
Abstract
Rickettsiales are a lineage of obligate intracellular Alphaproteobacteria, encompassing important human pathogens, manipulators of host reproduction, and mutualists. Here we report the discovery of a novel Rickettsiales bacterium associated with Paramecium, displaying a unique extracellular lifestyle, including the ability to replicate outside host cells. Genomic analyses show that the bacterium possesses a higher capability to synthesise amino acids, compared to all investigated Rickettsiales. Considering these observations, phylogenetic and phylogenomic reconstructions, and re-evaluating the different means of interaction of Rickettsiales bacteria with eukaryotic cells, we propose an alternative scenario for the evolution of intracellularity in Rickettsiales. According to our reconstruction, the Rickettsiales ancestor would have been an extracellular and metabolically versatile bacterium, while obligate intracellularity would have evolved later, in parallel and independently, in different sub-lineages. The proposed new scenario could impact on the open debate on the lifestyle of the last common ancestor of mitochondria within Alphaproteobacteria.
Similar content being viewed by others


Introduction
The “Bergey’s Manual of Systematics of Archaea and Bacteria” states that Rickettsiales (Alphaproteobacteria) “multiply only inside [eukaryotic] host cells” [1]. Obligate intracellularity indeed represents the main common feature of this bacterial order, considering that their host range, metabolic capabilities, and spectrum of effects on the hosts can vary greatly [2,3,4,5]. Indeed Rickettsiales encompass highly diverse representatives, including human pathogens [6,7,8], reproductive parasites of arthropods [3], mutualists of nematodes [3, 9] or arthropods [10], and several bacteria associated with unicellular eukaryotes, producing undisclosed consequences on their hosts [5]. Additionally, Rickettsiales are well known to evolutionary biologists, since several studies suggested them as the sister group of mitochondria [11,12,13], although this point is highly debated [14, 15].
A common trait of most Rickettsiales is the ability to perform horizontal transmission, testified by lack of congruence in host and symbiont phylogenies [2, 6, 7, 16,17,18,19,20,21] and experimental host transfers [20, 22,23,24]. Thus, Rickettsiales are “host-dependent”, but, with few exceptions [25], not “host-restricted”, because they are not exclusively vertically transmitted. The life cycle of most Rickettsiales can be ideally subdivided into two phases. The main phase is intracellular, during which all life functions, including replication, are exerted. During the other, facultative, phase, the bacteria can survive temporarily in the extracellular environment and can enter new host cells [24, 26,27,28]. According to our best knowledge, these “transmission forms”, sometimes displaying altered morphology [29,30,31], have never been reported to replicate.
Until recently, a sensu lato definition of Rickettsiales was employed, however recent studies [13, 32,33,34,35,36] repeatedly indicated that this group is paraphyletic. Thus, in this work we will follow the sensu stricto definition by Szokoli and co-authors [37] of Rickettsiales, i.e. including only the three families Rickettsiaceae, Anaplasmataceae, “Candidatus (Ca.) Midichloriaceae” (from now on, Midichloriaceae), and placing the other sensu lato members in the evolutionary independent order Holosporales. While Rickettsiales sensu stricto form a strongly supported monophyletic group (e.g. [13, 17, 35, 37, 38]), the members of the three families can be distinguished by multiple features, including the host cell entrance mechanism and the presence or absence of a host-derived membrane envelope in the intracellular phase [5, 26, 27]. Regardless of these differences, the transition to an obligate intracellular state has always been assumed to have occurred only once in Rickettsiales evolution, before the separation of the three families [7, 24, 39, 40]. Thus, this trait has been considered as directly inherited from their common ancestor.
Here we present the first report of an Rickettsiales bacterium, “Ca. Deianiraea vastatrix” (from now on Deianiraea vastatrix; Taxonomic Description in Supplementary Material 1), displaying an unprecedented extracellular and putatively parasitic lifestyle in the interaction with the ciliate protist Paramecium primaurelia. According to the obtained novel morphological, phylogenetic and genomic data, we re-evaluated traditional hypotheses on the evolution of Rickettsiales, which always implied an “intracellularity early”, dating back to the common ancestor. Thus, based on the here presented findings, we propose an alternative scenario, named “intracellularity late”, under which we suggest that intracellularity could have evolved in parallel and independently along multiple Rickettsiales sub-lineages from an extracellular, yet host-associated, ancestor.
Materials and methods
Paramecium strain origin and maintenance
The monoclonal Paramecium strain CyL4-1 was isolated on 29 September 2014 from a water sample (9‰ salinity) collected from a wastewater stream in Larnaca, Cyprus (34.91°N, 33.60°E). It was initially maintained in a lettuce infusion inoculated with Enterobacter aerogenes (gradually adapted to 0‰). In order to maintain it in the laboratory, a transfer procedure was performed every 2–3 days, i.e., single viable cells of Paramecium were picked up and transferred into a Petri dish containing 250 μl of fresh lettuce infusion inoculated with E. aerogenes and supplemented with 0.8 μg/ml β-sitosterol (Merck, Darmstadt, Germany).
For living cell observations, paramecia were analysed using differential interference contrast (DIC) with a Leica 6000 microscope (Leica Microsystems, Wetzlar, Germany) equipped with a digital camera DFC 500.
Transmission electron microscopy (TEM) and atomic force microscopy (AFM)
For TEM, Paramecium cells were processed and observed as described previously [41], except that 0.1 M phosphate buffer was used for fixation.
For AFM, living cells were placed on a cover slip and air-dried. The surface topography was registered by means of NTEGRA AURA microscope in a semi-contact mode.
Deianiraea shift assays
To check the ability of Deianiraea to colonise the cell surface of other Paramecium strains and affect them, cells of the strain CyL4-1, preliminary confirmed to be abnormally shaped, unable to move, and densely covered by Deianiraea bacteria, were placed in Petri dishes containing 250 μl of lettuce infusion inoculated with food bacteria, ~50 Paramecium cells for each dish. The Deianiraea-free CyL4-1* sub-replicate of the original strain plus six additional naïve strains were then selected (Supplementary Material 2), and each added in duplicate to these Petri dishes, 25–30 cells per dish. As a control, the target cells were transferred in dishes containing only lettuce infusion inoculated with food bacteria. Cells were fed once a week with 50 μl of fresh medium inoculated with food bacteria. The dishes were checked every 4 days for the presence of living motile cells and for the signs of the developing colonisation by Deianiraea for a period of 40 days.
Molecular characterisation of Paramecium and Deianiraea
About 80–100 CyL4-1 cells were individually picked up, washed through several passages in sterile water and fixed with 70% ethanol. DNA was extracted with the Nucleospin Plant II kit (Macherey-Nagel), following the protocol for mycelium. All PCR reactions were performed as previously described [42]. For the molecular characterisation of Paramecium, 18S rRNA gene, ITS and COI gene were amplified and directly sequenced [42] (Supplementary Material 3).
Characterisation of Deianiraea was performed by PCR amplification of the almost complete 16S rRNA gene with universal bacterial primers and sequencing with internal primers, as previously described [43].
Fluorescence in situ hybridisation (FISH)
Paramecium cells were processed and observed as described previously [41], with the following modifications: fixation was performed in 4% paraformaldehyde in PBS, and the hybridisation buffer contained 15% formamide. Specific probes for Deianiraea were newly designed, and their specificity was assessed by search on the Ribosomal Database Project (RDP) [44] and on SILVA [45] (Supplementary Material 4).
16S rRNA phylogenetic analysis
Maximum likelihood (ML) and Bayesian inference (BI) phylogenetic analyses on the partial 16S rRNA gene sequence of Deianiraea were performed with ARB [46], as described previously [47]. The sequences of 33 other Rickettsiales sensu stricto [37] sequences were selected, plus 10 other Alphaproteobacteria as outgroup. 16S rRNA gene identity values were calculated on the alignment employed for phylogeny (Supplementary Material 5 and 6).
