
Abstract
Variations in the gut microbiome have been associated with changes in health state such as Crohn’s disease (CD). Most surveys characterize the microbiome through analysis of the 16S rRNA gene. An alternative technology that can be used is flow cytometry. In this report, we reanalyzed a disease cohort that has been characterized by both technologies. Changes in microbial community structure are reflected in both types of data. We demonstrate that cytometric fingerprints can be used as a diagnostic tool in order to classify samples according to CD state. These results highlight the potential of flow cytometry to perform rapid diagnostics of microbiome-associated diseases.
Similar content being viewed by others


Introduction
Variations in the gut microbiome have been associated with changes in health state, such as obesity, inflammatory bowel diseases and diabetes [1,2,3]. Characterization of the microbiome is mostly done through analysis of the 16S rRNA gene. Because sequence-based surveys are becoming standardized, microbiome analysis shows great potential to be included in precision medicine [4]. Yet, sequence-based surveys are still budget limited and time intensive [5, 6].
Flow cytometry is a single-cell technology, able to measure up to thousands of individual cells in mere seconds. When applied to microbial communities, both morphological and physiological characteristics are recorded for every cell [7]. The aggregation of these cellular characteristics describes the status of a microbial community, which can be summarized by creating a cytometric fingerprint. This cytometric fingerprint, in turn, can be used to quantify community dynamics in order to relate it to space, time, or other external variables, such as a case and control status. Because of the strong connection between the microbiome and human health [8, 9], cytometric fingerprints have the potential to be used as a diagnostic tool to rapidly identify microbiome-associated diseases [10]. As such, they have been used to quantify changes in microbial community composition to study colitis in murine models [11], and therefore can serve as an information-rich alternative to quantify microbial diversity [12,13,14].
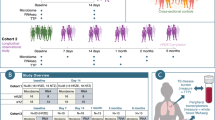
In this study, we reanalyzed the recently published data of a disease cohort containing samples diagnosed with Crohn’s disease (CD) (n = 29) and a healthy control (HC) group (n = 66). All samples have been analyzed independently by both flow cytometry and 16S rRNA gene amplicon sequencing [15]. The original study suggested a clear difference in microbial community composition between CD and HC samples based on 16S rRNA gene sequencing. In this work, we set forth to demonstrate that these differences are reflected in the cytometry data as well, and in addition, compare the predictive power of both technologies in a straightforward way. We used PhenoGMM, an adaptive cytometric fingerprinting strategy based on Gaussian mixture models, to cluster individual cells in operational groups [16]. This results in a relative cell count contingency table that describes samples by groups of phenotypically similar cells instead of grouped sequences. Random Forest classification was applied to classify unseen samples according to the disease state based on both types of data (Fig. 1). 16S rRNA gene amplicon sequencing resulted in a perfect identification (average area under the ROC curve (AUROC) = 1.0), with flow cytometry doing marginally worse (mean AUROC = 0.94). To assess the reproducibility of the workflow, flow cytometry train and test samples were additionally split according to the day at which they were measured (i.e., day 1, 2, or 3). The average AUROC resulted in values between 0.87 and 0.96 (SI Fig. 1).

The test set was created using leave-pair-out cross-validation. a Average receiver operating characteristic (ROC) curve (black) and standard deviation (SD, gray) for cytometry data. b Average ROC curve (black) and standard deviation (SD, gray) for 16S rRNA gene sequencing data. c Summary of the area under the ROC (AUROC) curves for both types of data, this time based on pairwise averaging of the test set. Each black dot represents the AUROC for an individual run, along with a visualization of the median. A boxplot displays the first and third quartile and the median line. Whiskers extend from the quartiles to 1.5 times the interquartile range.
We quantified the within-sample diversity in terms of richness (D0) and evenness (D2) for both types of data. Cytometric diversity, based on similarly grouped cells, was moderately correlated with taxonomic diversity based on genus abundances (Spearman’s rS(\(D_{0}^{\mathrm{16S}}, D_{0}^{\mathrm{FCM}}\)) = 0.30, P = 3.3 × 10−3; rS(\(D_{2}^{\mathrm{16S}}, D_{2}^{\mathrm{FCM}}\)) = 0.31, P = 2.5 × 10−3). Both taxonomic diversity (Fig. 2a) and cytometric diversity (Fig. 2b) were statistically significant markers in function of CD vs. HC, in which both the richness and evenness of gut microbiota were significantly lower for CD compared to HC samples (Mann–Whitney U test: P < 1 × 10−4). We also assessed which cytometric groups captured significant changes according to the disease state; 132 contained significantly more cell counts for CD than HC, while 103 groups contained significantly more cell counts for HC than CD (Mann–Whitney U test, adjusted P < 0.05 after Benjamini–Hochberg correction). The locations of these groups revealed a clear structure (Fig. 2c). In other words, structural differences in microbial community composition between healthy and disease state were captured by the cytometric fingerprints, which could be summarized in terms of the cytometric diversity.

