
Abstract
Prochlorococcus cells are the numerically dominant phototrophs in the open ocean. Cyanophages that infect them are a notable fraction of the total viral population in the euphotic zone, and, as vehicles of horizontal gene transfer, appear to drive their evolution. Here we examine the propensity of three cyanophages—a podovirus, a siphovirus, and a myovirus—to mispackage host DNA in their capsids while infecting Prochlorococcus, the first step in phage-mediated horizontal gene transfer. We find the mispackaging frequencies are distinctly different among the three phages. Myoviruses mispackage host DNA at low and seemingly fixed frequencies, while podo- and siphoviruses vary in their mispackaging frequencies by orders of magnitude depending on growth light intensity. We link this difference to the concentration of intracellular reactive oxygen species and protein synthesis rates, both parameters increasing in response to higher light intensity. Based on our findings, we propose a model of mispackaging frequency determined by the imbalance between the production of capsids and the number of phage genome copies during infection: when protein synthesis rate increase to levels that the phage cannot regulate, they lead to an accumulation of empty capsids, in turn triggering more frequent host DNA mispackaging errors.
Similar content being viewed by others


Introduction
With an estimated population of 3 × 1027 cells on Earth, Prochlorococcus is the most numerically abundant phytoplankton species in the global oceans [1]. Distributed throughout the euphotic zone in mid-latitude habitats, Prochlorococcus is responsible for fixing an estimated 4 billion tons of carbon each year, playing a central role in ocean food webs. Prochlorococcus genomes are highly streamlined, an adaptation for growth in oligotrophic surface waters [2]. Different ocean regions contain hundreds to thousands of Prochlorococcus coexisting subpopulations [3, 4]. Horizontal gene transfer (HGT) has had a large impact on Prochlorococcus evolution, both in terms of generating genomic variability in genomic islands [5] or in reinforcing genetic similarity in closely related cells [6, 7]. Furthermore, ways of exchanging genetic information among cells are particularly critical for streamlined organisms such as Prochlorococcus; its absence could lead to extinction in the face of environmental change [8].
Notably, Larkin et al. revealed that the drivers of Prochlorococcus fine-scale genomic diversity are multiple, complex, encompass biotic and abiotic factors, and remain elusive [9]. As shown recently by Arevalo et al. [7], HGT events leading to homologous recombination between chromosomal segments result in cohesive ‘gene-flow units’ (group of recombinogenic cells) in the environment, which align impressively well with phylogenetically defined ecological populations [7]. This critical observation raises the possibility that the frequency at which those HGT events occur in the environment, cumulated over evolutionary timescales, could participate in explaining the current spatial disparity of fine-scale diversity in wild Prochlorococcus populations.
Modes of HGT available to Prochlorococcus include lipid-bound vesicles, which contain DNA and are known to be abundant in the oceans [10, 11], natural transformation, but only in some LLIV clades that contain the necessary genes for competence [12], and transduction via phage capsids. The latter is one of the canonical modes of HGT in bacteria [13,14,15] and is thought to occur at significant frequencies in aquatic ecosystems [16,17,18]. Given the abundance of cyanophages (phages that infect cyanobacteria) that infect Prochlorococcus in the oceans (easily reaching 105 to 106 phages mL−1) [19, 20], which represent a notable fraction of the total viral population in the euphotic zone [21], it appears that transduction should be occurring. Surprisingly, however, this likely source of major genetic mixing within marine picocyanobacteria has barely been investigated. The only experimental evidence in that direction was performed by Clokie et al., who showed that the Synechococcus myovirus S-PM2 is capable of packaging its host chromosomal DNA at low frequency [22]. Thus, we focused this study on exploring and quantifying this phenomenon.
Specialized (reviewed in [23, 24]) and lateral [25] transduction in bacteria are strictly mediated by lysogenic phages, as they enter and leave their host chromosome. Prophages, the hallmarks of lysogeny, however, have not been observed in the hundreds of Prochlorococcus isolates or single-cell genomes from the wild [26], suggesting that the vast majority of phages infecting Prochlorococcus are strictly lytic. This leaves generalized transduction—the random mispackaging of host DNA into capsids and its subsequent injection and recombination in a recipient cell—as the dominant route of phage-mediated HGT in these bacteria.
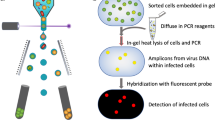
To explore the potential of Prochlorococcus lytic cyanophages to be vectors of generalized transduction, we quantified the mispackaging of fragments of host DNA during infection of Prochlorococcus MED4 by three cyanophages with different morphotypes belonging to the sipho- (P-HS2), myo- (P-HM2), and podovirus (P-SSP7) families (Fig. 1a). Our goal was to determine the frequency of mispackaging in the different morphotypes and begin to explore the environmental factors that might influence mispackaging events in the wild.

a Electron micrograph of the three cyanophages infecting Prochlorococcus MED4 used in this study. The scale bar is 50 nm. b Cartoon illustrating the experimental method. A cyanophage infection typically results in the production of a small fraction of cyanophage capsids having mispackaged host DNA, represented in red. c Detection of host DNA mispackaging inside cyanophage capsids under standard laboratory conditions, at a constant light intensity of 45 µmol Q m−2 s−1 (see ‘Materials and methods’ for details). The frequency of a gene mispackaging during infection is expressed in gene copies per million phages. Each symbol is the mean of three parallel infections for a given locus (Table 1). The colored dotted line is the mean value of all loci and all replicates. The three colors for the different phage are arbitrary—for ease of visualization. d Impact of growth light intensity on mispackaging during infection. The data 45 µmol Q m−2 s−1 are from (c), replotted for comparison. The dotted line at 0.5 gene copies per million phages marks the detection limit of the assay. The four colors for the different light intensities are arbitrary—for ease of visualization. The p values indicated between the lowest to highest light intensity samples were calculated by a Kruskal–Wallis test followed by a Dunn test. The graphs are replotted in Supplementary Fig. 1 to highlight locus to locus variations.