16S rRNA gene amplicon database screening
With ARB, the whole set of sequences belonging to each of the four Rickettsiales families sensu stricto were selected from the SSURef NR99 128 Silva database [45]. After filtering out chimeras with DECIPHER [48] and manual inspection, the sequences were grouped in clusters with ≥93% identity. The final set of Rickettsiales included a member for each cluster (the sequence with the lower average divergence from the same cluster) (Supplementary Material 7). Thus, the sequences from this set were queried on the integrated microbial NGS (IMNGS) platform [49], selecting >200 bp hits with 90% minimal identity. For each family, the retrieved hits were merged, filtering out those shared by two or more families. Operational taxonomic units (OTUs) for each family were retrieved with UCLUST [50] with 97% identity, and assigned to the respective prevalent environmental origin according to IMNGS categories.
Genome sequencing and assembly
The sequencing and assembly procedure is described in detail in the Supplementary Material 8–12. DNA was extracted from ∼100 cells of Paramecium covered by Deianiraea as described above. In order to reach sufficient DNA quantity, the extract was subjected to whole-genome amplification (WGA) with the REPLI-g Single Cell Kit (Qiagen). The WGA product was processed through a Nextera XT library and sequenced on an Illumina HiSeq X machine by Admera Health (South Plainfield, NJ, USA), producing 37,100,964 pairs of 150 bp reads.
After a preliminary assembly using SPAdes 3.6 [51], the blobology pipeline was applied [52] to separate contigs belonging to Deianiraea from those from Paramecium and other free-living bacteria. Namely, contigs with coverage ≥100× and best megablast hit with Bacteria or no hit were selected. This set of contigs was manually checked and revised. Thus, the reads mapping on the final selection of contigs were reassembled separately, obtaining a total of 12 contigs. According to the putative connections among contigs in the assembly graph, PCR assays were performed to join all the contigs (Supplementary Material 13) into a single circular chromosome (1,205,153 bp).
Genomic analyses
The Deianiraea genome was annotated using Prokka [53], followed by manual curation. Secreted proteins and transmembrane domains were predicted with SignalP [54] and TMHMM [55].
Presence of insertion sequences and prophages were predicted with IS finder [56] and PHAST [57], respectively, and comparing the obtained results with the annotation. The origin of the identified mobile elements was investigated by inspecting blastp hits on the whole NCBI nr protein, and on selected subsections of nr based on taxonomy.
Phylogenomic analyses
A previously published, curated dataset of 120 highly conserved orthologs was employed [36]. A representative taxon selection was chosen among Rickettsiales sensu stricto, other Alphaproteobacteria, and, as outgroup, other Proteobacteria (100 total organisms). The amino acidic sequences of Deianiraea orthologs were identified by reciprocal blastp hits followed by careful manual inspection. Then, they were added to the original alignment [36] with MAFFT [58] by keeping the originally aligned positions, and the site masking by Parks and co-authors [36] was employed (34,747 final positions).
Thus, after substitution model selection with ProtTest [59], ML and BI phylogenies were estimated for each set, respectively, with RaxML [60] with 1000 pseudoreplicates, and with MrBayes [61] with three runs each with one cold and three heated chains, iterating until convergence (i.e. average standard deviation of split frequencies below 0.01).
Metabolic prediction and comparison with other Rickettsiales
Metabolic pathways reconstruction was performed employing Pathway Tools [62], and KEGG [63], followed by manual inspection.
Cluster of orthology groups (COGs) were identified using the NCBI pipeline [64] for Deianiraea and other 11 representative Rickettsiales sensu stricto. The repertoire of COGs and functional categories was directly compared among those organisms, to identify Deianiraea peculiar traits.
The biosynthetic pathways for amino acids of Deianiraea were manually reconstructed in detail (Supplementary Materials 13–15).
Horizontal gene transfer (HGT) tests
In order to evaluate if the Deianiraea genes assigned to the COGs absent in all other Rickettsiales were a result of HGT, blastp hits on the whole NCBI nr protein database and on selected subsections based on taxonomy were inspected. “Informative HGT-candidate genes” (i.e. those with identity with best hits ≥ 50%, and with at least 5% difference in the identities with the best hits on whole nr and on Alphaproteobacteria-only) were selected for phylogenetic analyses. Briefly, orthologs were identified on selected genomes, and processed as described above (“Phylogenomic analyses”), in order to perform single-gene ML phylogenies.
For each amino acid biosynthetic pathway peculiar of Deianiraea (Supplementary Material 13), the ML phylogenetic analyses were performed on concatenated alignments, and compared with the respective organismal phylogeny. Additionally, compositional variations were tested respect to the whole Deianiraea gene set as described previously [40].
Results
A novel putative bacterial parasite of Paramecium
A monoclonal culture of P. primaurelia cells (strain CyL4-1) was obtained from a sample of wastewater collected in Larnaca—Cyprus. After one month of laboratory cultivation, many dead cells were registered. Microscopic observations revealed an abnormal phenotype: shortened ovoid-rounded shape and immotility caused by a nearly complete loss of cilia, which in paramecia fully cover the cell and are employed for locomotion and feeding (Fig. 1a, b; Supplementary Material 16d). The deciliated areas of the cell surface were covered by a dense layer of tightly packed intermingled bacteria (Figs. 1a and 2a; Supplementary Material 16c), in a fashion reminiscent of the arrangement of episymbiotic bacteria covering the surface of ciliates from oxygen-depleted environments [65, 66]. The thickness of the overlay looked roughly proportional to the level of degeneration in the shape of the ciliates. These bacteria appeared morphologically uniform, presenting the typical double-membrane cell structure of Gram-negatives, and were slightly curved rods (1.6–1.7 × 0.1–0.2 μm). Bacterial cells appeared to be narrowing on one end, with a sharp apical tip, which sometimes took directly contact with the external side of the Paramecium cell membrane (Fig. 2b, c). The bacterial cytoplasm was electron-dense, containing numerous ribosomes. Within the bacterial population, a small portion of dividing cells was regularly observed (Fig. 2d). In Paramecium, neither the intracellular ciliary basal bodies were altered in number and structure, nor the external cell membrane was damaged in its integrity. However, other features potentially linked to the pathological state were observed (Supplementary Material 16a, b). Moreover, no intracellular bacteria were ever observed aside from those in food vacuoles.

