
Abstract
Cobalamin (vitamin B12) is an essential enzyme cofactor for most branches of life. Despite the potential importance of this cofactor for soil microbial communities, the producers and consumers of cobalamin in terrestrial environments are still unknown. Here we provide the first metagenome-based assessment of soil cobalamin-producing bacteria and archaea, quantifying and classifying genes encoding proteins for cobalamin biosynthesis, transport, remodeling, and dependency in 155 soil metagenomes with profile hidden Markov models. We also measured several forms of cobalamin (CN-, Me-, OH-, Ado-B12) and the cobalamin lower ligand (5,6-dimethylbenzimidazole; DMB) in 40 diverse soil samples. Metagenomic analysis revealed that less than 10% of soil bacteria and archaea encode the genetic potential for de novo synthesis of this important enzyme cofactor. Predominant soil cobalamin producers were associated with the Proteobacteria, Actinobacteria, Firmicutes, Nitrospirae, and Thaumarchaeota. In contrast, a much larger proportion of abundant soil genera lacked cobalamin synthesis genes and instead were associated with gene sequences encoding cobalamin transport and cobalamin-dependent enzymes. The enrichment of DMB and corresponding DMB synthesis genes, relative to corrin ring synthesis genes, suggests an important role for cobalamin remodelers in terrestrial habitats. Together, our results indicate that microbial cobalamin production and repair serve as keystone functions that are significantly correlated with microbial community size, diversity, and biogeochemistry of terrestrial ecosystems.
Similar content being viewed by others


Introduction
Cobalamin (vitamin B12), once referred to as “nature’s most beautiful cofactor” [1], plays an important role as a coenzyme involved in the synthesis of nucleotides and amino acids, in addition to carbon processing and gene regulation within all domains of life [2, 3]. Despite widespread metabolic dependency on cobalamin, only a relatively small subset of bacteria and archaea are capable of its production [4,5,6]. Cobalamin is present across natural systems in several chemical forms that differ in their upper ligand, including the enzymatically active forms of adenosylcobalamin (Ado-B12), methylcobalamin (Me-B12), hydroxocobalamin (OH-B12), and the inactivated form cyanocobalamin (CN-B12), of which the upper ligands are interchangeable through both enzymatic and abiotic processes [4, 7]. Cobalamin biosynthesis requires more than 30 enzymatic steps, via aerobic or anaerobic pathways [8,9,10] (Fig. 1), and represents a high genomic and metabolic burden for microbial producers.

Cobalamin biosynthesis and transport pathways, separating anaerobic and aerobic pathways with early and late incorporation of cobalt, respectively. Adapted from Doxey et al. [15] and Fang et al. [86]. Sequence homology and functional equivalency between aerobic and anaerobic pathway enzymes are indicated by horizontal arrows. Colors denote the groups of profile HMMs (see TIGR or PFAM HMM numbers adjacent to corresponding pathway steps) that contribute to various stages of cobalamin synthesis and/or salvage transport
Previous research on cobalamin production and its environmental significance has focused on marine systems where many eukaryotic primary producers are limited by the availability of this short-lived cofactor [11], demonstrating a significant role for cobalamin in controlling marine microbial community composition and productivity [11,12,13,14,15,16,17,18,19,20,21,22]. Metagenomic, whole genome, and biochemical analyses revealed that taxa affiliated with Proteobacteria and Thaumarchaeota are major marine cobalamin producers [15, 19], whereas marine Cyanobacteria produce pseudocobalamin, a closely related compound with a lower ligand substituted by adenine [19]. Recent understanding of marine cobalamin and pseudocobalamin was furthered by methodological advances enabling the direct measurement of cobalamin in environmental samples [23]. Together, the availability of metagenomic data and advances in analytical chemistry techniques provide an ideal framework for exploring microbial cobalamin production, consumption, exchange, and interdependencies in terrestrial ecosystems.
Soils harbor high densities of microbial biomass and are among the most diverse microbial community ecosystems on Earth [24,25,26]. The majority of soil biogeochemical processes are mediated by microorganisms [27] and the sustainability of agricultural soils relies on microbial communities that help mediate nutrient supplies to crops [28]. Therefore, elucidating factors that influence soil microbial diversity, activity, and physiology help understand controls on terrestrial biogeochemical functions [29, 30]. As a cofactor required by a majority of microorganisms [31], cobalamin availability and distribution in soils are constrained by microbial producers, which may have profound and unexplored impacts on terrestrial biogeochemical cycles. Because cobalamin-dependent enzymes include ribonucleotide reductase [32], methyltransferases [33], and reductive dehalogenases [34], cobalamin availability governs a wide range of microbial processes, such as DNA replication and repair [35, 36], regulation of gene expression [37], amino acid synthesis [38], CO2 fixation [39], recycling of carbon to the tricarboxylic acid (TCA) cycle [13], and aromatic compound detoxification [40].
Nearly seventy years ago, microbial cultivation efforts showed that a high proportion of cultured soil bacteria rely on exogenous cobalamin [41,42,43], with the implication that a cohort of soil microorganisms must serve as in situ sources of this essential cofactor. Since these early studies, there has been a near-complete lack of research into the microbiology of soil cobalamin production, presumably due to methodological limitations. To address this knowledge gap, we identify and quantify representative marker genes encoding enzymes that are broadly distributed throughout the cobalamin-producing pathway in soil metagenomes with profile hidden Markov models (HMMs) and relate the distribution and taxonomy of genes encoding cobalamin biosynthesis proteins to genes encoding cobalamin transport and salvage proteins. We adapt a selection of representative marker genes (cob/cbi/bluB) for the cobalamin biosynthesis pathway from a previous study of cobalamin production in the marine environment (Fig. 1; Supplementary information S1) [14]. In addition, we survey for the cobalamin transporter gene, btuB, encoding a TonB-dependent outer membrane cobalamin receptor and transporter [44, 45], with a basic architecture similar to that of iron siderophore transporters [46]. In order to examine the use of cobalamin, we also analyzed the distribution of several genes encoding cobalamin-dependent enzymes [2]. Because many microorganisms require cobalamin as a cofactor, we tested the hypothesis that there would be a correlation between the relative abundances of cobalamin consumers and producers/remodelers. In addition to metagenomic analyses, we quantified and characterized the standing stock of in situ cobalamin in representative soil samples and assessed potential links between cobalamin concentration and soil microbial community abundance.
