
Abstract
Diatoms are important contributors to marine primary production and the ocean carbon cycle, yet the molecular mechanisms that regulate their acclimation and adaptation to temperature are poorly understood. Here we use a transcriptomic approach to investigate the molecular mechanisms associated with temperature acclimation and adaptation in closely related colder- and warmer-adapted diatom species. We find evidence that evolutionary changes in baseline gene expression, which we termed transcriptional investment or divestment, is a key mechanism used by diatoms to adapt to different growth temperatures. Invested and divested pathways indicate that the maintenance of protein processing machinery and membrane structure, important short-term physiological mechanisms used to respond to temperature changes, are key elements associated with adaptation to different growth temperatures. Our results also indicate that evolutionary changes in the transcriptional regulation of acetyl-CoA associated pathways, including lipid and branched chain amino acid metabolism, are used by diatoms to balance photosynthetic light capture and metabolism with changes in growth temperature. Transcriptional investment and divestment can provide a framework to identify mechanisms of acclimation and adaption to temperature.
Similar content being viewed by others

Introduction
Ocean temperatures are projected to increase over the coming century, with dramatic consequences for the marine biosphere [1]. Ocean warming has profound effects on marine phytoplankton, primary and export production, and the capacity of the ocean to act as a carbon sink [2,3,4,5,6,7]. Diatoms are responsible for about 20% of global net primary production and play important roles in carbon flow through marine food webs and in biogeochemical cycling [8]. Evidence from the field indicates temperature changes may lead to changes in diatom biogeography [9,10,11] and affect plankton phenology and coupling between trophic levels [12]. Model projections for the year 2100 suggest widespread restructuring of phytoplankton communities in the subtropics and North Atlantic [13, 14], but considerable uncertainty remains due to the evolutionary capacity of microbes [10, 15]. Little is known about the molecular consequences of changing temperature in diatoms and the variability of these responses across taxa adapted to different temperature regimes.
Diatoms are found throughout the ocean in habitats spanning the full range of observed temperatures. Each individual species and even strains within species have their own characteristic temperature performance curve [16], reflecting evolutionary constraints and past adaptations to its environmental regime [17]. Species with higher optimum growth temperatures generally grow faster under ideal conditions than those with lower optimum growth temperatures [17, 18]. Increasing temperature above a species’ growth optimum usually leads to a dramatic decrease in growth rate, while an increase in temperature below the optimum has a much more gradual impact on growth rate [16, 18]. Although the genomes of several model diatoms have been sequenced [19,20,21], the molecular bases underlying these different thermal optima are largely unknown. Organisms employ a range of molecular and physiological responses to changes in temperature including shifts in fatty acid composition [22, 23], the ratio of RNA to protein [24, 25], the use of heat-shock proteins [26], and changes in photosynthetic pigments and proteins like Rubisco in the case of photoautotrophs [27, 28]. Heat tolerance in corals is associated with constitutive changes in the expression of genes that control these physiological responses to temperatures like the use of heat shock, an adaptation commonly termed frontloading [29]. We do not know the extent to which these mechanisms are employed by diatoms for thermal adaptation. Transcriptomics has been widely used to investigate the responses of nonmodel phytoplankton to environment changes [30], and with careful experimental design, comparative transcriptomics can identify molecular mechanisms of acclimation and adaptation [29, 31].
Chaetoceros is a diverse and abundant marine centric planktonic diatom genus [32, 33]. Here we applied transcriptomics to two closely related Chaetoceros sp. with distinct biogeographies and optimal growth temperature (see “Materials and methods”) to study the molecular mechanisms associated with temperature acclimation and adaptation in marine diatoms. Specifically, we tested whether these closely related diatoms exhibited similar transcriptional responses to sub- and supra-optimal growth temperatures or have divergent responses consistent with evolutionary change in gene expression associated with the evolution of different temperature optima. Furthermore, we use comparative transcriptomics to identify genes and pathways associated with temperature acclimation and adaptation in Chaetoceros.
Materials and methods
Culture conditions and sampling
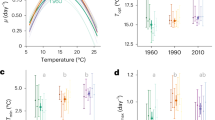
Two strains of Chaetoceros sp., CCMP158, and CCMP160 were acquired from the National Center for Marine Algae and Microbiota. CCMP158 was isolated from St. Margarets Bay, Nova Scotia, Canada (44.57 °N 64 °W) and maintained under 14 °C in the culture collection since 1982, while CCMP160 was isolated from the Bay of Marseilles, France (43.3 °N 5.4 °E), and maintained under 24 °C in the culture collection also since 1982. We grew the two species in f/2 + Si medium [34] under continuous light of 389 ± 27 (SD) μmol photons m−2 s−1. CCMP158 was maintained under 16 °C, and CCMP160 was maintained under 25 °C for 1–2 months before the experiment. Cultures were then acclimated in mid-exponential growth phase in triplicate at six additional temperatures ranging from 6 to 30 °C (average SD = 0.28 and average CV = 2%) for a minimum of nine generations (9–56 generations for CCMP158 and 16–42 generations for CCMP160, an average of three replicates) to determine the temperature performance curve. Growth rates were determined from chlorophyll a (chl a) fluorescence [35]. Subsequently, the two species were cultured for cell composition and transcriptomic analyses. These cultures were sampled after 27–49 generations (an average of three replicates) in acclimated growth at three experimental temperatures (see below). The experimental temperatures (Fig. 1) were where the maximum growth rate was achieved (Tmax), as well as suboptimal (Tsub, 12 °C for CCMP158 and 19 °C for CCMP160) and supraoptimal temperatures (Tsup, 22 °C for CCMP158 and 28 °C for CCMP160) where growth rate was ~50% of Tmax (except Tsup of CCMP160, where growth was 86% of Tmax because CCMP160 was not able to grow at 29 °C under our experiment conditions). The growth rate typically varied between 0.5% and 12% over the course of the experiment, with no evidence of temporal trend in growth rate. We propose that different Tmax of the two species indicate that they are evolutionarily adapted to different temperature regimes; CCMP158 whose Tmax is 16 °C is defined as the colder-adapted species, while the CCMP160 whose Tmax is 25 °C is defined as the warmer-adapted species. Cell counting was performed using Sedgewick–Rafter chamber (Wildco, Buffalo, NY, USA).