Results and discussion
Host DNA mispackaging frequency in cyanophage capsids
Using six different loci on the Prochlorococcus MED4 chromosome (see positions in Table 1), we quantified the level of mispackaging of MED4 DNA in the capsids of the three different cyanophages (Fig. 1a, Table 2) during infection (Fig. 1b, c). The packaging strategy of the podovirus is T7-like [27], the myovirus’ is likely T4-like headful packaging [28] while the siphovirus’ is unknown, but 490 bp direct terminal repeats suggest a T7-like packaging mechanism [29]. Host DNA was detected in the capsids of all three cyanophages (Fig. 1c). The levels of mispackaging are low compared to those reported for high transducing phages in Staphylococcus aureus [30] for example, but comparable to levels (~10 gene copies per million phages) reported for the marine Synechococcus cyanophage S-PM2 [22].
To place these mispackaging frequencies in perspective—podoviruses (T7-like cyanophages) that infect Prochlorococcus have been measured at concentration ranging from 3 to 7 × 105 mL−1 in a typical seawater sample from their habitat [20]—which means that the frequencies we observed for podovirus P-SSP7 would extrapolate to a potential of up to ~13–30 Mbp of encapsidated host DNA per mL of seawater, enough to encode 13,000–30,000 genes—roughly a 10−4 fraction of total Prochlorococcus DNA (see calculation in ‘Materials and methods’). Thus, even relatively low frequencies of DNA mispackaging inside capsids can represent a significant amount of Prochlorococcus genetic information available for HGT in the global ocean at any one time.
The difference we observe from one cyanophage to another probably reflects the different types of molecular machinery they use for packaging their genome inside capsids; among the three P-HM2’s packaging system appears to be the least prone to mispackaging errors. Of note, phage DNA packaging systems are not necessarily linked to phage morphotype [29]. To the best of our knowledge, all podoviruses infecting marine picocyanobacteria use a T7-like packaging mechanism [31, 32], and although all described cyanomyoviruses are T4-like [28, 33], their higher diversity makes it harder to ascertain that they share the same packaging mechanism. The siphovirus morphotype is by far the one encompassing the greatest overall biological diversity [34,35,36], and further studies are required to unveil their packaging mechanisms.
Effect of cell physiology on mispackaging
We next tested if different environmental factors, known to directly affect Prochlorococcus physiology, could have an impact on the mispackaging level of the three phages. We first grew the cells at different light intensities, a variable that not only influences the growth rate of Prochlorococcus [37] but also influences infection dynamics [38,39,40]. In fact, many cyanophages carry photosynthesis genes in their genomes to boost light-harvesting during infection [41,42,43]; like their hosts, cyanophage genes show daylight cycle transcriptional rhythms [38, 44]; and some cyanophages, including the myovirus P-HM2 used in this study, possess light-specific attachment mechanisms to ensure infection during daylight [38, 45].
Increasing growth light intensity significantly increased the mispackaging frequency—by orders of magnitude—for the podo- and siphovirus but did not impact the myovirus (Fig. 1d). The light-induced changes were large compared to the locus to locus variations in mispackaging frequency we observed in the different phages (Supplementary Fig. 1). Because the growth rate of the cultures also increases with light intensity [37] it is difficult to separate the effect of light vs growth rate. Examining the mispackaging frequency as a function of growth rate revealed, however, that growth rate alone cannot explain the results (Supplementary Fig. 2); mispackaging was greatest at the highest light intensity that was slightly inhibitory for growth. Thus, we hypothesized that mispackaging in the podo- and siphovirus might be caused by a physiological property related to the growth light intensity. We tested two such physiological parameters: reactive oxygen species (ROS) toxicity, and the rate of protein synthesis.
The role of reactive oxygen species in mediating differential levels of mispackaging inside capsids
ROS are abundant in the surface ocean [46,47,48,49], and because it lacks the ability to produce catalase, Prochlorococcus is highly sensitive and dependent on other species to detoxify them [48, 50,51,52,53]. The light intensities used in our experiments did, for the most part, result in an increase in intracellular ROS relative to cells kept in the dark (Supplementary Fig. 3).
We thus tested different ways of modulating the ROS toxicity in the culture medium and looked at the impact on mispackaging frequency. To increase intracellular ROS levels, we used the herbicide paraquat, which is known to capture electrons from photosystem I resulting in ROS production [54, 55]. A paraquat treatment at 20 µM at the time of infection triggered an increase in the level of mispackaging inside P-SSP7, but the effect remained below statistical significance (Fig. 2a). To decrease intracellular ROS levels, we tested a coculture of Prochlorococcus MED4 grown over many generations with the ‘helper’ heterotrophic Alteromonas macleodii strain MIT1002—known to reduce ROS and Prochlorococcus oxidative stress via its catalase activity [48, 50, 56]—and compared it with the axenic control. The coculture showed a dramatic decrease in the level of mispacking inside P-SSP7 capsids (Fig. 2b). Taken together, these results did not reject the hypothesis that oxidative stress plays a role in the frequency of mispackaging; more experiments are needed to confirm their role.

a Increased oxidative stress (ROS) induced by Paraquat. b Decreased oxidative stress induced by the presence of Alteromonas, a ‘helper bacteria’ known to reduce oxidative stress when co-cultured with Prochlorococcus [50]. The ROS increase experiments were performed at 45 µmol Q m−2 s−1, to start at a moderate level of ROS inside cells, and the ROS scavenging experiments at 100 µmol Q m−2 s−1, to compare the effect with a control at an elevated ROS level. The p values were calculated in each graph using a Mann–Whitney U test.
Overall, the results suggest that ROS could play a role in mispackaging of host DNA inside cyanophage capsids, helping explain the positive correlation between light intensity and the mispackaging level. That the helper strain, Alteromonas, could entirely mitigate the impact of high light intensity on mispackaging frequency was somewhat surprising. While at first glance this makes extrapolating our results to field conditions—where Prochlorococcus is surrounded by other bacteria—challenging, there are several things to consider. First, Prochlorococcus cells in the upper meter of the surface oceans can experience irradiance levels well above the maximum light level used in this study so the ROS detoxification challenges under those conditions could exceed those encountered in our cultures. Second, copiotrophic bacteria like Alteromonas are not the dominant bacteria in Prochlorococcus’ habitat. And finally, other sources of ROS exist in the wild, such as rainfall or the production by other microbes [52, 57]. Thus although mispackaging could be alleviated by the presence of Alteromonas in extremely dense laboratory cultures, this does not mean that mispackaging does not occur in the wild.
