
Abstract
Ammonia-oxidizing archaea (AOA) of the phylum Thaumarchaeota are widespread in marine and terrestrial habitats, playing a major role in the global nitrogen cycle. However, their evolutionary history remains unexplored, which limits our understanding of their adaptation mechanisms. Here, our comprehensive phylogenomic tree of Thaumarchaeota supports three sequential events: origin of AOA from terrestrial non-AOA ancestors, colonization of the shallow ocean, and expansion to the deep ocean. Careful molecular dating suggests that these events coincided with the Great Oxygenation Event around 2300 million years ago (Mya), and oxygenation of the shallow and deep ocean around 800 and 635–560 Mya, respectively. The first transition was likely enabled by the gain of an aerobic pathway for energy production by ammonia oxidation and biosynthetic pathways for cobalamin and biotin that act as cofactors in aerobic metabolism. The first transition was also accompanied by the loss of dissimilatory nitrate and sulfate reduction, loss of oxygen-sensitive pyruvate oxidoreductase, which reduces pyruvate to acetyl-CoA, and loss of the Wood–Ljungdahl pathway for anaerobic carbon fixation. The second transition involved gain of a K+ transporter and of the biosynthetic pathway for ectoine, which may function as an osmoprotectant. The third transition was accompanied by the loss of the uvr system for repairing ultraviolet light-induced DNA lesions. We conclude that oxygen availability drove the terrestrial origin of AOA and their expansion to the photic and dark oceans, and that the stressors encountered during these events were partially overcome by gene acquisitions from Euryarchaeota and Bacteria, among other sources.
Similar content being viewed by others


Introduction
The phylum Thaumarchaeota, encompassing all ammonia-oxidizing archaea (AOA), represents up to ~30% of the prokaryotic community in the marine water column [1, 2] and up to 3% in the soil [3, 4], playing a major role in the global nitrogen cycle [5,6,7,8]. They are prevalent in seawater, soil, marine sediment, freshwater sediment and hot springs among other habitats [6, 7, 9]. While members of Thaumarchaeota remain largely uncultivated, genomic sequences from pure cultures, enrichment cultures, uncultivated single cells, and metagenomic assemblies are becoming increasingly available from all of these environments, providing an opportunity to study metabolic adaptation of this important group during its evolution and niche expansion.
Knowledge of the evolutionary history of Thaumarchaeota could provide insights into the mechanisms underlying their colonization and adaptation to new environments. Phylogenetic trees based on alignments of either single marker genes (including 16S rRNA and amoA genes) or concatenated conserved protein-coding genes indicate that the deepest lineages of AOA members of Thaumarchaeota largely consist of the organisms inhabiting terrestrial thermal environments [6, 10, 11], suggesting a thermophilic ancestor for AOA [12,13,14,15]. Gene acquisition through lateral gene transfer (LGT) from Bacteria to Thaumarchaeota was proposed to have facilitated the transition of Thaumarchaeota from thermal environments to temperate waters, though this conclusion was initially based on phylogenetic analyses of a limited number of taxa and genes [16]. A later study of 545 thaumarchaeotal fosmid clones from metagenomic libraries derived from samples of deep Mediterranean waters suggested that 24% of the gene families in these Thaumarchaeota were acquired from Bacteria [17]. If validated, such massive inter-domain transfers would have fundamental implications for the evolutionary history of Thaumarchaeota. More recently, some uncultivated non-AOA members of Thaumarchaeota were discovered in hot springs [18,19,20], anoxic peat [21], aquifer sediments [22], and acidic forest soils [23], and they consistently represent the deepest lineages in the Thaumarchaeota phylogenomic trees [18, 20, 21].
To gain deeper insight into the evolutionary history of Thaumarchaeota, we constructed a phylogenomic tree of Thaumarchaeota using sequences from phylogenetically and ecologically diverse lineages. We also evaluated the impact of LGT from Bacteria on thaumarchaeotal niche expansions at various evolutionary stages by integrating genomic data from Bacteria and other Archaea clades. Our analyses showed strong coincidences of the origin and habitat expansions of Thaumarchaeota with the availability of molecular oxygen in these habitats, and a potential role for acquisition of genes from Bacteria as drivers of these major evolutionary transitions.
Materials and methods
Phylogenomic tree construction and molecular dating analysis
Amino acid sequences from 77 single-copy gene families were concatenated for maximum-likelihood (ML) phylogenomic tree construction with IQ-Tree (v1.6.2) [24]. Each of these families was largely shared by 64 AOA Thaumarchaeota, 17 non-AOA Thaumarchaeota, 13 Aigarchaeota, 13 Bathyarchaeota, 21 Crenarchaeota, and 38 Euryarchaeota. The complete list of genomes used is provided in Table S1 and metadata for the single amplified genomes (SAGs) and metagenome-assembled genomes (MAGs) [25] used is provided in Table S2.
Next, a Bayesian estimate of the Thaumarchaeota divergence time on the ML concatenated tree was carried out with MCMCTree [26], using as calibration several temporal constraints (Table S3) like the timing of the archaeal root and the timing of diversification of a few crenarchaeotal lineages. Either a recent root of 3.8–2.7 billion years ago (Gya) [27] or an ancient root of 4.38–3.46 Gya [28] was employed. The time constraints of crenarchaeotal diversification events were derived from distinguishing features for the different lineages. For example, both Thermoproteales and Sulfolobales likely originated after the Great Oxygenation Event (GOE), because they use oxygen as a terminal electron acceptor [29]. Given the uncertainty of these calibrations, we used different combinations of these two types of constraints in the dating analyses. There have been other constraints that have been published for Euryarchaeota [29], but these constraints were not used here because whether or not the root of Archaea is placed within Euryarchaeota remains highly controversial [30,31,32,33]. Further technical details are provided in the Supplementary Methods.