Results
Soil metagenomic survey of cobalamin producers
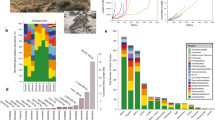
Using selected profile HMMs (Fig. 1; Supplementary material S1) and translated nucleic acid sequences, we analyzed 155 soil metagenomes from diverse geographical locations and land use types (Supplementary material S1) to assess the contribution of different taxa to cobalamin-production potential via cobalamin biosynthesis (cob/cbi/bluB) gene taxonomic profiles (Fig. 2). Sequences affiliated with the Proteobacteria phylum dominated the genetic potential to produce cobalamin, contributing 45.9% of cobalamin biosynthesis genes across all soil metagenomes, followed by Actinobacteria (24.9%), Firmicutes (6.2%), Acidobacteria (5.1%), and Thaumarchaeota (2.9%) (Fig. 2a). These phyla contributed similarly to each of the cobalamin biosynthesis gene groups that are defined based on the cobalamin biosynthesis pathway (Fig. 1; Supplementary material S1): corrin ring biosynthesis protein coding genes (“Group A”; seven genes), final synthesis and repair protein coding genes (“Group B”; four genes), and 5,6-dimethylbenzimidazole (DMB) synthase coding gene (“Group C”; one gene; bluB) (Fig. 2b–d) [47, 48]. Soil type influenced the composition of potential cobalamin-producing taxa (permutational multivariate analysis of variance, p < 0.01), and cobalamin biosynthesis gene relative abundance from each major potential cobalamin-producing phylum (Proteobacteria, Actinobacteria, Firmicutes, Acidobacteria, and Thaumarchaeota) varied significantly among different soil types (ANOVA; p < 0.01; Fig. S1). On average, proteobacterial cobalamin biosynthesis genes dominated, accounting for ~55% of cobalamin synthesis genes detected in forest, herb, and wetland soil metagenomes, whereas proteobacterial proportions were lowest in desert soils (~20%; Fig. S1).

Taxonomic composition among all 155 soil metagenomes for a all 12 cobalamin biosynthesis genes, b corrin ring biosynthesis genes, c final synthesis and repair genes, and d the DMB synthesis and activation genes. Only the top five phyla are shown individually, other phyla are collectively denoted as “others”
Several genera were associated with cobalamin synthesis based on the average relative abundance of HMM hits. For example, Streptomyces contributed the most to overall group A cobalamin biosynthesis genes (4.2%), followed by Bradyrhizobium (3.2%) and Nitrososphaera (2.8%) (Fig. S2A). Classifications of Bradyrhizobium (3.1%), Acidobacterium (2.9%), and Candidatus Koribacter (2.5%) were detected as the top three genera contributing to final synthesis and repair (Fig. S2B). The synthesis of DMB was attributed primarily to Bradyrhizobium (6.1%), Mycobacterium (4.8%), Streptomyces (4.5%), and Nitrososphaera (2.1%) (Fig. S2C). When evaluating the corresponding rpoB gene abundances for these same genera, 23.3% of the total soil microbial community, based on HMM hits to metagenomic sequence data, encoded corrin ring biosynthesis, 26.3% encoded final synthesis and repair, and 56.7% were associated with DMB production (Table 1).
Among all sampled metagenomes, 26 genera from 5 phyla (i.e., Proteobacteria, Actinobacteria, Firmicutes, Nitrospirae, and Thaumarchaeota) were associated with all 12 representative cobalamin biosynthesis genes (hereafter referred to as “complete”; Fig. S3). Together, these microorganisms with the complete genomic potential to produce cobalamin constituted 8.7 ± 2.3% of the global soil microbial community based on metagenome datasets, estimated by comparison to rpoB gene relative abundances of these same genera (Fig. 3 and S3). Of these, only three genera exceeded 1% average abundance (Solirubrobacter, 2.0%; Bradyrhizobium, 1.2%; Streptomyces, 1.1%). The number of genera with the potential for complete cobalamin synthesis showed a relationship with soil type (one-way ANOVA, p < 0.001, Fig. S4).

Proportion of genera encoding three cobalamin-dependent genes (i.e., mutA, metH, and rsmB) and those encoding the complete cobalamin biosynthesis pathway. Relative abundances were calculated as ratios to total rpoB gene abundances across all 155 soil metagenomes
Soil cobalamin producers and transporters
Across soil metagenomes, cobalamin-production and transport potential was evaluated by the relative abundance of cob/cbi/bluB and btuB genes, respectively. When examining the relationship between the relative abundance of cobalamin-producing enzyme coding genes (cob/cbi and bluB) and the cobalamin transporter protein coding gene (btuB) at the genus level across soil metagenomes, we observed mutual exclusion (permutation test p < 0.001) of these two gene complements for both rare biosphere members (Fig. 4a–c) and intermediate abundance taxa (between 0.1 and 5%; Fig. 4d–f). Thus, genera that were more represented among the cobalamin-synthesis enzyme coding gene pool were less well represented among the cobalamin transporter gene pool and vice versa. In general, dominant taxa encoded cobalamin transport potential while lacking genes associated with cobalamin production (Fig. 4g–i). However, there were several dominant (<10) genera that were affiliated with both cobalamin production and transport genes (Fig. 4g–i). The rare biosphere, as a whole, contributed to over half of the btuB gene abundance across all metagenomes, although these rare taxa collectively showed more cobalamin-producing potential than transport potential individually (Table 2).

