
Abstract
An enigmatic uncultured member of Firmicutes, Candidatus Desulforudis audaxviator (CDA), is known by its genome retrieved from the deep gold mine in South Africa, where it formed a single-species ecosystem fuelled by hydrogen from water radiolysis. It was believed that in situ conditions CDA relied on scarce energy supply and did not divide for hundreds to thousand years. We have isolated CDA strain BYF from a 2-km-deep aquifer in Western Siberia and obtained a laboratory culture growing with a doubling time of 28.5 h. BYF uses not only H2 but also various organic electron donors for sulfate respiration. Growth required elemental iron, and ferrous iron did not substitute for it. A complex intracellular organization included gas vesicles, internal membranes, and electron-dense structures enriched in phosphorus, iron, and calcium. Genome comparison of BYF with the South African CDA revealed minimal differences mostly related to mobile elements and prophage insertions. Two genomes harbored <800 single-nucleotide polymorphisms and had nearly identical CRISPR loci. We suggest that spores with the gas vesicles may facilitate global distribution of CDA followed by colonization of suitable subsurface environments. Alternatively, a slow evolution rate in the deep subsurface could result in high genetic similarity of CDA populations at two sites spatially separated for hundreds of millions of years.
Similar content being viewed by others


Introduction
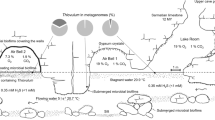
The pioneering studies estimating the deep biosphere as 40–50% of the world biomass [1] have drawn attention to subsurface biodiversity. There is a general opinion that subsurface microorganisms in situ relied on scarce energy sources and have low metabolic rates and very long generation times that span hundreds to thousands of years [2]. The later models recalculated the biomass of the continental subsurface as 23–31 Pg of carbon [3] and showed that the deep ecosystems are not always scarce in energy [4]. A composite genome of Candidatus Desulforudis audaxviator (CDA) has been retrieved from the fracture waters in the 2.8-km-deep Mponeng gold mine in South Africa where this strain, MP104C, comprised >99.9% of the microbial community [5]. Genomic data indicated that CDA is a chemolithoautotroph growing by hydrogenotrophic sulfate respiration [5]. All attempts to cultivate CDA were unsuccessful. The molecular signatures of CDA have been detected in several continental subsurface-related ecosystems in South Africa [2, 6, 7], North America (GenBank KF939343), and Europe [8, 9]. We have detected CDA in a 2-km-deep subsurface thermal aquifer in Western Siberia [10]. The aquifer residing in early Cretaceous sediments (150–100 Ma) was broached by an oil-exploration borehole designated 1-R, with a depth of 2.56 km [11]. The borehole was drilled about 60 years ago and was never used to inject anything from the surface to the aquifer. The artesian borehole overflows with thermal (40–45 °C) water, which was suggested to be of meteoric origin with a minor component of connate marine water on the basis of the 18O and 2H stable isotope composition [12]. The chloride:bromide mass ratio of the water is very close to seawater, suggesting that the aquifer salinity (Cl− 843 mg/L) may have its origin in relict ocean water. The groundwater from the borehole is highly reduced (Eh from −341 to −279 mV) and contains H2S (0.64–0.88 mg/L) and traces of methane. Previously, we have studied the composition of the microbial community of this aquifer and found that it mostly consisted of sulfate-reducing Firmicutes and Deltaproteobacteria as well as members of the phyla Chlorofexi, Ignavibacteriae, Riflebacteriae, and Aminicenantes, while archaea were nearly absent [10]. In particular, the frequency of CDA in the community during 2 years of monitoring varied from 8 to 14%. Here we report on the culturing and isolation of CDA in pure culture from the 1-R borehole water and its characterization and comparative analysis of the genomes of this isolate and the South African strain MP104C. The revealed morphological and genetic features provide basis to speculate on evolution of CDA and the possible way of its distribution among geographically distant deep subsurface environments.