Steady-state growth rate (d–1) of Chaetoceros CCMP158 (open circles) and CCMP160 (filled circles) as a function of temperature. The temperature response curve \(\mu = e^{ - b(t - t_0)^2}\) (line) was fit using brms [79] and added to each growth curve, along with the predicted uncertainty interval of the response (shaded regions). CCMP160 did not grow at 29.6 °C and that data point was not included in the regression model
Molecular biochemistry and photophysiology
For biochemical analyses, 50 mL of culture was collected by filtration at low pressure (5 PSI) on 0.8 μm polycarbonate filters, except samples for chl a for which Whatman GF/F filters were used. Protein and RNA were extracted and quantified by colorimetry and fluorometry, respectively, as described in [36]. Total cellular RNA was quantified using the method Berdalet et al. [37] with bead milling used during the extraction. Cellular chl a concentration was determined by methanol extraction and spectrophotometry [38]. Parameters of photosystem II photochemistry were measured using Fast repetition rate fluorometry (Photon Systems Instruments Fluorometer FL3500, Drasov, Czech Republic). Measurements were taken using a blue single-turnover saturating flash train after 30 s intervals of red + blue actinic light increasing in intensity from 0 to 254 μmol photons m−2 s−2. Nonphotochemical quenching (NPQ) is calculated as (Fm−Fm′)/Fm′ [39] using Fm′ value under 254 μmol photons m−2 s−2. Cell volume was estimated from linear dimensions of images of cells taken at 200× magnification following Hillebrand et al. (1999) using ImageJ (http://imagej.nih.gov/ij/).
Transcriptomics
Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Venlo, The Netherlands) following the manufacturer instructions. mRNA was sequenced using Illumina HiSeq 2000 by the Genome Quebec Innovation Centre using the TruSeq mRNA stranded library preparation protocols for 100 bp paired-end reads. The quality of the raw reads was checked using FASTQC version 3. PhiX contamination [40] was removed before assembly. Raw reads were then mapped to the PhiX genome using Bowtie2 version 2.2.9 (–local –very-sensitive) [41] and the unmapped reads were outputted using samtools version 1.3.1 [42]. The first 12 nt from the 5′ end of the reads [43] and the 3′ adapter fragments were removed using cutadapt version 1.10 [44]. Sequences between 25 nt and 80 nt were reserved and paired using Trimmomatic version 0.36 [45]. Unpaired reads were included with the left reads of the paired fragments for assembly. Assemblies were conducted for each species. Left reads and right reads from all samples were merged and normalized using Trinity’s version 2.2.0 [46] in silico normalization. Normalized left and right reads were then assembled into contigs using Trinity’s paired-end mode using default parameters according to [47]. Sequences reported by the contamination screen during National Center for Biotechnology Information (NCBI) TSA (transcriptome shotgun assembly) sequence database submission were removed from the transcriptome. Cleaned and trimmed reads of each sample were mapped to the corresponding assembled transcriptomes of each species using Bowtie2 version 2.2.9 (-a –rdg 6,5 –rfg 6,5 –score-min L,−0.6,−0.4 –no-mixed). Transcript abundance estimates were obtained with eXpress version 1.5.1 [48]. Genes with counts > 0 in at least two of three replicates at any temperature condition were used in differentially expressed gene analysis with DESeq2 [49] using the script from Trinity. Genes at Tsub or Tsup for each species were identified as differentially expressed if they had a log-fold change different from zero as compared to their expression level at Tmax with a false discovery rate adjusted p-value < 0.01.