Evaluating the role of LGT from Bacteria in Thaumarchaeota evolution
LGT events from Bacteria to Thaumarchaeota have been proposed to be crucial to the evolution and adaptation of Thaumarchaeota [15,16,17, 34]. We used two complementary approaches to infer potential LGT events. First, we inferred potential LGT events using the gene presence/absence pattern, which is independent of gene phylogeny. For example, LGT events may have occurred during the lifestyle transition from non-AOA to AOA if a gene family was absent from non-AOA members but prevalent among AOA members. LGT events may have also occurred during habitat expansion (from terrestrial to shallow ocean and from shallow ocean to deep ocean) if a gene family was absent from the members in the old habitat but present universally among the members in the new habitat. Once a gene family of interest was identified as a potential LGT based on the presence/absence pattern, the putative Bacteria origin of this gene family was inferred through ML phylogenetic analysis using IQ-Tree. The second method detected potential LGT events by systematically analyzing ML gene trees of 1346 gene families that we mapped to archaeal Clusters of Orthologous Groups (arCOGs) database [35]. Many genomes of Thaumarchaeota, Aigarchaeota, and Bathyarchaeota were not available when we performed this part of the analysis; thus, a reduced genomic data set of 63 AOA Thaumarchaeota, 14 Crenarchaeota, 32 Euryarchaeota, and 19 Bacteria were used. In addition, a large collection of fosmid clone sequences putatively derived from Thaumarchaeota [17] were included. Further technical details are provided in the Supplementary Methods.
Results and discussion
The evolutionary origin of ammonia-oxidizing Thaumarchaeota coincided with the Great Oxidation Event
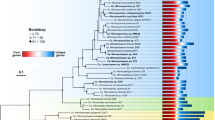
Our Thaumarchaeota phylogenomic tree (Fig. 1) identifies four main groups evolving sequentially, including the basal group, the terrestrial group, the shallow-water group, and the deep-water group, which is congruent with the branching order of these major groups estimated in previous studies [18]. The hallmark genes responsible for ammonia oxidation [36], the amoABC genes encoding the ammonia monooxygenase (AMO), were completely missing in the basal group but universally present in the terrestrial and marine groups of Thaumarchaea (Fig. 2), confirming previous findings in which the basal group and the remaining groups were non-AOA and AOA, respectively [18, 20, 21, 23]. The phylogenomic tree also displayed that the basal, the terrestrial, and the shallow-water groups are paraphyletic and the deep-water group as monophyletic (Fig. 1). This is again fully consistent with previous phylogenetic inferences in which Thaumarchaeota originated from non-AOA members [20, 21], that AOA evolved from non-AOA [18], and that marine AOA evolved from terrestrial AOA. Within the marine Thaumarchaeota lineages (Fig. 1), the deep-water group clustered with the two Candidatus Nitrosopelagicus brevis strains enriched from shallow water sampled at 25 m depth [37, 38] and with two Candidatus Nitrosopelagicus MAGs reconstructed from water sampled at 100 m depth [39].

Evolutionary timeline of Thaumarchaeota estimated using MCMCTree on top of the rooted ML concatenated tree consisting of AOA Thaumarchaeota (the deep-water group, the shallow-water group, and the terrestrial group), and the non-AOA Thaumarchaeota (the basal group), Aigarchaeota, Bathyarchaeota, Crenarchaeota, and Euryarchaeota. Nodes with known time constraints were marked with brown ovals. Nodes showing the non-AOA Thaumarchaeota origin (Node 1), the AOA Thaumarchaeota origin (Node 2), and major habitat expansions (Nodes 3 and 4) are labeled, with the flanking horizontal gray bars representing the posterior 95% confidence intervals (CI). The two vertical gray bars represent the timing of great oxygenation event (GOE, 2330 Mya) and multiple deep ocean oxygenation events in the overall anoxic Ediacaran (635–560 Mya) ocean. The schematic representation of oxygen evolutionary history in the atmosphere, the shallow and deep ocean is placed at the bottom and adapted from [96]. The ancient atmospheric oxygen concentration represented by the percent of present atmospheric level is shown as a vertical bar with red-to-orange gradient with the percent value placed above the bar. For the purpose of illustration, the height of the bars representing oxygen concentration is not proportional to the corresponding data above the bar

The phyletic pattern of the ecologically relevant genes in 81 Thaumarchaeota and 13 Aigarchaeota. The phylogenomic tree shown in the left was pruned from the full phylogenomic tree used to predict the chronogram shown in Fig. 1, and the phylogenetic groups were colored according to Fig. 1 (with color scheme shown in a box at the top-left corner). The nodes with a bootstrap value >95% were marked in black solid dots. The estimated completeness of each genome was placed near the taxon name. The potential gene gains based on the gene presence/absence pattern and based on gene tree topology (resulting from the analyses of 1346 gene trees) were separated by a vertical dashed line. The solid and open circles represent the presence and absence of the genes, respectively. amoABC ammonia monooxygenase subunit A, B, and C, fnr ferredoxin-NADP+ reductase; mut cobalamin-dependent methylmalonyl-CoA mutase, abfD 4-hydroxybutyryl-CoA dehydratase, Carboxylase* biotin-dependent acetyl-CoA/propionyl-CoA carboxylase, HP/HB cycle hydroxypropionate/hydroxybutyrate cycle, kefA K+ transporter, aprAB adenylylsulfate reductase, subunit A, B, narI nitrate reductase gamma subunit, narJ nitrate reductase molybdenum cofactor assembly chaperone, porABDG pyruvate ferredoxin oxidoreductase subunit alpha, beta, delta and gamma, cydA cytochrome bd ubiquinol oxidase subunit I (anaerobic), codhA/B acetyl-CoA decarbonylase/synthase complex subunit alpha and epsilon (component of the Wood–Ljungdahl pathway for anaerobic carbon fixation), uvrABC excinuclease ABC subunit A, B and C (for UV-induced DNA lesion repair), pstACS phosphate transport system, speE spermidine synthase, ferritin-like, DNA-binding ferritin-like protein, ynaK redox-sensitive bicupin protein, ectA l-2,4-diaminobutyric acid acetyltransferase, ectB diaminobutyrate-2-oxoglutarate transaminase, ectC l-ectoine synthase, ectD ectoine hydroxylase. The full names of the genes involved in vitamin (riboflavin, biotin, cobalamin) syntheses are not listed but available in a previous study on Nitrosopumilus maritimus SCM1 [36]
The Bayesian relaxed clock model implemented in MCMCTree [26] dated the origin of all non-AOA lineages (Node 1 in Fig. 1) at 2631 million years ago (Mya) (posterior 95% confidence interval (CI): 2765−2518 Mya), and the origin of all AOA lineages (Node 2 in Fig. 1) around 2165 Mya (95% CI: 2285−2060 Mya). Therefore, the estimated origins of non-AOA members and AOA members of Thaumarchaeota were well separated by the GOE, the latter being recently constrained to approximately 2330 Mya based on the analysis of multiple sulfur isotope records [40]. These results suggest that the origin of terrestrial AOA and the first major ecological and metabolic diversification within Thaumarchaeota were closely associated with the first major oxygenation event on Earth. The fact that the known terrestrial and marine AOA cultures are all oxygen dependent [41,42,43,44,45,46] further supports this hypothesis.