Relationship between genes coding for cobalamin transport (btuB) and synthesis enzymes (cob/cbi/bluB) across 155 soil metagenomes. Color denotes the number of genera occurring with a given cob/cbi/bluB and btuB gene relative abundance. a–c represent a genus from the rare biosphere (rpoB gene relative abundances were below 0.1%), d–f intermediate abundance taxa (rpoB gene relative abundance between 0.1 and 5%), and g–i dominant taxa (rpoB gene relative abundance over 5%). Mutual exclusion between the cobalamin transporter encoding gene (btuB) and cobalamin synthesis enzyme genes (cob/cbi/bluB) were tested with 105 permutations, and significant (p < 0.05) for each gene group and either rare biosphere or intermediate abundance taxa. From top to bottom: Group A, corrin ring biosynthesis; Group B, final synthesis and repair; Group C, 5,6-dimethylbenzimidazole (DMB) synthesis
Genes encoding cobalamin-dependent enzymes (methionine synthase, metH; methylmalonyl-CoA mutase, mutA; ribosomal small subunit methyltransferase, rsmB) were also surveyed among all 155 soil metagenomes to evaluate potential demand for cobalamin by genera that either produce (with enzymes encoded by cob/cbi and bluB genes) or transport (with transporter encoded by btuB gene) this cofactor (Supplementary material S1). Based on global soil metagenomes, genera affiliated with selected cobalamin-dependent enzyme genes (i.e., metH, rsmB, and mutA) accounted for over 70% of the total rpoB-encoding community (Fig. 3). The total encoded potential for cobalamin use, measured as the total abundance of genes encoding cobalamin-dependent enzymes, was significantly greater (paired t-test, p < 0.001) than the encoded potential for cobalamin production and transport (i.e., sum of cob/cbi/bluB and btuB gene abundance; Fig. S5).
Soil cobalamin measurements
To further investigate differences in cobalamin production, we measured cobalamin and DMB concentrations in an independent set of 40 soil samples. Representative soil samples were collected from different environments and processed to quantify cobalamin concentrations from both water-leachable and non-water-leachable (i.e., intracellular and/or mineral-bound cobalamin) extracts (Supplementary material S2). Total cobalamins (sum of both water-leachable and non-water-leachable cobalamin) ranged between 0.06 and 6.84 pmol g−1 dry soil across all 40 soil samples tested, with an average of 1.19 pmol g−1 dry soil (Fig. 5, Supplementary material S2), and with >90% of extractable cobalamins obtained quantitatively and reproducibly with a single extraction (Supplementary material S3; Supplementary information Materials and Methods). Within the total cobalamins pool, the water-leachable fraction generally accounted for a small proportion in all samples (10.1 ± 11.9%; Fig. 5), indicating a strong association between cobalamins and the soil matrix and/or microbial biomass.

Concentration of total cobalamin (sum of both water-leachable and non-water-leachable cobalamin) and 5,6-dimethylbenzimidazole (DMB) measured in soils collected from the Canadian MetaMicroBiome Library (CM2BL) [82], the rare Charitable Research Reserve [83], and the Craibstone pH plots [84]. Water-leachable cobalamin and DMB only accounted for a small portion of total cobalamin and DMB. 1AT: Arctic Tundra; 2ATN: Arctic Tundra; 4TS: Tar sand; 5BF: Boreal coniferous forest; 6TD: Temperate deciduous forest; 7TR: temperate rain forest; 8NP: Northern peatlands; 9WLM: Wetland soil; 10AS: Agricultural soil (soy); 11AW: Agricultural soil (wheat); 13CO: Compost. AA: rare active agricultural site; D03: rare decommissioned agricultural (since 2003) site; D07: rare decommissioned agricultural (since 2007) site; D10: rare decommissioned agricultural (since 2010) site; CA: rare forest site; HB: rare forest site; IW: rare forest site. A full list of the samples and their details can be found in Supplementary material S2.xlsx
Similar to total cobalamin, the cobalamin lower ligand (DMB) also dominated in the non-water-leachable pool that contained an average of 82.4 ± 31.8% of the total extractable DMB ligand. The average concentration of DMB was higher than that of total cobalamin (Fig. 6a Supplementary material S2). In one sample, the concentration of DMB was ~40 times that of total cobalamin (rare Charitable Research Reserve IW soil, 15–30 cm). The presence of DMB in soil samples has been reported in a grove soil and creek bank soil [49], and these previous values fall in the range of DMB concentration measured in this study. The presence of DMB concentrations in excess of cobalamin in tested soil samples was consistent with soil metagenomic data (albeit from different soil samples) showing that the DMB biosynthesis gene (group C cobalamin biosynthesis) was more abundant than the other two cbi/cob gene groups (Fig. 6b; Tukey’s HSD p < 0.001). Within the cobalamin pool (OH-, CN-, Ado-, and Me-B12), we observed OH-B12 as the dominant cobalamin form (relative to CN-, Ado-, and Me-B12) based on its concentration in the total pool (sum of both water-leachable and non-water-leachable) (Supplementary material S2), consistent with previous findings from marine systems [19, 23, 50].

Measured total cobalamin (sum of both water-leachable and non-water-leachable cobalamin) and 5,6-dimethylbenzimidazole (DMB) concentrations in 40 soil samples (a), and cobalamin biosynthesis gene summary for 155 soil metagenomes (b). Cobalamin-producing genes in b were grouped as: Group A, corrin ring biosynthesis; Group B, final synthesis and repair; Group C, 5,6-dimethylbenzimidazole (DMB) synthesis. The 155 soil metagenomes are independent from the 40 soil samples used for cobalamin measurement. The center line in the box plot represents the median value
We tested for links between microbial diversity, DNA concentration, and cobalamin concentrations in the 40 soil samples analyzed for cobalamin chemistry (Supplementary material S2). When correlating total cobalamin concentration potential with diversity indices for 16S rRNA gene data for all 40 soil samples, significant relationships were observed between cobalamin concentration and Chao1 indices (Spearman’s rank correlation rho = 0.48, p < 0.01), richness (observed OTUs; rho = 0.46, p < 0.01), and Shannon indices (rho = 0.42, p < 0.01). Soil extracted DNA concentrations, a rough estimate of biomass, correlated positively with total cobalamin concentration (Fig. S6; R2 = 0.57, p < 0.01).
Discussion
Our analysis of terrestrial metagenomes shows that soil-dwelling bacterial taxa with the complete cobalamin-producing pathway represent, on average, less than 10% of the total microbial community based on comparisons to rpoB gene relative abundances (Fig. 3). Thus, the data suggest that de novo biosynthesis of cobalamin is a function that is carried out by a relatively small cohort of bacteria and archaea, presumably supplying this essential nutrient to other soil microorganisms, including dominant taxa. Different cohorts of microorganisms with relatively small abundances across diverse soil samples have the genetic potential to carry out this function. This is consistent with cobalamin producers influencing and potentially regulating the growth of other community members by shouldering the high metabolic cost of producing cobalamin [16, 51,52,53]. Our data indicate that putative cobalamin producers identified in this study serve a “keystone function” in their respective environments. Akin to the notion of keystone species, where individual species exert a disproportionately large effect on the community as a whole, relative to their abundance [54], a keystone function serves a role that is more important than the collective abundances of the genes/species that carry out that function within the community. We argue that in microbial communities, where phylogenetic diversity far exceeds functional diversity, keystone functions may be more relevant as an ecological concept than keystone species.