Microscopy images of P. primaurelia CyL4-1 covered by extracellular Deianiraea bacteria (a, c–f) and of control uninfected host cells (b). a Differential interference contrast (DIC) of a heavily infected CyL4-1 cell, displaying an atypical shortened and ovoid shape. The Paramecium cell surface is covered by a bacterial multilayer (white arrowhead). As shown in the enlarged inset on the right (corresponding to the square in the main picture) the ciliature is highly reduced, with only few remnants. b DIC picture of control uninfected host cells. c Fluorescence in situ hybridisation microscopy image of the surface of a Deianiraea-covered CyL4-1 cell. Deianiraea cells are visible by the green signal of the FITC (fluorescein isothiocyanate)-labelled universal bacterial probe EUB338 [103]. d–f Multichannel microscopy images showing a transversal plan from a FISH experiment on a CyL4-1 cell at an advanced stage of invasion by the putative parasite. In (d) the blue signal from DAPI (4′,6-diamidino-2-phenylindole) highlights the Paramecium fragmented macronucleus in autogamy (green arrowhead) and the bacterial cells. In (e) the green signal of FITC-labelled EUB338 is shown; in (f) the red signal of the Deianiraea-specific probe Deia_416 labelled with Cy3. In (d–f), the Deianiraea cells covering the external surface of the host cell on its outline are fluorescently stained (white arrowheads). In (e), food bacteria in a digestive vacuole are also visible (red arrowhead), without any corresponding signal from the specific probe in (f). Bars stand for 10 μm

Transmission electron microscopy images of Deianiraea bacteria on the surface of P. primaurelia CyL4-1 cells. a Portion of Paramecium cell surrounded by a huge number of extracellular Deianiraea bacteria, forming multiple layers. Bacterial cells appear electron dense, irregularly arranged, and sectioned in variably oriented planes. b–d Details of Deianiraea cells at higher magnification. In (b) and (c) longitudinal sections of some Deianiraea cells are shown. The bacterial side proximal to the Paramecium cell progressively narrows, ending up in a sharp and slightly curved apical tip, which takes direct contact with the external side of the Paramecium cell membrane (black arrowheads). In (d) a dividing Deianiraea cell, with the division septum highlighted by a black arrow. The few residual Paramecium cilia are evidenced by black asterisks in (b, d) sectioned from variable angles. Scale bars stand for, respectively, 1 μm (a) and 200 nm (b–d)
The extracellular bacteria attached to the Paramecium surface propagated rapidly within the culture, spreading the abnormal phenotype to other ciliate cells. Survival of the ciliate population was possible only by operating regular (every 2–3 days) transfers of Paramecium cells into fresh medium containing food bacteria, thereby diluting the load of epibiotic bacteria. Untreated (i.e. non-transferred) host cells invariably died within maximum 4 days. This procedure allowed laboratory maintenance for nearly 11 months of sub-replicates of the original Paramecium cultures harbouring the bacteria, never demonstrating any sign of bacterial presence inside the Paramecium cells (except within food vacuoles) in DIC, FISH with specific probes, or TEM observations.
The observed behaviour led us to hypothesise a probable parasitic effect of the surface-attached bacteria. To investigate this aspect, we obtained a sub-replicate culture (sub-strain CyL4-1*), in which, through prolonged regular transferring, we managed to completely remove the surface-attached bacteria. This sub-strain was successfully maintained in culture with no sign of the altered phenotype. Strain CyL4-1* and six other laboratory Paramecium strains were tested for susceptibility to the putative parasitic bacteria in experimental shift assays. Sub-strain CyL4-1* cells died within the 4th day of the experiment, while two others were affected, showing the same phenotype as in the original CyL4-1 strain albeit more slowly than CyL4-1*. The remaining four strains were fully untouched (Supplementary Material 2).
Deianiraeaceae, a new family of order Rickettsiales
A single almost full-length 16S rRNA gene sequence (1431 bp) was obtained from CyL4-1 strain by PCR with universal bacterial primers followed by direct sequencing. The sequence showed the highest similarities with uncultured sequences of Rickettsiales-related bacteria, with best blastn hit against a bacterium from nephridia of the earthworm Eiseniella neapolitana (82.7% identity; JX644354 [67]). The use of newly designed specific probes in FISH experiments demonstrated that the characterised sequence belonged to the extracellular, putatively parasitic, bacterium (named Deianiraea vastatrix, Taxonomic description in Supplementary Material 1) and clearly showed that it constituted the entire external layers around the Paramecium cells (Fig. 1c–f; Supplementary Materials 17, 18). Conversely, Deianiraea was absent in food vacuoles, and never found inside the ciliate cells, confirming the extracellularity of this bacterium.
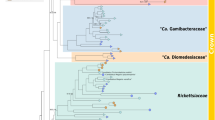
Phylogenetic analyses show that Deianiraea is a member of Rickettsiales sensu stricto, and belongs to a novel divergent family-level clade with 12 other uncharacterised database sequences. This clade is fully supported in the analyses (100 ML; 1.00 BI; Fig. 3; Supplementary Material 19) and will be referred from now on as Deianiraeaceae (Taxonomic Description in Supplementary Material 1). Within Rickettsiales, Deianiraeaceae formed a strongly supported group together with the families Anaplasmataceae and Midichloriaceae (97 ML; 1.00 BI). Although the precise relationships among those three lineages were unresolved, they were all equally supported. Overall, the 16S rRNA gene sequence identity within Deianiraeaceae and compared to other Rickettsiales are, respectively, 77.4–100% and 75.9–86.0% (Supplementary Material 6).

Bayesian phylogenetic tree on 1398 16S rRNA gene positions on 34 bacteria of the order Rickettsiales with the GTR+I+G substitution model. Posterior probabilities (after 1,000,000 generations) and maximum likelihood bootstrap values (with 1000 pseudo-replicates) of each internal tree nodes are shown on branches (values below 0.85|70 were omitted; full trees shown in Supplementary material 19). The newly characterised Deianiraea, putative parasite of P. primaurelia CyL4-1 is evidenced in bold. The four Rickettsiales families (Rickettsiaceae, Anaplasmataceae, Midichloriaceae, and the herein newly proposed Deianiraeaceae) are highlighted by coloured boxes. The outgroup, composed by other 10 Alphaproteobacteria, is shown collapsed as a trapezoidal shape. The scale bar stands for estimated 0.10 sequence divergence. On the right side, for each family the pie chart shows the proportion of prevalent environmental origins (according to the IMNGS categories) of the OTUs obtained after IMNGS database queries with the representative sequence set of the family. The relative areas of the charts are proportional to the total amount of OTUs of the respective families. Only environmental origins that are the prevalent for at least 3% of total OTUs assigned to one family were displayed
The sequences of other representatives of Deianiraeaceae belong to uncharacterised bacteria, mostly from aquatic environments, in several cases associated with diverse eukaryotes [67,68,69,70,71]. Other members of the clade were found in association to terrestrial animals, including, notably, on human skin of dermatitis-affected subjects [72, 73] (Fig. 3).
In order to evaluate the distribution and abundance of members of the novel family, a screening was performed on the IMNGS platform [49]. The family Rickettsiaceae displayed the highest total number of retrieved OTUs, followed by Deianiraeaceae, Midichloriaceae, and Anaplasmataceae (Fig. 3; Supplementary Material 20). In terms of environmental origin, Rickettsiaceae and Anaplasmataceae displayed similar patterns, with soil as the prevalent source (>20% OTUs), and several other OTUs from different water origins. On the contrary, Midichloriaceae and Deianiraeaceae OTUs derived mostly from aquatic environments, with the prevalence of seawater or marine for Midichloriaceae and freshwater for Deianiraeaceae.
Genome features
The genome of Deianiraea is formed by a single circular replicon, ~1.2 Mbp long. Its size and GC content (32.9%) are consistent with the range of known Rickettsiales. The overall coding percentage is ~92.2%, the highest among sequenced Rickettsiales (Supplementary Material 21). The genome includes 1129 ORFs, 34 tRNAs, and a single rRNA operon (split with 16S rRNA gene in one locus and 23S-5S rRNA genes in another, as in all Rickettsiales).
A limited number of mobile elements or their remnants was found: three transposases (two complete and one partial), two putative incomplete prophages (17 total phage genes), plus 13 other phage genes scattered in the genome (Supplementary Materials 22 and 23). The best blastp hits of the respective CDSs on NCBI nr proteins were quite divergent (Supplementary Materials 22 and 23), preventing a clear identification of their evolutionary relatedness to mobile sequences from other organisms.
Phylogenomic analyses were performed on a previously determined set of highly conserved protein coding genes [36]. The retrieved results confirmed 16S rRNA gene analysis, in particular the affiliation of Deianiraea to Rickettsiales sensu stricto and the monophyly of the group composed by Anaplasmataceae, Midichloriaceae and Deianiraea (Fig. 4; Supplementary Material 24). In addition, unlike the tritomy shown in the 16S phylogeny, these phylogenomic analyses indicated a sister relationship of Deianiraea to Anaplasmataceae. Moreover, none of the analysis retrieved a sister relationship of Holosporales with respect to Rickettsiales sensu stricto and, in turn (Supplementary Material 24), Holosporales did not appear monophyletic, in agreement with recent phylogenomic trees [36].