Accumulation of empty capsids during infection may lead to a higher mispackaging frequency
The rate of protein synthesis is another physiological parameter directly influenced by growth light intensity. In the cyanobacterium Synechocystis sp. PCC 6803, for example, there is a general upregulation of the translational machinery with increasing light intensity, which keeps increasing even under conditions of photoinhibition, presumably due to increased turnover of proteins subject to photodamage [58]. Moreover, Puxty et al. recently described that increased light levels during infection of a Synechococcus myovirus resulted in faster capsid production, while the phage DNA synthesis remained unchanged [40]. Such a decoupling between phage protein and phage DNA synthesis rates, modulated by light intensity, is likely to be a general feature of cyanophages all of which essentially rely on light energy influx to power the infection process [38]. Thus, we hypothesized that if protein translation rates increase to levels that the phage cannot regulate, this could result in an accumulation of empty capsids during infection, increasing the likelihood of host DNA mispackaging events. Indeed, whatever the mechanism by which the host DNA gets loaded inside an empty capsid, the accumulation of latent empty capsids should in turn increase the mispackaging frequency, as long as host DNA is present. Data for marine Synechococcus myovirus Syn9 [59, 60] Prochlorococcus myovirus P-HM2 [39, 60] and podovirus P-SSP7 [61] show that host DNA degradation happens quickly, within a few hours of infection, but host DNA is never entirely depleted, suggesting that leftover fragments remain available for mispackaging inside capsids until the cell lyses.
Since DNA damage is a consequence of ROS toxicity [49, 62], ROS serves to augment the imbalance between phage protein synthesis (capsid production) and phage DNA synthesis. Kolowrat et al. showed that long-term acclimation of Prochlorococcus to chronic high light and UV exposure leads to a delay in chromosome replication, probably caused by DNA lesions and replication fork arrests which slow DNA synthesis [63]. Thus, whether by increasing protein translation rate (effect of higher light intensity), or decreasing the phage DNA replication efficiency (effect of ROS), this would lead to an imbalance between phage DNA replication and capsid production leading to the accumulation of latent empty capsids waiting for phage genome replication.
We first explored this ‘empty capsid’ hypothesis by inhibiting protein translation and DNA synthesis via the introduction of sublethal doses [64] of the antibiotics chloramphenicol and ciprofloxacin, respectively. Both antibiotics are expected to impact the phage infection but via inhibition of two different host metabolic activities essential for phage particle production: protein translation (chloramphenicol) and DNA synthesis (ciprofloxacin) [65]. We added the antibiotics at the time of infection with the podovirus and observed that, according to our prediction, inhibiting protein translation tended to decrease the frequency of mispackaging while inhibiting DNA synthesis tended to increase it (though the effect was barely significant) (Fig. 3a).

a Impact of protein (chloramphenicol) or DNA synthesis (ciprofloxacin) inhibition on mispackaging frequency during infection of MED4 by the podovirus. The p values were calculated by a Kruskal–Wallis test followed by a Dunn test. The trend of ciprofloxacin and chloramphenicol effect followed our predictions (ciprofloxacin increased the mispackaging frequency, chloramphenicol decreased it), though below statistical significance compared to the control, but their effect was significant when compared to each other. b Fraction (%) of empty capsids in the lysate at two light levels. c Representative electron micrograph of each cyanophage lysate revealing full (blue arrow) and empty (red arrow) capsids. White arrows show glycogen granules.
As a more direct way to test the hypothesis, we used negative stain transmission electron microscopy to evaluate the proportion of full and empty capsids from the different treatments (Fig. 3b, c, Supplementary Figs. 4, 5); more specifically, we measured ratios of full to empty capsids during infection by the three phages at two light intensities (Fig. 3b). The fraction of empty capsids rose from ~10 to ~50% for the podovirus with an increase in light intensity—supporting the idea that empty capsids should accumulate at higher light intensities (Fig. 3b). A similar trend was seen in the siphovirus. In contrast, the fraction of empty capsids for the myovirus did not change significantly as a function of light intensity and was roughly 30–40% under both conditions—consistent with lack of light intensity-dependent mispackaging for this phage (Fig. 1d).
The different behavior in response to light of the podo and siphovirus compared to the myovirus—both in terms of mispackaging and full/empty capsid ratios—cannot be explained solely from their different DNA packaging mechanisms (Table 2). It could at least partly be explained by auxiliary metabolic genes (AMGs) encoded in their genome (Table 2). The myovirus P-HM2 genome encodes for 11 different AMGs, including a CP12 homolog, which inhibits the Calvin cycle activity, and a TalC homolog, which reroutes resources to the pentose phosphate pathway for nucleotide production [60], while the podovirus P-SSP7 encodes only four AMGs including TalC, and the siphovirus P-HS2 encodes none (though it possesses other unknown ways to influence the host metabolism) [34]. Thus, the myovirus—better equipped to ‘control’ the host metabolism—might be more resilient in the face of different metabolic regimes imposed by the light level, for example.
To explore this aspect further, we evaluated the three cyanophage’s fitness—as measured by the production of extracellular phage genome copies per infected cell (related to burst size) and the length of the lytic cycle—at two different light levels (Table 3). Indeed, there was reason to suspect that mispackaging of host DNA inside capsids, and the accumulation of empty capsids during infection, might impact overall phage fitness. The results are in accordance with our prediction since the myovirus infection parameters did not vary in function of light, while it changed widely for both the podovirus and siphovirus. Comparing the podovirus and siphovirus patterns, however, we see distinct differences. While the length of the lytic cycle decreased for both under higher light, the production of extracellular phage DNA decreased for the podovirus and increased for the siphovirus. This result was unexpected, given their similar responses to light in terms of host DNA mispackaging and the proportion of empty capsids. This suggests that mispackaging is independent of cyanophage fitness.