All genes were grouped into nine possible patterns of differential expression at Tsub and Tsup relative to Tmax (Table 1). Genes that are downregulated only at Tsup in the colder-adapted species and only at Tsub in the warmer-adapted species were described as potentially invested. Similarly, genes that are upregulated only at Tsup in the colder-adapted species and only at Tsub in the warmer-adapted species were described as potentially divested. The assembled transcriptomes and predicted coding regions from TransDecoder version 3.0.0 (http://transdecoder.github.io) were annotated with blastx and blastp against the Swiss-prot database release 2016.08 [50, 51] using BLAST+ [52]. KEGG [53] identification of each annotated gene was obtained from both the UniProt database and the GhostKOALA service [54]. KEGG pathways and Brite categories were selected for KEGG enrichment analyses from the following categories: metabolism, genetic information processing, environmental information processing, and cellular processes. Pathways with less than ten annotated KEGG orthology identifiers (KO IDs) in either species were removed from calculation. A pathway or Brite category was considered significantly enriched in differentially regulated and invested/divested KO IDs using a hypergeometric distribution test if the probability of obtaining the observed number, m, or more, of differentially regulated KO IDs in that pathway or Brite was low. We used a test of p < 0.05 with a false discovery rate correction [55], where
and N is the total number of KO IDs, n is the number of KO IDs in the selected pathway, and M is the total number of differentially regulated KO IDs.
All the raw read files (see Supplementary dataset 1 for SRA accessions), assembled transcriptomes [TSA accession: GHAM00000000 (CCMP158) and GHAN00000000 (CCMP160)], and gene expression data (GEO accession: GSE125205) are available through NCBI (BioProject: PRJNA495610).
Results
Influence of growth temperature on physiology, biochemical composition, and cell size
The colder-adapted species CCMP158 (Tmax = 16 °C) had a lower growth rate and lower temperature of maximum growth rate (Tmax) than the warmer-adapted species CCMP160 (Tmax = 25 °C) (Fig. 1 and Table 2). The growth rates under Tsub of each species (12 °C for CCMP158 and 19 °C for CCMP160) and Tsup of CCMP158 (22 °C) were ~50% of their maximum growth rates, while under Tsup for CCMP160 (28 °C), the growth rate was 86% of its maximum (Fig. 1 and Table 2). The two species differ in their photosynthetic physiology and biochemical composition at their optimal growth temperatures as well as in their acclimated response to sub- and supra-optimal growth temperatures. NPQ was significantly higher in the colder-adapted species (0.22–0.3) than the warmer-adapted species (0.1–0.16), but there were no obvious trends with growth temperature within either species (Table 2). The chl a content per cell volume (fg µm−3) increased in CCMP158 with temperature but varied very little with the temperature in CCMP160 (Table 2). The chl a content per cell volume was higher in the warmer-adapted species than the colder-adapted species under Tsub and Tmax but there was no difference at Tsup (Table 2). The changes in RNA and protein content per volume were not statistically significant with temperature in either species except for the significant increase of RNA content per cell volume in CCMP160 from Tsub to Tmax (Table 2). The RNA and protein content per volume was higher in CCMP160 than in CCMP158 at corresponding Tmax and Tsub but not Tsup (Table 2). RNA:Protein was significantly lower at Tmax in CCMP158 than at Tmax for CCMP160 and decreased in CCMP158 but increased in CCMP160 with growth temperature, but this trend was not statistically significant (p < 0.05). The cell volume of the colder-adapted species was nearly 2.5 times larger than the warmer-adapted species at their respective Tmax. The cell volume of CCMP158 increased with decreasing temperature, while the cell volume of CCMP160 increased with increasing temperature, but these trends were not statistically significant (p < 0.05).
Gene identification and annotation
The de novo assembly yielded 42,048 and 27,580 genes in CCMP158 and CCMP160, respectively (bioinformatics summary, see Supplementary Table 1 and Supplementary dataset 1). A total of 24.1% of the genes in CCMP158 and 25.8% of the genes in CCMP160 were assigned KEGG annotations. A total of 3569 KO IDs and 4913 OrthoMCL groups are common to both species (Supplementary Fig. 1a, b), and 2032 KO IDs and 3583 OrthoMCL groups are shared with the model diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum (Supplementary Fig. 1a, b). The taxonomy results from GhostKOALA show no significant addition of bacterial, archaeal, and fungal sequences to the two de novo assembled transcriptomes (Supplementary Fig 1c).
Differential gene expression in response to temperature
Overall, 7% of assembled genes were differentially expressed under Tsup and 3% were differentially expressed under Tsub (Supplementary Table 1) in CCMP158. In contract, in CCMP160, 3% of assembled genes were differentially expressed under Tsup and 7% were differentially expressed under Tsub (Supplementary Table 1). The expression of all the assembled genes are grouped into nine different patterns across Tsub and Tsup in each species (Table 1). In the colder-adapted CCMP158, the majority (67.4%) of differentially expressed genes were differentially expressed only at the higher temperature (Tsup), while a similar majority (67.8%) of differentially expressed genes were differentially expressed exclusively at the lower temperature (Tsub) in the warmer-adapted CCMP160 (Table 1). We define these differentially expressed genes as potentially invested or divested.