Bayesian phylogenomic tree of representative Rickettsiales and other Alphaproteobacteria. Inference was based on the concatenated protein alignment (34,747 characters) from 120 highly conserved orthologs [36] with the LG+I+G+F evolutionary model. Maximum-likelihood bootstrap values (with 1000 pseudo-replicates) and posterior probabilities (after 100,000 generations) of internal tree nodes are shown on branches (fully supported values of 100|1.00 were omitted). The newly characterised Deianiraea putative parasite of P. primaurelia CyL4-1 is evidenced in bold. The representatives of each of the four families of Rickettsiales (Rickettsiaceae, Anaplasmataceae, Midichloriaceae, and Deianiraeaceae) are evidenced in red on the right side. The three clades including other 49 total Alphaproteobacteria (two clades) and the outgroup (composed by 17 other Proteobacteria), are shown collapsed as trapezoidal shapes (full trees are shown in Supplementary material 24c, d). The scale bar stands for estimated 0.1 sequence divergence
Predicted Deianiraea metabolic features
Most of the Deianiraea ORFs (916) found a COG assignment, in total 749 distinct COGs were identified, of which 108 corresponding to more than a single ORF (Supplementary Material 25). Most of the predicted metabolic and cellular features, including number and proportion of different functional categories, are similar to other Rickettsiales (Supplementary Materials 26 and 27).
In terms of carbohydrate and energetic metabolism, Deianiraea is predicted to perform complete gluconeogenesis and the non-oxidative pentose-phosphate pathway (except transaldolase). The gene sets for pyruvate dehydrogenase, Krebs cycle and oxidative phosphorylation are present (the same repertoire of other Rickettsiales, with the exception cytochrome c and its oxidase and reductase, which are missing in Deianiraea). A tlc ATP/ADP translocase, homologue to those of other Rickettsiales, was identified as well.
Deianiraea is also equipped with biosynthetic pathways for the main structural components of cell inner membrane, cell wall and outer membrane. These traits are shared with many other Rickettsiales, with the significant exception of Anaplasmataceae and Orientia, which lack lipopolysaccharide (LPS) and, partially or completely, peptidoglycan. No capsule synthesis is present in Deianiraea, as compared to “Ca. Jidaibacter acanthamoeba” [24, 74].
In compliance with other Rickettsiales, there is an overall paucity of other biosynthetic pathways in Deianiraea. In particular, the potential for nucleotide synthesis is scarce, and only some cofactors can be produced (biotin, hem, ubiquinone, folate and Fe–S clusters). As in other Rickettsiales, the activity of multiple membrane transporters may complement the metabolic deficiencies of Deianiraea. Contrarily to other recently sequenced Rickettsiales genomes [24, 40, 75], no flagellar or chemotaxis genes were found.
A total of 84 COGs (~11.2%) of Deianiraea resulted absent in the other analysed Rickettsiales (Supplementary Material 28). Among these COGs, the prominent peculiarities were genes involved in the biosynthesis of amino acids and in cell adhesion. Notably, consistently with microscopy observations (Fig. 2), Deianiraea is also the only Rickettsiales member possessing a bacterial cytoskeletal component, namely a bactofilin family protein possibly involved in asymmetrical cell division and cell polarity [76].
Amino acid metabolism
Deianiraea has the predicted capability to synthesise the vast majority (16 out of 20) of proteinogenic amino acids, a striking difference with respect to all known Rickettsiales (Fig. 5; Supplementary Materials 13–15). Most importantly, the biosynthesis of eight of those amino acids is a unique competence of Deianiraea among Rickettsiales, raising the total number of amino acids that can be synthesised by at least one Rickettsiales member from 10 to 18. Deianiraea is by far the richest Rickettsiales in terms of amino acids biosynthesis, with a total of 56 genes devoted to amino acid production, while no other member of the order outreaches 30 genes (Fig. 5) [11, 24, 40, 75, 77].

Reconstrution of ancestral Rickettsiales features according to the “intracellularity late” hypothesis. a An idealised evolutionary tree of the four Rickettsiales families, is shown. The drawings and notes summarise selected features of each family on the right, and the inferred features of the ancestor at the root of the tree. In the drawings, flagella are shown if present in at least one member of the family, while vacuolar membrane is shown as a solid line if present for all members of the family (Anaplasmataceae), or a dashed line if present only for some members (Midichloriaceae). “Cell wall and outer membrane” and “Biosynthetic pathways” indicate the degree of completeness of, respectively, peptidoglycan and lipopolysaccharide biosynthesis or the synthesis of amino acids, cofactors, and nucleotides: “++” stands for complete in most known representatives, “+” for (at most) rich in most known representatives, “−” for (at most) scarce in most known representatives. Solid and dashed bacterial outlines in the drawing correspond to different degrees of “Cell wall and outer membrane”. “Flagella” and “Extracellular replication” indicate, respectively, the presence of flagella/flagellar genes or of an extracellular replication respect to the host cells (alike Deianiraea): “++” stands for present in most known representatives, “+” for present in some known representatives, “−” for absent in all known representatives. b Summary of the amino acids biosynthetic abilities in the Rickettsiales lineages. Columns stand for single amino acids, rows for organisms or families. Filled circles indicate that the corresponding biosynthetic pathway is present in the respective organism or family (presence in a single family member is the sufficient condition). The number in each circle indicate the total number of distinct ortholog genes involved in the pathway found in the respective organism or family (Supplementary material 14). The pathway presence in the Proto-Rickettsiales ancestor was inferred based on the presence in at least one current Rickettsiales. Total numbers of genes per organism or family are indicated on the right, and differ from the sum of the rows due to the presence of genes shared by multiple pathways (Supplementary material 14)
Interaction with Paramecium
Deianiraea is equipped with several secretion systems, which may mediate the interaction with Paramecium. These include most of the Rickettsiales typical set [78], namely Sec and Tat translocons, and type IV secretion system, the latter overall compliant with the canonical rvh set [79] (no VirB7 homologue was found, a possible false negative due to the short length of this gene and its high sequence divergence [80]).
On the other side, no components of secretion systems widely present in other Rickettsiales, namely the Apr/Hly type I secretion system proteins and Sca-like type V system autotransporter proteins [76], were found.
Additionally, the core gene set for type II secretion system [81], was identified. This structure has an equivalent complete counterpart among Rickettsiales only in “Ca. Jidaibacter”. Moreover, two ORFs, arranged in a putative operon, were recognised as secreted effectors (COG3210) of a two-partner type V secretion system, and include hemagglutinin repeats (Supplementary Material 25). Proteins of this family are involved in cell adhesion and toxicity towards eukaryotes or other bacteria, and frequently occur in operons [82]. The two proteins share extremely low (<32%) sequence identities with homologues from other bacteria, preventing to infer their function more precisely. In close proximity (but reverse oriented), the gene for the corresponding outer-membrane transporter was found (COG2831, Supplementary Material 25). No homologues of adhesins or invasins typical of other Rickettsiales were found.
Fifty-three ORFs were predicted to possess a signal peptide and no transmembrane helix, a group that may include secreted effectors (Supplementary Material 29). Interestingly, some of them present eukaryotic-like domains potentially involved in interaction with Paramecium, such as ankyrin and tetratricopeptide repeats. Additionally, three MORN repeat proteins (COG4642) were identified in the Deianiraea-exclusive set (Supplementary Material 28). This domain is also potentially involved in host cell adhesion and invasion [83].