Based on previous surveys of putative cobalamin-producing phyla [15, 19], Proteobacteria were detected as numerically abundant cobalamin producers in both marine [15] and soil environments (Supplementary material S1). The phyla Cyanobacteria and Bacteroidetes/Chlorobi are hypothesized to be abundant pseudocobalamin and/or cobalamin sources in aquatic environments [15, 19], but they did not contribute a significant proportion of genes that encode cobalamin synthesis enzymes. Instead, Actinobacteria, Firmicutes, and Acidobacteria numerically dominated phyla in soil metagenomes that affiliated with gene sequences coding for enzymes that catalyze cobalamin biosynthesis. Despite being abundant cobalamin producers in marine environments (e.g., contributing over 80% of the cobalamin genes in some samples) [15, 19], thaumarchaeotal cobalamin synthesis genes were relatively rare within sampled soil metagenomes (Fig. 2). Nevertheless, because all known thaumarchaeotal cultures produce cobalamin [55], and the per cell cobalamin concentrations for members of the Thaumarchaeota in exponential phase can be orders of magnitude higher than for members of the Proteobacteria [19], Thaumarchaeota may be a more important source of soil cobalamin than gene counts alone would suggest [56]. Similarly, although Proteobacteria dominated cobalamin synthesis gene affiliations, this was not necessarily the phylum that contributed most to cobalamin biosynthesis because the genes may not be functional and there is no fixed relationship between gene presence and cellular quotas for cobalamin.
Our results show that, collectively, rare biosphere (<0.1% relative abundance) and intermediate abundance (0.1–5.0% relative abundance) genera are equally likely to be cobalamin producers (~50%) but the most abundant soil genera (>5% relative abundance) generally lack genes coding for cobalamin synthesis (Table 2). Because rare taxa have been demonstrated to mediate nutrient cycles [57] and plant production [58], our results suggest that future experiments should investigate the collective importance of rare cobalamin-producers to microbial community function more broadly. Based on our metagenome analysis, it is still not clear whether, within the same genus, cobalamin biosynthesis can be completed via a collaboration among species within that genus. However, a recent whole genome study of bacteria and Thaumarchaeota demonstrated that when a genome contains genes necessary for corrin biosynthesis, it is likely to also have genes for DMB biosynthesis and activation [19]. Although taxonomic resolution is limited at the species level, a similar issue also exists when exploring cobalamin production and/or transport at the genus level. We observed that several abundant genera showed a high proportion of both cobalamin synthesis and transport potentials. For example, Sphingomonas, an abundant genus, affiliated with >20% of soil btuB and cob/cbi/bluB genes in some soil metagenomes. However, when retrieving representative genomes and examining them at the species level, we noticed either a lack of an annotated btuB gene in the genomes or a btuB gene with a low annotation score, whereas cobalamin synthesis genes are present in different species, suggesting that individual species probably synthesize or transport cobalamin, but not both. A caveat is that taxonomic assignment for a functional gene requires increased resolution at lower taxonomic ranks, in this case at the species level, to avoid masking by a higher rank. Horizontal gene transfer can also complicate taxonomic classification, which might cause a cobalamin biosynthesis gene to be attributed to the wrong species, and consequently overestimate certain genera.
In addition to measuring cobalamin concentrations in soils, we measured DMB, which has no other known function in cells beyond serving as the lower ligand of cobalamin. We found relatively high concentrations of DMB compared with cobalamin. Group C cobalamin biosynthesis gene abundances were higher than group A and B cobalamin biosynthesis gene abundances (Fig. 6), suggesting greater biosynthesis potential of DMB compared with cobalamin. Because the bluB gene codes for the aerobic pathway for DMB synthesis, and is only reported for Gram-negative bacteria and members of the Thaumarchaeota [19, 48, 59], it is possible that other microorganisms could employ the anaerobic pathway encoded by bzaABCDE genes [59] and not be included in our analysis. Therefore, our estimate of microorganisms with the potential to produce DMB represents a lower limit. Free DMB would offer the potential for cobalamin-like compounds (i.e., pseudocobalamin) present in terrestrial environments to be remodeled into cobalamin. Microorganisms capable of transforming cobalamin-like compounds to cobalamin play a critical role in maximizing the impact of cobalamin production. For example, the purple bacterium, Rhodobacter sphaeroides, has been shown to remodel pseudocobalamin with amidohydrolase (CbiZ) and cobinamide-phosphate synthase (CbiB) [20, 60]. It is also possible that free DMB is released during cobalamin degradation, as has been seen in gut bacteria [61], or that DMB is inherently more stable than cobalamin. Nevertheless, our data suggest the possibility that DMB could be readily exchanged among different microorganisms through cross feeding, enabling subsequent remodeling within cells, as has been hypothesized for the gut microbiome [6].
Soil nitrogen cycle microorganisms, which are important for soil primary production, either produce or rely on cobalamin. We found that Nitrospira spp., which are diverse and abundant nitrite-oxidizing bacteria [62], were detected in soil metagenomes with the genetic capacity for complete cobalamin synthesis, in agreement with previous reports of genes encoding cobalamin-producing enzymes in the Candidatus Nitrospira defluvii genome [63]. Our data demonstrated that Nitrospira sequences were associated with ~0.5% of all cobalamin biosynthesis genes across 155 soil metagenomes (Fig. S3), spanning cold desert, desert, forest, grassland, and other terrestrial environments. Genome analysis demonstrated the presence of cobalamin-dependent enzymes involved in porphyrin synthesis and methyl-accepting chemotaxis, suggesting a requirement for this cofactor [2, 63]. The presence of these cobalamin-dependent nitrogen cycling microorganisms that also produce cobalamin suggests links between cobalamin-production and the aerobic nitrogen cycle in diverse soil biomes. This points to a possible mechanism for the loss of cobalamin-producing genes that is mediated by the ubiquity of microorganisms that catalyze N-cycle transformations and simultaneously produce cobalamin for broader community benefit.