Identification of metabolic pathways associated with acclimation and adaptation to different temperatures
In the colder-adapted species, glycolysis/gluconeogenesis and the citrate cycle were upregulated, while chaperones and folding catalysts, as well as protein processing in the endoplasmic reticulum (ER) were downregulated under Tsub relative to Tmax (Table 3, KO counts and p-values, see Supplementary dataset 2). Cell cycle, DNA repair, and recombination proteins were upregulated, while amino acid related enzymes, photosynthesis, photosynthesis proteins, porphyrin and chlorophyll metabolism, fatty acid biosynthesis, lipid biosynthesis protein, chaperones and folding catalysts, proteasome, ribosome, RNA transporter, and translation factors were downregulated at Tsup relative to Tmax. By contrast, in the warmer-adapted species, the spliceosome and ribosome biogenesis were upregulated and fatty acid degradation, valine, leucine and isoleucine degradation, chaperones and folding catalysts, proteasome, and peptidases were downregulated under Tsub relative to Tmax (Table 3). The ubiquitin system and protein processing in the ER were upregulated, while lysine biosynthesis was downregulated under Tsup relative to Tmax. Several of these pathways exhibited patterns of expression that are consistent with temperature-induced evolutionary investment or divestment (Table 3). All the pathways that were downregulated under Tsup except chaperones and folding catalysts in the colder-adapted species were identified as potentially invested. Biosynthesis of unsaturated fatty acids and proteasome [PATH:ko03050] were also potentially invested in the colder-adapted species. The cell-cycle pathway [PATH:ko04111] was divested in the colder-adapted species (Table 3). Divested genes were mainly regulatory proteins/protein complexes of the S and mitotic phases of the cell cycle (Supplementary Table 3 and Supplementary Fig 2). All the pathways that were downregulated under Tsub in the warmer-adapted species were invested. The spliceosome and ribosome biogenesis were divested in the warmer-adapted species.
Discussion
Life has adapted to thrive under much of the range of ambient temperatures found on the Earth, but most species have specialized to live at a narrow subset of Earth’s broader temperature range [6]. Species with higher optimal growth temperatures generally have higher intrinsic growth rates under optimal conditions. Exposure to colder suboptimal or warmer supraoptimal growth temperatures induces energetic, metabolic, and redox imbalances that trigger a series of intracellular stresses and compensatory responses. We used a comparative transcriptomic analysis of two closely related Chaetoceros sp. with different optimal growth temperatures (Tmax) to identify molecular mechanisms associated with both temperature acclimation and adaptation, as well as the molecular basis for the general decreasing maximum growth rates with decreasing temperatures in diatoms.
Transcriptional profiles after acclimation to T sub and T sup are affected by investment and divestment in each species
We verified the quality of our de novo assembly by showing that a majority of the predicted proteins from the two Chaetoceros species were homologous at the protein level and were shared with the model diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum (Supplementary Fig. 1). Most of the genes differentially expressed in the colder-adapted CCMP158 arise at Tsup relative to Tmax, while most of the genes differentially expressed in the warmer-adapted CCMP160 arise at Tsub relative to Tmax (Table 1 and Supplementary Table 1). We hypothesize that these differences in transcriptional responses across species reflect evolutionary changes in gene expression after long-term exposure to different temperature regimes. There are nine possible patterns that can describe the differential expression of a gene across Tsub and Tsup within each species (including no differential expression), each with a different biological interpretation (Table 1). Two of these patterns are increases or decreases in gene expression levels as a function of temperature regardless of the overall metabolic rates of the whole organism, indicating that temperature is the dominant factor altering expression. Differential expression to compensate for changes in enzyme activity is a potential cause of this pattern. Two more patterns of differential expression can be recognized as being positively or negatively related to decreasing growth rates at Tsub and Tsup. These growth rate driven patterns could reflect downregulation of cell-cycle related genes or an upregulation of general stress response genes at Tsub and Tsup. The four remaining patterns are differential expressions at only one temperature relative to the optimum. These patterns could arise from an elevated or depressed expression of genes at the optimum temperature that is beneficial at a colder or warmer temperature. These four expression patterns in which a species seems constitutively poised for one direction of temperature change may reflect an adaptive response in overall gene expression we refer to here as investment or divestment, similar to the concept of frontloading [29, 56]. For example, a species adapted to colder temperatures may have elevated expression of genes related to protein synthesis (a potentially beneficial response to lower biosynthesis rates at colder temperatures) at its optimum temperature relative to its ancestor. This elevated expression may result in no further increase in expression at even colder temperatures and a decrease in expression at relatively warmer temperatures. A chi-squared test suggests that the patterns of differential expression are independent between the Tsub and Tsup treatments within each species. If temperature adaptation changes gene expression, the patterns of differential expression would be different across the two species that are adapted to different temperatures. A proportionality test suggests that six out of eight differential expression patterns are significantly different across the two species (Supplementary Table 2), but the strongest differences by a large margin are in the investment and divestment categories (Table 1). These counts differ between species indicating these patterns are strongly related to temperature adaptation. Therefore, we propose that diatoms may adapt to different temperature regimes partially through constitutive changes in the gene expression under their Tmax, reducing the need for additional transcriptomic responses to further increases or decreases in temperature. This hypothesis is consistent with the observed high levels of differential expression under supraoptimal but not under suboptimal temperatures in the colder-adapted species and in contrast the high levels of differential expression under sub-optimal but not supra-optimal growth temperature in the warm-adapted species.
