
Abstract
Understanding how biological patterns translate into functional processes across different scales is a central question in ecology. Within a spatial context, extent is used to describe the overall geographic area of a study, whereas grain describes the overall unit of observation. This study aimed to characterize the snake skin microbiota (grain) and to determine host–microbial assemblage–pathogen effects across spatial extents within the Southern United States. The causative agent of snake fungal disease, Ophidiomyces ophiodiicola, is a fungal pathogen threatening snake populations. We hypothesized that the skin microbial assemblage of snakes differs from its surrounding environment, by host species, spatial scale, season, and in the presence of O. ophiodiicola. We collected snake skin swabs, soil samples, and water samples across six states in the Southern United States (macroscale extent), four Tennessee ecoregions (mesoscale extent), and at multiple sites within each Tennessee ecoregion (microscale extent). These samples were subjected to DNA extraction and quantitative PCR to determine the presence/absence of O. ophiodiicola. High-throughput sequencing was also utilized to characterize the microbial communities. We concluded that the snake skin microbial assemblage was partially distinct from environmental microbial communities. Snake host species was strongly predictive of the skin microbiota at macro-, meso-, and microscale spatial extents; however, the effect was variable across geographic space and season. Lastly, the presence of the fungal pathogen O. ophiodiicola is predictive of skin microbial assemblages across macro- and meso-spatial extents, and particular bacterial taxa associate with O. ophiodiicola pathogen load. Our results highlight the importance of scale regarding wildlife host–pathogen–microbial assemblage interactions.
Similar content being viewed by others


Introduction
Understanding biologically meaningful patterns and the scale at which they occur is a central theme in ecology [1, 2]. Incorporating scale (spatial and/or temporal) into an experimental design may allow community ecologists to make predictions or answer applied questions about a system. Macro-ecologists have described distributional structuring of communities [3, 4]; however, host-associated microbial systems seem to lack predictable distributional patterns following the most classical examples [5]. Elucidating the mechanisms that underlie observed patterns across micro- to macroscales, and how they change within the scale of description, was explored here across a microbial system in the southern United States.
Extent and grain are factors used to elucidate ecological spatial scale-based patterns [1]. Scale is a function of both extent, or the overall area encompassed by a study, and grain size is the unit of measurement [1]. Using regional measurements of extent to predict localized effects may confound the interpretation of ecological phenomena. For example, a fine-scale study (small extent) may observe patterns at a fine grain size but produce little understanding of effects across a landscape. On the other hand, studying a broad extent may result in the failure to capture fine grain biological processes, which influence landscape phenomena [1, 6, 7]. In this regard, research incorporating multiple focal scales may help explain how ecological patterns translate into functional processes. Incorporating spatial considerations into hypothesis tests about emerging wildlife pathogens is especially important for understanding how fine to broad landscape patterns may predict host–pathogen–microbial assemblage associations. For example, Allender et al. [8] found that the skin microbiota of the endangered Eastern Massasaugua Rattlesnake (Sistrurus catenatus) differed in the presence of the snake fungal disease (SFD) pathogen. However, their study included a single species and encompassed four sites near a single lake in Illinois, USA. Our study took a similar approach to Allender et al. [8] but sampled across a greater host diversity and at different spatial extents.
Numerous species are currently facing global population declines due to a plethora of synergistic factors including anthropogenic-mediated habitat destruction, climate change, and emerging infectious pathogens [9,10,11,12,13]. Although habitat destruction and fragmentation are in part explanatory for species declines, emerging fungal pathogens are of concern for amphibians, reptiles, and bats [10, 12, 13]. Amphibians are suffering declines due to a mycotic infection caused by a chytrid species of fungus Batrachochytrium dendrobatidis [14]. This fungus has caused population declines to more than 520 species of frogs, toads, salamanders, newts, and caecilians [15] in biodiversity hotspots globally (e.g., Central America and Australia) [16,17,18,19]. Snakes have been affected by the fungal pathogen Ophidiomyces ophiodiicola, which has been linked to a syndrome diagnosed as SFD [20, 21]. Population declines due to O. ophiodiicola have yet to be clearly documented. However, O. ophiodiicola has been suggested as present in a declining population of Timber Rattlesnakes (Crotalus horridus [22]) and has been confirmed in a threatened population of Eastern Massasauga Rattlesnakes (S. catenatus [23]).
Although the fungus O. ophiodiicola has recently been determined as the causative agent of SFD [21, 24], there is limited information regarding the characteristics and life history of this pathogen. Recently, a series of in vitro experiments demonstrated the wide range of environmental parameters for O. ophiodiicola and the ability of this fungus to utilize complex carbon, nitrogen, and sulfur resources [25]. The authors hypothesized that O. ophiodiicola primarily infects snakes and can persist in the soil, although researchers have yet to find evidence in a natural system.
The microbiome functions as a part of the innate immune system in vertebrates [26,27,28]. The host microbiome may confer disease resistance from pathogenic fungi by producing antifungal metabolites, outcompeting the fungus for space, or by stabilizing the microbial community to drive defense efficacy [27]. The skin microbial assemblage of snakes remains relatively unstudied and little is known regarding the composition and diversity of bacteria that compose the snake skin microbial assemblage; thus, its characterization is critical to better understanding host–pathogen–microbial assemblage interactions across space and time.
We hypothesized that the snake skin microbial assemblage differed from environmental microbial assemblages, by host species, geographic region, season, and across micro- to macroscales. We postulated that the microbiota of snakes correlates with the presence of O. ophiodiicola, and that particular bacteria may correlate with higher pathogen loads. Our objectives were to: (1) determine whether environmental microbes, host species, geographic region, and/or season was predictive of snake microbial assemblages; (2) assess fine to broad scale spatial patterns of snake skin microbial assemblages; (3) determine whether the presence of a fungal pathogen is predictive of snake skin microbial assemblages; and (4) correlate particular bacterial taxa with O. ophiodiicola pathogen load.