Synthesis
This is the first report of the encapsidation of Prochlorococcus DNA in the capsids of cyanophages. While the myovirus showed a relatively low mispackaging level regardless of conditions, the podo- and siphoviruses showed significant levels of mispackaging at higher light intensities, representing a potential vector for horizontal gene exchange in Prochlorococcus by transduction. Our working model proposes that the dependence of mispackaging on light intensity may involve ROS production and/or an imbalance between DNA and protein synthesis during infection (Fig. 4)—a tendency that the myovirus P-HM2 might be able to mitigate thanks to tighter control of the host metabolism via AMGs in its genome.

Higher light levels produce higher intracellular ROS levels and push the host metabolism toward faster protein translation and/or slower DNA synthesis which causes an imbalance in the production rate of capsids and cyanophage genome copies in favor of capsids—a tendency that the myovirus P-HM2 might be able to mitigate thanks to a tighter control of the host metabolism via auxiliary metabolic genes (AMGs) in this cyanophage’s genome. The higher number of latent empty capsids during infection would then increase the likelihood of host DNA mispackaging events.
Interestingly, the underlying mechanisms we describe behind the mispackaging of host DNA inside cyanophage capsids are likely to extend beyond cyanophages. In particular, we observed that mispackaging can be impacted either positively or negatively by antibiotics, a result that could be relevant to the field of phage therapy [66, 67] which is often used in conjunction with antibiotics. In such instances, lateral transfer of DNA through phage capsids could have the unwanted effect of spreading antibiotic resistance genes.
Our results showing an effect of light irradiance on the physiology of infection has potentially significant spaciotemporal implications in the ocean ecosystem: is there greater mispackaging frequency, and production of empty capsids, at the surface compared to deeper in the water column? Are they higher overall in the summer compared to the winter? Are they higher around the equator? Each of these questions deserve dedicated investigations. Higher mispackaging frequencies likely lead locally to a higher flux of HGT, in turn impacting the distribution of picocyanobacteria population diversity [4, 9]. Higher production of empty capsids could impact the dynamics of particulate organic matter, to which phage particles contribute significantly [68].
Many unknowns remain to be studied before we can quantify with any certainty the amount of transduction happening among Prochlorococcus cells in the marine environment. For example, only a subset of cyanophage capsids delivers their content, and when they do, multiple barriers exist to prevent the recombination of the incoming exogenous DNA in the recipient cell [13, 14, 69].
Future directions to advance our understanding of transduction in the marine environment will involve the direct measurement of host DNA encapsidation in environmental samples [70,71,72], or if ways to prepare cyanophage capsids loaded with known and traceable DNA are developed, the direct analysis of transduction events [16].
Materials and methods
Culture conditions
Axenic Prochlorococcus MED4 cells were grown under constant light flux (45 μmol photons m−2 s−1 unless otherwise specified) at 24 °C in natural seawater-based Pro99 medium containing 0.2-µm-filtered Sargasso Seawater, amended with Pro99 nutrients (N, P, and trace metals) as previously described [73]. Growth was monitored using bulk culture fluorescence measured with a 10 AU fluorometer (Turner Designs). The presence of heterotroph contaminants was tested as previously described [73], by diluting a small amount of the Prochlorococcus culture in three different marine purity test broth. Periodically, the potential presence of contaminants was also examined by flow cytometry with Sybr Green.
For the coculture, A. macleodii strain MIT1002 [74] (maintained in ProMM medium—Pro99 medium, as above, plus lactate, pyruvate, glycerol, acetate, and Va vitamins [75]) was spun down and washed twice in Pro99 medium to minimize carryover of trace organic compounds prior to being added to the Prochlorococcus MED4 cultures. The coculture was acclimated to constant light flux along with axenic cultures used for comparison.
Encapsidated DNA extraction
We have set up a protocol to purify Prochlorococcus cyanophage-encapsidated DNA free of any host DNA contamination, based on published procedures [22, 30, 70]. Briefly, we omitted the cesium chloride gradient step to optimize the DNA yield per sample, and the potential presence of vesicle-encapsulated DNA was removed by chloroform treatment [11].
In total, 30 mL cultures of exponentially growing Prochlorococcus MED4 cells were infected with cyanophage P-SSP7, P-HS2, or P-HM2 at a multiplicity of infection (MOI) ~0.1. The MOI had no effect on the mispackaging frequency (Supplementary Fig. 6). Cultures were preacclimated to the specified light level for several transfers prior to infection. After complete lysis, the lysates were centrifuged at 7000 g for 20 min and filtered on 0.2 µm Steriflip filters (MilliporeSigma, Burlington, MA, USA). Lysates were then concentrated using 15 mL Amicon centrifugal concentrators 100 kDa cutoff (MilliporeSigma) down to a volume of ~2 mL. Samples were treated with 10% volume of chloroform, and washed two times in SM buffer (50 mM Tris pH = 7.5, 100 mM NaCl, 8 mM MgSO4) using a 4 mL Amicon centrifugal concentrators 100 kDa cutoff (MilliporeSigma). Of note, the lysate volume was never concentrated below 0.5 mL during washes, as this resulted in significant losses. As a control for digestion, 0.3 µg of pUC19 plasmid DNA (NEB, Ipswich, MA, USA) was added in the sample. The nonencapsidated DNA was then removed by a 1 h incubation at 37 °C with 4 U TURBO DNase enzyme (Thermo-Fisher Scientific), 1X TURBO DNase Buffer, 0.1 mg mL−1, RNase A (Thermo-Fisher Scientific) and 1X cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA); After 1 h, 4 U TURBO DNase enzyme was added and incubated for another hour at 37 °C. Finally, the encapsidated DNA was extracted using a standard phenol-chloroform extraction, followed by purification on AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). Samples were eluted with 50 µL of MilliQ water and used for qPCR quantification.