HGT evaluation
The possibility that the 98 proteins assigned to the 84 Deianiraea-only COGs were the result of HGT was investigated by inspecting sequence identity of blastp hits across multiple taxonomic ranges (Supplementary Materials 15 and 28), and, in selected cases (six genes), phylogenetic analyses. In all cases, the high sequence divergence allowed us to exclude a recent HGT event from a known donor, preventing to robustly trace the recent evolutionary history of those Deianiraea genes (Supplementary Material 30). On the other side, some of the genes (21) were found to have putative homologues in some Rickettsiales (other than those employed in the COG classification).
The genes assigned to Deianiraea-only COGs involved in amino acids biosynthetic pathways were subjected to additional and more detailed analyses including phylogenies on concatenated protein sequences, statistical tests based on GC content and codon adaptation index (CAI), and careful comparative analyses of the inferred pathways (Supplementary Materials 13–15). Overall, because of high sequence variation with respect to any other organism and compositional consistence with respect to the other Deianiraea ORFs, no clear recent HGT was identified (Supplementary Material 15), supporting the inference that these genes could have been vertically inherited from the Rickettsiales ancestor.
Discussion
The order Rickettsiales encompasses a wide range of human pathogens, mutualist symbionts, parasites, and manipulators of the host reproduction [2, 7,8,9,10]. In this plethora of lifestyles, the intracellular localisation has always been marked as a constant, since this condition is necessary for their cell division. Parsimoniously, such trait has always been considered as inherited from the ancestor of the order [7, 24, 39, 40].
Herein, we present Deianiraea vastatrix, the first extracellular Rickettsiales bacterium. This bacterium has been observed attached to the surface of the unicellular eukaryote P. primaurelia, but never entering the host cell (Figs. 1 and 2). Deianiraea replicates in this extracellular location, covering the Paramecium cells, and being the probable cause of observed morphological alterations and lethal effects. On the other side, it is not yet determined whether in other conditions or interacting with other organisms this bacterium could be able to invade host cells and become facultatively intracellular, thus it cannot be formally defined as “strictly extracellular”. The extracellular interaction of Deianiraea and Paramecium is reminiscent of the one between the phylogenetically unrelated Vampirovibrio chlorellavorus (Melainabacteria) and Chlorella unicellular algae [84]. However, Vampirovibrio has a much wider metabolic repertoire, including lytic enzymes putatively involved in digestion of host cells.
On the other side, the molecular bases of the interaction between Deianiraea and its host are not disclosed, but several genes in the genome of this bacterium provide hints on the possible underlying mechanisms, namely the presence of secretion systems and adhesion molecules. A distinctive two-partner type V secretion system likely enables Deianiraea to release effectors bearing hemagglutinin repeats, with potential adhesive and toxic effects [82]. A type II secretion system, rare in Rickettsiales, may be involved in adhesion and motility as well, considering that its components are homologous of pili [85]. Additionally, given the limited biosynthetic repertoire of Deianiraea, it probably acquires the necessary small precursor molecules directly from the Paramecium host, exploiting a relatively wide set of membrane transporters, as other Rickettsiales. The transport of small molecules and secreted proteins could be exerted extracellularly (e.g. interacting with proteins on the surface of eukaryotic cells), and, in addition, could work in a hypothetical, though never observed, intracellular phase.
In synthesis, Deianiraea presents a unique extracellular lifestyle, distinct from all other Rickettsiales. This novel finding raises the question whether Deianiraea has reverted to extracellularity from an obligate intracellular ancestor or extracellularity was the ancestral state of the last common ancestor of the order. The two opposed scenarios may be called “intracellularity early” and “intracellularity late”. The traditional “intracellularity early” hypothesis could be seen as more parsimonious, however it implies the reversal of Deianiraea to extracellularity. At present, it seems difficult to evaluate how many and which changes and gains-of-function would have been necessary for such a complex and, to our knowledge unprecedented, evolutionary process. On the other side, the alternative “intracellularity late” hypothesis can be newly inferred based on the novel and distinctive extracellular lifestyle of Deianiraea, and, as detailed below, may find additional support considering its phylogenetic position within Rickettsiales, its biosynthetic peculiarities, but also the diversity of “intracellularity” among all other Rickettsiales.
From a phylogenetic point of view (Figs. 3 and 4), Deianiraea is a member of a new family clade of Rickettsiales, named Deianiraeaceae, with an estimated diversity comparable to the other three families of Rickettsiales. While no information is available on other Deianiraeaceae bacteria, in many cases their origin, namely the skin of aquatic animals or humans [68,69,70, 72], allows us to wonder whether they may be extracellular as well, and whether this trait could have been directly inherited from the ancestor.
Moreover, Deianiraea displays genes encoding for the biosynthetic pathways of 16 amino acids, a repertoire much larger than that of any other known Rickettsiales (Fig. 5) [11, 24, 40, 77]. Due to the absence of the Deianiraea-only amino acids biosynthesis gene sets in all other sequenced Rickettsiales, to the low level of identity with available orthologs and to the consequently limited phylogenetic signal, it is difficult to evaluate whether they are ancestral or the result of an ancient HGT event. In the future, the sequencing of additional phylogenetically related genomes may allow to obtain a more complete picture. At present, considering also the compositional homogeneity with the rest of the genome, we can assert that there is no evidence of recent HGT for any of the pathways analysed (Supplementary Material 15). This scenario is consistent with the expectations in the case they were indeed inherited vertically from the last common ancestor of Rickettsiales (Proto-Rickettsiales), which could have been more biosynthetically capable than previously thought (up to 18 amino acids) (Fig. 5).
While all known Rickettsiales, except Deianiraea, are intracellular, most of them are able to perform horizontal transmission. The process and molecular recognition machinery used to enter host cells, as well as the consequent intracellular behaviour and location, are not conserved among the Rickettsiales lineages, and can differ even within the same family [26, 27, 86]. Rickettsiaceae enter host cells by inducing phagocytosis, and typically escape the phagosomal membrane, residing freely in the host cytoplasm and exploiting host cytoskeleton for movement [26, 86,87,88]. Anaplasmataceae hijack the phagocytic process, modifying the host vacuole for their survival and proliferation, exploiting the intra-vacuolar niche [27, 89,90,91,92]. Midichloriaceae are more diverse, encompassing both bacteria enclosed in host-derived membranes and others dwelling free in the cytoplasm [24, 40, 93]. Such observed differences could be indicative of an independent parallel evolution towards the strictly obligate intracellular lifestyle.
Following the “intracellularity late” hypothesis, we inferred the main features of the last common ancestor of Rickettsiales (Proto-Rickettsiales), based both on previous knowledge and on novel findings (Fig. 5). The Proto-Rickettsiales would have been extracellular, though probably displaying a pre-adaptation to the interaction with eukaryotic cells, such as type IV secretion systems [80] or ATP/ADP translocases [94], which could have been instrumental in the successive parallel evolution towards strictly intracellular lineages. Possibly, the Proto-Rickettsiales could have been already capable of temporary invasion of host cells, as a facultative intracellular bacterium. Considering that most Rickettsiales (including Deianiraea) are found in aquatic environments [5] (Fig. 5), the Proto-Rickettsiales was likely an aquatic bacterium as well [7, 17, 19, 95,96,97]. It probably had a rather high metabolic and functional complexity, consistent with the extracellular and likely eukaryotic-associated lifestyle (Fig. 5), which could be reflected in the diversity of the extant descendants, as well as their generally conserved ability to colonise new hosts. The Proto-Rickettsiales could have been rich in biosynthetic pathways, in particular for amino acids (present work), but also for cofactors and nucleotides (as in multiple current Anaplasmataceae [77]), and was possibly capable to survive in low-oxygen conditions [40]. It could have been provided with a robust structure made by functional peptidoglycan and LPS (present work [24, 40, 98]), could synthesise capsule [24], possessed pili (present work and Schulz et al. [24]), flagella [24, 40, 75, 99, 100], and could be able to perform chemotaxis [75]. During the course of evolution, the association of its descendants to eukaryotic cells would have become tighter and tighter, with a parallel reduction in genome size and functional complexity, finally leading, in most known lineages, to loss of capability to reproduce extracellularly, and to consequent adaptation to the obligate strict intracellular state. Such “intracellularity late” scenario would have additional consequences on evolutionary inferences on mitochondrial origin. Indeed, there is an open debate on whether mitochondria and Rickettsiales are sister groups [11,12,13] or not [14, 15]. Assuming that the Proto-Rickettsiales was in fact an extracellular bacterium, even in case mitochondria and Rickettsiales truly shared a common ancestor, it would become evident that such ancestor should have been extracellular as well. Consequently, their intracellular lifestyles would have been acquired independently, much similarly to what would have happened if Rickettsiales and mitochondria were unrelated [15]. Thus, the hypothetical common ancestor of mitochondria and Rickettsiales could be envisioned as a bacterium with a degree of metabolic independence, and possibly equipped with the machinery to interact with other microorganisms.