Methods
Field collections
During the field seasons of 2015–2017, samples were collected from 35 sites across six states in the Southern United States: Alabama, Arkansas, Florida, Georgia, Tennessee, and Texas (Fig. 1). We defined “macroscale” patterns of microbial assemblages as those observed in the Southern United States. Sampling emphasis focused on four Tennessee ecoregions (“mesoscale”), incorporating 28 sites across the state (“microscale”), to provide a finer scale understanding of the snake skin microbial assemblage (Fig. 1). All snake individuals encountered were captured and handled with clean nitrile gloves and transient microbes were removed by rinsing with 100 mL of sterile deionized water [28, 29] autoclaved for 2 h [30]. A single skin swab was collected using a sterile rayon-tipped swab (Puritan, VWR cat #10808-146) for later DNA extraction. Each snake was swabbed in a way to standardize “grain size” by taking 15 swab strokes along a 15 cm length of the middle-third body section of the animal, encompassing the ventral, dorsal, and lateral body sites. No snakes were collected or sampled that failed to exceed swabbing length.

Host species, geography, season, and pathogen presence are predictive of the microbiome across macro- to microspatial scales. Snake sampling sites and presence of O. ophiodiicola are marked with colored circles across the map. Statistically significant explanatory variables from adonis models are listed with the corresponding coefficient of determination (R2)
Environmental sampling
To assay the environment for O. ophiodiicola, soil samples (n = 48) were procured from the exact point of snake capture, excluding high traffic roads/paths. Soil samples were obtained by aseptically swabbing the soil in concentric circles not exceeding 30 cm in diameter with a rayon-tipped sterile applicator. Samples were immediately stored in dry 2 mL microcentrifuge tubes. To assay the environment for O. ophiodiicola, water samples (n = 29) were collected at the nearest accessible water source (e.g., stream bank adjacent to point capture) in proximity to captured aquatic snakes by aseptically filtering 500 mL of water through a Thermo Scientific Nalgene Analytical Test Funnel (CN 145-2020) with a measured pore size of 0.2 μm. Water was drawn through the filter by a peristaltic pump and the filter paper was aseptically placed into a dry 2 mL centrifuge tube. All three sample types (skin, soil, and water) were immediately stored at −10 °C in a CoolFreeze CF-25 vehicle freezer and transferred directly to a lab freezer at −20 °C until further processing.
DNA extraction and quantitative PCR
DNA was extracted from filter paper (1/4 slice), skin, and soil swab samples using the Qiagen DNeasy PowerSoil HTP 96 kit, per the manufacturer’s standard protocol. A single DNA extraction negative control blank was extracted on each plate of 96 samples or as a single preparation when samples exceeded the 96-well plate. All negative controls were screened using quantitative PCR (qPCR) and/or high-throughput DNA sequencing. To minimize cross-contamination, DNA extraction, qPCR, and library setup were done in separate AirClean Systems AC600 (AirClean Systems, Creedmoor, NC) dedicated to these processes. Each workstation had a dedicated set of pipettes that were routinely autoclaved before experimentation. DNA was concentrated to ≈20 µl using a Thermo Fisher Savant DNA SpeedVac. The molecular presence of O. ophiodiicola in all skin and environmental samples was detected using qPCR. The qPCR assay as described by Bohuski et al. [31] was used to amplify the internal transcribed spacer region of the rRNA gene of O. ophiodiicola. Each 96-well qPCR sample plate was processed with a positive control and two no-template negative control reactions. Sample reactions were run in triplicate and consisted of a 10 μL volume containing 5 μL IDT 2× Primetime MasterMix, 0.4 μL of IDT forward primer (10 μM), 0.4 μL of IDT reverse primer (10 μM), 0.1 μL (20 μM) IDT probe, 2.1 μL PCR-grade water, and 2 μL of DNA template. Thermocycling conditions consisted of a 3 min cycle at 95 °C, followed by 50 cycles of 95 °C for 10 s and 60 °C for 30 s. A positive test was confirmed by an exponential phase appearing at a Ct < 39 in each reaction. Samples showing incomplete evidence by testing positive for one or two replicates during the first run were reanalyzed on a separate plate. Samples still showing ambiguous results (one or two positive reactions) after the second round of amplification were considered positive [32,33,34], whereas samples not amplifying in triplicate on the second plate were considered negative. To calculate pathogen load from resulting Ct values, a single standard curve was run using a serial dilution of 1 × 1010 − 1 amplicon copies of a synthetic gBlocks (Integrated DNA Technologies) fragment identical to the target qPCR region in Bohuski et al. [31]. The standard curve was used to generate the formula, y = −0.2915 × +11.094, where “x” is average Ct value of each sample, and this was used to calculate the log copy number (herein, “pathogen load”) of the ITS region of the rRNA gene of O. ophiodiicola per qPCR reaction. All Ct values and resulting pathogen load values are reported in Supplementary Data File 1.
High-throughput sequencing and bioinformatics analyses
A subset of 168 skin swabs, ten soil samples from TN, and ten water samples from TN were selected for high-throughput sequencing. These samples represented a cross-sectional view of samples collected across 35 field sites and all three years. Sequencing was performed according to the Illumina 16S Metagenomic Sequencing Library Preparation protocol on the Illumina MiSeq instrument in two separate sequencing runs. The V4 region of the 16S rDNA was amplified using dual indexed fusion primers as described by Kozich et al. [35]. Samples were cleaned using Ampure XP magnetic beads after both the initial PCR and index PCR step. After amplification and indexing, PCR products were quantified on a Qubit fluorometer 3.0, per the manufacturer’s protocol, and visualized for amplicon size (≈450 bp) on an Agilent 2100 Bioanalyzer according to the DNA 1000 protocol, and then normalized. After library quality control and quantification, the library was loaded on an Illumina MiSeq v3 flow cell and sequenced using a 500-cycle reagent kit (paired-end 2 × 300 reads). A total of 13,908,315 raw data sequence reads were obtained during run one and 10,137,882 in run two. Data were processed per the MiSeq SOP described by Kozich et al. [35] using mothur v1.39.5. After assembling paired-end reads into contigs, sequences were removed from the analysis if they had fewer than 249 bp or >253 bp, contained homopolymers in excess of eight nucleotides, or contained any ambiguous base calls. Of these, unique sequences were aligned to the SILVA v.123 bacterial reference database [36] curated to the V4 region of interest, pre-clustered allowing for two nucleotide differences, and chimeras removed per the mothur UCHIME algorithm [37]. Remaining alignments were classified into taxonomic lineage using classify.seqs at an 80% cutoff value, and lineages identified as Archaea, Eukaryota, chloroplast, mitochondria, and unknown were removed [35]. Sequences passing all quality control metrics were clustered into operational taxonomic units (OTUs) using cluster.split at 97% sequence similarity [38]. Rare OTUs appearing five times or fewer throughout the dataset were removed [39]. In addition, 64 OTUs appearing in the negative control DNA extraction blanks (1,376 reads) were removed from the dataset. A total of 8,654,686 sequence reads were kept after filtering and utilized in all downstream analyses. Ten skin swab samples were removed (n = 178 total samples retained) due to inadequate coverage after data filtering and curation in mothur. To normalize, we ran summary.single in mothur to compute sample coverage, then subsampled at 2,101 sequence reads per sample, and used this dataset in statistical analyses. Code to reproduce the bioinformatics analysis is provided in the Supplementary “mothur code” File.