Invested and divested pathways include known physiological mechanisms associated with temperature adaptation
Protein translation, especially the initiation of translation by ribosomes, is slowed down by cooler temperatures [57, 58]. The diatoms Fragilariopsis cylindrus and Thalassiosira pseudonana have both shown short-term compensatory increases in ribosomal density and genes involved in ribosome production under low temperature [25]. Here we find that growth temperature influences the expression of genes associated with translation and protein synthesis very differently across the two species. The investment of aminoacyl-tRNA biosynthesis, ribosome proteins, RNA transporters, and translation factors (Table 3) in the colder-adapted species possibly reflects a high baseline expression of these pathways to better compensate for lower translation efficiency at cooler Tmax. Amino acid related enzymes (enzymes for tRNA synthesis) were also invested (Table 3), consistent with the investment of aminoacyl-tRNA biosynthesis. In contrast, there is an apparent divestment of ribosome biogenesis and the spliceosome in the warmer-adapted species (Table 3). Divestment of ribosome biogenesis in the warmer-adapted species not only reduces material and energy expenditure, but may also moderate polypeptide processing demand and thereby reduce proapoptotic ER stress, which is defined as the disruption of ER function and increased load of unfolded proteins in the ER lumen [59]. Investment of chaperones and folding catalysts also helps to reduce ER stress (Table 3). The investment of peptidases and proteasomes can facilitate the removal of misfolded proteins in the ER and support overall faster protein turnover under warmer temperatures [60] (Table 3). These changes in baseline expression related to protein metabolism indicate that future proteomics studies would greatly complement our transcriptomic findings and help reveal their ultimate physiological function.
Cooler temperatures have been hypothesized to increase the ratio of P-rich rRNA relative to nitrogen-rich proteins in phytoplankton, leading to large-scale biogeographic patterns in particulate N:P in the sea surface [25]. However, the colder-adapted species in our study has a lower RNA to protein ratio than the warmer-adapted species (Table 2). This is due to the higher RNA content per unit volume in the warmer-adapted species that is consistent with its faster growth rate [61]. Our results indicate that RNA:Protein in phytoplankton may not be a simple function of sea-surface temperature, in part due to the resource allocation strategies associated with adaptation to cold temperatures.
Colder temperatures slow down protein turnover rates and reduce membrane fluidity, which is reflected by the compensatory investment in photosynthesis proteins and biosynthesis of unsaturated fatty acids in the cold adapted species (Table 3). The diatom T. pseudonana appears to compensate for the high protein turnover requirements associated with the photosynthetic protein complex PSII by maintaining a large pool of inactive PSII repair cycle intermediates except PsbA, the core protein of this complex [62]. Cold-adapted phytoplankton appear to use a comparable strategy to cope with slower protein turnover by maintaining higher photosynthetic protein quotas and more stable photosynthetic pigment content compared to closely related, warm-adapted species [63,64,65]. Similarly, we find the investment of the PSII proteins PsbD, PsbC, and PsbB but not PsbA (Supplementary Table 4 and Supplementary Fig. 3), investment of proteosomal and chl a biosynthesis pathways (Table 3), and a more stable cellular chl a quota (Table 2) in the colder-adapted CCMP158. These features suggest a compensatory enhanced pool of PSII repair cycle intermediates in CCMP158 when PSII turnover is slowed at its lower Tmax [66]. Such cold adaptations are also accompanied by constitutively higher NPQ [63, 64] that may be needed to dissipate the greater excess light energy associated with photosynthesis at colder temperatures [67], which may explain the constitutively higher NPQ we observed in CCMP 158.
An investment of biosynthesis of unsaturated fatty acids in the colder-adapted species may facilitate the maintenance of membrane fluidity under cooler temperatures by modifying the saturation state of the fatty acids in the lipid bilayers [23, 68].
Regulation of acetyl-CoA metabolism is essential for diatom temperature adaptation
Light absorption, excitation energy transfer, and primary photochemistry are essentially independent of temperature [69], therefore in photosynthetic organisms, a change in temperature alters metabolic balance [3, 7] and will vary the ratio of central metabolites such as NADPH:NADP+ and actyl-CoA:CoA (CoA: coenzyme A). We find evidence of investment in fatty acid metabolism in both species and branched chain amino acid (BCAA, valine, leucine, and isoleucine) metabolism in CCMP160 (Table 3) that indicates that actyl-CoA metabolism plays a key role in temperature adaptation in diatoms. Lipids are an electron sink and can reduce the redox stress caused by imbalanced NADPH:NADP+ [70]. An undersupply of acetyl-CoA, which is the central metabolite for fatty acids and BCAA metabolism, will reduce rates of cell division [71,72,73], while excessively high level of cytosolic acetyl-CoA can result in the overacetylation of cytosolic proteins and consequent inhibition of enzyme activity [74]. We hypothesize that invested pathways associated with fatty acid and BCAA metabolism play a key role in temperature adaptation in diatoms. Under cooler conditions, higher rates of fatty acid biosynthesis help consume excess cytosolic acetyl-CoA resulting from the decreased nuclear acetyl-CoA requirements when growth rates are slow [74] and excess NADPH when temperature-independent, light-limited photosynthetic rate exceeds the rate of NADPH consumption by the temperature-dependent dark reactions. In contrast, under warmer conditions, photosynthetic carbon fixation may not meet cellular biosynthetic demands of acetyl-CoA because metabolic rate can exceed the temperature-independent photosynthetic rate. On the other hand, intensive histone acetylation resulting in excessive nuclear CoA may result in programmed cell death at high concentration [72]. The degradation of fatty acids and BCAA can supplement acetyl-CoA production and may support higher growth rates in the warmer-adapted species.