The molecular dating analyses were calibrated with the age of the ancient Archaea root (4.38−3.46 Gya) and the diversification time of several Crenarchaeota lineages. This ancient Archaea root was used as a calibration in a recent study estimating the age of methanogens [28], but it was challenged by an independent study [47], which estimated that Archaea emerged much later (<3.4 Ga). The use of the diversification time of several Crenarchaeota lineages as calibrations is problematic, as these ages are secondary calibrations derived from ancestral state reconstructions [48, 49]. For these reasons, another meaningful way to calibrate the timing of these events is to use the more recent Archaea root (3.8−2.7 Gya), which has been employed in multiple studies [27, 50], as the sole calibration. Interestingly, this new analysis led to nearly identical estimates of the timing of all four major nodes in the Thaumarchaeota phylogeny (Fig. S1) (Nodes 1 and 2 discussed in the above, and Nodes 3 and 4 discussed in the following). Other calibration alternatives, including the use of the ancient Archaea root as the sole calibration and the use of the recent Archaea root along with the Crenarchaeota diversification as the calibration, yielded reasonably older and younger ages of all nodes under discussion, respectively (Fig. S1).
Metabolic changes upon the origin of the ammonia-oxidizing Thaumarchaeota correlate with oxygen availability
In addition to amoABC mentioned above, several potentially important pathways related to electron transport and energy production were likely acquired to facilitate the transition of Thaumarchaeota from an anaerobic to aerobic lifestyle. For example, the gene family encoding a multicopper oxidase was found universally in AOA but rarely in non-AOA thaumarchaeota. This gene family is represented by multiple copies in most AOA thaumarchaeota genomes, including the model marine AOA Nitrosopumilus maritimus SCM1 [36], and they might be involved in electron transfer during ammonia oxidation. During ammonia oxidation, ammonia is first oxidized to hydroxylamine, which is further oxidized to nitrite [51]. While it is well established that the first step was catalyzed by AMO, the enzyme and electron carrier associated with the oxidation of hydroxylamine to nitrite remains unknown. One hypothesis is that the multicopper oxidases are involved in the second step [6, 36, 52]. Furthermore, the gene encoding ferredoxin-NADP+ reductase (fnr) is prevalent among the AOA members but completely missing in the non-AOA Thaumarchaeota (Fig. 2). FNR mainly participates in photosynthesis, where it transfers an electron from ferredoxin to NADP+ to form NADPH [53]. In nonphotosynthetic organisms, FNR works in reverse to provide reduced ferredoxin to various metabolic pathways [54], including the oxidative stress response [55].
The biosynthetic pathways of vitamins B2 (riboflavin), B7 (biotin), and B12 (cobalamin) were prevalent among AOA members, but completely missing from the non-AOA members of the Thaumarchaeota (Fig. 2), whereas those of vitamins B1 (thiamine) and B6 (pyridoxine) were commonly found in both AOA and non-AOA members (data not shown). The ability to synthesize vitamins B1, B2, B6, and B12 has been demonstrated in several pure cultures of the marine AOA of the genus Nitrosopumilus [56, 57], and the genes for biotin synthesis were shown to be functional in Candidatus Nitrososphaera gargensis [58]. Vitamins are often used as cofactors in some enzymes, including biotin-dependent enzymes such as acetyl-CoA/propionyl-CoA carboxylase [59, 60] and cobalamin-dependent enzymes such as methionine synthase (metH), methylmalony-CoA mutase (mut), and ribonucleotide reductase (rnr) [61]. Among these, acetyl-CoA/propionyl-CoA carboxylase and methylmalony-CoA mutase are known to be associated with inorganic carbon fixation via the autotrophic hydroxypropionate/hydroxybutyrate (HP/HB) cycle [62], which has been shown to be highly energy-efficient in the presence of oxygen in N. maritimus SCM1 [63]. Consistent with their tolerance of oxygen, these two genes are commonly found in all AOA groups, but largely missing in non-AOA thaumarchaeota (Fig. 2). A similar phyletic pattern was found for another key enzyme of the HP/HB cycle, 4-hydroxybutyryl-CoA dehydratase (abfD) involved in the formation of crotonyl-CoA from 4-hydroxybutyryl-CoA.
The successful transition from anaerobic non-AOA to aerobic AOA was also accompanied by the loss of genes involved in anaerobic metabolism. For example, the genes encoding adenylylsulfate reductase (aprAB) and nitrate reductase (narIJ) were found in non-AOA members only (Fig. 2), suggesting that some of the non-AOA members may use sulfate/nitrate as an electron acceptor in anaerobic respiration and gain energy through dissimilatory sulfate/nitrate reduction [20, 64]. Other genes (Fig. 2) involved in anaerobic energy production and restricted to non-AOA included four subunits of pyruvate:ferredoxin oxidoreductase (porABDG), a subunit of cytochrome bd-type terminal oxidase (cydA), and two subunits of the acetyl-CoA decarbonylase/synthase complex (codhAB). Pyruvate:ferredoxin oxidoreductase contains [4Fe-4S] clusters and thiamine pyrophosphate (TPP), catalyzing the reversibly oxidative decarboxylation of pyruvate to form acetyl-CoA in most anaerobes [65]. Cytochrome bd may serve as a high-affinity oxidase to support energy-requiring processes under microaerobiosis and to protect anaerobic processes from inhibition by oxygen [66]. Finally, acetyl-CoA decarbonylase/synthase is part of the Wood−Ljungdahl pathway for inorganic carbon fixation under anaerobic conditions and prevalent in Aigarchaeota and Bathyarchaeota, two sister clades of Thaumarchaeota that are strictly or predominantly anaerobes [18, 67, 68]. These lines of evidence support the previous conclusion that the non-AOA thaumarchaeota are anaerobes [18], suggesting that the oxygen requirement of the AOA thaumarchaeota and the metabolism of ammonia oxidation were evolutionary innovations rather than vertically inherited traits.