Statistical analyses—comparison of the skin with environment
All analyses and graphing were conducted in mothur v1.39.5 and R version 3.5.1 using the packages vegan [40], plyr [41], dplyr [42], ggplot2 [43], rcompanion [44], car [45], and gridExtra [46]. To test whether the snake skin microbial assemblages differed from the environment, the skin, soil, and water bacterial OTU beta diversity was calculated using the vegdist function to generate a Bray–Curtis dissimilarity matrix representing sample-to-sample pairwise distances, and these distances were further analyzed using the metaMDS function to generate a non-metric multidimensional scaling (nMDS) ordination. The adonis function was used to perform a permutational multivariate analysis of variance using 999 permutations on the Bray–Curtis dissimilarity matrix, to determine whether skin, soil, or water were explanatory variables for OTU assemblages.
Comparisons of the skin microbial assemblages across spatial extents
As the skin assemblage of snakes differed from the environmental microbial assemblages (see Results below), we used an indicator analysis (indicator values > 30, p < 0.05) to select a subset of 56 OTUs that were descriptive of the variation in the snake skin microbial assemblages across all collected skin samples. To understand the effect of spatial scale on the snake skin microbial assemblages, we subdivided our dataset into three categories for analysis including the following: (1) broad-scale patterns (macroscale) in the Southern United States (n = 158 samples); (2) mesoscale patterns across four geographically distinct Tennessee ecoregions (n = 124 samples); (3) microscale patterns of between site variation within each Tennessee ecoregion (Fig. 1). For the macroscale dataset, all samples were coded as occurring in a geographic region (Alabama, Arkansas, Florida, Georgia, Tennessee, or Texas). Tennessee ecoregion (mesoscale) factors included the blue ridge mountains, interior plateau, ridge and valley, and Southwestern Appalachians. Microscale categories included four sites within the blue ridge mountains (n = 16 skin samples), 13 sites within the interior plateau (n = 73 skin samples), and 8 sites within the Southwestern Appalachians (n = 32 skin samples). Site-based differences within the ridge and valley ecoregion were not analyzed due to a small sample size effect (three sites, three samples). The adonis function was used to perform a permutational multivariate analysis of variance using 999 permutations on the Bray–Curtis dissimilarity matrix of 56 indicator OTUs to test whether the explanatory variables including host species, geographic location, season, year, O. ophiodiicola status (positive/negative), or interactions between these factors were predictive of microbiota diversity at micro-, meso-, and macroscales. We also evaluated alpha diversity as a tentative variable component of snake skin microbial assemblages by comparing inverse Simpson diversity values for the explanatory variables host species and O. ophiodiicola status (positive/negative) using a Kruskal–Wallis test across micro-, meso-, and macroscales.
Host life history and the skin microbiota
“Ecomode” is defined as modal categories (e.g., arboreal, aquatic, fossorial, and terrestrial) of habitat use in species that do not necessarily display convergent morphology [47]. To determine whether snake life history was predictive of microbial assemblages, all snake species were categorized by ecomode including aquatic, arboreal, fossorial, or terrestrial, and an nMDS ordination was run using the metaMDS function on Bray–Curtis dissimilarity matrix values for each micro- to macroscale dataset of the 56 previously described indicator OTUs. The adonis function in vegan was used to perform a permutational multivariate analysis of variance using 999 permutations on the Bray–Curtis dissimilarity matrix of indicator OTUs to test whether the explanatory variables including ecomode, O. ophiodiicola status, or an interaction between these factors was predictive of microbiome diversity at micro-, meso-, and macroscales. We assessed the alpha diversity of ecomode categories using a Kruskal–Wallis test on inverse Simpson diversity values across micro-, meso-, and macroscales.
Pathogen load and the skin microbiota
To understand the structure of the microbiota in the presence of a fungal pathogen across space, we performed indicator analyses (indicator values > 30, p < 0.05) in mothur to select OTUs descriptive of the skin microbial assemblage in the presence/absence of O. ophiodiicola independently across macro-, meso-, and microscales. We conducted a constrained analysis of proximities using the capscale function in vegan to model the continuous explanatory variable, O. ophiodiicola pathogen load (qPCR results), on Bray–Curtis dissimilarity values from microbial assemblages of O. ophiodiicola-positive/negative snakes. Both O. ophiodiicola pathogen load/reaction and OTU relative abundances were cube root transformed before the capscale analysis. We modeled the correlation between O. ophiodiicola pathogen load and microbial assemblages for 16 indicator OTUs at the macroscale, 17 OTUs at the mesoscale, 12 OTUs for the interior plateau (microscale), and 12 OTUs for the Southwestern Appalachian mountain ecoregion (microscale). We did not model the described associations for the ridge and valley ecoregion, as we only collected one O. ophiodiicola positive snake from this location. The complete list of samples processed, taxonomic identifications, and statistical models is presented in Supplementary Tables 1–3. See Grisnik et al. [48] for information about clinical signs for a subset of snakes used in these analyses.
Results
Distribution of O. ophiodiicola
We detected the presence of O. ophiodiicola DNA on 43 snake individuals from 13 species and at 19 field sites. Of all soil samples collected (n = 48), two locations on the landscape tested positive for DNA of O. ophiodiicola at Waterloo Falls, TN (Ct = 36.3, copies/rxn = 3.2) and Edgar Evins State Park, TN (Ct = 37.3, copies/rxn = 1.6) [Supplementary Data File 1]. Positive soil samples correlated with point capture locations for O. ophiodiicola-positive animals. Of all water samples (n = 29), zero tested positive for O. ophiodiicola DNA.