Statistical differences were assessed using a Mann–Whitney U test. Each boxplot displays the first and third quartile and the median line. Whiskers extend from the quartiles to 1.5 times the interquartile range. Points that lie outside this range are visualized as outliers. a Within-sample diversity based on genus abundances as derived from 16S rRNA gene amplicon sequencing. b Within-sample diversity based on cytometric fingerprinting. c Location of the means of each flow cytometric operational group in the FL1-H–FSC-H scatterplot. Groups are annotated whether they contain significantly more cell counts for CD than HC (CD > HC), significantly less cell counts for CD than HC (CD < HC) or whether differences were not significant (NS), P-values were corrected for multiple testing using a Benjamini–Hochberg correction.
It is important to emphasize that it is not the aim of this study to benchmark our flow cytometry results with those of 16S rRNA gene sequencing. Both technologies generate different data types and have their own distinct methodological and computational biases, especially in the case of complex matrices such as fecal material [17]. In the studied data set, samples for flow cytometry analysis were prepared differently than those for the marker gene analysis because flow cytometry analysis aims to measure individual bacterial cells. As such, our computational workflow considered the successfully extracted and nucleic acid-stained single-cell fraction of the community, and missed any residual particle-associated cells and extracellular DNA. Marker gene analysis considers bulk DNA derived from both particle-attached cells, dead and living cells, free DNA, and is sensitive to DNA extraction efficiencies and gene copy number variations [18]. These baseline differences in methodological biases can be further amplified by the level of sample homogenization, which has been the subject of intense research [19, 20]. Many disease states, including CD, are also associated with differences in fecal moisture content, which may affect both cell and DNA extraction efficiencies and therefore necessitates further scrutiny and optimization of flow cytometry and marker gene analysis of fecal samples [21]. However, given these possible biases, we observed a consistent outcome for both approaches, which supports recent research showing that methodological variability caused by sample storage conditions outweighs sample homogenization variability [20].
Flow cytometry has become a vital part of clinical diagnostics [22], which is reflected by the fact that most major hospitals have a flow cytometry instrument at their disposal. This makes its application to the human microbiome readily available, yet cytometric analyses of the human microbiome are rarely considered. An exception is a recent analysis of different stages of chronic kidney disease [23] or the study of a simplified intestinal microbial community [24]. Although flow cytometry does not allow to inspect the genetic make-up of the microbial community, it may enable rapid (i.e., within hours) and affordable (i.e., $214 per day for the analysis of 100 samples [25]) screening of microbiome-associated diseases.
Materials and methods
Sample and data collection
Flow cytometric and sequencing data from the disease cohort were retrieved from the original study by Vandeputte et al. [15]. The cohort consists of 29 patients diagnosed with CD vs. 66 HC samples. The CD cohort is fully described in [26], HC samples were taken from the Flemish Gut Flora Project [27].
In brief, frozen aliquots of fecal material were analyzed by flow cytometry. Before freezing, samples were first mechanically homogenized. Next, aliquots were diluted 100,000 times in physiological solution and filtered using a sterile syringe filter with a pore size of 5 μm. 1 ml of the microbial suspension was stained with 1 μl Sybr Green I (1:100 dilution in dimethylsulfoxide; shaded 15 min incubation at 37 °C; 10,000 concentrate). Flow cytometric measurements were performed using an Accuri C6 (BD Biosciences), according to the protocol of Prest et al. [28]. Forward and side scatter were collected and used for further analysis, along with fluorescence information collected by the FL1 (533/30 nm) and FL3 (>670 nm) detectors. The flow rate was set at 14 μl per minute and the acquisition rate did not exceed 10,000 events per second. A threshold value of 2000 was applied on the FL1 channel. Instrument settings were identical for all samples, and measured twice at three different days, resulting in six replicate samples per patient. The coefficient of variation (CV) regarding the total cell counts per sample (n = 6) is lower than 0.66, with an average CV of 0.23 (SI Fig. 2).
Frozen aliquots of faecal material were also analyzed by 16S rRNA gene amplicon sequencing. Taxa were identified at the genus level based on similar 16S rRNA genes and the genus table was used as reported by Vandeputte et al. [15]. Procedures and data analysis are fully described in the original publication.
Analysis
The flow cytometry data were analyzed according to the following steps, which are laid out in more detail below: (1) preprocessing of the data (background and noise removal), (2) deriving cytometric fingerprints using a Gaussian mixture model, (3) performing patient status classification, and (4) calculating cytometric diversity metrics.
