
Abstract
Thaumarchaeota are responsible for a significant fraction of ammonia oxidation in the oceans and in soils that range from alkaline to acidic. However, the adaptive mechanisms underpinning their habitat expansion remain poorly understood. Here we show that expansion into acidic soils and the high pressures of the hadopelagic zone of the oceans is tightly linked to the acquisition of a variant of the energy-yielding ATPases via horizontal transfer. Whereas the ATPase genealogy of neutrophilic Thaumarchaeota is congruent with their organismal genealogy inferred from concatenated conserved proteins, a common clade of V-type ATPases unites phylogenetically distinct clades of acidophilic/acid-tolerant and piezophilic/piezotolerant species. A presumptive function of pumping cytoplasmic protons at low pH is consistent with the experimentally observed increased expression of the V-ATPase in an acid-tolerant thaumarchaeote at low pH. Consistently, heterologous expression of the thaumarchaeotal V-ATPase significantly increased the growth rate of E. coli at low pH. Its adaptive significance to growth in ocean trenches may relate to pressure-related changes in membrane structure in which this complex molecular machine must function. Together, our findings reveal that the habitat expansion of Thaumarchaeota is tightly correlated with extensive horizontal transfer of atp operons.
Similar content being viewed by others

Introduction
The ammonia-oxidizing archaea (AOA) are among the most abundant and ubiquitous microorganisms on Earth [1,2,3]. First identified as the dominant ammonia-oxidizing population in the upper ocean and soils [4, 5], subsequent studies also demonstrated a major presence in acidic and geothermal habitats [6,7,8]. Detailed molecular surveys of environmental distribution, based primarily on amplicon sequencing of the amoA coding for the alpha subunit of ammonia monooxygenase (AMO), further extended their habitat range to include the hadopelagic zone of the oceans [9].
Following the initial description of Nitrosopumilus maritimus SCM1 [10, 11], analysis of the physiology of AOA has been aided by cultivation of additional ecotypes, including neutrophilic Nitrososphaera species and obligately acidophilic Candidatus (Ca.) Nitrosotalea devanaterra Nd1 isolated from soils [12,13,14], and thermophilic Nitrosocaldus species enriched from geothermal habitats [15,16,17]. Among open ocean populations, amoA-based surveys point to a depth-dependent partitioning of distinct AOA ecotypes, including shallow (water column A) and deep marine groups (water column B) in the upper ocean [4, 18, 19] as well as a clade encompassing Nitrosopumilus species in the hadal zone below 6000 m [9, 20]. Although the deep ocean AOA ecotypes are presumably obligate or facultative piezophiles, adaptation to extreme hydrostatic pressure has not yet been explicitly considered as a major factor driving allopatric diversification.
Despite a habitat range that encompasses an extraordinarily wide range of physical/chemical conditions, all cultured AOA appear to rely solely on the energy generated from oxidation of ammonia that some AOA can also produce from urea or cyanate [11,12,13,14, 21]. AOA derive energy for anabolic processes by coupling ammonia oxidation to a copper-centric electron transfer system for generating a proton motive force (PMF) for ATP synthesis via an integral membrane ATPase [22]. The evolutionarily diverse archaeal ATPases (designated A-type ATPases) are functionally similar to the eukaryotic and bacterial F-type ATPases that catalyze ATP synthesis using a PMF [23]. However, they are structurally more similar to the vacuolar-type (V-type) ATPases of eukaryotes and some bacteria that function as proton pumps driven by ATP hydrolysis [23, 24]. Significant variation in subunit composition, structure, and mechanism of the archaeal variants is thought to confer adaptive advantage in the variety of extreme environments that archaea inhabit [25]. Since AOA are united by a single autotrophic metabolism dependent on ATP generation via a PMF, we examined the possible role that variation in ATPase composition and structure might play in their adaptive radiation into environments of low pH and high pressure.
Stable isotope probing (SIP) with 13C-labeled CO2 was used to selectively recover DNA from metabolically active AOA in acidic soils, significantly expanding the genomic representation of Thaumarchaeota active in acidic environments through the assembly of two novel genomes. Comparative analysis of these and available archaeal genomes and genomic fragments was then used to explore the evolutionary diversification of ATPases in relationship to archaeal habitats of varying pH and hydrostatic pressure. This revealed that the radiation of Thaumarchaeota into low-pH and high-pressure environments was clearly correlated with horizontal transfer of an operon encoding the same variant ATPase. The functional significance of this variant to adaptation to low pH is supported by our observations showing (i) increased transcription of the ATPase variant genes by an acid-tolerant AOA following transition from neutral to acidic growth conditions and (ii) higher growth rates at low pH of an Escherichia coli (E. coli) genetically modified to express the archaeal variant.
Materials and methods
Site description and soil sampling
Soil samples for DNA-SIP were collected at the long-term experimental field of the Tea Research Institute of the Chinese Academy of Agricultural Sciences (30°14′N, 120°09′E), Zhejiang Province, China [26, 27] in August 2011. Soil cores (20 cm long) were retrieved from a forest soil with a pH of 5.31 (site 1, FS) and tea orchard soil with a pH of 3.75 (site 1, TS). The latter was converted from the forest soil in 1974 [26, 27]. Both soils were 80 m apart and classified as quaternary red earth Ultisols. The sampling region was characterized by a subtropical wet monsoon climate with a mean annual temperature of 17 °C and a mean annual rainfall of 1533 mm. The tea orchard field plot received approximately 600 kg ha−1 urea-N, 280 kg ha−1 potassium sulfate, and 1125 kg ha−1 organic fertilizer (mainly rapeseed cake) annually since 1974 [26]. The 20 cm cores were collected in triplicate, homogenized, sieved through a 2.0 mm sieve, and stored at −20 °C until further analysis. Detailed soil properties are listed in Table S1.
For additional metagenomic comparisons, a second set of soil samples was collected from an acidic natural forest soil with pH 4.35 (site 2) in Yingtan City, Jiangxi Province of southern China (site 2, 24°29′N, 113°34′E) in June 2012 [28]. This soil was mainly covered with secondary evergreen broad-leaf camphor (Cinnamomum camphora L.) trees [28]. Three soil cores were collected, homogenized, sieved through a 2.0-mm sieve, and stored at −20 °C for further analysis.