The timeline of major habitat expansion of ammonia-oxidizing Thaumarchaeota
Our molecular dating analysis suggests that the transition of the AOA thaumarchaeota from the terrestrial habitat to shallow ocean water (Node 3 in Fig. 1) occurred approximately 1017 Mya (95% CI: 1084−959 Mya). This estimate agrees with the results of a previous study that estimated the divergence of Thaumarchaeota group I.1a (corresponding to the marine groups) from the group I.1b (corresponding to the terrestrial group, except that the genus Candidatus Nitrosotenuis is a member of the terrestrial group in the present study but was assigned to group I.1a previously) later than 950 Mya. This previous inference was based on an LGT event involving the gene encoding the DnaJ-Ferredoxin fused protein, from the common ancestor (LGT donor) of green algae and land plants to the common ancestor (LGT receptor) of Thaumarchaeota groups I.1a and I.1b [69]. The underlying principle for estimating diversification time based on LGT is that the donor must have existed before the receptor, and thus the diversification time of the donor serves as a constraint for the timing of the receptor diversification. Rather than linking the invasion of the shallow ocean by Thaumarchaeota to a geological event, we asked why it did not occur earlier. We conclude that this is because the Earth underwent a “boring billion” around 1800−800 Mya, during which the atmospheric oxygen levels were around 0.1% of the present atmospheric level (Fig. 1) and the surface ocean was largely as anoxic as the deep ocean [70, 71].
To overcome the salinity barrier during the transition from the land to ocean habitats, Thaumarchaeota may have acquired genes to regulate osmotic pressure. For example, the gene kefA encoding K+ transporter is universally present in marine Thaumarchaeota but largely missing from the terrestrial group (Fig. 2), consistent with the fact that transporting K+ across cell membranes is necessary for prokaryotes to respond and adapt to osmotic pressure [72, 73]. Furthermore, we estimated that the expansion of marine AOA from shallow to deep-water habitats (Node 4 in Fig. 1) occurred approximately 643 Mya (95% CI: 701–588 Mya), coinciding with multiple events of deep sea oxygenation embedded in an overall anoxic Ediacaran ocean (ca. 635−560 Mya) [74]. The coincidence of deep ocean oxygenation and expansion of Thaumarchaeota to deep water is consistent with the potential oxygen requirement for the deep-water group of Thaumarchaeota [75]. Although members of the deep-water group of Thaumarchaeota have not been cultivated, the metabolic profile of the deep-water group, including ammonia oxidation, electron transfer and vitamin synthesis, aligns well with the shallow-water group and the terrestrial group but differs fundamentally from the basal group, strongly suggesting that members of the deep-water group required oxygen for growth. Interestingly, the transition of marine AOA from shallow to deep water seems to have coincided with the loss of the uvr system encoding excinuclease for repairing ultraviolet (UV) light-induced DNA lesions and the loss of the pst system for phosphate scavenging under phosphorus limitation, as these genes are completely missing in all deep-water group members but consistently present in all other AOA thaumarchaeota (Fig. 2). These gene losses are consistent with the physicochemical conditions in the deep ocean where UV light is absent and phosphate is replete [76, 77].
Potential bacterial and archaeal gene transfers to Thaumarchaeota
Several studies proposed that LGT from Bacteria to Thaumarchaeota occurred frequently and played a major role in Thaumarchaeota adaptation [15,16,17, 34]. However, these studies did not resolve the relative timing of the LGT events, and thus were not able to link them to ancient evolutionary events such as the origin of AOA thaumarchaeota and their subsequent expansion. As discussed in the above sections, the universal presence of a gene family in the terrestrial and marine AOA but completely or largely absent from the non-AOA thaumarchaeota (34 out of 45 gene families shown in Fig. 2) strongly suggests acquisition of the gene occurred during the evolutionary transition from non-AOA to AOA. Likewise, the universal presence of a gene family in the marine AOA but completely or largely absent from the terrestrial AOA (11 out of 45 gene families shown in Fig. 2) strongly suggests acquisition of the gene occurred during the evolutionary transition from terrestrial AOA to marine AOA. Our phylogenetic analyses imply that during the transition from non-AOA to AOA, 11 of 34 gene families were potentially acquired from Euryarchaeota (Fig. S2), two from Crenarchaeota (Fig. S3), the sources of 14 were unresolved (Fig. S4), five were exclusive to AOA (amoABC, cbtA and kefA), and the remaining two were consistent with a pattern of vertical inheritance (Fig. S5). Among the 17 genes responsible for cobalamin synthesis shown in Fig. 2, nine were potentially acquired from Euryarchaeota (Fig. S2A-I), one from Crenarchaeota (Fig. S3A), one exclusively from Thaumarchaeota, the remaining six with unresolved sources (Fig. S4A–F). Among the three genes involved in riboflavin synthesis in AOA, two were potentially imported from Euryarchaeota (Fig. S2K) and Crenarchaeota (Fig. S3B), the remaining one with an unresolved source (Fig. S4J). The inconsistent sources of potential LGT may be explained by insufficient sampling of prokaryotic genomes in our analysis, or by the independent acquisition of component genes from multiple sources during the expansion of Thaumarchaeota. We did not find a strong evidence for LGT from Bacteria to Thaumarchaeota among these gene families.
The identification of LGT events based on gene presence and absence is not perfect, as potential LGT events within gene families shared by all groups of Thaumarchaeota cannot be captured. We therefore performed extensive phylogenetic analyses of 1346 gene families from arCOGs [78, 79]. Each arCOG selected for this analysis was broadly represented by genes from Thaumarchaeota, Crenarchaeota, Euryarchaeota, and Bacteria. Bacterial genes were automatically removed from 261 arCOGs as a result of quality check of the alignments using trimAl [80], suggesting that these bacterial genes may not be true orthologs or that they are too divergent to align well. In 689 of the remaining 1085 arCOGs (Table S4), thaumarchaeotal members comprise one or more well-supported monophyletic group that are more closely related to crenarchaeotal or euryarchaeotal genes compared to bacterial genes (Fig. S6A), suggesting that thaumarchaeotal genes in these families were not likely acquired from Bacteria. There was variable support for LGT from Bacteria to Thaumarchaeota in the remaining arCOGs, but few of these genes mapped to ancestral lineages present at the origin of AOA thaumarchaeota prior to their expansion into major ocean niches.