In recent years, an increasing amount of research was produced on neglected members of the order Rickettsiales (e.g. [24, 38, 41, 74, 75, 101, 102]), which, contrarily to those thoroughly studied in the past, are not of direct medical or veterinary concern. Such investigations represented a major contribution to understand the phylogenetic and genomic diversity of Rickettsiales and are gradually reshaping our interpretation of their evolution. In this framework, the discovery of Deianiraea has the potential to represent a tipping point for the evolutionary views on Rickettsiales, providing the first supporting evidence for the novel idea that obligate intracellularity in this order would have evolved multiple times in parallel and independently from an extracellular ancestor. Obviously, with the available data it is still premature to conclude which hypothesis is preferable between the “intracellularity early” and “intracellularity late” in Rickettsiales. In this regard, future research on other under-investigated lineages of Rickettsiales, in particular Deianiraeaceae, may definitely lead to a more complete understanding of the processes that have led to the evolution and diversification of extant Rickettsiales.
Data availability
Sequence data have been deposited in GenBank with the accession codes: P. primaurelia CyL4-1 partial 18S-ITS1-5.8S-ITS2-28S—MH185950,P. primaurelia CyL4-1 partial cytochrome oxidase subunit I gene—MH188082, Deianiraea vastatrix CyL4-1 partial 16S rRNA gene—MH197138; Deianiraea vastatrix CyL4-1 genome—CP028925.
References
Dumler JS, Walker DH. Rickettsiales. In: Whitman WB, editor. Bergey’s manual of systematics of archaea and bacteria. John Wiley & Sons, Ltd; 2015.
Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. Proc R Soc B. 2006;273:2097–106.
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51.
Montagna M, Sassera D, Epis S, Bazzocchi C, Vannini C, Lo N, et al. “Candidatus Midichloriaceae” fam. nov. (Rickettsiales), an ecologically widespread clade of intracellular alphaproteobacteria. Appl Environ Microbiol. 2013;79:3241–8.
Castelli M, Sassera D, Petroni G. Biodiversity of “non-model” Rickettsiales and their association with aquatic organisms. In: Thomas S, editor. Rickettsiales—biology, molecular biology, epidemiology, and vaccine development. p. 59–91 (Springer International Publishing, Cham, Switzerland, 2016).
Kocan KM, De La Fuente J, Blouin EF, Garcia-Garcia JC. Anaplasma marginale (Rickettsiales: Anaplasmataceae): recent advances in defining host–pathogen adaptations of a tick-borne rickettsia. Parasitology. 2004;129:S285–S300.
Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM. Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009;7:6.
Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, et al. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev. 2013;26:657–702.
Taylor MJ, Bandi C, Hoerauf A. Wolbachia endosymbionts of filarial nematodes. Adv Parasitol. 2005;60:245–84.
Hosokawa T, Koga R, Kikuchi Y, Meng XY, Fukatsu T. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci USA. 2010;107:769–74.
Andersson SGE, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UCM, Podowski RM, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–43.
Fitzpatrick DA, Creevey CJ, McInerney JO. Genome phylogenies indicate a meaningful α-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol Biol Evol. 2006;23:74–85.
Wang Z, Wu M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci Rep. 2015;5:7949.
Esser C, Ahmadinejad N, Wiegand C, Rotte C, Sebastiani F, Gelius-Dietrich G, et al. A genome phylogeny for mitochondria among α-proteobacteria and a predominantly eubacterial ancestry of yeast nuclear genes. Mol Biol Evol. 2004;21:1643–60.
Martijn J, Vosseberg J, Guy L, Offre P, Ettema TJG. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature. 2018;557:101–5.
Epis S, Sassera D, Beninati T, Lo N, Beati L, Piesman J, et al. Midichloria mitochondrii is widespread in hard ticks (Ixodidae) and resides in the mitochondria of phylogenetically diverse species. Parasitology. 2008;135:485–94.
Driscoll T, Gillespie JJ, Nordberg EK, Azad AF, Sobral BW. Bacterial DNA sifted from the Trichoplax adhaerens (Animalia: Placozoa) genome project reveals a putative rickettsial endosymbiont. Genome Biol Evol. 2013;5:621–45.
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a One Health perspective. Trends Parasitol. 2012;28:437–46.
Schrallhammer M, Ferrantini F, Vannini C, Galati S, Schweikert M, Görtz HD, et al. “Candidatus Megaira polyxenophila” gen. nov., sp. nov.: considerations on evolutionary history, host range and shift of early divergent rickettsiae. PLoS ONE. 2013;8:e72581.
Senra MVX, Dias RJP, Castelli M, Silva-Neto ID, Verni F, Soares CAG, et al. A house for two-double bacterial infection in Euplotes woodruffi Sq1 (Ciliophora, Euplotia) sampled in southeastern Brazil. Microb Ecol. 2016;71:505–17.
Matsuura Y, Kikuchi Y, Meng XY, Koga R, Fukatsu T. Novel clade of alphaproteobacterial endosymbionts associated with stinkbugs and other arthropods. Appl Environ Microbiol. 2012;78:4149–56.
Braig HR, Guzman H, Tesh RB, O’Neill SL. Replacement of the natural Wolbachia symbiont of Drosophila simulans with a mosquito counterpart. Nature. 1994;367:453–5.
Caspi-Fluger A, Inbar M, Mozes-Daube N, Katzir N, Portnoy V, Belausov E, et al. Horizontal transmission of the insect symbiont Rickettsia is plant-mediated. Proc R Soc B. 2012;279:1791–6.
Schulz F, Martijn J, Wascher F, Lagkouvardos I, Kostanjšek R, Ettema TJG, et al. A Rickettsiales symbiont of amoebae with ancient features. Environ Microbiol. 2016;18:2326–42.
Casiraghi M, Anderson TJ, Bandi C, Bazzocchi C, Genchi C. A phylogenetic analysis of filarial nematodes: comparison with the phylogeny of Wolbachia endosymbionts. Parasitology. 2001;122:93–103.
Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6:375–86.
Rikihisa Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat Rev Microbiol. 2010;8:328–39.
Rasgon JL, Gamston CE, Ren X. Survival of Wolbachia pipientis in cell-free medium. Appl Environ Microbiol. 2006;72:6934–7.
Philip RN, Casper EA. Serotypes of spotted fever group rickettsiae isolated from Dermacentor andersoni (Stiles) ticks in western Montana. Am J Trop Med Hyg. 1981;30:230–8.