DNA-SIP-based metagenomic analysis of AOA
To determine the pH response of AOA in soils, 10 g of TS and FS soil samples were suspended in 50 ml 10 mM phosphate buffer adjusted to pH 4.5 and pH 7.0, amended with 200 μg 15N-urea (15N atom, 98% excess) g−1 dry weight soil (d.w.s.), and incubated in the dark for 7 days at 25 °C (Fig. S1). Ammonia oxidation activity was monitored by production of 15N-labeled nitrate plus nitrite as previously described [29]. Growth of AOA and ammonia-oxidizing bacteria (AOB) were determined by real-time PCR quantification of AOA and AOB amoA gene copies as described before [30] (Table S2). To recover AOA genomic DNA, DNA-SIP microcosms were constructed at native acidic pH as previously described [31]. Three microcosm-labeling treatments were performed: 13CO2-labeled, 12CO2 (control), and 13CO2+C2H2 (control). Each microcosm was amended with 100 μg urea-N g−1 d.w.s. weekly and incubated at 25 °C in the dark. Total DNA was extracted at day 56 using a FastDNA Spin Kit for Soil (MP Biomedicals, Cleveland, OH, USA) and subjected to isopycnic density gradient centrifugation [31]. Fourteen DNA gradient fractions were recovered from each 5.1 ml ultracentrifugation tube, and the DNA was separated from CsCl by PEG6000 precipitation [32] and dissolved in TE buffer for the following metagenomic analysis. More details can be found in Supplementary Information.
Multiple displacement amplification (MDA) of the 13C-labeled DNA (fractions 4–6, 1.730–1.740 g ml−1) from DNA-SIP microcosms were carried out using the REPLI-g Single Cell Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol (Fig. S2). The 13C-labeled DNA before and after MDA were analyzed by metagenomic sequencing on Illumina HiSeq and/or PacBio RS II platforms. HiSeq reads and PacBio reads of the 13C-DNA mainly fell into two bins of ~40% and 70% GC content. Phylogenetic analysis suggested that reads representing AOA genomes were found in the low GC cluster. Two AOA genomes, namely, FS and TS, were assembled by Velvet from the low GC bins [33]. Genome completeness and contamination were estimated using comparison of 145 lineage-specific marker genes of archaea by CheckM [34]. The average amino acid identity (AAI) [35] was calculated as in Herbold et al. [36]. The gene annotation and comparative genomic analysis can be found in Supplementary Information.
Phylogenetic analyses
Inference of phylogenetic relationships among AOA and archaea was based on analysis of concatenated alignments of 36 and 122 conserved archaeal phylogenetic marker proteins [37,38,39] from 50 (meta)genomes of AOA and 158 selected representative genomes of Euryarchaeota and the archaeal superphyla TACK, Asgard, and DPANN (Table S3). Marker proteins in each genome were identified using hidden Markov models. Each protein was individually aligned using hmmalign [40]. The concatenated alignment was trimmed by TrimAL [41] with flag “-gt 0.5 -cons 50 -w 3.” Then maximum likelihood trees were built using IQ-TREE [42] with the best-fit model of “LG+I+G4” followed by extended model selection with FreeRate heterogeneity and 1000 times ultrafast bootstrapping [43].
The corresponding phylogenetic tree of ATPases was constructed based on the concatenation of seven subunits (A, B, D, E, F, I, and K) that were present in all selected archaeal genomes. ATPase genes were aligned by MAFFT [44] and the alignments were trimmed by TrimAL [41] with “-gappyout” flag. Inferences of maximum likelihood tree were achieved by IQ-TREE with “-MFP -bb 1000” flag for best-fit model selection and 1000 times ultrafast bootstrapping [43]. Phylogenies of amoA genes, aspartate carbamoyltransferase regulatory subunit (PyrI) genes, and thiosulfate/3-mercaptopyruvate sulfurtransferase genes flanking the atp operons of AOA were also constructed using IQ-TREE [42] as described above.
Physiological experiments and transcription analyses
In order to determine relative transcription activity of V-ATPase of Ca. Nitrosocosmicus oleophilus strain MY3 at different pH, MY3 was incubated in artificial fresh water medium adjusted to pH 5.2 and 5.5 by HOMOPIPES buffer and 7.5 by HEPES buffer with the addition of 500 μM NH4Cl at 25 °C, respectively [45]. Each pH treatment had three replicates. Ammonium and nitrite concentrations were determined using a Skalar SAN Plus segmented flow autoanalyzer (Skalar, Breda, the Netherlands) as described before [31]. To determine growth of MY3, SYBR Gold-stained cells were directly counted on 0.2 µm polycarbonate GTTP membranes (Merck Milipore, Germany) by fluorescence microscopy [45]. Total RNA was extracted from 500 ml of exponentially growing cultures grown at pH 5.5 and 7.5 using the RNeasy Mini Kit according to manufacturer’s protocol (Qiagen, Germany). cDNA was synthesized from the total RNA using the SuperScript First Strand synthesis system (Invitrogen, San Diego, CA) with RNaseOUT solution (40 U μl−1, Invitrogen) according to the manufacturer’s instruction. The quantity of RNA and cDNA were determined by a Nanodrop ND-100 UV-vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE). Transcript copy numbers of the genes of ntpA (encoding ATPase subunit A), 16S rRNA, amoA, 4-hydroxybutyrl-CoA dehydratase, and methylmalonyl-CoA mutase large subunit were quantified by real-time PCR using the iQ SYBR Green Supermix Kit on a C1000 CFX96 real-time PCR system (Bio-Rad). Finally, the cDNA transcripts of each gene were normalized to 1 ng of RNA for comparison between pH 5.5 and 7.5. More details of pressure experiments can be found in Supplementary Information.
pH-dependent growth experiments with E. coli expressing the thaumarchaeotal V-type atp operon
The V-type atp operon from the genome of Ca. Nitrosotalea okcheonensis CS was synthesized by Sangon Biotech (Shanghai, China) after codon optimization for E. coli (see the sequence in Supplementary Information), inserted into the NdeI-XhoI site of plasmid pET29a(+), and transformed into E. coli BL21(DE3) [46]. A single positive colony from LB agar plate with 50 μg ml−1 kanamycin was picked and inoculated into 100 ml of LB medium. It was incubated at 37 °C with shaking at 200 rpm until an OD600 of ~1.8 was reached. Then 0.3 ml of the culture was transferred into 100 ml of M9 medium (Na2HPO4, 6.78 g l−1; KH2PO4, 3 g l−1; NaCl, 0.5 g l−1; NH4Cl, 1 g l−1; MgSO4, 0.12 g l−1; CaCl2, 0.01 g l−1; glucose, 4 g l−1) with 15 μg ml−1 yeast extract, 50 μg ml−1 kanamycin, and 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at pH 4.2, 4.5, 5.0, and 7.0, respectively. These cultures were incubated at 37 °C with shaking at 200 rpm. The growth of E. coli were determined by optical density (OD) measurements at 600 nm every ~2–3 h. E. coli BL21(DE3) containing the empty vector pET29a(+) was used as the negative control. Each treatment was performed in three replicates.
