZG, Liang F. Distribution of ARGs and MGEs among glacial soil, permafrost, and sediment using metagenomic analysis. Environ Pollut. 2018;234:339–46.
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8:251–9.
Green JK, Konings AG, Alemohammad SH, Berry J, Entekhabi D, Kolassa J, et al. Regionally strong feedbacks between the atmosphere and terrestrial biosphere. Nat Geosci. 2017;10:410–4.
Li J, Cao JJ, Zhu YG, Chen QL, Shen FX, Wu Y, et al. Global survey of antibiotic resistance genes in air. Environ Sci Technol. 2018;52:10975–84.
McEachran AD, Blackwell BR, Hanson JD, Wooten KJ, Mayer GD, Cox SB, et al. Antibiotics, bacteria, and antibiotic resistance genes: aerial transport from cattle feed yards via particulate matter. Environ Health Perspect. 2015;123:337–43.
Xie JW, Jin L, Luo XS, Zhao Z, Li XD. Seasonal disparities in airborne bacteria and associated antibiotic resistance genes in PM2.5 between urban and rural sites. Environ Sci Technol Lett. 2018;5:74–79.
Kellogg CA, Griffin DW. Aerobiology and the global transport of desert dust. Trends Ecol Evol. 2006;21:638–44.
Maring H, Savoie DL, Izaguirre MA, Custals L, Reid JG. Mineral dust aerosol size distribution change during atmospheric transport. J Geophys Res Atmos. 2003;108:1–5.
Hatakeyama S, Takami A, Sakamaki F, Mukai H, Sugimoto N, Shimizu A, et al. Aerial measurement of air pollutants and aerosols during 20–22 March 2001 over the East China Sea. J Geophys Res Atmos. 2004;109:D13304.
Błaś M, CichałaKamrowska K, Sobik M, Polkowska Z, Namieśnik J. Conditions controlling atmospheric pollutant deposition via snowpack. Environ Rev. 2010;18:87–27.
Christner BC, Morris CE, Foreman CM, Cai R, Sands DC. Ubiquity of biological ice nucleators in snowfall. Science. 2008;319:1214–1214.
Saleeby SM, Cotton WR, Lowenthal D, Borys RD, Wetzel MA. Influence of cloud condensation nuclei on orographic snowfall. J Appl Meteorol Clim. 2009;48:903–22.
Fontes T, Li PL, Barros N, Zhao PJ. Trends of PM2.5 concentrations in China: a long term approach. J Environ Manag. 2017;196:719–32.
Wang YS, Yao L, Wang LL, Liu ZR, Ji DS, Tang GQ, et al. Mechanism for the formation of the January 2013 heavy haze pollution episode over central and eastern china. Sci China Earth Sci. 2014;57:14–25.
Sun YL, Jiang Q, Wang ZF, Fu PQ, Li J, Yang T, et al. Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013. J Geophys Res Atmos. 2014;119:4380–98.
D’Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, et al. Antibiotic resistance is ancient. Nature. 2011;477:457–518.
Tripathi V, Cytryn E. Impact of anthropogenic activities on the dissemination of antibiotic resistance across ecological boundaries. Essays Biochem. 2017;61:11–21.
Li J, Osada K. Preferential settling of elongated mineral dust particles in the atmosphere. Geophys Res Lett. 2007;34:251–70.
Draxler RR, Hess GD. Description of the HYSPLIT4 modeling system. NOAA Tec Mem ERL ARL-224. Sci Rep UK. 1997;28:197–9.
Draxler R, Stunder B, Rolph G HYSPLIT4 user’s guide, version 4. NOAA Tech Mem ERL ARL, Report. 2012.
Li YJ, Lee BP, Su L, Fung JCH, Chan CK. Seasonal characteristics of fine particulate matter (PM) based on high- resolution time-of-flight aerosol mass spectrometric (HR-ToF-AMS) measurements at the HKUST Supersite in Hong Kong. Atmos Chem Phys. 2015;15:37–53.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. Qiime allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Li B, Yang Y, Ma LP, Ju F, Guo F, Tiedje JM, et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015;9:2490–502.