Mispackaging quantification by qPCR
DNA samples were quantified using PicoGreen® (Thermo-Fisher Scientific, Waltham, USA) and quantitative PCR reactions were performed with the QuantiTect Probe PCR Kit (Qiagen, Hilden, Germany), using 1 ng of extracted phage DNA in 25 µL reaction volume. Primers for the six amplicons from the Prochlorococcus MED4 genome used in all experimental conditions to evaluate mispackaging frequency are listed in Table 1. Amplification was carried out in a CFX96 thermocycler (Bio-Rad, Hercules, CA, USA), cycling conditions were 15 min at 95 °C, followed by 40 cycles of denaturing for 15 s at 94 °C and annealing for 30 s at 55 °C. In each run, the DNA sample was compared to a serial dilution of MED4 genomic DNA from 1 × 10−9 g to 1 × 10−12 g. The number of encapsidated gene copies was calculated with the formula:
GCN: gene copy number in the PCR reaction; C: measured concentration from standard curve (ng/µL); V: volume of PCR reaction (µL); M: molecular weight of host chromosome (g/mol); Na: Avogadro’s number; k: number of gene repeats in the genome. The results are then expressed in gene copies per million phages, by dividing the GCN value with the number cyanophage genome copies inside 1 ng of cyanophage DNA, times 1 million. According to qPCR control reactions containing no input template, values below 0.5 gene copies per million phages could be noise (set as the detection limit).
To confirm the complete removal of nonencapsidated DNA, the same qPCR reaction was performed using primers M13F (gtaaaacgacggccagt) and M13R (caggaaacagctatgac) on each sample. If traces of the pUC19 plasmids were detected, samples were considered contaminated and discarded.
Extrapolation to the natural environment
To extrapolate our results for the podovirus to the natural environment we began with the following assumptions:
-
(1)
The value obtained with P-SSP7 applies to all Prochlorococcus-infecting podoviruses, measured at concentrations ranging from 300,000 to 700,000 per mL of seawater [20].
-
(2)
‘Mispackaged’ cyanophage capsids are filled with host DNA to capacity, that is ~45 kb, the average podovirus genome size [31].
-
(3)
The entire Prochlorococcus chromosome can be mispackaged at the mean value we obtained for the 6 loci we tested. So considering the mean value of 25.97 gene copies per million phages at 45 µmol Q m−2 s−1 (a conservative value, given that under laboratory conditions, mispackaging reached an average of 87.75 gene copies per million phages at 145 µmol Q m−2 s−1), we extrapolate a mispackaging frequency of 25.97 chromosome copies per million phage capsids in the environment.
A million phage capsids contain 45 kb × 106 = 45,000 Mbp of DNA so that given the assumptions, 25.97 chromosome copies per million phage capsids results in a ratio of phage:host encapsidated DNA of 45,000 Mbp/(25.97 × (Prochlorococcus genome size)) = 1045. In other words, 1 out of 1045 capsids is filled with host DNA. Considering 300,000 capsids per mL of seawater, that equates to ~13 Mbp of host encapsidated DNA per mL of seawater. Considering 700,000 capsids, it goes up to ~30 Mbp.
Obviously, this estimation is subject to vast uncertainty, but it serves as an indication of how mispackaging values in gene copies per million phages can relate to the context of Prochlorococcus and its phages in the wild.
Measurement of intracellular ROS
Intracellular ROS levels were measured via H2DCFDA assay (Thermo-Fisher Scientific), a cell-permeant that is oxidized to the fluorescent DCF form in the presence of ROS. Fluorescence per cell was measured on an Influx flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA); cells were excited with a blue 488 nm laser and analyzed for chlorophyll fluorescence (692/40 nm) and DCF (530/40 nm). Calculations of relative fluorescence per cell were done by normalizing red chlorophyll fluorescence per cell to 2 µm reference fluorescent beads (catalog no. 18604; Polysciences, Warrington, PA, USA) as previously described [76]. All flow cytometry data were analyzed using FlowJo version 10.6.1 (FloJo, LLC, Ashland, OR, USA). Triplicate Prochlorococcus MED4 cultures acclimated to the different light levels were harvested by centrifugation and resuspended in fresh Pro99 medium before the addition of reconstituted H2DCFDA to a final concentration of 30 µM. Cells were incubated in the dark at room temperature for 30 min, then exposed to their growth light intensity for 30 min, and samples were immediately run on the flow cytometer.
One-step method for cyanophages growth curve experiment
Infection dynamics were assayed using qPCR to enumerate both intracellular and extracellular cyanophage GCN [60, 61]. Triplicate mid-exponential cultures acclimated to high light (145 μmol Q m−2 s−1) and low light (50 μmol Q m−2 s−1) were infected at MOI ~1. To ensure the same MOI was used for a given phage at the two light levels, cell abundances were measured prior to infection and the addition of the cyanophage stock was adjusted accordingly. After 1 h, cultures were diluted 1:10 in fresh Pro99 medium, to stop further infection. Samples were taken immediately after dilution of the infected cultures and then at 2, 5, 8, 11, 14, 17, 20, 23, 26, and 31 h. In total, 200 µL samples were aliquoted into 96-well MultiScreen-HTS GV filter plate with 0.22 µm filters (MilliporeSigma, MSGVN2210) and filtered using a MultiScreen-HTS Vacuum Manifold (MilliporeSigma) into 96-well polystyrene microplates. Filtrates were used for qPCR quantification of extracellular cyanophage genome copies, while filters were used for qPCR quantification of intracellular cyanophage genome copies. Quantitative PCR conditions were the same as above, using primers gaacacttccgcccttacct and ctgcaacgaaagggaattgt for P-SSP7; cgtagagaaggtggcagagg and gaccttccgatgttaaattgc for P-HM2; gaattgctccaatcgtcgtt and cagctcgtgaaaacatcgaa for P-HS2.