Strong positive correlations between extracted DNA and cobalamin concentrations (Fig. S6) imply an importance for cobalamin supply in overall soil microbial community size. Although DNA extract yields from soil samples have been used to represent bacterial biomass [64], we acknowledge that the efficiency of extraction might also be influenced by soil type [65, 66] and DNA from dead cells as so-called relic DNA [67, 68]. However, we note that previous work found no correlation between intracellular cobalamin and microbial biomass in marine samples [19, 50]. We showed that the highest cobalamin concentration in a gram of soil can be two orders of magnitude greater than those measured in a liter of seawater [19], which is likely due to the lower amount of microbial biomass in 1 l of seawater relative to a gram of soil [69]. Although release and uptake mechanisms are not well understood, lower, but quantitatively significant, concentrations of water-leachable relative to non-water-leachable cobalamin in soils suggest that cobalamin is likely efficiently scavenged and relatively stable in soil. It is also possible that a portion of cobalamin in soil is mineral-bound and unavailable to microorganisms.
Cobalamin may be exuded into the soil matrix by living cells or released by cell lysis. In either case, cobalamin producers may play a role that is consistent with the Black Queen hypothesis [70]. Because most bacteria and archaea lack the ability to biosynthesize cobalamin, and thus depend on the small population of cobalamin producers for this cofactor (Fig. 3), cobalamin may play an important role in shaping and supporting soil microbial communities. Microorganisms with genes that encode the cobalamin-dependent methionine synthase (metH) alone accounted for ~90% of all microbial community members, almost eightfold more abundant than microbial cobalamin producers. Indeed, Giovannoni proposed that cofactor production by a subset of microbes in a community could be “the most widespread and important example of metabolic outsourcing” [16]. In the marine environment, organic compounds are hypothesized to be exchanged for cobalamin between bacteria and algae [11, 71,72,73]. The significant positive correlation between cobalamin concentration and diversity indices implies that a greater amount of cobalamin producers but lower amount of cobalamin consumers might foster a more diverse soil microbial community.
As novel cobalamin analogs are being discovered, it is likely that our understanding of how this cofactor relates to soil microbial ecology will continue to expand. For example, nitrocobalamin (NO2-cobalamin) was recently discovered in a marine ammonia-oxidizing archaea isolated from an oxygen deficient zone and other low oxygen water samples [56]. Because soil depth influences oxygen availability, future research will target this form of cobalamin in subsurface soil samples and better understand its relevance for AOA within aquatic and terrestrial habitats. In addition, future research should examine cobalamin transport in more detail. A recent study reported that the human gut microbiome contains corrinoid (a group of compounds containing corrin rings; cobalamin being one corrinoid) transporter families that can preferentially capture distinct corrinoids [4]. It would be intriguing to see if soil microorganisms are equipped with similar transport systems. Although relatively abundant in the marine environment [19], psuedocobalamin (i.e., a cobalamin analog with adenine as a lower ligand instead of DMB) was only detected in one soil sample (13CO, CM2BL; data not shown). Differences in cobalamin patterns between terrestrial and marine habitats might underpin distinct cobalamin usage mechanisms and consequently metabolisms among microorganisms.
Pioneering studies on cobalamin as a growth factor for soil bacteria in the 1950s observed bacteria that did not grow on nonselective media could recover when supplemented with soil extracts [41,42,43], and more than half of isolates that could only grow with a soil extract supplement also grew with cobalamin amendment alone [41], implicating cobalamin as a significant microbial growth factor in soil. Our current metagenomic and biochemical perspectives provide strong evidence for an important role played by cobalamin-producing taxa in relation to the much larger overall microbial community in terrestrial environments. Lower ligand remodeling mechanisms are likely to be common among soil microorganisms given the presence of higher concentrations of free DMB than total cobalamin, and a higher relative abundance of DMB synthase encoding genes than those encoding corrin biosynthesis enzymes in soil metagenomes. This study, by quantifying soil cobalamin and identifying the bacteria and archaea that potentially contribute to cobalamin synthesis, transport, dependence, and remodeling in soils, strongly implies that cobalamin producers help maintain an abundant and diverse terrestrial microbial community that, in turn, plays a critical role in the broader health of terrestrial ecosystems.
Materials and Methods
Soil metagenomes analysis
We analyzed 155 soil metagenomes available from the Metagenomics RAST Server (MG-RAST; accession numbers listed in Supplementary material S1), ensuring a diverse selection of soil habitats including grassland, agriculture, forest, desert, cold desert, wetland, pasture, herb, and tundra. A set of 12 genes and associated profile HMMs were selected to represent both aerobic and anaerobic cobalamin biosynthesis pathways (Fig. 1; Supplementary material S1), minimizing a potential pathway-specific bias, as described previously for the analysis of marine metagenomes [15, 19]. Marker genes for cobalamin production were classified into three categories (Fig. 1 and Supplementary material S1): group A, corrin ring biosynthesis; group B, final synthesis and repair; group C, DMB synthesis [11, 19]. Taxa included in any of the three groups must show the presence of all marker genes within that group from the same soil metagenome and the taxa with complete cobalamin synthesis pathways were assigned based on the presence of all 12 cobalamin biosynthesis genes in the same taxonomic group from the same soil metagenome. Total microbial communities associated with each metagenome were estimated by quantifying a single-copy gene for the RNA polymerase beta subunit (rpoB) [74,75,76]. The cobalamin transporter gene, btuB [45, 77], was selected as a marker for evaluating cobalamin uptake potential. Three cobalamin-dependent enzyme coding genes, metH, mutA, and rsmB [2] were included to further explore cobalamin utilization potential. The input HMM profiles (Supplementary material S1) for cob/cbi, bluB, btuB, metH, mutA, and rsmB genes were retrieved from TIGRFAM [78] or Pfam [79], and the phylogenetic marker, rpoB, from FunGene [80]. Metagenomic matches to cobalamin genes were identified, quantified, and taxonomically annotated using MetAnnotate [81]. The detailed MetAnnotate setup are summarized in Supplementary information Materials and Methods.