-
(1)
Preprocessing: all channels were transformed by f(x) = asinh(x). A fixed gating strategy, different from the original publication, was applied to all samples to remove noise in the FL1-SSC space (see SI Figs. 3 and 4). Additional automated denoising was performed using the FlowAI package (v1.4.4., target channel: FL1, changepoint detection: 150) [29].
-
(2)
Cytometric fingerprinting: each patient was characterized by six replicate samples. The FSC-A, SSC-A, FL1-A, and FL3-A channels were included in the analysis. Cytometric fingerprints were determined using a Gaussian mixture model. After train and test set creation (see below), 400 mixtures were fitted to 26,784 cells, based on a concatenation of data from 93 training patient times 288 cells per patient to determine the fingerprint template. The number of cells was determined by the replicate that contains the lowest number of cells in the entire data set; this amounted to 48 cells, and therefore, 288 cells per patient. Next, the fitted mixture model was used to assign cells to specific mixtures per patient, including test samples. In this step, replicate samples were subsampled to the lowest number of cells available for that specific patient, and then pooled. The number of included cells was between 288 and 39,414 cells per patient (SI Fig. 5), with a median value of 6078 cells. Finally, cell fractions were determined, resulting in a contingency table of relative cell counts per patient. This methodology, called PhenoGMM [16], is available as a wrapper function in the R package PhenoFlow [14].
-
(3)
Patient status classification: the cell count contingency table was compared to the genus abundance table to perform classification according to the patient status (CD vs. HC). Train and test sets were created using a leave-pair-out cross-validation strategy [30]. One CD sample and one HC were randomly left out as test samples, with the remainder used as samples for training. The training set was used to derive cytometric fingerprints and train a Random Forest classifier with 400 trees [31]. The hyperparameters were optimized with the remaining training set using stratified tenfold cross-validation and a random grid search [32]. Hundred random combinations of hyperparameter values were evaluated with the AUROC as a performance metric. The maximum number of variables that were considered at an individual split for a decision tree was randomly drawn from {1, …, K}, in which K denotes the number of mixtures or genera, and the minimum number of samples for a specific leaf was randomly drawn between 1, …, 5. Cross-validation, Random Forest classification, and performance evaluation were performed using the scikit-learn machine learning library [33]. ROC curves were created based on pooled predictions of the test set. AUROC values were calculated after averaging predictions per test pair [30]. Twenty-nine different test pairs were created in such a way that each CD sample was left out once. Two additional steps were carried out to address the robustness of the analysis. First, the whole procedure was repeated ten times per test pair. Second, to address variability between measurements at different days, train and test samples were split according to the day at which they were measured and the procedure was repeated.
-
(4)
Microbial diversity: the within-sample diversity was calculated based on relative cell and genera abundances for all samples. This was done using the Hill numbers [34], defined as D0 = S and D2 = 1/(ΣSi=1 pi2). S denotes the number of genera or non-empty mixtures and pi the relative abundance of genus or mixture i. Correspondence between taxonomic diversity and cytometric diversity was assessed using Spearman’s correlation with SciPy’s spearmanr() function [35]. Statistical differences between CD and HC cytometric and taxonomic diversity and cytometric groups were assessed according to a Mann–Whitney U test, using SciPy’s mannwhitneyu() function. P-values for the latter were adjusted using a Benjamini–Hochberg correction, by means of the multipletests() function from the statsmodels package [36].
Data availability
The genus table can be accessed as supporting information to the original publication [15]. Denoised raw flow cytometry data can be accessed via FlowRepository (ID:FR-FCM-ZYVH). Code and data to reproduce the analysis supporting the paper can be accessed via https://github.com/prubbens/PhenoGMM_CD.
References
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–5.
Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92.
Larsen N, Vogensen FK, Van Den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLOS ONE. 2010;5:e9085.
Kuntz TM, Gilbert JA. Introducing the microbiome into precision medicine. Trends Pharmacol Sci. 2017;38:81–91.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–32.
van Dorst J, Bissett A, Palmer AS, Brown M, Snape I, Stark JS, et al. Community fingerprinting in a sequencing world. FEMS Microbiol Ecol. 2014;89:316–30.
Müller S, Nebe-Von-Caron G. Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev. 2010;34:554–87.
Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. 2017;356:j831.
Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24:392–400.
Koch C, Müller S. Personalized microbiome dynamics—cytometric fingerprints for routine diagnostics. Mol Aspects Med. 2018;59:123–34.
Zimmermann J, Hübschmann T, Schattenberg F, Schumann J, Durek P, Riedel R, et al. High-resolution microbiota flow cytometry reveals dynamic colitis-associated changes in fecal bacterial composition. Eur J Immunol. 2016;46:1300–3.