Our phylogenetic analysis suggests that the thaumarchaeotal gene clusters in 32 arCOGs (Table S5) are embedded in the bacterial lineages (Fig. S6B & C), supporting their transfer from Bacteria to Thaumarchaeota. Among these arCOGs are 15 gene families where the embedded thaumarchaeotal lineages contain members of both marine and terrestrial groups (Fig. S6B), suggesting that gains of these bacterial genes potentially occurred at the MRCA of all AOA Thaumarchaeota lineages. One example is the gene family arCOG00050, encoding a spermidine synthase (speE, Fig. S7A) responsible for spermidine synthesis. Apart from its known function in cell growth and proliferation, spermidine also functions as a scavenger of free radicals, protecting DNA from reactive oxygen species (ROS) damage [81]. A second example is arCOG01101 (Fig. S7B), encoding a DNA-binding ferritin-like protein, which is involved in the protection of DNA from oxidative stress by using ferroxidase activity to deplete ferrous iron (Fe2+) and hydrogen peroxide [82,83,84]. A third example is the arCOG02935 (Fig. S7C), which encodes a redox-sensitive bicupin protein (YnaK) belonging to the pirin family. Experimental evidence from studies in Escherichia coli shows that this family is sensitive to oxidative stress and suggests that it may have a role in cellular redox regulation [85]. Since the origin of AOA coincided with the GOE, the recruitment of these gene families from Bacteria may have helped them overcome the oxidative stress imposed by the GOE, facilitating the adaptation of Thaumarchaeota from the anoxic to oxic habitats.
The remaining 17 arCOGs embedded in Bacteria gene clusters contain exclusively marine members (Fig. S6C), suggesting that these gene gains potentially occurred at the MRCA of marine Thaumarchaeota, and thus likely facilitated the expansion of this lineage to the marine environment. For example, arCOG00915 (ectB, Fig. S8) in this group is a key member of the operon ectABCD responsible for the biosynthesis of ectoine, a prominent member of the compatible solutes [86]. Its role in osmotic stress resistance has been demonstrated recently in the model thaumarchaeote Nitrosopumilus maritimus SCM1 [87]. In fact, the remaining three genes (ectACD) show identical phyletic patterns to ectB (Fig. 2) and the ectC was inferred to be derived from Bacteria via LGT in a previous study [87]. While these gene trees favor the hypothesis that these gene acquisitions occurred at the branch leading to the MRCA of all AOA or the MRCA of marine AOA, these hypotheses remain to be confirmed, as these genes show patchy distributions among the lineages of AOA sampled (Fig. 2). We were not able to map any LGT event specifically to the ancestral branch giving rise to the deep-water group of Thaumarchaeota. This is consistent with the conclusion that the deep-water group members evolved from the shallow-water group, and that members of the latter were already equipped with antioxidative capability.
More gains of bacterial gene may have occurred after the thaumarchaeotal transition into the ocean, which may facilitate the adaptation of their descendant lineages to particular micro-niches. Several phylogenetic patterns support this idea. For example, we found 86 arCOGs each containing many thaumarchaeotal members, among which only a few Thaumarchaeota genes clustered with bacterial genes, versus the majority that form sister groups with Crenarchaeota or Euryarchaeota lineages (Fig. S6D). This distribution suggests that these few thaumarchaeotal genes were acquired recently from Bacteria. In contrast, another 268 arCOGs each contain few (i.e., less than five) thaumarchaeotal genes. Among these are 121 arCOGs where the thaumarchaeotal genes are exclusively derived from organisms inhabiting thermal springs: Candidatus Nitrososphaera gargensis Ga9.2 [41], the single cell AB-179-E04 [19], and the marine sponge associate Cenarchaeum symbiosum A [88] (Fig. S6E), explaining in part that N. gargensis Ga9.2 (2.83 Mbp) and C. symbiosum A (2.05 Mbp) have larger genomes than the pelagic members (1.60–1.70 Mbp). In fact, the inflated genome of N. gargensis Ga9.2 has been considered to be the result of extensive gene duplication and LGT after it diverged from other Thaumarchaeota lineages [52]. Among the remaining 147 arCOGs where Thaumarchaeota genes are rare, 58 contain Thaumarchaeota clustered with Bacteria (Fig. S6F). A parsimonious explanation for this might be that these LGT events occurred at the tips of the tree rather than at the MRCA of marine Thaumarchaeota, and thus have a limited impact on the ancient transition to life in the pelagic ocean.
Several genes, while acquired at the tips of the phylogeny, may also have contributed to the adaptation of recently evolved Thaumarchaeota lineages. An example is the restriction modification system (RM), which may protect marine Thaumarchaeota from viral infection or other foreign DNA invasion [89]. This includes a type I RM system including the restriction subunit (arCOG00878), methyltransferase subunit (arCOG02632) and S subunit (arCOG02626), that were potentially recently acquired from Bacteria (Table S4). Lastly, the thaumarchaeotal genes in the remaining 89 arCOGs are intermingled with Euryarchaeota and Crenarchaeota lineages in the tree, suggesting possible LGT from both major archaeal clades.
The extensive taxonomic sampling performed in the present study allowed identification of important vertically inherited genes previously thought to be acquired from Bacteria. One example is dnaK (arCOG03060), also known as Hsp70, which is a heat shock chaperone protein expressed universally to protect cells from heat shock, among other stresses [90]. The thaumarchaeotal dnaK gene has been considered an important example of LGT from Bacteria, and its role was hypothesized to facilitate Thaumarchaeota adaptation to the pelagic environment [16, 69]. We found that this gene (arCOG03060, Fig. S9) is widespread among Thaumarchaeota members and that the marine and terrestrial members form a monophyletic group, except that the gene from Candidatus Nitrosopumilus salaria BD31 is embedded within bacterial lineages. Because the Bathyarchaeota members of dnaK form a sister lineage to the thaumarchaeotal members (Fig. S9), which is congruent with the concatenated evolutionary tree of these two archaeal phyla, the dnaK must be an ancestral trait found in the MRCA of these phyla instead of LGT from Bacteria to Thaumarchaeota.
Concluding remarks
Genome sequences from phylogenetically diverse Thaumarchaeota lineages occupying distinct niches in terrestrial and marine biomes have only recently become available. A few of them are derived from organisms occupying crucial phylogenetic positions. These include the two SAGs [19, 91] and 15 MAGs located at the base of Thaumarchaeota (known as non-AOA) [20,21,22, 39], and two surface ocean Thaumarchaeota enrichment cultures Candidatus Nitrosopelagicus brevis CN25 [38] and U25 [37] and two Candidatus Nitrosopelagicus MAGs [39] clustering with the deep-water group. These phylogenetic branches are critical in providing an accurate estimate of the timing of the origin of ammonia-oxidizing Thaumarchaeota and their subsequent major niche expansions using genome-based molecular dating analysis. Our study also greatly benefited from the recently available genome sequences of the phylogenetically diverse Aigarchaeota [18, 92] and Bathyarchaeota [67, 93,94,95]. The genome sequences of sister groups of Thaumarchaeota make it possible to validate key LGT events (e.g., speE and ectB) from Bacteria to Thaumarchaeota and to rule out other putative LGT events (e.g., dnaK). In conclusion, our study places Thaumarchaeota evolution in the context of major geological events on the ancient earth. A major finding is that oxygen availability was one of the most important drivers underlying the evolution of Thaumarchaeota at geological timescales. Although the expansion of Thaumarchaeota into new niches was facilitated by LGT of crucial genes from Bacteria, our analysis suggests that the overall impact of this process on the evolution of Thaumarchaeota is far less than previously reported.