Materials and methods
Groundwater sampling
The geographical location and geology of the deep borehole, its water characteristics, and sampling procedures were described previously [12]. The borehole is located in the town of Byelii Yar in the Tomsk region of Western Siberia (Supplementary Fig. 1). Briefly, CDA has been isolated from water flowing out from the borehole 1-R under pressure and collected on August 04, 2014 in sterile plastic bottles. The outflow was sampled several times during 2013–2018. Water temperature, pH, and Eh were measured at the wellhead using pH-meter HI 8314 (Hanna Instruments, Vöhringen, Germany) with appropriate electrodes. The water sample was fixed with 2.4% Zn-acetate in proportion of 1:5 for hydrogen sulfide determination. H2S was measured colorimetrically with the methylene blue method [13] in triplicate using a Smart Spec Plus spectrophotometer (Bio-Rad Laboratories, Hercules, CA).
Enrichment and pure culture isolation
The water sample for CDA isolation was collected at the borehole 1-R on August 04, 2014. The initial enrichment was set up in freshwater Widdel and Bak (WB) medium [14] that contained (per liter) 4 g Na2SO4, 0.2 g KH2PO4, 0.25 g NH4Cl, 1 g NaCl, 0.4 g MgCl2·6H2O, 0.5 g KCl, 0.113 g CaCl2, 2 mL of vitamin solution, 1 mL of microelement solution, and 1 mL each of Na2SeO3 (final concentration 23.6 µM) and Na2WO4 (24.2 µM) solutions [14]. Medium was adjusted to pH 8.0 with NaHCO3 and Na2S·9H2O (2 mL of Na2S·9H2O stock solution; 48 g/L of Na2S·9H2O per liter of WB basal medium) was used as a reducing agent. Each cultivation vial received an iron wire (100% Fe) as previously described [15]. In all, 7.5 mM formate was used as electron donor for the initial enrichments. Culture vials (12 mL) were filled to the top, closed, and sealed by aluminum caps. No redox indicator was used, since it is conceivable that resazurin might inhibit growth of sulfate-reducing bacteria (SRB). We used medium blackening, microscopic observations, and H2S measurements as indicators of sulfidogenic growth. The vials with sterile medium, zero-valent iron wire, and 3 mm glass beads (1/4 of the vial volume) were inoculated on-site with 2.5 mL water sample per vial by sterile syringes and incubated at 50 °C. Culture purity was checked microscopically and by separation and sequencing of 16S rRNA gene fragments in a denaturing gradient gel electrophoresis (PCR-DGGE) as previously described [16]. It was also confirmed by the lack of growth on aerobic Plate Count Agar (per liter: 9.0 g agar, 10 g glucose, 5 g tryptone, 2.5 g yeast extract) and Anaerobic Agar (Becton Dickinson, Franklin Lakes, NJ) media and finally by genome sequencing.
Characterization of CDA morphology and physiology
Cell morphology was observed by phase contrast microscopy using an Axio Imager A1 microscope, by transmission electron microscopy (TEM) of ultrathin sections, as described previously [17]. For TEM, the biomass from a 1-L batch culture grown for 14 days was harvested by centrifugation at 20,000 × g for 10 min. For elemental mapping, ultrathin sections were prepared with a Leica UC7 ultramicrotome (Leica Microsystems, Vienna). The sections were examined with a transmission electron microscope JEM-2100 (JEOL) equipped with X-Max EDS detector for elemental mapping (Oxford Instruments, Abingdon, U.K.).
The growth experiments at different pH values, NaCl concentrations, and incubation temperatures were carried out in modified WB medium with 7.5 mM formate and 2 mM acetate mixture as an electron donor and carbon source, respectively. Bicarbonate buffer was excluded from basal WB media to allow for pH adjustments, and the pH was adjusted with 0.5 M H2SO4 or 2 M NaOH to the desired pH values. The pH measurements were taken at the incubation temperature. Growth was analyzed in the pH range from 6.0 to 10.0 at the optimum temperature of 55 °C. Growth was tested at different incubation temperatures in the range from 37 to 70 °C in 12 mL headspace-free test tubes. Growth was determined by microscopic cell counts in triplicate samples. Specific growth rates were calculated from the cell counts during the exponential phase of growth. Further physiological experiments were conducted at the optimum pH of 8.0 and temperature of 55 °C. The optimum salinity for growth was determined in the range of 0–1.1% NaCl.