Molecular basis for lower maximum growth rates under lower temperature
Generally, species adapted to higher temperatures have higher growth rates. The molecular mechanism(s) that links growth rate and temperature optima are largely unknown. In the colder-adapted species we find a divestment of genes associated with the cell-cycle pathway [PATH:ko04141] (Supplementary Table 3 and Supplementary Fig. 2). Most of the divested genes are associated with S and mitotic phases of the cell cycle (Supplementary Table 3 and Supplementary Fig. 2). DNA replication licensing factor MCM3 (K02541) (MCM) is part of the minichromosome maintenance complex (MCM complex) that is required for the initiation and regulation DNA replication during the S phase [75]. The cohesion complex, condesin complex, anaphase-promoting complex subunit 8 (APC8), which is part of the anaphase promoting complex/cyclosome (APC/C complex), and S-phase kinase-associated protein 1, which is the essential component of the SCF (SKP1-CUL1-F-box protein) ubiquitin ligase complex, are associated with the separation of sister chromatids [76]. The relatively low baseline expression of these cell-cycle genes and encoded proteins may lengthen the G2 phase before mitosis and contribute to larger cell volumes in CCMP158 (Table 2), as previously reported in yeast [77]. The divested genes associated with DNA synthesis and mitosis indicates a molecular basis of the lower maximum growth rate of the colder-adapted species under its optimal growth temperature (Fig. 1 and Table 2). However, more work is required to determine if this is a general mechanism.
We verified our results for differential expression between the two species under their respective Tmax using a pseudo-transcriptome generated from all the diatom transcriptomes deposited in the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) [78] (Supplementary Methods). The pathway analysis reveals that the expression level of protein biosynthesis and photosynthesis related genes are higher in CCMP158 than CCMP160 (Supplementary Table 5), which is consistent with an investment in protein biosynthesis and photosynthesis in CCMP158 and a divestment in ribosome and ribosome biogenesis in CCMP160. Meanwhile, the higher expression level of cell-cycle related pathways in CCMP160 is consistent with the divestment of cell-cycle related pathway in CCMP158 (Supplementary Table 5). Although investment in the proteasome was seen in both species, the expression levels of proteasome related genes are higher in CCMP158 than CCMP160. The enrichment of fatty acid biosynthesis in the genes with higher expression in CCMP158, and the enrichment of fatty acid and BCAA degradation in the genes with higher expression in CCMP160 are not significant, however, the distribution of observed counts of KO IDs in these pathways are consistent with the investment of fatty acid biosynthesis in CCMP158 and investment of fatty acid and BCAA degradation in CCMP160 (Supplementary Table 5). These results further support our hypothesis that temperature adaptation changes the baseline expression level of key genes and pathways to maintain metabolic homeostasis.
In conclusion, our results indicate that diatoms have evolved different optimal growth temperatures via changes in baseline expression (investment and divestment) of several key pathways including amino acid, carbohydrate, energy, and lipid metabolism, protein folding sorting and degradation, cell growth and death, and transcription and translation (Table 3). As a result of evolutionary change in gene expression, the colder- and warmer-adapted species have largely different transcriptional responses to growth under sub- and supra-optimal temperatures relative to their individual optimal temperatures for growth. In aggregate, these results indicate that short-term acclimation to temperature will be species specific and shaped by evolutionary history. Future studies with lab-evolved strains could be used to improve our understanding of the evolution and fitness costs associated with transcriptional investment/divestment.
References
Pachauri RK, Allen MR, Barros VR, Broome J, Cramer W, Christ R, et al. Climate change 2014: synthesis report. Contribution of working groups i, ii and iii to the fifth assessment report of the intergovernmental panel on climate change. Geneva, Switzerland: Intergovernmental Panel on Climate Change; 2014. p. 151.
Doney SC. Oceanography: plankton in a warmer world. Nature. 2006;444:695.
García-Carreras B, Sal S, Padfield D, Kontopoulos D-G, Bestion E, Schaum C-E, et al. Role of carbon allocation efficiency in the temperature dependence of autotroph growth rates. Proc Natl Acad Sci USA. 2018;115:201800222.
Laws EA, Falkowski PG, Smith WO Jr, Ducklow H, McCarthy JJ. Temperature effects on export production in the open ocean. Glob Biogeochem Cycles. 2000;14:1231–46.
López-Urrutia Á, San Martin E, Harris RP, Irigoien X. Scaling the metabolic balance of the oceans. Proc Natl Acad Sci USA. 2006;103:8739–44.
Pörtner H-O, Karl DM, Boyd PW, Cheung W, Lluch-Cota SE, Nojiri Y, et al. Ocean systems. Climate change 2014: impacts, adaptation, and vulnerability. Part A: global and sectoral aspects. contribution of working group II to the fifth assessment report of the intergovernmental panel on climate change. Cambridge: Cambridge University Press; 2014. p. 411–484.
Regaudie-de-Gioux A, Duarte CM. Temperature dependence of planktonic metabolism in the ocean. Glob Biogeochem Cycles. 2012;26:GB1015.
Tréguer P, Bowler C, Moriceau B, Dutkiewicz S, Gehlen M, Leblanc K, et al. Influence of diatom diversity on the ocean biological carbon pump. Nat Geosci. 2017.