Comparison of snake skin microbial assemblages with the environment
Analysis of the three sample types indicated that the skin microbial assemblage of individual snakes hosted between 25 and 824 OTUs (mean = 250, SD = 187, n = 158), whereas individual soil samples (range = 382–991 OTUs, mean = 786, SD = 173, n = 10) and water samples (range = 582–1,294 OTUs, mean = 966, SD = 230, n = 10) hosted more species-rich communities. To determine how well we sampled each community based on the rarefied data, we calculated sequence sample coverage for all samples (55% outlier, range = 61–99%, mean = 89%, SD = 8.7%). We determined that the snake skin microbial assemblage was distinct from the surrounding environment (F2, 177 = 4.01, p < 0.001, R2 = 5.4%; Fig. 2a, Supplementary Table 1). Although the beta diversity of skin, soil, and water differed, the three community types shared 1,029 OTUs (Fig. 2b). Furthermore, 23 of the 56 OTUs selected by the indicator analysis as describing significant variation in the snake skin microbial assemblage are shared with the environment (Fig. 2c).

a Beta diversity patterns of the snake skin microbial assemblage compared with environmental microbiota from both soil and water, visualized using a non-metric multidimensional scaling ordination. b Venn diagram showing overlap of OTUs between skin (red), soil (green), and water (blue) samples. c Venn diagram showing results from indicator analysis selecting for OTUs that describe variation in the snake skin microbial assemblage
Comparisons of the skin microbial assemblages across spatial extents
At the macroscale (Southern United States), host snake species (F20, 157 = 4.22, p < 0.005, R2 = 33.7%), geography (F4, 157 = 2.27, p < 0.005, R2 = 3.6%), season (F5, 157 = 2.19, p < 0.005, R2 = 4.4), and O. ophiodiicola status (F1, 157 = 2.33, p < 0.005, R2 = 0.9%) were predictive of the snake skin microbial assemblage (Fig. 1, Supplementary Table 1). The effect of season on the skin microbiota differed by snake species (F19, 157 = 1.48, p < 0.005, R2 = 11.3%) and this varied further by O. ophiodiicola status (F9, 157 = 1.23, p < 0.05, R2 = 4.4%). Season also had a differential effect on the skin microbial assemblage across geographic space (F1, 157 = 1.78, p < 0.05, R2 = 0.7%). Both geographic location (F5, 157 = 1.29, p < 0.05, R2 = 2.6%) and sampling year (F7, 157 = 1.23, p < 0.05, R2 = 3.4%) had a varied effect on independent host species’ microbial assemblages. At the macroscale, alpha diversity differed by host species (χ2 [20] = 61.4, p < 0.001) but not O. ophiodiicola status (χ2 [1] = 1.26, p > 0.05) [Supplementary Table 1].
At the mesoscale (Tennessee ecoregion), host snake species (F16, 123 = 3.78, p < 0.005, R2 = 31.9%), geography “ecoregion” (F3, 123 = 3.23, p < 0.005, R2 = 5.1%), season (F5, 123 = 1.82, p < 0.005, R2 = 4.8%), and O. ophiodiicola status (F1, 123 = 1.90, p < 0.05, R2 = 0.9%) were predictive of the skin microbial assemblages. We determined that geography “ecoregion” (F13, 123 = 1.44, p < 0.005, R2 = 9.8%), season (F14, 123 = 1.26, p < 0.005, R2 = 9.3%), and O. ophiodiicola status (F7, 123 = 1.22, p < 0.05, R2 = 4.4%) all have a differential effect on independent host snake species microbial assemblages [Supplementary Table 1]. At the mesoscale, alpha diversity varied by host species (χ2 [16] = 44.4, p < 0.001) but not O. ophiodiicola status (χ2 [1] = 0.67, p > 0.05) [Supplementary Table 1].
At the microscale, host snake species was predictive of the skin assemblage for the interior plateau (F14, 72 = 2.93, p < 0.005, R2 = 37.4%), Southwestern Appalachians (F7, 31 = 3.60, p < 0.005, R2 = 45.4%), and blue ridge mountains (F6, 15 = 1.44, p < 0.05, R2 = 51.8%). Both geography “site” and season were predictive of the skin microbial assemblage for the interior plateau (geography: F11, 72 = 1.48, p < 0.005, R2 = 14.8%, season: F4, 72 = 1.49, p < 0.01, R2 = 5.4%) and Southwestern Appalachians (geography: F6, 31 = 1.37, p < 0.05, R2 = 14.8%, season: F3, 31 = 2.24, p < 0.005, R2 = 12.1%) but not the blue ridge mountains (geography: F1, 15 = 0.98, p > 0.05, season: F1, 15 = 0.70, p > 0.05). The effect of season on the skin microbial assemblage differed by host species (F7, 72 = 1.47, p < 0.005, R2 = 9.4) within the interior plateau. There were no statistically significant interactions between explanatory variables for the Southwestern Appalachians or blue ridge mountain ecoregions (Supplementary Table 1). The presence of O. ophiodiicola was not predictive of skin microbial assemblages for the interior plateau (F1, 72 = 1.19, p > 0.05) or Southwestern Appalachians (F1, 31 = 1.22, p > 0.05) and was not tested in the blue ridge mountains due to small sample sizes. Alpha diversity differed by host species within the interior plateau (χ2 [14] = 26.4, p < 0.05) and Southwestern Appalachian ecoregions (χ2 [7] = 14.8, p < 0.05) but not the blue ridge mountains (χ2 [6] = 10.6, p > 0.05). Alpha diversity did not differ by O. ophiodiicola status within the interior plateau (χ2 [1] = 0.18, p > 0.05) or Southwestern Appalachian ecoregions (χ2 [1] = 3.2, p > 0.05).
Host life history and the skin microbiota
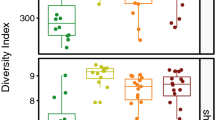
Snake ecomode was strongly predictive of skin microbial assemblages at the macro-, meso-, and microscales (interior plateau; Fig. 3). At the macroscale, ecomode (F3, 157 = 8.10, p < 0.005, R2 = 13.3%) was predictive of microbial assemblages and O. ophiodiicola presence had a variable effect within independent ecomodes (F3, 157 = 1.73, p < 0.005, R2 = 2.8%)[Supplementary Table 1]. Similarly, at the mesoscale (Tennessee ecoregions), ecomode (F3, 123 = 6.32, p < 0.005, R2 = 13.3%) was predictive of skin microbial assemblages and this effect differed by O. ophiodiicola status within independent ecomodes (F3, 123 = 1.59, p < 0.005, R2 = 3.3%). Likewise, at the microscale (interior plateau), ecomode (F3, 72 = 4.56, p < 0.005, R2 = 15.9%) was predictive of the skin microbial assemblage and O. ophiodiicola status had a differing effect within independent ecomodes (F3, 72 = 1.57, p < 0.05, R2 = 5.5%). Ecomode was also predictive of the skin microbial assemblage at the microscale within the Southwestern Appalachians (F3, 31 = 2.63, p < 0.005, R2 = 21.8%) but not in the blue ridge mountain ecoregion (F3, 15 = 1.27, p > 0.05).