Soil cobalamin measurement
Soil samples for cobalamin and DMB measurements (Supplementary material S2) were composed of a collection of soils from three different projects: Canadian MetaMicroBiome Library (CM2BL) soils covering wide range of soil biomes across Canada [82], two distinctive land-uses along soil profiles (rare Charitable Research Reserve, Cambridge, Ontario) [83], and a pH gradient (4.5–7.5) of agricultural soils (Scottish Agricultural College, Craibstone, Scotland) [84]. Both water-leachable cobalamins and DMB, and non-water-leachable cobalamins and DMB were extracted and measured from all soil samples using a method modified from that developed by Heal et al. [19]. Detailed processes and 16S rRNA gene data sources are summarized in Supplementary information Materials and Methods.
Soil DNA was used for microbial community size estimation in these soils, and was extracted according to the manufacturer protocols with the PowerSoil DNA Isolation Kit (MO BIO, Carlsbad, CA), and quantified with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA).
Data analysis and visualization
Soil metagenome data were transformed into sample-based relative abundance (dividing gene reads of each taxa over total gene reads in the corresponding soil metagenome) before statistical analysis. All statistical analyses were carried out using R (V 3.2.3). Analysis of variance (ANOVA) was performed to test the effect of environmental factors on gene abundance or cobalamin concentration, followed by post-hoc Tukey HSD test. A Shapiro–Wilk normality test was used to check normality assumption prior to correlation analysis. Spearman’s rank correlations were employed to compare cobalamin concentrations and microbial biomass, and a simple linear regression was used to test for a significant linear relationship. The influence of soil types on cobalamin-producing phyla composition was evaluated by permutational multivariate analysis of variance (adonis) with the vegan (V 2.4.3) package in R. In order to test the relationship between cobalamin-producing and consuming taxa in the soil metagenomes studied, Spearman correlation coefficients between the relative abundance of cob/cbi/bluB and btuB genes were calculated and compared to the coefficient through 105 permutations to determine significance values. The phyloseq (V 1.14.0) [85] R package was used to preprocess OTU tables and calculate diversity indices.
References
Stubbe J. Binding site revealed of nature’s most beautiful cofactor. Science. 1994;266:1663–4.
Romine MF, Rodionov DA, Maezato Y, Anderson LN, Nandhikonda P, Rodionova IA, et al. Elucidation of roles for vitamin B12 in regulation of folate, ubiquinone, and methionine metabolism. Proc Natl Acad Sci USA. 2017;114:E1205–E1214.
Roth J, Lawrence J, Bobik T. Cobalamin (coenzyme B12): synthesis and biological significance. Annu Rev Microbiol. 1996;50:137–81.
Degnan PH, Barry NA, Mok KC, Taga ME, Goodman AL. Human gut microbes use multiple transporters to distinguish vitamin B12 analogs and compete in the gut. Cell Host Microbe. 2014;15:47–57.
Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genom. 2009;10:78.
Seth EC, Taga ME. Nutrient cross-feeding in the microbial world. Front Microbiol. 2014;5:350.
Cheng Z, Yamamoto H, Bauer CE. Cobalamin’s (vitamin B12) surprising function as a photoreceptor. Trends Biochem Sci. 2016;41:647–50.
Blanche F, Cameron B, Crouzet J, Debussche L, Thibaut D, Vuilhorgne M, et al. Vitamin B12: how the problem of its biosynthesis was solved. Angew Chem Int Ed. 1995;34:383–411.
Roth JR, Lawrence JG, Rubenfield M, Kieffer-Higgins S, Church GM. Characterization of the cobalamin (vitamin B12) biosynthetic genes of Salmonella typhimurium. J Bacteriol. 1993;175:3303–16.
Raux E, Schubert HL, Roper JM, Wilson KS, Warren MJ. Vitamin B12: Insights into biosynthesis’s mount improbable. Bioorg Chem. 1999;27:100–18.
Bertrand EM, McCrow JP, Moustafa A, Zheng H, McQuaid JB, Delmont TO, et al. Phytoplankton–bacterial interactions mediate micronutrient colimitation at the coastal Antarctic sea ice edge. Proc Natl Acad Sci USA. 2015;112:9938–43.
Sañudo-Wilhelmy SA, Gómez-Consarnau L, Suffridge C, Webb EA. The role of B vitamins in marine biogeochemistry. Annu Rev Mar Sci. 2014;6:339–67.
Bertrand EM, Allen AE. Influence of vitamin B auxotrophy on nitrogen metabolism in eukaryotic phytoplankton. Front Microbiol. 2012;3:375.
Bertrand EM, Saito MA, Jeon YJ, Neilan BA. Vitamin B12 biosynthesis gene diversity in the Ross Sea: the identification of a new group of putative polar B12 biosynthesizers. Environ Microbiol. 2011;13:1285–98.
Doxey AC, Kurtz DA, Lynch MD, Sauder LA, Neufeld JD. Aquatic metagenomes implicate Thaumarchaeota in global cobalamin production. ISME J. 2015;9:461–71.
Giovannoni SJ. Vitamins in the sea. Proc Natl Acad Sci USA. 2012;109:13888–9.
Panzeca C, Tovar-Sanchez A, Agustí S, Reche I, Duarte CM, Taylor GT, et al. B vitamins as regulators of phytoplankton dynamics. Eos Trans Am Geophys Union. 2006;87:593–6.
Tang YZ, Koch F, Gobler CJ. Most harmful algal bloom species are vitamin B1 and B12 auxotrophs. Proc Natl Acad Sci USA. 2010;107:20756–61.
Heal KR, Qin W, Ribalet F, Bertagnolli AD, Coyote-Maestas W, Hmelo LR, et al. Two distinct pools of B12 analogs reveal community interdependencies in the ocean. Proc Natl Acad Sci USA. 2017;114:364–9.
Helliwell KE, Lawrence AD, Holzer A, Kudahl UJ, Sasso S, Kräutler B, et al. Cyanobacteria and eukaryotic algae use different chemical variants of vitamin B12. Curr Biol. 2016;26:999–1008.
Frischkorn KR, Haley ST, Dyhrman ST. Coordinated gene expression between Trichodesmium and its microbiome over day–night cycles in the North Pacific Subtropical Gyre. ISME J. 2018;12:997–1007.