Labruna MB, Whitworth T, Horta MC, Bouyer DH, McBride JW, Pinter A, et al. Rickettsia species infecting Amblyomma cooperi ticks from an area in the state of São Paulo, Brazil, where Brazilian spotted fever is endemic. J Clin Microbiol. 2004;42:90–98.
Sunyakumthorn P, Bourchookarn A, Pornwiroon W, David C, Barker SA, Macaluso KR. Characterization and growth of polymorphic Rickettsia felis in a tick cell line. Appl Environ Microbiol. 2008;74:3151–8.
Muñoz-Gómez S, Hess S, Burger G, Lang BF, Susko E, Slamovits CH, et al. An updated phylogeny of the Alphaproteobacteria reveals that the parasitic Rickettsiales and Holosporales have independent origins. eLife 2019;8:e42535.
Georgiades K, Madoui MA, Le P, Robert C, Raoult D. Phylogenomic analysis of Odyssella thessalonicensis fortifies the common origin of Rickettsiales, Pelagibacter ubique and Reclimonas americana mitochondrion. PLoS ONE. 2011;5:e24857.
Ferla MP, Thrash JC, Giovannoni SJ, Patrick WM. New rRNA gene-based phylogenies of the Alphaproteobacteria provide perspective on major groups, mitochondrial ancestry and phylogenetic instability. PLoS ONE. 2013;8:e83383.
Hess S, Suthaus A, Melkonian M. “Candidatus Finniella” (Rickettsiales, Alphaproteobacteria), novel endosymbionts of viridiraptorid amoeboflagellates (Cercozoa, Rhizaria). Appl Environ Microbiol. 2016;82:659–70.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumell PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004.
Szokoli F, Castelli M, Sabaneyeva E, Schrallhammer M, Krenek S, Doak TG, et al. Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (“Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Appl Environ Microbiol. 2016;82:7236–47.
Floriano AM, Castelli M, Krenek S, Berendonk TU, Bazzocchi C, Petroni G, et al. The genome sequence of “Candidatus Fokinia solitaria”: insights on reductive evolution in Rickettsiales. Genome Biol Evol. 2018;10:1120–6.
Darby AC, Cho NH, Fuxelius HH, Westberg J, Andersson SG. Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 2007;23:511–20.
Sassera D, Lo N, Epis S, D’Auria G, Montagna M, Comandatore F, et al. Phylogenomic evidence for the presence of a flagellum and cbb3 oxidase in the free-living mitochondrial ancestor. Mol Biol Evol. 2011;28:3285–96.
Szokoli F, Sabaneyeva E, Castelli M, Krenek S, Schrallhammer M, Soares CAG, et al. “Candidatus Fokinia solitaria”, a novel “stand-alone” symbiotic lineage of Midichloriaceae (Rickettsiales). PLOS ONE. 2016;11:e0145743.
Lanzoni O, Fokin SI, Lebedeva N, Migunova A, Petroni G, Potekhin A. Rare freshwater ciliate Paramecium chlorelligerum Kahl, 1935 and its macronuclear symbiotic bacterium “Candidatus Holospora parva”. PLOS ONE. 2016;11:e0167928.
Boscaro V, Petroni G, Ristori A, Verni F, Vannini C. “Candidatus Defluviella procrastinata” and “Candidatus Cyrtobacter zanobii”, two novel ciliate endosymbionts belonging to the “Midichloria clade”. Microb Ecol. 2013;65:302–10.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 2013;41:D590–D596.
Westram R, Bader K, Prüsse E, Kumar Y, Meier H, Glöckner FO, et al. ARB: a software environment for sequence data. In de Bruijn FJ, editor. Handbook of molecular microbial ecology I: metagenomics and complementary approaches. p. 399–406 (John Wiley & Sons, Hoboken, New Jersey, 2011).
Sabaneyeva E, Castelli M, Szokoli F, Benken K, Lebedeva N, Salvetti A, et al. Host and symbiont intraspecific variability: The case of Paramecium calkinsi and “Candidatus Trichorickettsia mobilis”. Eur J Protistol. 2018;62:79–94.
Wright ES, Yilmaz LS, Noguera DR. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol. 2012;78:717–25.
Lagkouvardos I. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep. 2016;6:33721.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol. 2012;19:455–77.
Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013;4:237.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–71.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–6.
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–52.
Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34:2490–2.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Stamatakis A. Using RAxML to infer phylogenies. Curr Protoc Bioinform. 2015;51:6.14.1–6.14.14.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Karp PD, Latendresse M, Paley SM, Krummenacker M, Ong QD, Billington R, et al. Pathway tools version 19.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform. 2016;17:877–90.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62.
Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–9.
Modeo L, Fokin SI, Boscaro V, Andreoli I, Ferrantini F, Rosati G, et al. Morphology, ultrastructure, and molecular phylogeny of the ciliate Sonderia vorax with insights into the systematics of order Plagiopylida. BMC Microbiol. 2013;13:40.
Nitla V, Serra V, Fokin SI, Modeo L, Verni F, Sandeep BV, et al. Critical revision of the family Plagiopylidae (Ciliophora: Plagiopylea), including the description of two novel species, Plagiopyla ramani and Plagiopyla narasimhamurtii, and redescription of Plagiopyla nasuta Stein, 1860 from India. Zool J Linn Soc. 2018. https://doi.org/10.1093/zoolinnean/zly041
Davidson SK, Powell R, James S. A global survey of the bacteria within earthworm nephridia. Mol Phylogenet Evol. 2013;67:188–200.
Fraune S, Bosch TCG. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc Natl Acad Sci USA. 2007;104:13146–51.
Fraune S, Augustin R, Anton-Erxleben F, Wittlieb J, Gelhaus C, Klimovich VB, et al. In an early branching metazoan, bacterial colonization of the embryo is controlled by maternal antimicrobial peptides. Proc Natl Acad Sci USA. 2010;107:18067–72.
Bauermeister J, Ramette A, Dattagupta S. Repeatedly evolved host-specific ectosymbioses between sulfur-oxidizing bacteria and amphipods living in a cave ecosystem. PloS ONE. 2012;7:e50254.
Dishaw LJ, Flores-Torres J, Lax S, Gemayel K, Leigh B, Melillo D, et al. The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PloS ONE. 2014;9:e93386.
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–9.
Shinkai T, Enishi O, Mitsumori M, Higuchi K, Kobayashi Y, Takenaka A, et al. Mitigation of methane production from cattle by feeding cashew nut shell liquid. J Dairy Sci. 2012;95:5308–16.
Wang Z, Wu M. Complete genome sequence of the endosymbiont of Acanthamoeba strain UWC8, an amoeba endosymbiont belonging to the “Candidatus Midichloriaceae” family in Rickettsiales. Genome Announc. 2014;2:e00791–14.
Martijn J, Schulz F, Zaremba-Niedzwiedzka K, Viklund J, Stepanauskas R, Andersson SG, et al. Single-cell genomics of a rare environmental alphaproteobacterium provides unique insights into Rickettsiaceae evolution. ISME J. 2015;9:2373–85.
Kühn J, Briegel A, Mörschel E, Kahnt J, Leser K, Wick S, et al. Bactofilins, a ubiquitous class of cytoskeletal proteins mediating polar localization of a cell wall synthase in Caulobacter crescentus. EMBO J. 2010;29:327–39.
Dunning Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen J, et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006;2:e21.
Gillespie JJ, Kaur SJ, Rahman MS, Rennoll-Bankert K, Sears KT, Beier-Sexton M, et al. Secretome of obligate intracellular Rickettsia. FEMS Microbiol Rev. 2015;39:47–80.
Gillespie JJ, Phan IQ, Driscoll TP, Guillotte ML, Lehman SS, Rennoll-Bankert KE, et al. The Rickettsia type IV secretion system: Unrealized complexity mired by gene family expansion. Path Dis. 2016;74:ftw058.