Accession numbers of nucleotide sequences and genomes
The AOA metagenome assembled genomes (MAGs) obtained in this study have been deposited in NCBI under accession numbers RCMB00000000, RCMC00000000, and QURE00000000. Nucleotide sequences for 16S rRNA genes and amoA genes have been deposited in GenBank under accession numbers MF037823-MF037825 and MF041805-MF041807.
Results and discussion
13C-DNA-labeling of acidophilic Thaumarchaeota
15N-tracer experiments as well as archaeal amoA qPCR data suggested ammonia-oxidizing activity and growth of AOA in the acidic forest and tea orchard soils (Fig. S3 and Supplementary Information). Following labeling of urea-amended forest and tea orchard soils with 13C-labeled CO2 at in situ pH, DNA from two distinct Nitrosotalea-like AOA (FS and TS) sharing 87.0% amoA gene sequence identity was highly enriched in the 13C-DNA heavy fraction (Figs. S3c, d and S4). Addition of the inhibitor acetylene confirmed that incorporation of CO2 by AOA primarily depended on the energy generated from ammonia oxidation [47] (Fig. S3). Near complete MAGs of the forest type, FS (1.82 Mb, 89.66% completeness), and tea orchard type, TS (1.56 Mb, 82.85% completeness), were assembled from the 13C-labeled DNA metagenome (Table S4).
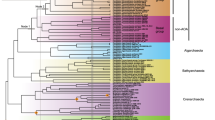
The AAIs between the FS and TS genomes and the four published Nitrosotalea genomes (Nd1, Nd2, CS, and SbT1) were 78.0–81.8% and 67.7–69.2%, respectively, and thus the newly recovered MAGs significantly increase the genomic representation of Ca. Nitrosotaleales [36] (Fig. S5). FS and TS shared 682 and 631 genes (91.7% and 84.9%), respectively, of the 743 core genome genes of AOA recently reported by Herbold et al. [36] (Fig. S6 and Tables S5–7) and possess a set of genes implicated by those authors in adaptation to low pH (Fig. S7 and Tables S8–11). These genes were partly acquired via horizontal gene transfer from acidophiles [36] and further studies are needed to explore exact function in acidophilic AOA. However, a feature not explored at depth in that study is our observation here that all Thaumarchaeota from acidic soils, including the new strains reported here (FS, TS, and AFS, a Nitrososphaera-like MAG recovered from another acidic soil, see Supplementary Information) and the four published Nitrosotalea strains as well as an acid-tolerant strain Ca. Nitrosocosmicus oleophilus MY3 regardless of their phylogenetic status [36, 45] (Figs. 1a and S8a), encode a specific energy-yielding ATPase (Figs. 1b and 2). This clade of ATPases forms a well-supported monophyletic group and is phylogenetically distinct from those encoded by neutrophilic soil Thaumarchaeota (Figs. 1b and S8d–i). This distribution strongly implies that variants of the energy-yielding ATPase are associated with niche specialization.

Comparative phylogeny of 36 conserved phylogenetic marker proteins and energy-yielding ATPases of ammonia-oxidizing archaea (AOA). All culture genomes, metagenome assembled genomes (MAGs), and single amplified genomes (SAGs) of AOA deposited in NCBI and JGI were collected, and only those containing the whole atp operons and at least 30 of the selected 36 and 100 of the selected 122 conserved phylogenetic marker proteins were used for comparative phylogenetic analysis (Figs. 3, S8, and S13). a Maximum likelihood species phylogenetic tree of AOA based on 36 concatenated phylogenetic marker proteins [IQ-TREE [42] with best-fit model LG+I+G4, 1000 ultrafast bootstraps]. “Nʹsphaerales” represents Nitrososphaerales and “Nʹtaleales” represents Nitrosotaleales. b Maximum likelihood tree of the ATPases based on the concatenation of the 7 subunits (A, B, D, E, F, I, and K) present in all AOA genomes [IQ-TREE [42] with best-fit model, 1000 ultrafast bootstraps]

Schematic illustration of gene operons encoding for energy-yielding ATPases of Thaumarchaeota and other selected archaeal representatives. Homologous genes are indicated by color and connected by lines. Arrows indicate the direction of transcription. The genes flanking the ATPase operons are also shown. “SCGC_AC_312_E17” represents marine Thaumarchaeota SCGC_AC_312_E17 in Fig. 1. “PyrB” represents aspartate carbamoyltransferase, “PyrI” represents aspartate carbamoyltransferase regulatory subunit, “sulfT” represents thiosulfate/3-mercaptopyruvate sulfurtransferase, “HA-Amt” represents high-affinity ammonia transporter, and “Cas4” represents CRISPR-associated exonuclease Cas4. The number in the brace indicates the number of genes that are not shown
Distribution of thaumarchaeotal ATPase variants in relationship to pH and hydrostatic pressure
The membrane rotary ATPases, including the A-type in archaea, V-type in eukaryotes and a few bacteria, and F-type in eukaryotes and bacteria, normally contain ~8−11 subunits, which form a hydrophilic catalytic A1/V1/F1 domain and a membrane-embedded A0/V0/F0 domain as well as one central and 1–3 peripheral stalks [25, 48, 49] (Table S12). The ATPase of acidophilic/acid-tolerant AOA is homologous to the proton-pumping V-ATPase of eukaryotes and the ion-pumping V-ATPase of Enterococcus hirae, each containing nine subunits encoded by a single operon structurally and phylogenetically distinct from A-ATPases of neutrophilic AOA [48] (Figs. 2 and 3). In contrast, only eight of the nine corresponding subunits of the A-ATPase of neutrophilic Thaumarchaeota have been identified by homology searches (Fig. 2). No clear homolog of subunit H (corresponding to subunit G in some studies of V-ATPases [49], thus termed as subunit H/G in this study) in neutrophilic AOA has yet been reported. For simplicity, we here refer to the ATPase variant present in acidophilic/acid-tolerant AOA (e.g., Nitrosotalea spp. and Nitrososphaera-like spp., such as AFS obtained from acidic soil) as V-type-like, and the variant present in cultured neutrophiles (e.g., cultured Nitrososphaera spp. [12, 14]) as A-type. Our comparative analyses clearly show that neutrophilic strains contain the A-type, whereas those retrieved from acidic environments share in common the V-type-like ATPase of acidophilic Nitrosotalea spp. (Figs. 1b, 2, and 3).