Walworth NG, Lee MD, Suffridge C, Qu P, Fu F, Saito MA, et al. Functional genomics and phylogenetic evidence suggest genus-wide cobalamin production by the globally distributed marine nitrogen fixer Trichodesmium. Front Microbiol. 2018;9:189.
Heal KR, Carlson LT, Devol AH, Armbrust EV, Moffett JW, Stahl DA, et al. Determination of four forms of vitamin B12 and other B vitamins in seawater by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2014;28:2398–404.
Roesch LFW, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–90.
Richter DD, Markewitz D. How deep is soil? BioScience. 1995;45:600–9.
Daniel R. The metagenomics of soil. Nat Rev Micro. 2005;3:470–8.
Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G. Microbial diversity and soil functions. Eur J Soil Sci. 2003;54:655–70.
Kennedy AC, Smith KL. Soil microbial diversity and the sustainability of agricultural soils. Plant Soil. 1995;170:75–86.
Bier RL, Bernhardt ES, Boot CM, Graham EB, Hall EK, Lennon JT, et al. Linking microbial community structure and microbial processes: an empirical and conceptual overview. FEMS Microbiol Ecol. 2015;91:fiv113.
Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–7.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 2003;278:41148–59.
Dickman S. Ribonucleotide reduction and the possible role of cobalamin in evolution. J Mol Evol. 1977;10:251–60.
Banerjee R. Chemistry and biochemistry of B12. New York: Wiley; 1999.
Janssen DB, Oppentocht JE, Poelarends GJ. Microbial dehalogenation. Curr Opin Biotechnol. 2001;12:254–8.
Blakley RL, Barker HA. Cobamide stimulation of the reduction of ribotides to deoxyribotides in Lactobacillus Leichmannii. Biochem Biophys Res Commun. 1964;16:391–7.
Blakley RL. Cobamides and ribonucleotide reduction: I. Cobamide stimulation of ribonucleotide reduction in extracts of Lactobacillus leichmannii. J Biol Chem. 1965;240:2173–80.
Ortiz-Guerrero JM, Polanco MC, Murillo FJ, Padmanabhan S, Elías-Arnanz M. Light-dependent gene regulation by a coenzyme B12-based photoreceptor. Proc Natl Acad Sci USA. 2011;108:7565–70.
Banerjee RV, Matthews RG. Cobalamin-dependent methionine synthase. FASEB J. 1990;4:1450–9.
Berg IA, Kockelkorn D, Buckel W, Fuchs G. A 3-Hydroxypropionate/4-Hydroxybutyrate autotrophic carbon dioxide assimilation pathway in Archaea. Science. 2007;318:1782–6.
Giedyk M, Goliszewska K, Gryko D. Vitamin B12 catalysed reactions. Chem Soc Rev. 2015;44:3391–404.
Lochhead AG, Thexton RH. Vitamin B12 as a growth factor for soil bacteria. Nature. 1951;167:1034–1034.
Lochhead AG, Burton M. Soil as a habitat of vitamin-requiring bacteria. Nature. 1956;178:144–5.
Lochhead AG, Burton MO, Thexton RH. A bacterial growth-factor synthesized by a soil bacterium. Nature. 1952;170:282–282.
Heller K, Mann BJ, Kadner RJ. Cloning and expression of the gene for the vitamin B12 receptor protein in the outer membrane of Escherichia coli. J Bacteriol. 1985;161:896.
Chimento DP, Mohanty AK, Kadner RJ, Wiener MC. Substrate-induced transmembrane signaling in the cobalamin transporter btuB. Nat Struct Mol Biol. 2003;10:394–401.
Buchanan SK, Smith BS, Venkatramani L, Xia D, Esser L, Palnitkar M, et al. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat Struct Biol. 1999;6:56–63.
Martens J-H, Barg H, Warren M, Jahn D. Microbial production of vitamin B12. Appl Microbiol Biotechnol. 2002;58:275–85.
Campbell GR, Taga ME, Mistry K, Lloret J, Anderson PJ, Roth JR, et al. Sinorhizobium meliloti bluB is necessary for production of 5, 6-dimethylbenzimidazole, the lower ligand of B12. Proc Natl Acad Sci USA. 2006;103:4634–9.
Crofts TS, Men Y, Alvarez-Cohen L, Taga ME. A bioassay for the detection of benzimidazoles reveals their presence in a range of environmental samples. Front Microbiol. 2014;5:592.
Suffridge C, Cutter L, Sañudo-Wilhelmy SA. A new analytical method for direct measurement of particulate and dissolved B-vitamins and their congeners in seawater. Front Mar Sci. 2017;4:11.
Lynch MDJ, Neufeld JD. Ecology and exploration of the rare biosphere. Nat Rev Micro. 2015;13:217–29.
Mas A, Jamshidi S, Lagadeuc Y, Eveillard D, Vandenkoornhuyse P. Beyond the black queen hypothesis. ISME J. 2016;10:2085–91.
Mills LS, Soulé ME, Doak DF. The keystone-species concept in ecology and conservation. BioScience. 1993;43:219–24.
Power ME, Tilman D, Estes JA, Menge BA, Bond WJ, Mills LS, et al. Challenges in the quest for keystones: identifying keystone species is difficult—but essential to understanding how loss of species will affect ecosystems. BioScience. 1996;46:609–20.
Qin W, Heal KR, Ramdasi R, Kobelt JN, Martens-Habbena W, Bertagnolli AD, et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int J Syst Evol Microbiol. 2017;67:5067–79.
Heal KR, Qin W, Amin SA, Devol AH, Moffett JW, Armbrust EV, et al. Accumulation of NO2-cobalamin in nutrient-stressed ammonia-oxidizing archaea and in the oxygen. Environ Microbiol Rep. 2018;10:453–7.
Pester M, Bittner N, Deevong P, Wagner M, Loy AA. ‘rare biosphere’microorganism contributes to sulfate reduction in a peatland. ISME J. 2010;4:1591–602.
Hol W, De Boer W, Termorshuizen AJ, Meyer KM, Schneider JH, Van Dam NM, et al. Reduction of rare soil microbes modifies plant–herbivore interactions. Ecol Lett. 2010;13:292–301.