Fruchterman T, Reingold E. Graph drawing by force-directed placement. Softw Pr Exp. 1991;21:1129–64.
Banerjee S, Schlaeppi K, van der Marcel MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16:567–76.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Takamatsu D, Osaki M, Sekizaki T. Chloramphenicol resistance transposable element tnss1 of streptococcus suis, a transposon flanked by is6-family elements. Plasmid. 2003;49:143–51.
Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62:725–74.
Aubert D, Naas T, Heritier C, Poirel L, Nordmann P. Functional characterization of IS1999, an IS4 family element involved in mobilization and expression of β-lactam resistance genes. J Bacteriol. 2006;188:6506–14.
Mao DQ, Luo Y, Mathieu J, Wang Q, Feng L, Mu Q, et al. Persistence of extracellular DNA in river sediment facilitates antibiotic resistance gene propagation. Environ Sci Technol. 2014;48:71–78.
Liu SS, Qu HM, Yang D, Hu H, Liu WL, Qiu ZG, et al. Chlorine disinfection increases both intracellular and extracellular antibiotic resistance genes in a full-scale wastewater treatment plant. Water Res. 2018;136:131–6.
Zhang Y, Snow DD, Parker D, Zhou Z, Li X. Intracellular and extracellular antimicrobial resistance genes in the sludge of livestock waste management structures. Environ Sci Technol. 2013;47:10206–13.
Hu JL, Zhao FZ, Zhang XX, Li K, Li CR, Ye L, et al. Metagenomic profiling of ARGs in airborne particulate matters during a severe smog event. Essays Biochem. 2017;61:11–21.
Xie JW, Jin L, He TT, Chen BW, Luo XS, Feng BH, et al. Bacteria and antibiotic resistance genes (ARGs) in PM2.5 from China: implications for human exposure. Sci Total Environ. 2018;615:1332–40.
Caffrey PF, Ondov JM, Zufall MJ, Davidson CI. Determination of size-dependent dry particle deposition velocities with multiple intrinsic elemental tracers. Environ Sci Technol. 1998;32:1615–22.
Harrison RM, Yin JX. Particulate matter in the atmosphere: which particle properties are important for its effects on health? Sci Total Environ. 2000;249:85–101.
Rodriguez-Mozaz S, Chamorro S, Mari E, Huerta B, Gros M, Sànchez-Melsió A, et al. Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 2015;69:234–42.
Su JQ, Wei B, Qu-Yang WY, Huang FY, Zhao Y, Xu HJ, et al. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ Sci Technol. 2015;49:7356–63.
Gillings MR. Integrons: past, present, and future. Microbiol Mol Biol Rev. 2014;78:257–77.
Zhang T, Li XY, Wang MF, Chen HX, Yang Y, Chen QL, et al. Time-resolved spread of antibiotic resistance genes in highly polluted air. Environ Int. 2019;127:333–9.
Attard E, Yang H, Delort AM, Amato P, Oschl U, Glaux C, et al. Effects of atmospheric conditions on ice nucleation activity of Pseudomonas. Atmos Chem Phys Discuss. 2012;12:9491–516.
Dulac F, Buat‐Ménard P, Arnold M, Martin D. Atmospheric input of trace metals to the western Mediterranean Sea: 1. Factors controlling the variability of atmospheric concentrations. J Geophys Res Atmos. 1987;92:8437–53.
Smith PN, McEachran AD, Wooten KJ, Blackwell BR. A preliminary evaluation of veterinary antibiotics, estrogens, in vitro estrogenic activity and microbial communities in airborne particulate matter collected near dairy production facilities. Aerobiologia. 2019;35:315–26.
Ekhaise FO, Isitor EE, Idehen O, Emoghene AO. Airborne microflora in the atmosphere of a hospital environment of University of Benin Teaching Hospital (UBTH), Benin city, Nigeria. World J Agric Sci. 2010;6:166–70.
Schmale Iii DG, Ross SD. Highways in the sky: scales of atmospheric transport of plant pathogens. Annu Rev Phytopathol. 2015;53:591–601.
Prata CorreiaJ. Airborne microplastics: consequences to human health? Environ Pollut. 2018;234:115–26.