References
Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409:507–10.
Teira E, Lebaron P, van Aken H, Herndl GJ. Distribution and activity of Bacteria and Archaea in the deep water masses of the North Atlantic. Limnol Oceanogr. 2006;51:2131–44.
Buckley DH, Graber JR, Schmidt TM. Phylogenetic analysis of nonthermophilic members of the kingdom Crenarchaeota and their diversity and abundance in soils. Appl Environ Microbiol. 1998;64:4333–9.
Torsten O, Drazenka S, Achim Q, Liza B-O, Christa S. Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol. 2003;5:787–97.
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB. Ubiquity and diversity of ammonia-oxidizing Archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA. 2005;102:14683–8.
Stahl DA, de la Torre JR. Physiology and diversity of ammonia-oxidizing archaea. Annu Rev Microbiol. 2012;66:83–101.
Stieglmeier M, Alves RJE, Schleper C. The Phylum Thaumarchaeota. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes: other major lineages of bacteria and the Archaea. Berlin Heidelberg: Springer; 2014. p. 347–62.
Wuchter C, Abbas B, Coolen MJL, Herfort L, van Bleijswijk J, Timmers P, et al. Archaeal nitrification in the ocean. Proc Natl Acad Sci USA. 2006;103:12317–22.
Nicol GW, Leininger S, Schleper C. Distribution and activity of ammonia-oxidizing Archaea in natural environments. In: Ward BB, Arp DJ, Klotz MJ, editors. Nitrification. Washington, DC: ASM Press; 2011. p. 157−78.
Groussin M, Gouy M. Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in Archaea. Mol Biol Evol. 2011;28:2661–74.
Alves RJE, Minh BQ, Urich T, von Haeseler A, Schleper C. Unifying the global phylogeny and environmental distribution of ammonia-oxidising archaea based on amoA genes. Nat Commun. 2018;9:1517.
Abby SS, Melcher M, Kerou M, Krupovic M, Stieglmeier M, Rossel C, et al. Candidatus Nitrosocaldus cavascurensis, an ammonia oxidizing, extremely thermophilic archaeon with a highly mobile genome. Front Microbiol. 2018;9:28.
Brochier-Armanet C, Gribaldo S, Forterre P. Spotlight on the Thaumarchaeota. ISME J. 2012;6:227–30.
Daebeler A, Herbold CW, Vierheilig J, Sedlacek CJ, Pjevac P, Albertsen M, et al. Cultivation and genomic analysis of “Candidatus Nitrosocaldus islandicus,” an obligately thermophilic, ammonia-oxidizing thaumarchaeon from a hot spring biofilm in Graendalur Valley, Iceland. Front Microbiol. 2018;9:193.
Lopez-Garcia P, Zivanovic Y, Deschamps P, Moreira D. Bacterial gene import and mesophilic adaptation in archaea. Nat Rev Microbiol. 2015;13:447–56.
López-García P, Brochier C, Moreira D, Rodríguez-Valera F. Comparative analysis of a genome fragment of an uncultivated mesopelagic crenarchaeote reveals multiple horizontal gene transfers. Environ Microbiol. 2004;6:19–34.
Deschamps P, Zivanovic Y, Moreira D, Rodriguez-Valera F, Lopez-Garcia P. Pangenome evidence for extensive interdomain horizontal transfer affecting lineage core and shell genes in uncultured planktonic thaumarchaeota and euryarchaeota. Genome Biol Evol. 2014;6:1549–63.
Hua Z-S, Qu Y-N, Zhu Q, Zhou E-M, Qi Y-L, Yin Y-R, et al. Genomic inference of the metabolism and evolution of the archaeal phylum Aigarchaeota. Nat Commun. 2018;9:2832.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7.
Beam JP, Jay ZJ, Kozubal MA, Inskeep WP. Niche specialization of novel Thaumarchaeota to oxic and hypoxic acidic geothermal springs of Yellowstone National Park. ISME J. 2014;8:938–51.
Lin X, Handley KM, Gilbert JA, Kostka JE. Metabolic potential of fatty acid oxidation and anaerobic respiration by abundant members of Thaumarchaeota and Thermoplasmata in deep anoxic peat. ISME J. 2015;9:2740–4.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219.
Weber EB, Lehtovirta-Morley LE, Prosser JI, Gubry-Rangin C. Ammonia oxidation is not required for growth of Group 1.1c soil Thaumarchaeota. FEMS Microbiol Ecol. 2015;91:fiv001.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Blank CE. An expansion of age constraints for microbial clades that lack a conventional fossil record using phylogenomic dating. J Mol Evol. 2011;73:188–208.
Wolfe JM, Fournier GP. Horizontal gene transfer constrains the timing of methanogen evolution. Nat Ecol Evol. 2018;2:897–903.
Blank CE. Phylogenomic dating—a method of constraining the age of microbial taxa that lack a conventional fossil record. Astrobiology. 2009;9:173–91.
Adam PS, Borrel G, Brochier-Armanet C, Gribaldo S. The growing tree of Archaea: new perspectives on their diversity, evolution and ecology. ISME J. 2017;11:2407.
Petitjean C, Deschamps P, Lopez-Garcia P, Moreira D. Rooting the domain archaea by phylogenomic analysis supports the foundation of the new kingdom Proteoarchaeota. Genome Biol Evol. 2014;7:191–204.
Raymann K, Brochier-Armanet C, Gribaldo S. The two-domain tree of life is linked to a new root for the Archaea. Proc Natl Acad Sci USA. 2015;112:6670–5.
Williams TA, Szöllősi GJ, Spang A, Foster PG, Heaps SE, Boussau B, et al. Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc Natl Acad Sci USA. 2017;114:E4602–11.
Brochier-Armanet C, Deschamps P, Lopez-Garcia P, Zivanovic Y, Rodriguez-Valera F, Moreira D. Complete-fosmid and fosmid-end sequences reveal frequent horizontal gene transfers in marine uncultured planktonic archaea. ISME J. 2011;5:1291–302.
Makarova KS, Wolf YI, Koonin EV. Archaeal Clusters of Orthologous Genes (arCOGs): An update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life. 2015;5:818–40.