Beta diversity patterns of the snake skin microbial assemblage compared by ecomode and visualized using a non-metric multidimensional scaling ordination. a Macroscale—Southern United States. b Mesoscale—Tennessee ecoregions. c Microscale—Southwestern Appalachians ecoregion. d Microscale—Interior plateau ecoregion. e Microscale—Blue ridge mountains ecoregion
Fungal pathogen load and the skin microbiota
The amount of the SFD pathogen, determined by number of ITS copies per reaction (pathogen load), was correlated with specific microbial taxa (Table 1). Five OTUs are explanatory of higher O. ophiodiicola pathogen load across macro-, meso-, and microscales (Fig. 4a, b, d). Similarly, one OTU (Otu00258) was predictive of higher O. ophiodiicola pathogen load across all four spatial scales tested (Fig. 4a–d). Two OTUs (Otu00125 and Otu00149) were correlated with O. ophiodiicola absence across both macro- and mesoscales (Fig. 4a, b).

Results depicting constrained analysis of proximities to model the continuous explanatory variable, O. ophiodiicola pathogen load (qPCR results), on Bray–Curtis dissimilarity values from skin microbial assemblages of O. ophiodiicola-positive/negative snakes. Taxonomic affiliation of OTU (e.g., Otu00006) labels is found in Table 1. Colored circles show the microbial assemblage of O. ophiodiicola-positive/negative individuals. Colored ovals show OTUs that correlate with pathogen load across macro-, meso-, and microscale spatial extents. The blue vector labeled “pathogen load” shows the direction of the relationship between higher fungal load, snake skin microbial assemblages, and indicator OTUs
Discussion
These results help to elucidate the distribution of O. ophiodiicola across the Southern United States, in particular with fine-scale resolution across Tennessee. Further, they illustrate that O. ophiodiicola DNA is found in the soil, the skin bacterial assemblage is unique to the host snake species, partially distinct from the environment, and variable between host niche. Specific to our objectives, these data demonstrate that (1) snake species was a significant and strong predictor of the skin microbial assemblage across regions of the Southern United States (macroscale), within-state ecoregions (mesoscale), and within independent ecoregions (microscale); (2) the fungal pathogen O. ophiodiicola was also a strong predictor of snake skin microbial assemblages for certain hosts across macro- and mesoscales but not at the microscale; (3) snake ecomode was indicative of the skin microbial assemblages across spatial scales with O. ophiodiicola having a differential effect on microbial assemblages by ecomode; and (4) pathogen load of O. ophiodiicola correlated with certain bacterial OTUs across spatial scales.
Host snake species was a significant predictor of the skin microbial assemblage across all scales, although the effect varied across both geographic location and season. From this result, we inferred that snake species are associated with taxon-specific skin bacterial assemblages and snake behavior may cause microbial shifts on a temporal (seasonal) basis across the landscape. Similar skin microbial assemblage patterns have been observed in both tropical and temperate amphibian species [49, 50] with similarities pertaining to antifungal metabolites that may protect the host against emerging fungal pathogens, such as Batrachochytrium salamandrivorans or B. dendrobatidis [28, 51,52,53,54,55,56,57]. Similar to our study, in snakes, Hill et al. [58] identified unique bacterial and fungal assemblages from the skin of North American Racers (Coluber constrictor) that differed from Timber Rattlesnakes (C. horridus). Snake microhabitat preference, seasonal physiological shifts, or environmental differences between spatial extents likely all contribute to the strong effect observed between host species and the skin microbial assemblages.
The fungal pathogen O. ophiodiicola was also strongly predictive of the host snake species skin microbial assemblages, although this effect was species specific and seasonal. Interestingly, the presence of O. ophiodiicola correlated with beta- but not alpha-diversity of the skin microbial assemblages across spatial scales. Perhaps this can be explained by the addition (via infection) of O. ophiodiicola to the skin creating a less stable environment, shifting beta- (turnover and colonization), but not alpha-diversity of microbial assemblages. This result is of conservation concern, as the host skin microbial assemblage may influence susceptibility to wildlife pathogens such as O. ophiodiicola. The presence of O. ophiodiicola coincides with changes in the abundance and composition of the native microbial assemblages found on snake skin. Similarly, Lemieux-Labonté et al. [59] and Allender et al. [8] found that both bacterial and fungal assemblages correlate with the presence of a pathogen in the bat and snake skin microbial assemblage, respectively. This illustrates an interesting parallel of disturbance, whether physical or pathogen mediated, which correlates with alterations in the skin microbial assemblage of a host. Moreover, this is one of the few studies to show that the presence of a wildlife pathogen on the skin of snakes may influence the host’s skin microbial assemblage. The question remains about whether O. ophiodiicola drives changes in skin microbial assemblages or a disturbance occurs, and the pathogen capitalizes. Alternatively, perhaps the skin microbiota respond to pathogenicity via a change in community composition. Equally likely are seasonal changes in behavior, immunological response, and thermoregulatory requirements being responsible for the differences in the skin microbial assemblages, and no direct relation to disease at all. Future lab-based manipulation of the snake skin microbial assemblages in the presence of O. ophiodiicola will permit tests of these hypotheses.