Gillespie JJ, Ammerman NC, Dreher-Lesnick SM, Rahman MS, Worley MJ, Setubal JC, et al. An anomalous type IV secretion system in Rickettsia is evolutionarily conserved. PLoS ONE. 2009;4:e4833.
Korotkov KV, Sandkvist M, Hol WGJ. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol. 2012;10:336–51.
Guérin J, Bigot S, Schneider R, Buchanan SK, Jacob-Dubuisson F. Efficiency and simplicity in the secretion of large proteins for bacteria–host and bacteria–bacteria interactions. Front Cell Infect Microbiol. 2017;7:148.
Manson McGuire A, Cochrane K, Griggs AD, Haas BJ, Abeel T, Zeng Q, et al. Evolution of invasion in a diverse set of Fusobacterium species. Mbio. 2014;5:e01864–14.
Soo RM, Woodcroft BJ, Parks DH, Tyson GW, Hugenholtz P. Back from the dead; the curious tale of the predatory cyanobacterium Vampirovibrio chlorellavorus. PeerJ. 2015;3:e968.
Mattick JS. Type IV pili and twitching motility. Annu Rev Microbiol. 2002;56:289–314.
Ge Y, Rikihisa Y. Subversion of host cell signaling by Orientia tsutsugamushi. Microbes Infect. 2011;13:638–48.
Cardwell MM, Martinez JJ. The Sca2 autotransporter protein from Rickettsia conorii is sufficient to mediate adherence to and invasion of cultured mammalian cells. Infect Immun. 2009;77:5272–80.
Gouin E, Egile C, Dehoux P, Villiers V, Adams J, Gertler F, et al. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature. 2004;427:457–61.
Mohan Kumar D, Yamaguchi M, Miura K, Lin M, Los M, Coy JF, et al. Ehrlichia chaffeensis uses its surface protein EtpE to bind GPI-anchored protein DNase X and trigger entry into mammalian cells. PloS Pathog. 2013;9:e1003666.
Renesto P, Dehoux P, Gouin E, Touqui L, Cossart P, et al. Identification and characterization of a phospholipase D‐superfamily gene in rickettsiae. J Infect Dis. 2003;188:1276–83.
Kahlon A, Ojogun N, Ragland SA, Seidman D, Troese MJ, Ottens AK, et al. Anaplasma phagocytophilum Asp14 is an invasin that interacts with mammalian host cells via its C terminus to facilitate infection. Infect Immun. 2013;81:65–79.
Seidman D, Ojogun N, Walker NJ, Mastronunzio J, Kahlon A, Hebert KS, et al. Anaplasma phagocytophilum surface protein AipA mediates invasion of mammalian host cells. Cell Microbiol. 2014;16:1133–45.
Vannini C, Ferrantini F, Schleifer KH, Ludwig W, Verni F, Petroni G. “Candidatus Anadelfobacter veles” and “Candidatus Cyrtobacter comes,” two new Rickettsiales species hosted by the protist ciliate Euplotes harpa (Ciliophora, Spirotrichea). Appl Environ Microbiol. 2010;76:4047–54.
Schmitz-Esser S, Linka N, Collingro A, Beier CL, Neuhaus HE, Wagner M, et al. ATP/ADP translocases: a common feature of obligate intracellular amoebal symbionts related to Chlamydiae and Rickettsiae. J Bacteriol. 2004;186:683–91.
Vannini C, Petroni G, Verni, Rosati G. A bacterium belonging to the Rickettsiaceae family inhabits the cytoplasm of the marine ciliate Diophrys appendiculata (Ciliophora, Hypotrichia). Microb Ecol. 2004;49:434–42.
Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, et al. Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PloS Genet. 2006;2:e76.
Kang YJ, Diao XN, Zhao GY, Chen MH, Xiong Y, Shi M, et al. Extensive diversity of Rickettsiales bacteria in two species of ticks from China and the evolution of the Rickettsiales. BMC Evol Biol. 2014;14:167.
Driscoll TP, Verhoeve VI, Guillotte ML, Lehman SS, Rennoll SA, Beier-Sexton M, et al. Wholly Rickettsia! Reconstructed metabolic profile of the quintessential bacterial parasite of eukaryotic cells. Mbio. 2017;8:e00859–17.
Vannini C, Boscaro V, Ferrantini F, Benken KA, Mironov TI, Schweikert M, et al. Flagellar movement in two bacteria of the family Rickettsiaceae: a re-evaluation of motility in an evolutionary perspective. PLoS ONE. 2014;9:e8771.
Kwan JC, Schmidt EW. Bacterial endosymbiosis in a chordate host: long-term co-evolution and conservation of secondary metabolism. PLoS ONE. 2013;8:e80822.
Castelli M, Serra V, Senra MVX, Basuri CK, Soares CAG, Fokin SI, et al. The hidden world of Rickettsiales symbionts: “Candidatus Spectririckettsia obscura,” a novel bacterium found in Brazilian and Indian Paramecium caudatum. Microb Ecol 2018. https://doi.org/10.1007/s00248-018-1243-8
Yurchenko T, Ševčíková T, Přibyl P, El Karkouri K, Klimeš V, Amaral R, et al. A gene transfer event suggests a long-term partnership between eustigmatophyte algae and a novel lineage of endosymbiotic bacteria. ISME J. 2018;12:2163–75.
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. Combination of 16S ribosomal-RNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol. 1990;56:1919–25.
Acknowledgements
This work was funded by the European Commission FP7-PEOPLE-2009-IRSES grant 247658 (project CINAR-PATHOBACTER) to GP, the Human Frontier Science Programme Grant RGY0075/2017 to DS, the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Programme (2018–2022)—Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia to DS, and, for the work of the group from St. Petersburg, by the RSF grant 16-14-10157 to AP. We would like to thank A. Oren for advice in bacterial nomenclature and species description. S. Gabrielli and S. Lometto are acknowledged for assistance in graphical artwork and in phylogenomic analyses, respectively. Cultures were maintained in Culture Collection of Microorganisms (RC CCM), Saint Petersburg State University.
Author information
Authors and Affiliations
Contributions
NL isolated the host cells and performed live experiments. ES performed TEM and fluorescence microscopy. KB performed AFM. OL, AP and GP performed molecular experiments and probe design. MC, OL and GP performed phylogeny. MC and OL performed IMNGS analyses. MC, AMF and DS performed genome assembly and analyses. MC, AMF, SG and DS performed metabolic and evolutionary analyses. General interpretation of the results and manuscript writing were performed by MC, DS and GP, and contributed by CB, LM, ES and AP. All the authors contributed to the interpretation and writing of the specific results, and to the discussions. GP planned and coordinated the whole project, with DS planning and coordinating the genomic analyses, and ES coordinating the microscopy.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Castelli, M., Sabaneyeva, E., Lanzoni, O. et al. Deianiraea, an extracellular bacterium associated with the ciliate Paramecium, suggests an alternative scenario for the evolution of Rickettsiales. ISME J 13, 2280–2294 (2019). https://doi.org/10.1038/s41396-019-0433-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0433-9
This article is cited by
-
Host association and intracellularity evolved multiple times independently in the Rickettsiales
Nature Communications (2024)
-
Symbionts of Ciliates and Ciliates as Symbionts
Indian Journal of Microbiology (2024)
-
The terrestrial isopod symbiont ‘Candidatus Hepatincola porcellionum’ is a potential nutrient scavenger related to Holosporales symbionts of protists
ISME Communications (2023)
-
Three-dimensional images reveal the impact of the endosymbiont Midichloria mitochondrii on the host mitochondria
Nature Communications (2023)
-
Single-cell Microbiomics Unveils Distribution and Patterns of Microbial Symbioses in the Natural Environment
Microbial Ecology (2023)