Growth was analyzed with the following electron donors: 4.5 mM succinate, 7.3 mM lactate, 7.5 mM each of malate and formate, 7 mM each of pyruvate and butyrate, 9 mM each of acetate and fumarate, 13.5 mM propionate, 2.5 mM benzoate, 1 mM palmitate, 0.011 mM cysteine, 0.008 mM alanine, 3 mM sucrose, 5 mM each of fructose and glucose, 25 mM ethanol, 17 mM propanol, 13.5 mM isobutanol, and 11 mM glycerol. Growth on 7.5 mM formate and H2 was also tested in the presence of 2 mM acetate. All the potential electron donors were tested in the presence of 28 mM sulfate. Carboxymethyl cellulose (100 mg per 12 mL cultivation tube), 1% gelatin, 1% choline, and 1% peptone (all from Sigma-Aldrich) were also tested as carbon sources and electron donors. Carbohydrate stock solutions were sterilized using polyethersulfone 0.22 µm Millex-GP filter units (Merck Millipore, Darmstadt). The use of H2 as electron donor was tested in 10 mL medium in 15 mL Hungate tubes under a headspace of H2+CO2 (80:20 v/v, 10 kPa). The tested electron acceptors were 2 mM and 20 mM sodium sulfite, 20 mM sodium thiosulfate, 10 mM fumarate, 2 mM sodium nitrite, 0.4 mM and 5 mM calcium nitrate, 20 mM Fe(III)NTA, and 2% elemental sulfur. For testing solid-phase electron acceptors, cultures received barite (BaSO4) or celestine (SrSO4) (50 mg in 12 mL medium). All the presumed acceptors were tested in the presence of 7.5 mM formate (plus 2 mM acetate). If growth was observed, the culture was subcultured at least five times in the presence of each electron donor and acceptor to confirm their utilization.
Genome sequencing and assembly
Genomic DNA was isolated from the cells in the late exponential growth phase (the cells were grown in 500 mL serum bottles) by the CTAB/NaCl method [18]. Complete genome of strain BYF was obtained using a combination of Illumina HiSeq2500 and MinIon (Oxford Nanopore) single molecule sequencing technologies. The sequencing of a TrueSeq DNA library generated 5,041,785 paired-end reads (2 × 100 nt), 1,008,357,000 bases in total. Sequencing primers were removed using Cutadapt v.1.8.3 [19] and low quality read regions (q < 33) were trimmed using Sickle v.1.33 (https://github.com/najoshi/sickle). Illumina reads were de novo assembled using SPAdes v.3.7.1 [20]. In addition, 27,199 reads with a total length of 38,233,188 bp were obtained on MinIon sequencer. Contigs assembled using Illumina reads were scaffolded using MinIon reads with the npScarf software [21]. Remaining gaps were closed manually using reference MP104C genome information. MinIon reads, Illumina reads, and contigs were mapped to the reference genome sequence using BWA 0.7 [22], Bowtie2 v.2.3.4.1 [23], and BLASTN, respectively. All mappings were visualized using IGV browser [24]. The contigs were ordered according to the reference genome and the existence of correctly aligned reads spanning adjacent contigs was verified. Consensus sequence was polished using Pilon v.1.22 [25] with Illumina reads mapping. Gene search and annotation were performed using the RAST server 2.0 [26], followed by manual correction. Clusters of regularly interspaced repeats (CRISPR) were identified by CRISPR Finder [27].
Single-nucleotide polymorphism (SNP) calling and identification of structural differences between the genomes of strains BYF and MP104C
Illumina reads obtained for BYF genome were mapped to CDA strain MP104C reference genome using Bowtie 2 v.2.3.4.1 [23] in end-to-end mode with default parameters. SNPs were detected using FreeBayes v.1.1.0 [28]. SNPs with >90% of reads supporting non-reference variant and a minimum 20× mapping depth were retained. Mobile elements and other repeat sequences were excluded from the analysis. The non-synonymous to synonymous substitution rate, or the dN/dS ratio, was calculated from the table of obtained SNPs. The same approach was used for identification of SNPs between the genome of strain BYF and the CDA genome BY57, assembled from the metagenome of groundwater collected at the borehole 1-R [10]. Corresponding Illumina reads were retrieved from the NCBI SRA database (SRX4031087).