In each biological replicate, the burst size was calculated as the total number of cyanophages produced during the lytic cycle (extracellular cyanophage GCN at the end of the lytic cycle minus extracellular cyanophage GCN at t0) divided by the number of infected cells at t0 (intracellular cyanophage GCN at t0). Note that cyanophage latent period lengths and burst sizes are difficult to establish and subject to significant uncertainty. Both parameters are better regarded as relative measures that are specific to the methods and conditions we used to obtain them.
Quantification of full and empty capsids fractions in cyanophage lysates by electron microscopy
Triplicate mid-exponential cultures acclimated to high light (145 μmol Q m−2 s−1) and low light (25 μmol Q m−2 s−1) were infected at MOI ~0.1. Once lysis of the culture was completed, samples were centrifuged at 7000 g for 20 min to remove bacterial debris and filtered onto 0.2 µm Steriflip filter units (MilliporeSigma). In total, 4 mL of each clarified lysate was subsequently ultracentrifuged at 32,000 RPM for 1 h, 4 °C in SW 60 Ti rotor (Beckman Coulter) to pellet cyanophage particles, which were resuspended in 50 µL of fresh and filtered Pro99 medium. In total, 10 μL drops of each suspension were then placed directly on glow discharged carbon-coated grids (EMS, USA) for 1 min. The grids were then blot-dried on filter paper, washed on a drop of ultrapure water, and negatively stained with 2% uranyl acetate in water. Specimens were examined on an FEI Tecnai T12 electron microscope operating at 80 kV at nominal magnifications of 18,500–48,000 and 1–3 μm defocus. Of note, P-HS2 particles were significantly aggregated after resuspension. To allow counting of this phage, Triton X-100 detergent (MilliporeSigma) was added to a final concentration of 0.1% for 1 h at room temperature before grid preparation.
The morphology of empty capsids (Fig. 3c, Supplementary Fig. 4) was consistent with examples from the literature [77,78,79,80], and could easily be differentiated from extracellular membrane vesicles (Supplementary Fig. 5). Empty capsids for the tailed cyanophages P-HM2 and P-HS2 were not tailed, consistent with the fact that they are immature capsids that have not been filled, and not capsids that delivered their content inside cells. A minimum of 100 particles (more frequently 200–400 particles) was counted on electron micrographs for each biological replicate to assess the full:empty capsid ratio in the lysate.
References
Flombaum P, Gallegos JL, Gordillo RA, Rincón J, Zabala LL, Jiao N, et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc Natl Acad Sci USA. 2013;110:9824–9.
Braakman R, Follows MJ, Chisholm SW. Metabolic evolution and the self-organization of ecosystems. Proc Natl Acad Sci USA. 2017;114:E3091–100.
Kashtan N, Roggensack SE, Rodrigue S, Thompson JW, Biller SJ, Coe A, et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science. 2014;344:416–20.
Kashtan N, Roggensack SE, Berta-Thompson JW, Grinberg M, Stepanauskas R, Chisholm SW. Fundamental differences in diversity and genomic population structure between Atlantic and Pacific Prochlorococcus. ISME J. 2017;11:1997–2011.
Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, DeLong EF, et al. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311:1768–70.
Berube PM, Rasmussen A, Braakman R, Stepanauskas R, Chisholm SW. Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. Elife. 2019;8:e41043.
Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF. A reverse ecology approach based on a biological definition of microbial populations. Cell. 2019;178:820–34.e14.
Bentkowski P, Van Oosterhout C, Mock T. A model of genome size evolution for prokaryotes in stable and fluctuating environments. Genome Biol Evol. 2015;7:2344–51.
Larkin AA, Blinebry SK, Howes C, Lin Y, Loftus SE, Schmaus CA, et al. Niche partitioning and biogeography of high light adapted Prochlorococcus across taxonomic ranks in the North Pacific. ISME J. 2016;10:1555–67.
Biller SJ, Schubotz F, Roggensack SE, Thompson AW, Summons RE, Chisholm SW. Bacterial vesicles in marine ecosystems. Science. 2014;343:183–6.
Biller SJ, McDaniel LD, Breitbart M, Rogers E, Paul JH, Chisholm SW. Membrane vesicles in sea water: heterogeneous DNA content and implications for viral abundance estimates. ISME J. 2017;11:394–404.
Taton A, Erikson C, Yang Y, Rubin BE, Rifkin SA, Golden JW, et al. The circadian clock and darkness control natural competence in cyanobacteria. Nat Commun. 2020;11:1688.
Popa O, Dagan T. Trends and barriers to lateral gene transfer in prokaryotes. Curr Opin Microbiol. 2011;14:615–23.
Popa O, Landan G, Dagan T. Phylogenomic networks reveal limited phylogenetic range of lateral gene transfer by transduction. ISME J. 2016;11:543–54.
Touchon M, Moura de Sousa JA, Rocha EP. Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr Opin Microbiol. 2017;38:66–73.
Jiang SC, Paul JH. Gene transfer by transduction in the marine environment. Appl Environ Microbiol. 1998;64:2780–7.
Kenzaka T, Tani K, Nasu M. High-frequency phage-mediated gene transfer in freshwater environments determined at single-cell level. ISME J. 2010;4:648–59.
Aminov RI. Horizontal gene exchange in environmental microbiota. Front Microbiol. 2011;2:158.
Sullivan MB, Waterbury JB, Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–51.
Baran N, Goldin S, Maidanik I, Lindell D. Quantification of diverse virus populations in the environment using the polony method. Nat Microbiol. 2017;340:1–11.
Parsons RJ, Breitbart M, Lomas MW, Carlson CA. Ocean time-series reveals recurring seasonal patterns of virioplankton dynamics in the northwestern Sargasso Sea. ISME J. 2012;6:273–84.
Clokie MRJ, Millard AD, Wilson WH, Mann NH. Encapsidation of host DNA by bacteriophages infecting marine Synechococcus strains. FEMS Microbiol Ecol. 2003;46:349–52.
Canchaya C, Fournous G, Chibani-Chennoufi S, Dillmann ML, Brüssow H. Phage as agents of lateral gene transfer. Curr Opin Microbiol. 2003;6:417–24.