Li WKW. Cytometric diversity in marine ultraphytoplankton. Limnol Oceanogr. 1997;42:874–80.
García FC, Alonso-Sáez L, Morán XAG, López-Urrutia Á. Seasonality in molecular and cytometric diversity of marine bacterioplankton: the re-shuffling of bacterial taxa by vertical mixing. Environ Microbiol. 2015;17:4133–42.
Props R, Monsieurs P, Mysara M, Clement L, Boon N. Measuring the biodiversity of microbial communities by flow cytometry. Methods Ecol Evol. 2016;7:1376–85.
Vandeputte D, Kathagen G, D’Hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–11.
Rubbens P, Props R, Kerckhof F-M, Boon N, Waegeman W. PhenoGMM: Gaussian mixture modelling of cytometry data enables efficient predictions of microbial biodiversity. biorXiv. 2019:641464. https://doi.org/10.1101/641464.
Gorzelak MA, Gill SK, Tasnim N, Ahmadi-Vand Z, Jay M, Gibson D. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PLOS ONE. 2015;10:e0134802.
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10:1–11.
Byrd DA, Chen J, Vogtmann E, Hullings A, Song SJ, Amir A, et al. Reproducibility, stability, and accuracy of microbial profiles by fecal sample collection method in three distinct populations. PLOS ONE. 2019;14:e0224757.
Liang Y, Dong T, Chen M, He L, Wang T, Liu X, et al. Systematic analysis of impact of sampling regions and storage methods on fecal gut microbiome and metabolome profiles. mSphere. 2020;5:1–13.
Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62.
Robinson JP, Roederer M. Flow cytometry strikes gold. Science. 2015;350:739–40.
Gryp T, Paepe KD, Vanholder R, Kerckhof F-M, Van Biesen W, Van de Wiele T, et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020;97:1230–42.
Schäpe SS, Krause JL, Engelmann B, Fritz-Wallace K, Schattenberg F, Liu Z, et al. The Simplified Human Intestinal Microbiota (SIHUMIx) shows high structural and functional resistance against changing transit times in in vitro bioreactors. Microorganisms. 2019;7:641.
Van Nevel S, Koetzsch S, Proctor CR, Besmer MD, Prest EI, Vrouwenvelder JS, et al. Flow cytometric bacterial cell counts challenge conventional heterotrophic plate counts for routine microbiological drinking water monitoring. Water Res. 2017;113:191–206.
Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut. 2016;65:1681–9.
Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–4.
Prest EI, Hammes F, Kötzsch S, van Loosdrecht MCM, Vrouwenvelder JS. Monitoring microbiological changes in drinking water systems using a fast and reproducible flow cytometric method. Water Res. 2013;47:7131–42.
Monaco G, Chen H, Poidinger M, Chen J, Magalhães JPD, Larbi A. FlowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics. 2016;32:2473–80.
Airola A, Pahikkala T, Waegeman W, Baets BD, Salakoski T. An experimental comparison of cross-validation techniques for estimating the area under the ROC curve. Comput Stat Data Anal. 2011;55:1828–44.
Breiman L. Random forests. Mach Learn. 2001;45:5–32.
Bergstra J, Bengio Y. Random search for hyper-parameter optimization. J Mach Learn Res. 2012;13:281–305.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30.
Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology. 1973;54:427–32.
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0–fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261–72.
Seabold S, Perktold J. Statsmodels: econometric and statistical modeling with Python. In: 9th Python in science conference. Austin, Texas, USA, 2010. https://conference.scipy.org/proceedings/scipy2010/pdfs/proceedings.pdf.
Acknowledgements
We thank Gunther Kathagen and Jeroen Raes for sharing the raw flow cytometry data. Part of the computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, the Hercules Foundation, and the Flemish Government department EWI. P.R. was supported by Ghent University (BOFSTA2015000501). W.W. received funding from the Flemish Government under the “Onderzoeksprogramma Artificielë Intelligentie (AI) Vlaanderen”.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Rubbens, P., Props, R., Kerckhof, FM. et al. Cytometric fingerprints of gut microbiota predict Crohn’s disease state. ISME J 15, 354–358 (2021). https://doi.org/10.1038/s41396-020-00762-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00762-4
This article is cited by
-
Latent dirichlet allocation for double clustering (LDA-DC): discovering patients phenotypes and cell populations within a single Bayesian framework
BMC Bioinformatics (2023)
-
Fast quantification of gut bacterial species in cocultures using flow cytometry and supervised classification
ISME Communications (2022)
-
An outlook on fluorescent in situ hybridization coupled to flow cytometry as a versatile technique to evaluate the effects of foods and dietary interventions on gut microbiota
Archives of Microbiology (2022)