Hazra AB, Han AW, Mehta AP, Mok KC, Osadchiy V, Begley TP, et al. Anaerobic biosynthesis of the lower ligand of vitamin B12. Proc Natl Acad Sci USA. 2015;112:10792–7.
Yi S, Seth EC, Men Y-J, Stabler SP, Allen RH, Alvarez-Cohen L, et al. Versatility in corrinoid salvaging and remodeling pathways supports corrinoid-dependent metabolism in Dehalococcoides mccartyi. Appl Environ Microbiol. 2012;78:7745–52.
Carkeet C, Dueker SR, Lango J, Buchholz BA, Miller JW, Green R, et al. Human vitamin B12 absorption measurement by accelerator mass spectrometry using specifically labeled 14C-cobalamin. Proc Natl Acad Sci USA. 2006;103:5694.
Daims H, Nielsen JL, Nielsen PH, Schleifer K-H, Wagner M. In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in wastewater treatment plants. Appl Environ Microbiol. 2001;67:5273–84.
Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Natl Acad Sci USA. 2010;107:13479–84.
Marstorp H, Guan X, Gong P. Relationship between dsDNA, chloroform labile C and ergosterol in soils of different organic matter contents and pH. Soil Biol Biochem. 2000;32:879–82.
Semenov M, Blagodatskaya E, Stepanov A, Kuzyakov Y. DNA-based determination of soil microbial biomass in alkaline and carbonaceous soils of semi-arid climate. J Arid Environ. 2018;150:54–61.
Leckie SE, Prescott CE, Grayston SJ, Neufeld JD, Mohn WW. Comparison of chloroform fumigation-extraction, phospholipid fatty acid, and DNA methods to determine microbial biomass in forest humus. Soil Biol Biochem. 2004;36:529–32.
Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland MS, Fierer N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol. 2016;2:16242.
Aoshima H, Kimura A, Shibutani A, Okada C, Matsumiya Y, Kubo M. Evaluation of soil bacterial biomass using environmental DNA extracted by slow-stirring method. Appl Microbiol Biotechnol. 2006;71:875–80.
Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–83.
Morris JJ, Lenski RE, Zinser ER. The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio. 2012;3:e00036–12.
Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature. 2005;438:90–3.
Cooper MB, Smith AG. Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr Opin Plant Biol. 2015;26:147–53.
Durham BP, Sharma S, Luo H, Smith CB, Amin SA, Bender SJ, et al. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Natl Acad Sci USA. 2015;112:453–7.
Case RJ, Boucher Y, Dahllöf I, Holmström C, Doolittle WF, Kjelleberg S. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol. 2007;73:278–88.
Dahllöf I, Baillie H, Kjelleberg S. rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl Environ Microbiol. 2000;66:3376–80.
Kembel SW, Wu M, Eisen JA, Green JL. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput Biol. 2012;8:e1002743.
Cadieux N, Kadner RJ. Site-directed disulfide bonding reveals an interaction site between energy-coupling protein TonB and BtuB, the outer membrane cobalamin transporter. Proc Natl Acad Sci USA. 1999;96:10673–8.
Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–3.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285.
Fish J, Chai B, Wang Q, Sun Y, Brown CT, Tiedje J, et al. FunGene: the functional gene pipeline and repository. Front Microbiol. 2013;4:291.
Petrenko P, Lobb B, Kurtz DA, Neufeld JD, Doxey AC. MetAnnotate: function-specific taxonomic profiling and comparison of metagenomes. BMC Biol. 2015;13:1–8.
Neufeld J, Engel K, Cheng J, Moreno-Hagelsieb G, Rose D, Charles T. Open resource metagenomics: a model for sharing metagenomic libraries. Stand Genom Sci. 2011;5:203–10.
Lu X, Seuradge BJ, Neufeld JD. Biogeography of soil Thaumarchaeota in relation to soil depth and land usage. FEMS Microbiol Ecol. 2017;93:fiw246–fiw246.
Kemp JS, Paterson E, Gammack SM, Cresser MS, Killham K. Leaching of genetically modified Pseudomonas fluorescens through organic soils: influence of temperature, soil pH, and roots. Biol Fertil Soils. 1992;13:218–24.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Fang H, Kang J, Zhang D. Microbial production of vitamin B12: a review and future perspectives. Micro Cell Factor. 2017;16:15.
Acknowledgements
We thank Noah Fierer for providing partial soil metagenomes, Michael Lynch and Jackson Tsuji for computational method suggestions, Norman Tran for assistance with data visualization, and Wei Qin for valuable discussions. The Canadian MetaMicroBiome Library (CM2BL), rare Charitable Research Reserve, and the Craibstone pH plots are thanked for soil samples; Graeme Nicol is thanked for sampling the Craibstone plots originally. Laura Carlson, Regina Lionheart, Haley Leatherman, and Yasemin Lopez assisted with cobalamin extraction and UPLC/MS analysis. This work was supported by grants from the Simons Foundation (SCOPE Award ID 329108, AEI; Award ID 598819, KRH) and NSF award OCE1046017 (AEI). KRH acknowledges NSF Graduate Research Fellowship Program (GRFP). ACD and JDN acknowledge Discovery Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Author information
Authors and Affiliations
Contributions
XL, KRH, ACD, AEI, and JDN conceived the study. XL and KRH performed the primary data analysis. ACD contributed to data analysis, and KRH, AEI, and JDN contributed to data interpretation and visualization. XL drafted the paper and incorporated revisions from all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, X., Heal, K.R., Ingalls, A.E. et al. Metagenomic and chemical characterization of soil cobalamin production. ISME J 14, 53–66 (2020). https://doi.org/10.1038/s41396-019-0502-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0502-0
This article is cited by
-
A framework for the targeted recruitment of crop-beneficial soil taxa based on network analysis of metagenomics data
Microbiome (2023)
-
Growth rate is a dominant factor predicting the rhizosphere effect
The ISME Journal (2023)
-
Inter-bacterial mutualism promoted by public goods in a system characterized by deterministic temperature variation
Nature Communications (2023)
-
Vitamin B12 is not shared by all marine prototrophic bacteria with their environment
The ISME Journal (2023)
-
Availability of vitamin B12 and its lower ligand intermediate α-ribazole impact prokaryotic and protist communities in oceanic systems
The ISME Journal (2022)