Walker CB, de la Torre JR, Klotz MG, Urakawa H, Pinel N, Arp DJ, et al. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc Natl Acad Sci USA. 2010;107:8818–23.
Carini P, Dupont Christopher L, Santoro Alyson E. Patterns of thaumarchaeal gene expression in culture and diverse marine environments. Environ Microbiol. 2018;20:2112–24.
Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR, et al. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci USA. 2015;112:1173–8.
Haroon MF, Thompson LR, Parks DH, Hugenholtz P, Stingl U. A catalogue of 136 microbial draft genomes from Red Sea metagenomes. Sci Data. 2016;3:160050.
Luo G, Ono S, Beukes NJ, Wang DT, Xie S, Summons RE. Rapid oxygenation of Earth’s atmosphere 2.33 billion years ago. Sci Adv. 2016;2:e1600134.
Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, et al. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci USA. 2008;105:2134–9.
Jung MY, Kim JG, Sinninghe Damste JS, Rijpstra WI, Madsen EL, Kim SJ, et al. A hydrophobic ammonia-oxidizing archaeon of the Nitrosocosmicus clade isolated from coal tar-contaminated sediment. Environ Microbiol Rep. 2016;8:983–92.
Qin W, Meinhardt KA, Moffett JW, Devol AH, Virginia Armbrust E, Ingalls AE, et al. Influence of oxygen availability on the activities of ammonia-oxidizing archaea. Environ Microbiol Rep. 2017b;9:250–6.
Bayer B, Vojvoda J, Offre P, Alves RJ, Elisabeth NH, Garcia JA, et al. Physiological and genomic characterization of two novel marine thaumarchaeal strains indicates niche differentiation. ISME J. 2016;10:1051–63.
Tourna M, Stieglmeier M, Spang A, Konneke M, Schintlmeister A, Urich T, et al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci USA. 2011;108:8420–5.
Zhalnina KV, Dias R, Leonard MT, Dorr de Quadros P, Camargo FAO, Drew JC, et al. Genome sequence of Candidatus Nitrososphaera evergladensis from Group I.1b enriched from Everglades soil reveals novel genomic features of the ammonia-oxidizing archaea. PLoS ONE. 2014;9:e101648.
Betts HC, Puttick MN, Clark JW, Williams TA, Donoghue PCJ, Pisani D. Integrated genomic and fossil evidence illuminates life’s early evolution and eukaryote origin. Nat Ecol Evol. 2018;2:1556–62.
Parham JF, Donoghue PCJ, Bell CJ, Calway TD, Head JJ, Holroyd PA, et al. Best practices for justifying fossil calibrations. Syst Biol. 2012;61:346–59.
Schenk JJ. Consequences of secondary calibrations on divergence time estimates. PLoS ONE. 2016;11:e0148228.
Blank CE. Not so old Archaea—the antiquity of biogeochemical processes in the archaeal domain of life. Geobiology. 2009;7:495–514.
Vajrala N, Martens-Habbena W, Sayavedra-Soto LA, Schauer A, Bottomley PJ, Stahl DA, et al. Hydroxylamine as an intermediate in ammonia oxidation by globally abundant marine archaea. Proc Natl Acad Sci USA. 2013;110:1006–11.
Spang A, Poehlein A, Offre P, Zumbragel S, Haider S, Rychlik N, et al. The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol. 2012;14:3122–45.
Spaans S, Weusthuis R, Van Der Oost J, Kengen S. NADPH-generating systems in bacteria and archaea. Front Microbiol. 2015;6:742.
Aliverti A, Pandini V, Pennati A, de Rosa M, Zanetti G. Structural and functional diversity of ferredoxin-NADP+ reductases. Arch Biochem Biophys. 2008;474:283–91.
Giró M, Carrillo N, Krapp AR. Glucose-6-phosphate dehydrogenase and ferredoxin-NADP(H) reductase contribute to damage repair during the soxRS response of Escherichia coli. Microbiology. 2006;152:1119–28.
Heal KR, Qin W, Ribalet F, Bertagnolli AD, Coyote-Maestas W, Hmelo LR, et al. Two distinct pools of B12 analogs reveal community interdependencies in the ocean. Proc Natl Acad Sci USA. 2017;114:364–9.
Qin W, Heal KR, Ramdasi R, Kobelt JN, Martens-Habbena W, Bertagnolli AD. et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int J Syst Evol Microbiol. 2017;67:5067–79.
Chow J, Danso D, Ferrer M, Streit WR. The Thaumarchaeon N. gargensis carries functional bioABD genes and has a promiscuous E. coli ΔbioH-complementing esterase EstN1. Sci Rep. 2018;8:13823.
Patton DA, Volrath S, Ward ER. Complementation of an Arabidopsis thaliana biotin auxotroph with an Escherichia coli biotin biosynthetic gene. Mol Gen Genet. 1996;251:261–6.
Satiaputra J, Shearwin KE, Booker GW, Polyak SW. Mechanisms of biotin-regulated gene expression in microbes. Synth Syst Biotechnol. 2016;1:17–24.
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 2003;278:41148–59.
Ishii M, Miyake T, Satoh T, Sugiyama H, Oshima Y, Kodama T, et al. Autotrophic carbon dioxide fixation in Acidianus brierleyi. Arch Microbiol. 1996;166:368–71.
Konneke M, Schubert DM, Brown PC, Hugler M, Standfest S, Schwander T, et al. Ammonia-oxidizing archaea use the most energy-efficient aerobic pathway for CO2 fixation. Proc Natl Acad Sci USA. 2014;111:8239–44.
Quatrini R, Appia-Ayme C, Denis Y, Jedlicki E, Holmes DS, Bonnefoy V. Extending the models for iron and sulfur oxidation in the extreme Acidophile Acidithiobacillus ferrooxidans. BMC Genom. 2009;10:394.
Chabrière E, Charon MH, Volbeda A, Pieulle L, Hatchikian EC, Fontecilla–Camps JC. Crystal structures of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nat Struct Biol. 1999;6:182.
Jünemann S. Cytochrome bd terminal oxidase. Biochim Biophys Acta Bioenerg. 1997;1321:107–27.
Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science. 2015;350:434–8.
Zhou Z, Pan J, Wang F, Gu J, Li M. Bathyarchaeota: globally distributed metabolic generalists in anoxic environments. FEMS Microbiol Rev. 2018;42:639–55.
Petitjean C, Moreira D, López-García P, Brochier-Armanet C. Horizontal gene transfer of a chloroplast DnaJ-Fer protein to Thaumarchaeota and the evolutionary history of the DnaK chaperone system in Archaea. BMC Evol Biol. 2012;12:226.