Comparative phylogeny of 36 conserved phylogenetic marker proteins and energy-yielding ATPases of Archaea domain. Only the genomes containing at least 30 of the selected 36 and 100 of the selected 122 conserved phylogenetic marker proteins as well as the whole atp operons were selected for comparative phylogenetic analysis (Figs. 1, S8, and S13). a Maximum likelihood species phylogenetic tree of archaea based on 36 conserved phylogenetic marker proteins [IQ-TREE [42] best-fit model LG+I+G4, 1000 ultrafast bootstraps]. Totally, 50 thaumarchaeotal genomes as well as 158 representative genomes of the phylum Euryarchaeota and superphyla TACK, Asgard, and DPANN were included. b Maximum likelihood tree of the ATPases based on the concatenation of the 7 major subunits (A, B, D, E, F, I, and K) present in all archaeal genomes [IQ-TREE [42] best-fit model, 1000 ultrafast bootstraps]. The V-ATPases from S. cerevisiae and E. hirae were also included in the phylogenetic analysis. The phylogenetic position of the S. cerevisiae V-ATPase (solid circles (●), dotted line) was inferred from the phylogeny of only subunits A and B, since its subunit compositions differed from archaeal ATPases. Some Archaeal branches whose positions in ATPase tree disagreed with their positions in the species tree are marked by stars
Our comparative analyses also revealed an unusual bifurcation of archaeal A-ATPase and V-ATPase variants with depth in the ocean. Prior ocean surveys revealed that marine AOA fell into three distinct phylogenetic clusters, termed Nitrosopumilus/Nitrosomarinus-like AOA, water column A (WCA), and water column B (WCB), across diverse oceanographic regions [4, 50] (Fig. 1a). Nitrosopumilus/Nitrosomarinus-like AOA are typically most abundant in coastal areas, while WCA and WCB represent two vertically segregated major phylotypes in the open ocean [18, 50] (Fig. 1a). WCA predominate in the epipelagic water and are less abundant below the upper mesopelagic zone [51, 52]. In contrast, WCB are found primarily below the euphotic zone, becoming dominant in the deeper mesopelagic and bathypelagic zones [51, 52]. Consequently, the WCA- and WCB-AOA have been thought to represent shallow and deep-adapted pelagic ecotypes, respectively. Notably, we found a very clear separation of these two ecotypes by distinct ATPase gene clusters (Figs. 1b and 2). The gene clusters encoding A-type ATPase occur in Nitrosopelagicus-like WCA-AOA (Figs. 1b and 2). In contrast, V-type-like ATPase gene clusters occur in WCB-AOA populations from the deeper ocean, sharing the conserved atp operon composition and organization with those of the acidophilic and acid-tolerant soil AOA (Figs. 1b and 2).
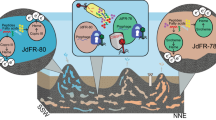
Marine AOA comprise nearly 40% of bacteria and archaea below 1000 m and are abundant in the deepest hadal zones of the oceans [1, 9]. A notable observation in the recent characterizations of the Challenger Deep was the greater representation of AOA ecotypes closely related to estuarine/coastal Nitrosopumilus species (genus Nitrosopumilus; [10]) between 6000 and 10,000 m [9, 20]. Similar observations were made in the Puerto Rico Trench at 8200 m, identifying Thaumarchaeota and SAR11 species as presumptive piezophilic populations at this depth [53]. These hadopelagic AOA share >98% 16S rRNA gene sequence identity and >80% whole-genome identity with N. maritimus SCM1 [20], a cultivated coastal marine AOA representative [10, 11]. The conserved A-type atp operons were found in both Nitrosopumilus-like hadopelagic and coastal marine AOA genomes (sharing >82.2% identity; Table S13). However, surprisingly, these deep sea variants also contain a complete gene cluster encoding the V-type-like ATPase present in acidophilic AOA (Figs. 1b and 2). The presence of a second ATPase thus appears to be related to the growth of Nitrosopumilus-like AOA in the hadopelagic environment.
Evidence and implication of functional significance
We experimentally evaluated the expression of the V-type-like ATPase in an AOA at different pH using Ca. Nitrosocosmicus oleophilus MY3 isolated from a coal-tar contaminated sediment [45]. This organism grows from pH 5.2 to 8.5 [45] (Figs. 4a and S9), enabling a comparative transcript analysis of the V-type-like ATPase relative to other genes of central metabolism at both circum-neutral and acidic pH. The relative transcript levels of genes coding for the 16S rRNA, the alpha subunit of the AMO, and enzymes in the pathway for CO2 fixation (4-hydroxybutyryl-CoA dehydratase and methylmalonyl-CoA mutase) remained relatively constant between pH 5.5 and 7.5 (Fig. 4b). In contrast, the relative transcript abundance for the gene encoding subunit A of the V-ATPase increased 3.5-fold at pH 5.5 relative to pH 7.5 (P < 0.01) (Fig. 4b). Since cultivated AOA species encode either an A-type or V-type ATPase, but not both, for generating ATP, the V-type of the acidophilic Thaumarchaeota apparently functions both in ATP synthesis and ATP-dependent proton pumping under low-pH conditions. A function in proton pumping is also consistent with a close phylogenetic association of the ATPase of acidophilic Thaumarcheaota and the proton/ion-pumping V-type ATPases of Saccharomyces cerevisiae and E. hirae [48] (Fig. 3b). A closer inspection of the amino acid sequences of the V-ATPases of the Thaumarchaeota suggests preferential selectivity for protons over sodium cations as the critical hydrophobic residues were detected around the ion-binding site in the primary structure of the vc/K rings (Supplementary Information, Fig. S10). Although a dual function of the thaumarchaeotal variant (ATP synthesis and ATP-dependent proton pumping) has yet to be experimentally verified, comparative structural analysis points to significant functional differences between the two types of ATPases found in Thaumarchaeota (Supplementary Information, Fig. S11).