Particular bacterial members of the snake skin microbiota correlated with fungal pathogen load across spatial scales. Two OTUs in the Actinobacteria, including Nocardioides sp. and an unknown taxon were predictive of O. ophiodiicola absence across macro- to meso- spatial scales. Interestingly, a species of Nocardioides was documented with antifungal activity on salamander skin [28]. From this, we may hypothesize that Nocardioides species are important members of the snake skin microbial assemblage with potential for antifungal functional activity across the spatial landscape. Two OTUs in the Bacteroidetes (Pedobacter sp. and Sphingobacterium sp.), two in the Proteobacteria (Devosia sp. and Paracoccus sp.), and one in the Actinobacteria (Arthrobacter sp.) correlated with higher O. ophiodiicola fungal loads across macro-, meso-, and micro-spatial scales. The Proteobacteria have been documented as a core members of the skin microbiota of a variety of Panamanian frogs [57], fishes [60], and Appalachian salamanders [61]. Proteobacteria account for a great range of diversity and function, including pathogens and free-living organisms [62]. Similar to our study, Allender et al. [8] found that Eastern Massasauga Rattlesnakes with SFD had an increased relative abundance of Paracoccus sp. and Sphingobacterium sp. The OTUs that we identified as Paracoccus sp. (Otu00006) and Sphingobacterium sp. (Otu00452) showed the strongest correlative pattern with high pathogen loads in O. ophiodiicola-positive individuals. Perhaps, most interestingly, the indicator OTUs (Pedobacter sp., Sphingobacterium sp., Devosia sp., Paracoccus sp., and Arthrobacter sp.) that correlated with fungal pathogen load were consistent across broad to fine spatial scales, possibly signaling their functional importance as taxa composing the skin microbial assemblage. Disease state was not incorporated into this study due to observation inconsistencies by multiple researchers, the possibility of inaccurately mistaking a lesion as SFD, and tentative development of detectable signs in a qPCR-positive snake after it was sampled and released. Accurate quantification of disease state is likely an important explanatory factor of the skin microbial assemblage and could be more accurately assessed in a controlled vivarium study.
Predictor variables of the snake skin microbiota differed between extents, illustrating the importance of incorporating scale into experimental design to predict biologically and ecologically meaningful patterns. For example, host snake species was predictive of the skin microbial assemblage across all extents, whereas geographic location was weakly predictive of microbial assemblages at a broad extent but stronger at the microscale (Fig. 1). Similar trends have been observed in bats, where the skin microbiota was significantly influenced by habitat (ecomode), host species, and between geographic regions (e.g., states [63]), demonstrating how regional environmental differences play a significant role in skin microbial assemblage composition. In addition, Loudon et al. [64] determined that salamander skin is a selective medium for specific bacterial taxa from their environment, highlighting the intricacies between environmental and species-specific microbial selection. We found that the snake skin microbiome was composed of many bacteria that exist in the surrounding environment, although structure and diversity varied and was largely host species specific. Perhaps, snake skin also serves as a selective medium for particular microbes from the environment as observed in this study, although testing for selective processes in microbial community ecology would require careful lab-based manipulation experiments. Ecomode was a predictor of skin microbial assemblages across all scales, except for the Blue Ridge Mountain ecoregion (likely due to limited sample size). To date, few studies have examined how fundamental niches among species may influence skin microbial assemblages. Price et al. [65] observed distinct bacterial community composition shifts in juvenile Green Sea Turtles (Chelonia mydas) when transitioning from neritic to pelagic habitats, suggesting both diet and environment may have a significant effect on microbial communities. Other studies have observed bacterial variation across plant habitat classes, e.g., leaves, stems, roots, soils (diagnosed as niches within a single organism) irrespective of genotype [66]. Among vertebrates, ecomode may be a novel predictor of observed differences in skin microbial assemblages. We found significant interactions between ecomode and snakes positive for O. ophiodiicola, indicating that the SFD pathogen may not have an equal effect among ecomodes across spatial comparisons. Understanding fungal pathogenicity by snake ecomode may help target surveillance efforts or how to better approximate required sample sizes during SFD studies in a given habitat. Future studies should examine whether shifts occur in the skin microbiota of snakes during different developmental stages, potentially elucidating periods of increased pathogen susceptibility.
SFD clearly represents a growing global conservation concern for snake species [67]. The perceived recent emergence of this fungal pathogen could be due to increased awareness of the disease, changing climatic variables, introduction of an exotic pathogen into a naive host population, or other uncharacterized factors. Nevertheless, snake species are of particular significance, as they are indicators of trophic dynamics and ecosystem health [68], and yield potential benefits to humans, such as the reduction of disease-carrying rodents [69].
This study characterized the skin bacterial assemblage of snakes and its interaction with the causative agent of SFD across the Southern United States. A more comprehensive dataset encompassing a continuous geographic range across a broader temporal period will aid in the development of models for at-risk snake populations. Predictive conservation-based models could yield targeted treatment or management techniques aimed at high-risk areas and aid in the preservation of species critical to ecosystem processes.
Data accessibility
All raw sequence data has been submitted to GenBank SRA under the accession number PRJNA531014. All R and mothur code have been made publicly accessible in the Supplemental Materials.
References
Wiens J. Spatial scaling in ecology. Funct Ecol. 1989;3:385–97.
Levin S. The problem of pattern and scale in ecology: The Robert H. MacArthur Award Lecture. Ecology. 1992;73:1943–67.
Hopkins M, Thurman C. The geographic structure of morphological variation in eight species of fiddler crabs (Ocypodidae: genus Uca) from the eastern United States and Mexico. Biol Jour of the Linn Soc. 2010;100:248–70.
Montesinos-Navarro A, Estrada A, Font X, Matias M, Meireles C, Mendoza M, et al. Correction: Community structure informs species geographic distributions. PLoS ONE. 2018;13:e0200556.
Nemergut D, Schmidt S, Fukami T, O’Neill S, Bilinski T, Stanish L, et al. Patterns and processes of microbial community assembly. Micro and Mol Biol Rev. 2013;77:342–56.
Huston M, DeAngelis D, Post W. New computer models unify ecological theory. BioSci. 1988;38:682–91.
Milne B. Measuring the fractal geometry of landscapes. Appl Math Comp. 1988;27:67–79.
Allender MC, Baker S, Britton M, Kent AD. Snake fungal disease alters skin bacterial and fungal diversity in an endangered rattlesnake. Sci Rep. 2018;8:12147.
Daszak P, Berger L, Cunningham A, Hyatt A, Green D, Speare R. Emerging infectious diseases and amphibian population declines. Emer Inf Dis. 1999;5:735–48.
Gibbons J, Scott D, Ryan T, Buhlmann K, Tuberville T, Metts B, et al. The global decline of reptiles, déjà vu amphibians. BioSci. 2000;50:653.
Wake D, Vredenburg V. Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc Natl Acad Sci USA. 2008;105:11466–73.
Whitfield S, Bell K, Philippi T, Sasa M, Bolanos F, Chaves G, et al. Amphibian and reptile declines over 35 years at La Selva, Costa Rica. Proc Natl Acad Sci USA. 2007;104:8352–6.
Vredenburg V, Knapp R, Tunstall T, Briggs C. Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc Natl Acad Sci USA. 2010;107:9689–94.