Li J, Zhang K, Zhang H. Adsorption of antibiotics on microplastics. Environ Pollut. 2018;237:460–7.
Horton AA, Dixon SJ. Microplastics: an introduction to environmental transport processes. Wiley Interdiscip Rev. Water. 2017;5:268.
Hu YJ, Lin J, Zhang SQ, Kong LD, Fu HB, Chen JM. Identification of the typical metal particles among haze, fog, and clear episodes in the Beijing atmosphere. Sci Total Environ. 2015;511:369–80.
Stepanauskas R, Glenn TC, Jaqoe CH, Tuckfield RC, Lindell AH, McArthur JV. Elevated microbial tolerance to metals and antibiotics in metal-contaminated industrial environments. Environ Sci Technol. 2005;39:3671–8.
Crutzen PJ. Geology of mankind. Nature. 2002;415:23.
Duce RA, Laroche J, Altieri KE, Arrigo KR, Baker AR, Capone DG, et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science. 2008;320:893–7.
Falkowski P, Scholes RJ, Boyle EA, Canadell JG, Canfield DE, Elser JJ, et al. The global carbon cycle: a test of our knowledge of earth as a system. Science. 2002;290:291–6.
Belousova E, Griffin W, O’Reilly SY, Fisher N. Igneous zircon: trace element composition as an indicator of source rock type. Contrib Miner Petr. 2002;143:602–22.
Marx SK, Kamber BS, Mcgowan HA. Scavenging of atmospheric trace metal pollutants by mineral dusts: inter-regional transport of Australian trace metal pollution to New Zealand. Atmos Environ. 2008;42:2460–78.
Magnani F, Mencuccini M, Borghetti M, Berbigier P, Berninger F, Delzon S, et al. The human footprint in the carbon cycle of temperate and boreal forests. Nature. 2007;447:849–51.
Tipple BJ, Pagani M. The early origins of terrestrial C4 photosynthesis. Annu Rev Earth Pl Sci. 2007;35:435–61.
Acknowledgements
First of all, we are very grateful to Dr. Zhiwu Wang for sampling in USA and Dr. Zhe Chen for sampling in Munich. This research was financially supported by the National Natural Science Foundation of China (Nos. 41322012, 41671471, and 91851204), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Z176), National Key R&D Program (2016YFA0602303), and special fund from the State Key Joint Laboratory of Environment Simulation and Pollution Control (Research Center for Eco-environmental Sciences, Chinese Academy of Sciences) (18Z02ESPCR). MSMJ is supported by SIAM 024002002. GBZ gratefully acknowledges the Program of the Youth Innovation Promotion Association (CAS).
Author information
Authors and Affiliations
Contributions
GBZ and YGZ designed the project. ZFW provided conceptual advice on atmospheric biogeochemical cycle, and JQS was responsible for ARG Molecular Ecology. XMW, TY, YQ, SYW, CW, FJ, BRL, CLL, HL, XEL, and XMW contributed to sample analysis. GBZ and YGZ wrote the manuscript with contributions from MG, MSMJ, and JQS. All authors discussed and interpreted the results and contributed to the manuscript. Correspondence and requests for materials should be addressed to GBZ (gbzhu@rcees.ac.cn), ZFW (Zifawang@mail.iap.ac.cn), and YGZ (ygzhu@rcees.ac.cn).
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, G., Wang, X., Yang, T. et al. Air pollution could drive global dissemination of antibiotic resistance genes. ISME J 15, 270–281 (2021). https://doi.org/10.1038/s41396-020-00780-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00780-2
This article is cited by
-
The antibiotic resistance reservoir of the lung microbiome expands with age in a population of critically ill patients
Nature Communications (2024)
-
The soil-microbe-plant resistome: A focus on the source-pathway-receptor continuum
Environmental Science and Pollution Research (2024)
-
Frequency of occurrence and habitat selection shape the spatial variation in the antibiotic resistome in riverine ecosystems in eastern China
Environmental Microbiome (2022)
-
Antibiotic resistance in the environment
Nature Reviews Microbiology (2022)
-
Dynamic distribution and potential transmission of antibiotic resistance genes in activated sludge
Applied Microbiology and Biotechnology (2022)