Growth and transcription of V-ATPase genes in strain Nitrosocosmicus oleophilus MY3 at different pH. a Strain MY3 was grown in artificial fresh water medium (AFM) containing 500 μM NH4Cl at pH 5.2, 5.5, and 7.5, respectively. Growth was monitored via nitrite accumulation. The initial cell density after inoculation was ~1.0 × 105 cells ml−1. b Transcript abundance of 16S rRNA, amoA, 4-hydroxybutyryl-CoA dehydratase, methylmalonyl-CoA mutase large subunit, and ATPase subunit A were determined by quantitative PCR (qPCR) after reverse transcription of total RNA extracted from MY3 at mid-exponential phase (marked with arrows) at pH 5.5 and 7.5, respectively. All qPCR results were normalized to 1 ng of RNA for comparison between pH 5.5 and 7.5. Error bars represent the standard errors of triplicate incubations. For each gene, significance difference in measured cDNA copy numbers between pH 5.5 and 7.5 was detected using a t test (**P < 0.01)
In order to further experimentally investigate the role of thaumarchaeotal V-ATPase in adaptation to acidic environments, the V-ATPase encoding operon of Ca. Nitrosotalea okcheonensis CS was (after codon optimization) expressed in E. coli. Interestingly, E. coli BL21(DE3) expressing the heterologous V-ATPase from an acidophilic AOA grew significantly faster than the negative control E. coli BL21(DE3) strain carrying the empty vector [pET29a(+)] in M9 medium at an initial pH of 4.5 and 5.0 (Fig. 5b, c). After ~12 h of incubation of E. coli BL21(DE3) [pET29a(+)+V-atp] in M9 media, the OD600 increased to 0.23 and 0.56 at initial pH values of 4.5 and 5.0, respectively (Fig. 5b, c). This was significantly higher (P < 0.01) than the negative control, reaching OD600 values of only 0.09 and 0.35 (Fig. 5b, c) at pH 4.5 and 5.0, respectively. Similar growth rates of both E. coli strains were observed at an initial pH of 7.0 for the first ~10 h of incubation (Fig. 5d). However, as the pH dropped to below ~6.0 at longer incubation times, the growth of E. coli harboring V-atp was slightly faster than that of the negative control (P > 0.05; Fig. 5d). This pH drop reflects the release of organic acids by E. coli during its growth on glucose in M9 media [54]. Decreasing pH values were also observed in the growth experiments with initial pH values of 4.5 and 5.0 (Fig. 5b, c). No growth was observed at an initial pH of 4.2 (Fig. 5a). These results strongly suggest an important role of V-ATPases in the adaptation of acidophilic/acid-tolerant AOA to low-pH environments, although more work is clearly needed to biochemically characterize the thaumarchaeotal V-ATPases.

Growth of E. coli BL21(DE3) [pET29a(+)+V-atp] and E. coli BL21(DE3) [pET29a(+)] in M9 media at different pH. In all, 0.3 ml of the mother culture with an OD600 of ~1.8 was inoculated into 100 ml of M9 medium with 50 μg ml−1 kanamycin and 0.25 mM IPTG at pH 4.2 (a), 4.5 (b), 5.0 (c), and 7.0 (d), respectively, and incubated at 37 °C with shaking at 200 rpm. Growth of E. coli and media pH were determined at ~2-h intervals. Error bars represent the standard errors of triplicate incubations. Error bars smaller than symbols are not shown.
Since pH variability is low in the well-buffered deep ocean, the V-ATPase in deep marine AOA more likely functions in adaptation to other characteristics specific to deep waters. A central feature of deep marine environments is elevated hydrostatic pressure [55, 56]. Many microorganisms that populate the deepest depths of the ocean are obligate piezophiles, requiring high hydrostatic pressures for optimal growth [55]. Notably, it has been shown that high pressure will decrease the pKa of many weak acids in biological systems, including carbonic acids, carboxylic acids, and dihydrogen phosphoric acid, which would release more free protons inside cells and decrease the intracellular pH [57, 58]. Thus the presence of V-ATPase with the function of proton pumping may offer fitness advantages for deep marine AOA under high-pressure conditions.
In the absence of a hadopelagic isolate, we could only examine the sensitivity of an A-ATPase-type coastal organism, N. maritimus SCM1, to high hydrostatic pressure (Fig. S12). Under the tested experimental conditions, cells were partially inhibited at 10 MPa (100 atmospheres) and completely inhibited at a 20 MPa (200 atmospheres) equivalent to the pressure at approximately 2000 m (Fig. S12), suggesting the inability of the coastal isolate to grow at greater depths. Given their environmental distribution, the A-type ATPase containing WCA-AOA are likely also piezosensitive.
Additional evidence for the possible function of the V-ATPase at elevated pressures come from studies of yeast, showing that high hydrostatic pressure promotes the dissociation of protons from carbonic acid and glycolytic intermediates [59]. The fungal V-ATPase was found to be associated with maintenance of pH homeostasis by pumping out of protons from the acidified cytoplasm into the vacuole [59]. In addition, a recent transcriptional study of the obligate deep sea piezophilic archaeon Pyrococcus yayanosii reported that the V-ATPase genes were among the most tightly regulated genes in response to changes in hydrostatic pressure, suggesting that they play a key role in piezophilic adaptation [60]. However, the isolation and characterization of additional marine AOA species, representing a broader range of ecotypes, will be essential to confirming a functional role of the V-type-like ATPase in adaptation to high-pressure environments.
Horizontal transfer of the atp operon among Thaumarchaeota
The evolutionary radiation of Thaumarchaeota into low-pH and high-pressure habitats is correlated with a common deviation between ATPase and speciation phylogenies (Figs. 1 and 3). Maximum likelihood speciation phylogenies for ammonia-oxidizing Thaumarchaeota and the whole Archaea domain were constructed using concatenated alignments of 36 [38] and 122 [39] conserved marker genes (Figs. 1a, 3a, and S13) from 50 genomes of AOA and 158 genomes of Euryarchaeota as well as three archaeal superphyla, respectively (Table S3). The species trees based on the two different marker gene sets are highly consistent with each other (Figs. 1a, 3a, and S13). A complementary ATPase phylogeny was constructed based on a concatenated alignment of shared homologous subunits among archaeal ATPases (Figs. 1b and 3b). There is good congruence between the A-ATPase and the concatenated marker gene phylogenies of AOA members affiliated to the neutrophilic soil/hot spring groups and estuarine/coastal/shallow marine groups (Fig. 3). In contrast, phylogenetic relationships inferred from V-type-like ATPase and concatenated 36/122 conserved proteins for acidophilic/acid-tolerant Thaumarchaeota and presumptive piezophilic/piezotolerant Thaumarchaeota are discordant (Fig. 3). The ATPases of the latter group were found to be well separated from the lineage of soil neutrophilic Thaumarchaeota (Fig. 3b) and exclusively recovered as a well-supported monophyletic sister group of acidophilic Euryarchaeota (Fig. 3b). The ATPases of the four reported acidophilic Nitrosotalea species and those identified in thaumarchaeotal metagenomes recovered from acidic environments also affiliate with this lineage [36, 61] (Fig. 3b). The same observation was made for the acidophilic Micrarchaeota, Parvarchaeota, and recently identified acidophilic Marsarchaeota [62,63,64], and their ATPase-based phylogeny is incongruent with the corresponding species tree, being affiliated within the acidophilic/acid-tolerant lineage ATPases of Thaumarchaeota as well as Thermoplasmatales (Fig. 3).