Garner T, Perkins M, Govindarajulu P, Seglie D, Walker S, Cunningham A, et al. The emerging amphibian pathogen Batrachochytrium dendrobatidis globally infects introduced populations of the North American bullfrog, Rana catesbeiana. Biol Lett. 2006;2:455–9.
Van Rooij P, Martel A, Haesebrouck F, Pasmans F. Amphibian chytridiomycosis: a review with focus on fungus-host interactions. Vet Res. 2015;46:137.
Berger L, Speare R, Daszak P, Green D, Cunningham A, Goggin C, et al. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci USA. 1998;95:9031–6.
Lips K, Reeve J, Witters L. Ecological traits predicting amphibian population declines in Central America. Conserv Biol. 2003;17:1078–88.
Lips K, Brem F, Brenes R, Reeve J, Alford R, Voyles J, et al. Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc Natl Acad Sci USA. 2006;103:3165–70.
Stuart S, Chanson J, Cox N, Young B, Rodrigues A, Fischman D, et al. Status and trends of amphibian declines and extinctions worldwide. Science. 2004;306:1783–6.
McBride M, Wojick K, Georoff T, Kimbro J, Garner M, Wang X, et al. Ophidiomyces ophiodiicola dermatitis in eight free-ranging timber rattlesnakes (Crotalus horridus) from Massachusetts. J Zoo Wildl Med. 2015;46:86–94.
Allender M, Baker S, Wylie D, Loper D, Dreslik M, Phillips C, et al. Development of snake fungal disease after experimental challenge with Ophidiomyces ophiodiicola in Cottonmouths (Agkistrodon piscivorous). PLoS ONE. 2015;10:e0140193.
Clark R, Marchand M, Clifford B, Stechert R, Stephens S. Decline of an isolated timber rattlesnake (Crotalus horridus) population: interactions between climate change, disease, and loss of genetic diversity. Biol Conserv. 2011;144:886–91.
Allender M, Dreslik M, Wylie S, Phillips C, Wylie D, Maddox C, et al. Chrysosporium sp. infection in Eastern Massasauga Rattlesnakes. Emerg Inf Dis. 2011;17:2383–4.
Lorch J, Lankton J, Werner K, Falendysz E, McCurley K, Blehert D. Experimental infection of snakes with Ophidiomyces ophiodiicola causes pathological changes that typify snake fungal disease. mBio. 2015;6:e01534–15.
Allender MC, Raudabaugh DB, Gleason FH, Miller AN. The natural history, ecology, and epidemiology of Ophidiomyces ophiodiicola and its potential impact on free-ranging snake populations. Fungal Ecol. 2015;17:187–96.
Scharschmidt T, List K, Grice E, Szabo R, Renaud G, Lee C, et al. Matriptase-deficient mice exhibit ichthyotic skin with a selective shift in skin microbiota. J Invest Dermatol. 2009;129:2435–42.
Bletz M, Loudon A, Becker M, Bell S, Woodhams D, Minbiole K, et al. Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol Lett. 2013;16:807–20.
Lauer A, Simon M, Banning J, André E, Duncan K, Harris R. Common cutaneous bacteria from the Eastern Red-Backed Salamander can inhibit pathogenic fungi. Copeia. 2007;2007:630–40.
Walker DM, Lawrence B, Esterline D, McAndrew M, Edelbrock J, Graham S, et al. A novel protocol for washing environmental microbes from amphibian skin. Herpetol Rev. 2015;46:349–53.
Gefrides L, Powell M, Donley M, Kahn R. UV irradiation and autoclave treatment for elimination of contaminating DNA from laboratory consumables. Forensic Sci Int Genet. 2010;4:89–94.
Bohuski E, Lorch J, Griffin K, Blehert D. TaqMan real-time polymerase chain reaction for detection of Ophidiomyces ophiodiicola, the fungus associated with snake fungal disease. BMC Vet Res. 2015;11:95.
Ellison SL, English CA, Burns MJ, Keer JT. Routes to improving the reliability of low-level DNA analysis using real-time PCR. BMC Biotechnol. 2006;6:33.
Pilliod DS, Goldberg CS, Arkle RS, Waits LP. Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples. Can. J. Fish. Aquat. Sci. 2013;70:1123–30.
Walker DM, Leys JE, Dunham KE, Oliver JC, Schiller EE, Stephenson KS, et al. Methodological considerations for detection of terrestrial small‐body salamander eDNA and implications for biodiversity conservation. Mol Ecol Resour. 2017;17:1223–30.
Kozich J, Westcott S, Baxter N, Highlander S, Schloss P. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–6.
Edgar R, Haas B, Clemente J, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 2011;77:3219–26.
Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjøller R, et al. Fungal community analysis by high‐throughput sequencing of amplified markers–a user’s guide. New Phytol. 2013;199:288–99.
Oksanen J, Guillaume Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. R package version 2.5-2. 2018; https://CRAN.R-project.org/package=vegan
Wickham H. The Split-Apply-Combine Strategy for Data Analysis. J Stat Softw. 2011;40:1–29. http://www.jstatsoft.org/v40/i01/ URL
Wickham H, François R, Henry L, Müller K. dplyr: A Grammar of Data Manipulation. R package version 0.7.6. 2018; https://CRAN.R-project.org/package=dplyr
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag; 2016.
Mangiafico S. rcompanion: Functions to support extension education program evaluation. R package version 2.0.10. 2019; https://CRAN.R-project.org/package=rcompanion
Fox J, Weisberg S. An {R} companion to applied regression. Second Edition. Thousand Oaks CA: Sage; 2011. http://socserv.socsci.mcmaster.ca/jfox/Books/Companion
Auguie B. gridExtra: Miscellaneous Functions for “Grid” Graphics. R package version 2.3. 2017; https://CRAN.R-project.org/package=gridExtra
Nicholson K. It is time for a new classification of anoles (Squamata: Dactyloidae). Auckland: Mag Press; 2012.
Grisnik M, Leys JE, Bryan D, Hardman RH, Miller DL, Cobb VA, et al. Host and geographic range of snake fungal disease in Tennessee, USA. Herpetol Rev. 2018;49:682–90.
McKenzie V, Bowers R, Fierer N, Knight R, Lauber C. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 2011;6:588–96.
Belden L, Hughey M, Rebollar E, Umile T, Loftus S, Burzynski E, et al. Panamanian frog species host unique skin bacterial communities. Front Microbiol. 2015;6:1171.