Irwin AJ, Nelles AM, Finkel ZV. Phytoplankton niches estimated from field data. Limnol Oceanogr. 2012;57:787–97.
Irwin AJ, Finkel ZV, Müller-Karger FE, Ghinaglia LT. Phytoplankton adapt to changing ocean environments. Proc Natl Acad Sci USA. 2015;112:5762–66.
Xiao W, Liu X, Irwin AJ, Laws EA, Wang L, Chen B, et al. Warming and eutrophication combine to restructure diatoms and dinoflagellates. Water Res. 2018;128:206–16.
Edwards M, Richardson AJ. Impact of climate change on marine pelagic phenology and trophic mismatch. Nature. 2004;430:881–84.
Barton AD, Irwin AJ, Finkel ZV, Stock CA. Anthropogenic climate change drives shift and shuffle in marine phytoplankton communities. Proc Natl Acad Sci USA. 2016;113:2964–69.
Thomas MK, Kremer CT, Klausmeier CA, Litchman E. A global pattern of thermal adaptation in marine phytoplankton. Science. 2012;338:1085–88.
O’Donnell DR, Hamman CR, Johnson EC, Kremer CT, Klausmeier CA, Litchman E. Rapid thermal adaptation in a marine diatom reveals constraints and trade-offs. Global change biology. 2018;24:4554–65.
Boyd PW, Rynearson TA, Armstrong EA, Fu F, Hayashi K, Hu Z, et al. Marine phytoplankton temperature versus growth responses from polar to tropical waters–outcome of a scientific community-wide study. PLoS ONE 2013;8:e63091.
Suzuki Y, Takahashi M. Growth responses of several diatom species isolated from various environments to temperature. J Phycol. 1995;31:880–88.
Eppley RW. Temperature and phytoplankton growth in the sea. Fish Bull. 1972;70:1063–85.
Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science. 2004;306:79–86.
Mock T, Otillar RP, Strauss J, McMullan M, Paajanen P, Schmutz J, et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature. 2017;541:36.
Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 2008;456:239–44.
Marr AG, Ingraham JL. Effect of temperature on the composition of fatty acids in Escherichia coli. J Bact. 1962;84:1260–67.
Suutari M, Liukkonen K, Laakso S. Temperature adaptation in yeasts: the role of fatty acids. Microbiology. 1990;136:1469–74.
Storch D, Lannig G, Pörtner HO. Temperature-dependent protein synthesis capacities in Antarctic and temperate (North Sea) fish (Zoarcidae). J Exp Biol. 2005;208:2409–20.
Toseland AD, Daines SJ, Clark JR, Kirkham A, Strauss J, Uhlig C, et al. The impact of temperature on marine phytoplankton resource allocation and metabolism. Nat Clim Change. 2013;3:979–84.
Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol. 1999;61:243–82.
Valentin K, Mock T. Photosynthesis and cold acclimation: molecular evidence from a polar diatom. J Phycol. 2004;40:732–41.
Young JN, Goldman JAL, Kranz SA, Tortell PD, Morel FMM. Slow carboxylation of rubisco constrains the rate of carbon fixation during Antarctic phytoplankton blooms. New Phycol. 2015;205:172–81.
Barshis DJ, Ladner JT, Oliver TA, Seneca FO, Traylor-Knowles N, Palumbi SR. Genomic basis for coral resilience to climate change. Proc Natl Acad Sci USA. 2013;110:1387–92.
Caron DA, Alexander H, Allen AE, Archibald JM, Armbrust EV, Bachy C, et al. Probing the evolution, ecology and physiology of marine protists using transcriptomics. Nat Rev Microbiol. 2017;15:6.
Kenkel CD, Matz MV. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat Ecol Evol. 2017;1:0014.
Leblanc K, Arístegui J, Kopczynska E, Marshall H, Peloquin J, Piontkovski S, et al. A global diatom database–abundance, biovolume and biomass in the world ocean. Earth System Science Data. 2012;4:149–65.
Tomas CR. Identifying marine phytoplankton. 1st ed. San Diego, US: Elsevier; 1997.
Guillard RRL, Ryther JH. Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt and Detonula confervacea Cleve. Can J Microbiol. 1962;8:229–239.
Brand LE. Genetic variability in reproduction rates in marine phytoplankton populations. Evolution. 1981;35:1117–27.
Liefer JD, Garg A, Fyfe MH, Irwin AJ, Benner I, Brown CM, et al. The macromolecular basis of phytoplankton C:N:P under nitrogen starvation. Front Microbiol. 2019;10:763.
Berdalet E, Roldán C, Olivar MP, Lysnes K. Quantifying RNA and DNA in planktonic organisms with SYBR green II and nucleases. Part A. Optimisation of the assay. Sci Mar. 2005;69:1–16.
Ritchie RJ. Consistent sets of spectrophotometric chlorophyll equations for acetone, methanol and ethanol solvents. Photosynth Res. 2006;89:27–41.
Krause G, Vernotte C, Briantais J-M. Photoinduced quenching of chlorophyll fluorescence in intact chloroplasts and algae. Resolution into two components. Biochim Biophys Acta Bioenerg. 1982;679:116–24.