Horizontal transfer of the entire atp operons (~8.9 kb in length) is the most parsimonious explanation for the presence of closely related V-type-like ATPases among distantly related archaeal taxa [65], including two distinctly related halophilic archaeal taxa, Halobacteriales and Nanohaloarchaeota (Fig. 3). Thus the phylogeny of acidophile V-type-like ATPase of these archaeal lineages tracks habitat, not organismal phylogeny. In contrast, the A-type ATPases of Thaumarchaeota were predominantly vertically inherited, following the general evolutionary history of speciation due to the congruence between A-ATPase and species tree (Fig. 3). Thermoplasmatales may have been the donor of V-ATPase in one or more of the other four archaeal phyla, because it has been reported that Thermoplasmatales exchanged dozens of other genes with archaeal partners in acidophilic environments [36, 66] (Fig. S14a), and their atp operons are flanked by genes encoding transposases and nucleases, key enzymes in mobilizing DNA fragments (Fig. 2). Our study provides evidence that horizontal transfer of V-type-like atp operon occurs widely in the Archaea domain, spanning the TACK and DPANN superphyla as well as Euryarchaeota phylum (Fig. 3), and the acquisition of V-ATPase via interphylum horizontal operon transfer (HOT) appears to play a significant role in the adaptive radiation of these archaeal species into a wide variety of habitats.
The AOA species tree was rooted with the A-type ATPase of the thermophilic Nitrosocaldales, suggesting that the last common ancestor of AOA harbored an A-type ATPase, and the V-type-like ATPase was later introduced into AOA by HOT (Fig. 1). An earlier analysis by Gubry-Rangin et al. [67] based on inferred changes in evolutionary rates also concluded that the split of the Nitrosotaleales/Nitrosopumilales lineage from the Nitrososphaerales lineage was coupled with expansion in low-pH environments. We postulate that, after the early divergence of the Nitrosotaleales/Nitrosopumilales lineage from the Nitrososphaerales, A- and V-ATPases have been frequently replaced in AOA via HOT (Fig. S14b). Notably, despite the frequent displacement of two distinct types of ATPases in Nitrosotaleales and Nitrosopumilales lineages (Fig. 1), their atp operons are always located between the genes encoding aspartate carbamoyltransferase and sulfurtransferase (Fig. 2). Both of these enzymes possess ATP-binding sites, and their enzyme activities are regulated in response to cellular ATP levels [68, 69], suggesting that they are possibly under the same transcriptional regulation as atp operons. Intriguingly, unlike the atp operons, the evolutionary histories of these neighboring genes are largely congruent with the AOA speciation history (Fig. S8b, c). Thus it appears that, in relatively recent HOT events, the thaumarchaeotal atp operons have undergone extensive in situ displacement with high precision between the conserved resident genes (Fig. 2). In this context, it is interesting to note that stand-alone Cas4 genes are located in the downstream flanking regions of atp operons, and these genes have been shown to be frequently recruited by archaeal viruses [70] (Fig. 2). Thus it is tempting to speculate that the diagnostic replacement of atp operons is associated with virus-mediated homologous recombination.
Our finding that single hadopelagic AOA species contain both types of ATPases (Fig. 2) is remarkable too, as it is strongly suggesting that extensive HOT of atp operons is a highly active and ongoing process within Thaumarchaeota. Consistent with the close phylogenetic relationship between Nitrosopumilus-like hadopelagic AOA and estuarine/coastal marine AOA, their A-type atp operons and flanking genes are highly conserved (Fig. S8b, c, Table S13). Thus A-ATPase is most likely the original ATPase type of hadopelagic AOA. In contrast, their V-type-like atp operons have unique neighboring genes (Fig. 2), suggesting that V-ATPase was acquired through relatively recent HOTs. The acquisition of V-ATPase is tightly correlated with the adaptive expansion of closely related Nitrosopumilus-like AOA populations from shallow waters to the high pressures of oceanic trenches. Unlike hadopelagic AOA, other AOA lineages only contain one type of ATPase (Fig. 2), thus in these AOA the acquisition of a variant ATPase via HOT was accompanied by the loss of their original type.
Overall, we show the occurrence of extensive interphylum HOT of V-ATPase across the Archaea domain, spanning the TACK and DPANN superphyla as well as Euryarchaeota phylum (Fig. 3). The acquisition of V-ATPase in ammonia-oxidizing Thaumarchaeota is closely associated with their adaptive radiation into acidic and higher-pressure habitats, which sheds new light on the major role of horizontal gene transfer in the evolutionary transitions of globally abundant AOA. Besides the HOT of V-ATPase, additional evolutionary processes associated with the energy-yielding ATPases were observed in multiple archaeal taxa. For instance, the position of Bathyarchaeota in the ATPase tree disagrees with their position in the species tree (Figs. 3 and S13). The ATPases of Desulfurococcales and Sulfolobales species lack subunit C [71], and Desulfurococcales members only contain seven subunits in their ATPase complex [71]. Furthermore, no canonical atp operon has been identified in most Nanoarchaeota in this study with the exception of Nanoarcheaum equitans that contains only five subunits [71, 72] (Fig. 3), and nearly all ATPases of other members in the DPANN superphylum were exclusively clustered with those of Euryarchaeota (Fig. 3), which might result from their severe genome reductions and symbiotic lifestyles with Euryarchaeota [64, 73]. The relationships between the high evolutionary dynamics of ATPases and the environmental adaptations of these archaea species remains a topic for future study. Finally, since our study has only emphasized the comparative phylogenetic analysis of a single gene cluster, we anticipate that the ongoing extensive molecular surveys and comprehensive comparative genomic studies of Thaumarchaeota will ultimately provide a much more complete mechanistic understanding of the evolution of some of the most successful organisms on Earth.
References
Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409:507–10.
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C. Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol. 2003;5:787–97.
Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2011;5:908–17.
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA. 2005;102:14683–8.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–9.
Nicol GW, Leininger S, Schleper C, Prosser JI. The influence of soil pH on the diversity, abundance and transcriptional activity of ammonia oxidizing archaea and bacteria. Environ Microbiol. 2008;10:2966–78.
Reigstad LJ, Richter A, Daims H, Urich T, Schwark L, Schleper C. Nitrification in terrestrial hot springs of Iceland and Kamchatka. FEMS Microbiol Ecol. 2008;64:167–74.
Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, et al. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci USA. 2008;105:2134–9.
Nunoura T, Takaki Y, Hirai M, Shimamura S, Makabe A, Koide O, et al. Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on Earth. Proc Natl Acad Sci USA. 2015;112:E1230–6.
Qin W, Heal KR, Ramdasi R, Kobelt JN, Martens-Habbena W, Bertagnolli AD, et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int J Syst Evol Microbiol. 2017;67:5067–79.
Könneke M, Bernhard AE, de la Torre J, Walker CB, Waterbury JB, Stahl DA. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature. 2005;437:543–6.
Tourna M, Stieglmeier M, Spang A, Konneke M, Schintlmeister A, Urich T, et al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci USA. 2011;108:8420–5.
Lehtovirta-Morley LE, Stoecker K, Vilcinskas A, Prosser JI, Nicol GW. Cultivation of an obligate acidophilic ammonia oxidizer from a nitrifying acid soil. Proc Natl Acad Sci USA. 2011;108:15892–7.
Palatinszky M, Herbold C, Jehmlich N, Pogoda M, Han P, von Bergen M, et al. Cyanate as an energy source for nitrifiers. Nature. 2015;524:105–8.
De La Torre JR, Walker CB, Ingalls AE, Könneke M, Stahl DA. Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ Microbiol. 2008;10:810–8.
Daebeler A, Herbold CW, Vierheilig J, Sedlacek CJ, Pjevac P, Albertsen M, et al. Cultivation and genomic analysis of “Candidatus Nitrosocaldus islandicus,” an obligately thermophilic, ammonia-oxidizing Thaumarchaeon from a hot spring biofilm in Graendalur Valley, Iceland. Front Microbiol. 2018;9:193.
Abby SS, Melcher M, Kerou M, Krupovic M, Stieglmeier M, Rossel C, et al. Candidatus Nitrosocaldus cavascurensis, an ammonia oxidizing, extremely thermophilic archaeon with a highly mobile genome. Front Microbiol. 2018;9:28.
Hallam SJ, Mincer TJ, Schleper C, Preston CM, Roberts K, Richardson PM, et al. Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol. 2006;4:520–36.
Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR, et al. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci USA. 2015;112:1173–8.
Wang Y, Huang JM, Cui GJ, Nunoura T, Takaki Y, Li WL, et al. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ Microbiol. 2019;21:716–29.
Kitzinger K, Padilla CC, Marchant HK, Hach PF, Herbold CW, Kidane AT, et al. Cyanate and urea are substrates for nitrification by Thaumarchaeota in the marine environment. Nat microbiol. 2019;4:234–43.
Stahl DA, de la Torre JR. Physiology and diversity of ammonia-oxidizing archaea. Annu Rev Microbiol. 2012;66:83–101.
Schafer G, Meyering-Vos M. The plasma membrane ATPase of archaebacteria. A chimeric energy converter. Ann N Y Acad Sci. 1992;671:293–309.
Gogarten JP, Kibak H, Dittrich P, Taiz L, Bowman EJ, Bowman BJ, et al. Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes. Proc Natl Acad Sci USA. 1989;86:6661–5.
Gruber G, Manimekalai MS, Mayer F, Muller V. ATP synthases from archaea: the beauty of a molecular motor. Biochim Biophys Acta. 2014;1837:940–52.
Han WY, Kemmitt SJ, Brookes PC. Soil microbial biomass and activity in Chinese tea gardens of varying stand age and productivity. Soil Biol Biochem. 2007;39:1468–78.
Lu L, Han WY, Zhang JB, Wu YC, Wang BZ, Lin XG, et al. Nitrification of archaeal ammonia oxidizers in acid soils is supported by hydrolysis of urea. ISME J. 2012;6:1978–84.
Huang R, Wu YC, Zhang JB, Zhong WH, Jia ZJ, Cai ZC. Nitrification activity and putative ammonia-oxidizing archaea in acidic red soils. J Soil Sediment. 2012;12:420–8.
Zhang JB, Zhu TB, Cai ZC, Müller C. Nitrogen cycling in forest soils across climate gradients in Eastern China. Plant Soil. 2011;342:419–32.
Wang BZ, Zhao J, Guo ZY, Ma J, Xu H, Jia ZJ. Differential contributions of ammonia oxidizers and nitrite oxidizers to nitrification in four paddy soils. ISME J. 2015;9:1062–75.
Lu L, Jia ZJ. Urease gene-containing Archaea dominate autotrophic ammonia oxidation in two acid soils. Environ Microbiol. 2013;15:1795–809.
Freitag TE, Chang L, Prosser JI. Changes in the community structure and activity of betaproteobacterial ammonia-oxidizing sediment bacteria along a freshwater-marine gradient. Environ Microbiol. 2006;8:684–96.
Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Konstantinidis KT, Tiedje JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258–64.
Herbold CW, Lehtovirta-Morley LE, Jung MY, Jehmlich N, Hausmann B, Han P, et al. Ammonia-oxidising archaea living at low pH: Insights from comparative genomics. Environ Microbiol. 2017;19:4939–52.
Guy L, Saw JH, Ettema TJ. The archaeal legacy of eukaryotes: a phylogenomic perspective. CSH Perspect Biol. 2014;6:a016022.
Spang A, Saw JH, Jorgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015;521:173–9.
Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:16170.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Jung MY, Kim JG, Damste JSS, Rijpstra WIC, Madsen EL, Kim SJ, et al. A hydrophobic ammonia-oxidizing archaeon of the Nitrosocosmicus clade isolated from coal tar-contaminated sediment. Environ Microbiol Rep. 2016;8:983–92.
Wang BZ, Guo P, Hang BJ, Li L, He J, Li SP. Cloning of a novel pyrethroid-hydrolyzing carboxylesterase gene from Sphingobium sp. strain JZ-1 and characterization of the gene product. Appl Environ Micro. 2009;75:5496–500.
Hyman M, Arp D. 14C2H2- and 14CO2-labeling studies of the de novo synthesis of polypeptides by Nitrosomonas europaea during recovery from acetylene and light inactivation of ammonia monooxygenase. J Biol Chem. 1992;267:1534–45.
Mulkidjanian AY, Makarova KS, Galperin MY, Koonin EV. Inventing the dynamo machine: the evolution of the F-type and V-type ATPases. Nat Rev Microbiol. 2007;5:892–9.
Oot RA, Huang LS, Berry EA, Wilkens S. Crystal structure of the yeast vacuolar ATPase heterotrimeric EGChead peripheral stalk complex. Structure. 2012;20:1881–92.