Harris R, James T, Lauer A, Simon M, Patel A. Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. EcoHealth. 2006;3:53–56.
Harris R, Lauer A, Simon M, Banning J, Alford R. Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis Aqua Org. 2009;83:11–16.
Ramsey J, Reinert L, Harper L, Woodhams D, Rollins-Smith L. Immune defenses against Batrachochytrium dendrobatidis, a fungus linked to global amphibian declines, in the South African Clawed Frog, Xenopus laevis. Infect Immun. 2010;78:3981–92.
Rollins-Smith L, Carey C, Longcore J, Doersam J, Boutte A, Bruzgal J, et al. Activity of antimicrobial skin peptides from ranid frogs against Batrachochytrium dendrobatidis, the chytrid fungus associated with global amphibian declines. Dev Comp Immunol. 2002;26:471–9.
Rollins-Smith L, Conlon J. Antimicrobial peptide defenses against chytridiomycosis, an emerging infectious disease of amphibian populations. Dev Comp Immunol. 2005;29:589–98.
Rollins-Smith L. The role of amphibian antimicrobial peptides in protection of amphibians from pathogens linked to global amphibian declines. Biochim Biophys Acta. 2009;1788:1593–9.
Becker M, Walke J, Cikanek S, Savage A, Mattheus N, Santiago C, et al. Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc R Soc B Biol Sci. 2015;282:20142881–20142881.
Hill A, Leys J, Bryan D, Erdman F, Malone K, Russell G, et al. Common cutaneous bacteria isolated from snakes inhibit growth of Ophidiomyces ophiodiicola. EcoHealth. 2017;15:109–20.
Lemieux-Labonté V, Simard A, Willis C, Lapointe F. Enrichment of beneficial bacteria in the skin microbiota of bats persisting with white-nose syndrome. Microbiome. 2017;5:115.
Chiarello M, Villéger S, Bouvier C, Bettarel Y, Bouvier T. High diversity of skin-associated bacterial communities of marine fishes is promoted by their high variability among body parts, individuals and species. FEMS Microbiol Ecol. 2015;91:fiv061.
Fitzpatrick B, Allison A. Similarity and differentiation between bacteria associated with skin of salamanders (Plethodon jordani) and free-living assemblages. FEMS Microbiol Ecol. 2014;88:482–94.
Stackebrandt E, Murray R, Truper H. Proteobacteria classis nov., a name for the phylogenetic taxon that includes the “purple bacteria and their relatives”. Int J Syst Bact. 1988;38:321–5.
Avena C, Parfrey L, Leff J, Archer H, Frick W, Langwig K, et al. Deconstructing the bat skin microbiome: Influences of the host and the environment. Front Microbiol. 2016;7:1753.
Loudon A, Venkataraman A, Van Treuren W, Woodhams D, Parfrey L, McKenzie V, et al. Vertebrate hosts as islands: dynamics of selection, immigration, loss, persistence, and potential function of bacteria on salamander skin. Front Microbiol. 2016;7:333.
Price J, Paladino F, Lamont M, Witherington B, Bates S, Soule T. Characterization of the juvenile green turtle (Chelonia mydas) microbiome throughout an ontogenetic shift from pelagic to neritic habitats. PLoS ONE. 2017;12:e0177642.
Cregger M, Veach A, Yang Z, Crouch M, Vilgalys R, Tuskan G, et al. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome. 2018;6:31.
Sutherland W, Aveling R, Brooks T, Clout M, Dicks L, Fellman L, et al. A horizon scan of global conservation issues for 2014. Trends Ecol Evol. 2014;29:15–22.
Lind A, Welsh H, Tallmon D. Garter snake population dynamics from a 16-year study: consideration for ecological monitoring. Ecol Appl. 2005;15:294–303.
Kabay E. Timber Rattlesnakes may reduce incidence of Lyme disease in the Northeastern United States. 98th ESA Annual Convention. Minneapolis, MN USA; 2013.
Acknowledgements
DMW thanks the three anonymous reviewers for their critical comments that helped to improve this manuscript. We acknowledge Aubree Hill, Isabel Papraniku, Chris Ogle, Chris Simpson, Daniel Istavanko, Joshua Campbell, Danny Bryan, Sean Graham, Calvin Hall, Robert Buck, Dylan Peters, Gabrielle Russell, Fantasia Erdman, Gabriel Kindl, Andrew Moore, John T. Kimrey, John Griffith, Vincent A. Cobb, Danny L. Bryan, Megan L. Leys, John Hewlett, Chris Fatzinger, Daniel Pinkston, Lisa Powers, and Brandon Edelbrock for helping to collect samples. This work was approved by IACUC 15-16-001, TDEC 2016-026, TWRA 3886, EVER-00535, AR 020520155, AL2014044693068680, AL2016021753868680, 29-WJH-16-184, SPR-0415-048. Funding and support for this research project was provided by Tennessee Wildlife Resources Agency as a State Wildlife Grant to DMW.
Author information
Authors and Affiliations
Contributions
DMW, JEL and MCA conceived the study. DMW, JEL, MG, AGP and CMM collected specimens and contributed to field work. JEL and MG completed all molecular work. DMW, JEL and CMM ran the bioinformatics and statistical analyses, and all authors wrote the manuscript and contributed equally to revisions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walker, D.M., Leys, J.E., Grisnik, M. et al. Variability in snake skin microbial assemblages across spatial scales and disease states. ISME J 13, 2209–2222 (2019). https://doi.org/10.1038/s41396-019-0416-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0416-x
This article is cited by
-
Captivity and Animal Microbiomes: Potential Roles of Microbiota for Influencing Animal Conservation
Microbial Ecology (2023)
-
Host microbiome responses to the Snake Fungal Disease pathogen (Ophidiomyces ophidiicola) are driven by changes in microbial richness
Scientific Reports (2022)
-
Ophidiomyces ophidiicola detection and infection: a global review on a potential threat to the world’s snake populations
European Journal of Wildlife Research (2022)
-
Occurrence of Microplastics in Herpetological Museum Collection: Grass Snake (Natrix natrix [Linnaeus, 1758]) and Dice Snake (Natrix tessellata [Laurenti, 1769]) as Model Organisms
Water, Air, & Soil Pollution (2022)
-
The presence of Pseudogymnoascus destructans, a fungal pathogen of bats, correlates with changes in microbial metacommunity structure
Scientific Reports (2021)