Penadés JR, Chen J, Quiles-Puchalt N, Carpena N, Novick RP. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 2015;23:171–8.
Chen J, Quiles-Puchalt N, Chiang YN, Bacigalupe R, Fillol-Salom A, Chee MSJ, et al. Genome hypermobility by lateral transduction. Science. 2018;362:207–12.
Berube PM, Biller SJ, Hackl T, Hogle SL, Satinsky BM, Becker JW, et al. Single cell genomes of Prochlorococcus, Synechococcus, and sympatric microbes from diverse marine environments. Sci Data. 2018;5:180154–11.
Sabehi G, Lindell D. The P-SSP7 cyanophage has a linear genome with direct terminal repeats. PLoS ONE. 2012;7:e36710.
Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035–56.
Casjens SR, Gilcrease EB. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol. 2009;502:91–111.
Mašlaňová I, Doškař J, Varga M, Kuntová L, Mužík J, Malúšková D, et al. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ Microbiol Rep. 2012;5:66–73.
Labrie SJ, Frois-Moniz K, Osburne MS, Kelly L, Roggensack SE, Sullivan MB, et al. Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ Microbiol. 2013;15:1356–76.
Huang S, Zhang S, Jiao N, Chen F. Comparative genomic and phylogenomic analyses reveal a conserved core genome shared by estuarine and oceanic cyanopodoviruses. PLoS ONE. 2015;10:e0142962–17.
Clokie MRJ, Millard AD, Mann NH. T4 genes in the marine ecosystem: studies of the T4-like cyanophages and their role in marine ecology. Virol J. 2010;7:291.
Frois-Moniz K. Host/virus interactions in the marine cyanobacterium Prochlorococcus. Massachusetts Institute of Technology; 2014.
Sullivan MB, Krastins B, Hughes JL, Kelly L, Chase M, Sarracino D, et al. The genome and structural proteome of an ocean siphovirus: a new window into the cyanobacterial ‘mobilome’. Environ Microbiol. 2009;11:2935–51.
Huang S, Wang K, Jiao N, Chen F. Genome sequences of siphoviruses infecting marine Synechococcus unveil a diverse cyanophage group and extensive phage-host genetic exchanges. Environ Microbiol. 2012;14:540–58.
Moore LR, Chisholm SW. Photophysiology of the marine cyanobacterium Prochlorococcus: Ecotypic differences among cultured isolates. Limnol Oceanogr. 1999;44:628–38.
Liu R, Liu Y, Chen Y, Zhan Y, Zeng Q. Cyanobacterial viruses exhibit diurnal rhythms during infection. Proc Natl Acad Sci USA. 2019;63:201819689–201814082.
Thompson LR, Zeng Q, Chisholm SW. Gene expression patterns during light and dark infection of Prochlorococcus by cyanophage. PLoS ONE. 2016;11:e0165375–20.
Puxty RJ, Evans DJ, Millard AD, Scanlan DJ. Energy limitation of cyanophage development: implications for marine carbon cycling. ISME J. 2018;12:1273–86.
Lindell D, Jaffe JD, Johnson ZI, Church GM, Chisholm SW. Photosynthesis genes in marine viruses yield proteins during host infection. Nature. 2005;438:86–89.
Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F, Chisholm SW. Transfer of photosynthesis genes to and from Prochlorococcus viruses. PNAS. 2004;101:11013–8.
Puxty RJ, Millard AD, Evans DJ, Scanlan DJ. Shedding new light on viral photosynthesis. Photosynth Res. 2015;126:71–97.
Demory D, Liu R, Chen Y, Zhao F, Coenen AR, Zeng Q, et al. Linking light-dependent life history traits with population dynamics for Prochlorococcus and cyanophage. mSystems. 2020;5:e00586–19.
Jia Y, Shan J, Millard A, Clokie MRJ, Mann NH. Light-dependent adsorption of photosynthetic cyanophages to Synechococcus sp. WH7803. FEMS Microbiol Lett. 2010;310:120–6.
Cooper WJ, Zika RG, Petasne RG, Plane JM. Photochemical formation of hydrogen peroxide in natural waters exposed to sunlight. Environ Sci Technol. 1988;22:1156–60.
Gerringa LJA, Rijkenberg MJA, Timmermans R, Buma AGJ. The influence of solar ultraviolet radiation on the photochemical production of H2O2 in the equatorial Atlantic Ocean. J Sea Res. 2004;51:3–10.
Morris JJ, Johnson ZI, Szul MJ, Keller M, Zinser ER. Dependence of the cyanobacterium Prochlorococcus on hydrogen peroxide scavenging microbes for growth at the ocean’s surface. PLoS ONE. 2011;6:e16805.
Ziegelhoffer EC, Donohue TJ. Bacterial responses to photo-oxidative stress. Nat Rev Microbiol. 2009;7:856–63.
Morris JJ, Kirkegaard R, Szul MJ, Johnson ZI, Zinser ER. Facilitation of robust growth of Prochlorococcus colonies and dilute liquid cultures by ‘helper’ heterotrophic bacteria. Appl Environ Microbiol. 2008;74:4530–4.
Morris JJ, Lenski RE, Zinser ER. The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio. 2012;3:e00036–12.
Zinser ER. Cross-protection from hydrogen peroxide by helper microbes: the impacts on the cyanobacterium Prochlorococcus and other beneficiaries in marine communities. Environ Microbiol Rep. 2018;10:1–35.
Mella-Flores D, Six C, Ratin M, Partensky F, Boutte C, Le Corguillé G, et al. Prochlorococcus and Synechococcus have evolved different adaptive mechanisms to cope with light and UV stress. Front Microbiol. 2012;3:285.
Blot N, Mella-Flores D, Six C, Le Corguillé G, Boutte C, Peyrat A, et al. Light history influences the response of the marine cyanobacterium Synechococcus sp. WH7803 to oxidative stress. Plant Physiol. 2011;156:1934–54.