Holland HD. The oxygenation of the atmosphere and oceans. Philos Trans R Soc Lond B Biol Sci. 2006;361:903–15.
Planavsky NJ, Reinhard CT, Wang X, Thomson D, McGoldrick P, Rainbird RH, et al. Low Mid-Proterozoic atmospheric oxygen levels and the delayed rise of animals. Science. 2014;346:635–8.
Rosen BP. Recent advances in bacterial ion transport. Annu Rev Microbiol. 1986;40:263–86.
Sleator RD, Hill C. Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol Rev. 2002;26:49–71.
Sahoo SK, Planavsky NJ, Jiang G, Kendall B, Owens JD, Wang X, et al. Oceanic oxygenation events in the anoxic Ediacaran ocean. Geobiology. 2016;14:457–68.
Swan BK, Chaffin MD, Martinez-Garcia M, Morrison HG, Field EK, Poulton NJ, et al. Genomic and metabolic diversity of marine group I Thaumarchaeota in the mesopelagic of two subtropical gyres. PLoS ONE. 2014;9:e95380.
Luo H, Zhang H, Long RA, Benner R. Depth distributions of alkaline phosphatase and phosphonate utilization genes in the North Pacific Subtropical Gyre. Aquat Microb Ecol. 2011;62:61–9.
Luo H, Tolar BB, Swan BK, Zhang CL, Stepanauskas R, Ann Moran M, et al. Single-cell genomics shedding light on marine Thaumarchaeota diversification. ISME J. 2014;8:732–6.
Makarova KS, Sorokin AV, Novichkov PS, Wolf YI, Koonin EV. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol Direct. 2007;2:33.
Wolf YI, Makarova KS, Yutin N, Koonin EV. Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct. 2012;7:46.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Khan AU, Mei YH, Wilson T. A proposed function for spermine and spermidine: protection of replicating DNA against damage by singlet oxygen. Proc Natl Acad Sci USA. 1992;89:11426–7.
Martinez A, Kolter R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol. 1997;179:5188–94.
Arnold AR, Barton JK. DNA protection by the bacterial ferritin Dps via DNA charge transport. J Am Chem Soc. 2013;135:15726–9.
Zeth K. Dps biomineralizing proteins: multifunctional architects of nature. Biochem J. 2012;445:297–311.
Gurmu D, Lu J, Johnson KA, Nordlund P, Holmgren A, Erlandsen H. The crystal structure of the protein YhaK from Escherichia coli reveals a new subclass of redox sensitive enterobacterial bicupins. Proteins. 2009;74:18–31.
Czech L, Hermann L, Stöveken N, Richter A, Höppner A, Smits S, et al. Role of the extremolytes Ectoine and Hydroxyectoine as stress protectants and nutrients: genetics, phylogenomics, biochemistry, and structural analysis. Genes. 2018;9:177.
Widderich N, Czech L, Elling FJ, Konneke M, Stoveken N, Pittelkow M, et al. Strangers in the archaeal world: osmostress-responsive biosynthesis of ectoine and hydroxyectoine by the marine thaumarchaeon Nitrosopumilus maritimus. Environ Microbiol. 2016;18:1227–48.
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y, Sugahara J, et al. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci USA. 2006;103:18296–301.
Ahlgren NA, Chen Y, Needham DM, Parada AE, Sachdeva R, Trinh V, et al. Genome and epigenome of a novel marine Thaumarchaeota strain suggest viral infection, phosphorothioation DNA modification and multiple restriction systems. Environ Microbiol. 2017;19:2434–52.
Young JC. Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol. 2010;88:291–300.
Plominsky AM, Trefault N, Podell S, Blanton JM, De la Iglesia R, Allen EE, et al. Metabolic potential and in situ transcriptomic profiles of previously uncharacterized key microbial groups involved in coupled carbon, nitrogen and sulfur cycling in anoxic marine zones. Environ Microbiol. 2018;20:2727–42.
Beam JP, Jay ZJ, Schmid MC, Rusch DB, Romine MF, Jennings Rd M, et al. Ecophysiology of an uncultivated lineage of Aigarchaeota from an oxic, hot spring filamentous ‘streamer’ community. ISME J. 2015;10:210.
He Y, Li M, Perumal V, Feng X, Fang J, Xie J, et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol. 2016;1:16035.
Lazar CS, Baker BJ, Seitz K, Hyde AS, Dick GJ, Hinrichs KU, et al. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ Microbiol. 2016;18:1200–11.
Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD, et al. Predominant archaea in marine sediments degrade detrital proteins. Nature. 2013;496:215–8.
Somero GN, Lockwood B, Tomanek L. Oxygen and metabolism. In: Somero GN, Lockwood B, Tomanek L, editors. Biochemical adaptation. Massachusetts: Sinauer Associates, Inc. Publishers; 2017.
Acknowledgements
We thank Professor Chuanlun Zhang and Professor Mary Ann Moran for their comments in the earlier versions of the manuscript. This research is supported by the National Key R&D Program of China (2018YFC0309800), the National Science Foundation of China (41776129), and the Hong Kong Research Grants Council Area of Excellence Scheme (AoE/M-403/16). Work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231. JTH’s participation was supported by NSF OCE 15-38677. We thank three anonymous reviewers for their useful comments. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the DHS or S&T. In no event shall DHS, NBACC, S&T or Battelle National Biodefense Institute have any responsibility or liability for any use, misuse, inability to use, or reliance upon the information contained herein. DHS does not endorse any products or commercial services mentioned in this publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ren, M., Feng, X., Huang, Y. et al. Phylogenomics suggests oxygen availability as a driving force in Thaumarchaeota evolution. ISME J 13, 2150–2161 (2019). https://doi.org/10.1038/s41396-019-0418-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0418-8
This article is cited by
-
Metagenomic characterization of a novel non-ammonia-oxidizing Thaumarchaeota from hadal sediment
Microbiome (2024)
-
Nitrogen and phosphorous acquisition strategies drive coexistence patterns among archaeal lineages in soil
The ISME Journal (2023)
-
Diversity and salinity adaptations of ammonia oxidizing archaea in three estuaries of China
Applied Microbiology and Biotechnology (2023)
-
Thermophilic Dehalococcoidia with unusual traits shed light on an unexpected past
The ISME Journal (2023)
-
Diverse ecophysiological adaptations of subsurface Thaumarchaeota in floodplain sediments revealed through genome-resolved metagenomics
The ISME Journal (2022)