Mukherjee S, Huntemann M, Ivanova N, Kyrpides NC, Pati A. Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand Genom Sci. 2015;10:18.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–79.
Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131–e131.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29:644.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494.
Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31.
Strimmer K. A unified approach to false discovery rate estimation. BMC Bioinform. 2008;9:303.
Gleason LU, Burton RS. RNA‐seq reveals regional differences in transcriptome response to heat stress in the marine snail Chlorostoma funebralis. Mol Ecol. 2015;24:610–27.
Farewell A, Neidhardt FC. Effect of temperature on in vivo protein synthetic capacity in Escherichia coli. J Bacteriol. 1998;180:4704–10.
Friedman H, Lu P, Rich A. Temperature control of initiation of protein synthesis in Escherichia coli. J Mol Biol. 1971;61:105–21.
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Investig. 2005;115:2656–64.
Ferguson DL, Guikema JA, Paulsen GM. Ubiquitin pool modulation and protein degradation in wheat roots during high temperature stress. Plant Physiol. 1990;92:740–46.
Sterner RW, Elser JJ. Ecological stoichiometry: the biology of elements from molecules to the biosphere. Princeton, NJ: Princeton University Press; 2002.
Wu H, Cockshutt AM, McCarthy A, Campbell DA. Distinctive PSII photoinactivation and protein dynamics in marine diatoms. Plant Physiol. 2011;156:111.178772.
Liefer JD, Garg A, Campbell DA, Irwin AJ, Finkel ZV. Nitrogen starvation induces distinct photosynthetic responses and recovery dynamics in diatoms and prasinophytes. PLoS ONE 2018;13:e0195705.
Ni G, Zimbalatti G, Murphy CD, Barnett AB, Arsenault CM, Li G, et al. Arctic Micromonas uses protein pools and non-photochemical quenching to cope with temperature restrictions on photosystem II protein turnover. Photosynth Res. 2017;131:203–20.
Szyszka B, Ivanov AG, Hüner NP. Psychrophily is associated with differential energy partitioning, photosystem stoichiometry and polypeptide phosphorylation in Chlamydomonas raudensis. Biochim Biophys Acta Bioenerg. 2007;1767:789–800.
Aro E-M, Hundal T, Carlberg I, Andersson B. In vitro studies on light-induced inhibition of photosystem II and D1-protein degradation at low temperatures. Biochim Biophys Acta Bioenerg. 1990;1019:269–75.
Hüner N, Dahal K, Kurepin LV, Savitch L, Singh J, Ivanov AG, et al. Potential for increased photosynthetic performance and crop productivity in response to climate change: role of CBFs and gibberellic acid. Front Chem. 2014;2:18.
Neidleman SL. Effects of temperature on lipid unsaturation. Biotechnol Genet Eng Rev. 1987;5:245–68.
Raven JA, Geider RJ. Temperature and algal growth. New Phytol. 1988;110:441–61.
Li X, Moellering ER, Liu B, Johnny C, Fedewa M, Sears BB, et al. A galactoglycerolipid lipase is required for triacylglycerol accumulation and survival following nitrogen deprivation in Chlamydomonas reinhardtii. Plant Cell. 2012;112:105106.
Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at gowth genes. Mol Cell. 2011;42:426–37.
Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metabol. 2015;21:805–21.
Shi L, Tu BP. Acetyl-CoA induces transcription of the key G1 cyclin CLN3 to promote entry into the cell division cycle in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2013;110:7318–23.
Weinert BT, Iesmantavicius V, Moustafa T, Schölz C, Wagner SA, Magnes C, et al. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol Syst Biol. 2014;10:716.
Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69.
Vodermaier HC. APC/C and SCF: controlling each other and the cell cycle. Curr Biol. 2004;14:R787–R796.
Moseley JB, Mayeux A, Paoletti A, Nurse P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature. 2009;459:857.
Keeling PJ, Burki F, Wilcox HM, Allam B, Allen EE, Amaral-Zettler LA, et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014;12:e1001889.
Bürkner P-C. brms: an R package for Bayesian multilevel models using Stan. J Stat Softw. 2017;80:1–28.
Acknowledgements
This work was supported by the NSERC Discovery (ZVF, AJI) and Canada Research Chairs (ZVF) programs. The authors acknowledge the contribution of scientists and staff of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for RNA library preparation and sequencing.
Author information
Authors and Affiliations
Contributions
JAK, YL, AJI, and ZVF designed research, JAK, YL, and JDL performed research and analyses, and JAK, YL, AJI, ZVF, and JDL wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Liang, Y., Koester, J.A., Liefer, J.D. et al. Molecular mechanisms of temperature acclimation and adaptation in marine diatoms. ISME J 13, 2415–2425 (2019). https://doi.org/10.1038/s41396-019-0441-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0441-9
This article is cited by
-
Temperature optima of a natural diatom population increases as global warming proceeds
Nature Climate Change (2024)
-
Microbial metabolites in the marine carbon cycle
Nature Microbiology (2022)
-
Variation in the fatty acid profiles of two cold water diatoms grown under different temperature, light, and nutrient regimes
Journal of Applied Phycology (2021)
-
Temperatures above thermal optimum reduce cell growth and silica production while increasing cell volume and protein content in the diatom Thalassiosira pseudonana
Hydrobiologia (2020)