Abrashev R, Krumova E, Dishliska V, Eneva R, Engibarov S, Abrashev I, et al. Differential effect of paraquat and hydrogen peroxide on the oxidative stress response in Vibrio Cholerae Non O1 26/06. Biotechnol Biotechnol Equip. 2011;25:72–6.
Lindell D. The genus Prochlorococcus, Phylum Cyanobacteria. Prokaryotes. 2014; 829–45.
Zinser ER. The microbial contribution to reactive oxygen species dynamics in marine ecosystems. Environ Microbiol Rep. 2018;10:412–27.
Zavřel T, Faizi M, Loureiro C, Poschmann G, Stühler K, Sinetova M, et al. Quantitative insights into the cyanobacterial cell economy. Elife. 2019;8:273.
Doron S, Fedida A, Hernández-Prieto MA, Sabehi G, Karunker I, Stazic D, et al. Transcriptome dynamics of a broad host-range cyanophage and its hosts. ISME J. 2016;10:1437–55.
Thompson LR, Zeng Q, Kelly L, Huang KH, Singer AU, Stubbe J, et al. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc Natl Acad Sci USA. 2011;108:E757–64.
Lindell D, Jaffe JD, Coleman ML, Futschik ME, Axmann IM, Rector T, et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature. 2007;449:83–6.
Imlay JA. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol. 2013;11:443–54.
Kolowrat C, Partensky F, Mella-Flores D, Le Corguillé G, Boutte C, Blot N, et al. Ultraviolet stress delays chromosome replication in light/dark synchronized cells of the marine cyanobacterium Prochlorococcus marinus PCC9511. BMC Microbiol. 2010;10:204.
Laurenceau R, Bliem C, Osburne MS, Becker JW, Biller SJ, Cubillos-Ruiz A, et al. Toward a genetic system in the marine cyanobacterium Prochlorococcus. Access Microbiol. 2020;2:acmi000107.
Abedon ST. Phage-Antibiotic combination treatments: antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics. 2019;8:182.
Gordillo Altamirano FL, Barr JJ. Phage therapy in the postantibiotic era. Clin Microbiol Rev. 2019;32:31–25.
Schmidt C. Phage therapy’s latest makeover. Nat Biotechnol. 2019;37:1–6.
Guidi L, Chaffron S, Bittner L, Eveillard D, Larhlimi A, Roux S, et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature. 2016;532:465–70.
Oliveira PH, Touchon M, Rocha EPC. Regulation of genetic flux between bacteria by restriction–modification systems. Proc Natl Acad Sci USA. 2016;113:5658–63.
Colomer-Lluch M, Jofre J, Muniesa M. Antibiotic resistance genes in the bacteriophage DNA fraction of environmental samples. PLoS ONE. 2011;6:e17549.
Brown-Jaque M, Calero-Cáceres W, Espinal P, Rodríguez-Navarro J, Miró E, González-López JJ, et al. Antibiotic resistance genes in phage particles isolated from human feces and induced from clinical bacterial isolates. Int J Antimicrob Agents. 2017;51:1–35.
Larrañaga O, Brown-Jaque M, Quirós P, Gómez-Gómez C, Blanch AR, Rodríguez-Rubio L, et al. Phage particles harboring antibiotic resistance genes in fresh-cut vegetables and agricultural soil. Environ Int. 2018;115:133–41.
Moore LR, Coe A, Zinser ER, Saito MA, Sullivan MB, Lindell D, et al. Culturing the marine cyanobacterium Prochlorococcus. Limnol Oceanogr Methods. 2007;5:353–62.
Biller SJ, Coe A, Martin-Cuadrado A-B, Chisholm SW. Draft genome sequence of Alteromonas macleodii strain MIT1002, isolated from an enrichment culture of the marine Cyanobacterium Prochlorococcus. Genome Announc. 2015;3:e00967–15.
Berube PM, Biller SJ, Kent AG, Berta-Thompson JW, Roggensack SE, Roache-Johnson KH, et al. Physiology and evolution of nitrate acquisition in Prochlorococcus. ISME J. 2014;9:1195–207.
Olson RJ, Chisholm SW, Zettler ER, Altabet MA, Dusenberry JA. Spatial and temporal distributions of prochlorophyte picoplankton in the North Atlantic Ocean. Deep Sea Res A. 1990;37:1033–51.
Cuervo A, Dans PD, Carrascosa JL, Orozco M, Gomila G, Fumagalli L. Direct measurement of the dielectric polarization properties of DNA. Proc Natl Acad Sci USA. 2014;111:E3624–30.
Fang P-A, Wright ET, Weintraub ST, Hakala K, Wu W, Serwer P, et al. Visualization of bacteriophage T3 capsids with DNA incompletely packaged in vivo. J Mol Biol. 2008;384:1384–99.
Shen PS, Domek MJ, Sanz-Garcia E, Makaju A, Taylor RM, Hoggan R, et al. Sequence and structural characterization of great salt lake bacteriophage CW02, a member of the T7-like supergroup. J Virol. 2012;86:7907–17.
Manning KA, Quiles-Puchalt N, Penadés JR, Dokland T. A novel ejection protein from bacteriophage 80α that promotes lytic growth. Virology. 2018;525:237–47.
Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:e144.
Acknowledgements
This study was supported in part by the Simons Foundation (Life Sciences Project Award IDs 337262, 509034SCFY17, 647135, and SCOPE Award ID 329108, to SWC), and an NSF-EDGE grant (1645061 to SWC). We thank Nicki Watson for her help setting up the protocol for TEM visualization of cyanophage capsids, Cameron Haase-Pettingell for her recommendations on how to concentrate and purify cyanophage particles, Debbie Lindell and Dror Shitrit for their recommendations on the best methods to measure cyanophage infection parameters, and Thomas Hackl for his help with mispackaging frequency calculations.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laurenceau, R., Raho, N., Forget, M. et al. Frequency of mispackaging of Prochlorococcus DNA by cyanophage. ISME J 15, 129–140 (2021). https://doi.org/10.1038/s41396-020-00766-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00766-0
This article is cited by
-
Metagenome-derived virus-microbe ratios across ecosystems
The ISME Journal (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}