Biller SJ, Mosier AC, Wells GF, Francis CA. Global biodiversity of aquatic ammonia-oxidizing archaea is partitioned by habitat. Front Microbiol. 2012;3:252.
Smith JM, Damashek J, Chavez FP, Francis CA. Factors influencing nitrification rates and the abundance and transcriptional activity of ammonia-oxidizing microorganisms in the dark northeast Pacific Ocean. Limnol Oceano. 2016;61:596–609.
Sintes E, De Corte D, Haberleitner E, Herndl GJ. Geographic distribution of archaeal ammonia oxidizing ecotypes in the Atlantic Ocean. Front Microbiol. 2016;7:77.
Leon-Zayas R, Novotny M, Podell S, Shepard CM, Berkenpas E, Nikolenko S, et al. Single cells within the puerto rico trench suggest hadal adaptation of microbial lineages. Appl Environ Micro. 2015;81:8265–76.
Song JM, Wei DZ. Production and characterization of cellulases and xylanases of Cellulosimicrobium cellulans grown in pretreated and extracted bagasse and minimal nutrient medium M9. Biomass Bioenerg. 2010;34:1930–4.
Fang J, Zhang L, Bazylinski DA. Deep-sea piezosphere and piezophiles: geomicrobiology and biogeochemistry. Trends Microbiol. 2010;18:413–22.
Tamburini C, Boutrif M, Garel M, Colwell RR, Deming JW. Prokaryotic responses to hydrostatic pressure in the ocean - a review. Environ Microbiol. 2013;15:1262–74.
Neuman RC, Kauzmann W, Zipp A. Pressure dependence of weak acid ionization in aqueous buffers. J Phys Chem. 1973;77:2687–91.
Weber G, Drickamer HG. The effect of high pressure upon proteins and other biomolecules. Q Rev Biophys. 1983;16:89–112.
Abe F, Horikoshi K. Analysis of intracellular pH in the yeast Saccharomyces cerevisiae under elevated hydrostatic pressure: a study in baro- (piezo-) physiology. Extremophiles. 1998;2:223–8.
Michoud G, Jebbar M. High hydrostatic pressure adaptive strategies in an obligate piezophile Pyrococcus yayanosii. Sci Rep. 2016;6:27289.
Beam JP, Jay ZJ, Kozubal MA, Inskeep WP. Niche specialization of novel Thaumarchaeota to oxic and hypoxic acidic geothermal springs of Yellowstone National Park. ISME J. 2014;8:938–51.
Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD, et al. Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci USA. 2010;107:8806–11.
Jay ZJ, Beam JP, Dlakic M, Rusch DB, Kozubal MA, Inskeep WP. Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats. Nat Microbiol. 2018;3:732–40.
Baker BJ, Tyson GW, Webb RI, Flanagan J, Hugenholtz P, Allen EE, et al. Lineages of acidophilic archaea revealed by community genomic analysis. Science. 2006;314:1933–5.
Hilario E, Gogarten JP. Horizontal transfer of ATPase genes-the tree of life becomes a net of life. Biosystems. 1993;31:111–9.
Golyshina OV, Toshchakov SV, Makarova KS, Gavrilov SN, Korzhenkov AA, La Cono V, et al. ‘ARMAN’ archaea depend on association with euryarchaeal host in culture and in situ. Nat Commun. 2017;8:60.
Gubry-Rangin C, Kratsch C, Williams TA, McHardy AC, Embley TM, Prosser JI, et al. Coupling of diversification and pH adaptation during the evolution of terrestrial Thaumarchaeota. Proc Natl Acad Sci USA. 2015;112:9370–5.
Gouaux JE, Stevens RC, Lipscomb WN. Crystal structures of aspartate carbamoyltransferase ligated with phosphonoacetamide, malonate, and CTP or ATP at 2.8-A resolution and neutral pH. Biochemistry. 1990;29:7702–15.
Leimkuhler S, Rajagopalan KV. A sulfurtransferase is required in the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor Z in Escherichia coli. J Biol Chem. 2001;276:22024–31.
Hudaiberdiev S, Shmakov S, Wolf YI, Terns MP, Makarova KS, Koonin EV. Phylogenomics of Cas4 family nucleases. BMC Evol Biol. 2017;17:232.
Lewalter K, Muller V. Bioenergetics of archaea: ancient energy conserving mechanisms developed in the early history of life. Biochim Biophys Acta. 2006;1757:437–45.
Mohanty S, Jobichen C, Chichili VPR, Velázquez-Campoy A, Low BC, Hogue CWV, et al. Structural basis for a unique ATP synthase core complex from Nanoarcheaum equitans. J Biol Chem. 2015;290:27280–96.
Huber H, Hohn MJ, Rachel R, Fuchs T, Wimmer VC, Stetter KO. A new phylum of archaea represented by a nanosized hyperthermophilic symbiont. Nature. 2002;417:63–7.
Acknowledgements
This work was supported by the National Science Foundation of China (41471208, 41430859, 41530857, and 91751204), Youth Innovation Promotion Association, CAS (2017363), Strategic Priority Research Program of the CAS (XDB15040000), and Innovation Project of the Institute of Soil Science, CAS (ISSASIP1636). DAS was supported by United States National Science Foundation Grants (MCB-0604448), Dimensions of Biodiversity Program (OCE-1046017), and the Genomic Science and Technology for Energy and the Environment grant (DE-SC0006869) from the United States Department of Energy. This work was supported by grants from the Simons Foundation (SCOPE Award ID 329108 to AEI), and WQ was supported by Simons Postdoctoral Fellowship in Marine Microbial Ecology (548565). MW and CH were supported by an ERC Advanced Grant (NITRICARE, 294343 to MW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Qin, W., Ren, Y. et al. Expansion of Thaumarchaeota habitat range is correlated with horizontal transfer of ATPase operons. ISME J 13, 3067–3079 (2019). https://doi.org/10.1038/s41396-019-0493-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0493-x
This article is cited by
-
Metagenomic characterization of a novel non-ammonia-oxidizing Thaumarchaeota from hadal sediment
Microbiome (2024)
-
Genomics of soil depth niche partitioning in the Thaumarchaeota family Gagatemarchaeaceae
Nature Communications (2023)
-
Nitrogen and phosphorous acquisition strategies drive coexistence patterns among archaeal lineages in soil
The ISME Journal (2023)
-
ATP synthase evolution on a cross-braced dated tree of life
Nature Communications (2023)
-
Effects of acidification on nitrification and associated nitrous oxide emission in estuarine and coastal waters
Nature Communications (2023)