The de novo assembled sequence of BYF genome was compared with MP104C reference genome using BLASTN search. Pairwise alignment of BYF and MP104C genomes was performed using Mauve v. 2.4.0 [29]. The average nucleotide identity (ANI) of the BYF and MP104C genomes was computed using the ani.rb script from Enveomics Collection [30].
Results
Isolation of CDA strain BYF
The chemistry of 1-R borehole outflow water has been reported in our previous studies [10, 12]. Analysis of samples collected at different times showed slight variation of the pH (from 7.9 to 8.5), temperature (from 38 to 45 °C), and redox potential (from −550 to −220 mV) (Supplementary Fig. S1). The sample used for CDA isolation was collected on August 4, 2014. In accordance with previous reports of CDA occurrence in fracture water or underground aquifers, we pondered that solid rock–water interaction phase may be important for cultivation of the organism. Therefore, we used glass beads to simulate the solid phase during cultivation in vials filled with liquid-modified WB medium and supplemented with zero-valent iron. A set of enrichments with 7.5 mM formate supplemented with zero-valent iron was set up to isolate CDA. The vials were inoculated directly from the borehole wellhead. Sulfidogenic growth appeared in formate-containing medium after 2 months of incubation at 50 °C and pH 8.0. Microscopic observations revealed different morphotypes, dominated by motile rods, with a minority of filamentous cells, cocci, and vibrios. The enrichment grew slowly with a lag phase of 1 month. The elongated rods, which had a morphotype previously reported for CDA, were enriched by multiple serial dilutions in formate-containing media at 50 °C. The PCR-DGGE analysis revealed that the filamentous cells were members of genus Chloroflexi (data not shown). They were eliminated from the enrichment by culture exposure at 90 °C for 20 min followed by multiple serial dilutions. The pure culture rod-shaped isolate was designated strain BYF after the name of the town Byelii Yar, where the borehole is located. The isolation of the strain in pure culture took >2 years due to its slow growth. Repeated subcultures of BYF for a year in formate-amended media adapted it to the laboratory conditions and decreased the lag phase to 10 days. The 16S rRNA gene sequences of strains BYF and MP104C were 100% identical. The absence of other phylotypes in culture was confirmed by microscopic observation, PCR-DGGE, and genome sequencing.
Morphology of strain BYF
Strain BYF appeared as motile slim rods 2.1–2.8-μm long and 0.3–0.4-μm wide (Fig. 1a, b) and grew slowly with the initial lag phase of a month. The cellular morphology changed when BYF switched to a faster growth after 1 year of repeated subcultures. In parallel, the size of the cells turned shorter (1.4–1.7 μm). TEM of ultrathin sections revealed a Gram-positive character of the cell wall without an outer membrane (Fig. 1c). Strain BYF had complex intracellular organization including numerous irregular-shaped electron-dense structures (Fig. 1c, d), mesosome-like structures (Fig. 1e–g), and bud-like structures (Fig.1h–j). Elemental mapping showed that these electron-dense structures were enriched in Ca, Fe, and P as compared to the cell cytoplasm (Fig. 2d–f). The electron-dense structures were visible under electron microscope magnification only and could not be tracked under phase contrast microscopy. We have observed them in all cells studied with electron microscopic cultivation conditions, which included cells in the exponential and late stationary growth phase and cells incubated with elevated concentrations of Fe, Cu, and Cd. Spore formation was a rare event and spores had irregular appearance in the cells (Fig. 3). Temperature, oxygen and metal stress, or use of various electron donors and acceptors did not facilitate spore formation. A characteristic feature of the cells with spores was concomitant occurrence of gas vesicles (Fig. 3a–c) and they were always absent in cells without spores.

Phase contrast and transmission electron microscopic micrographs of cells and ultra-thin sections of the strain BYF. a Slow-growing cells with lag phase of a month. The scale bar is 5 μm. b Slow-growing cells, ultra-thin section. c Fast-growing cells and electron-dense structures. d Cell wall and electron dense structure. e–g Mesosome-like structures. h–j Bud-like structures. eds, electron dense structures; cw, cell wall; m, mesosome; bls, bud-like structures. The scale bars are 0.2 μm

Electron-dense structures in strain BYF cells. Transmission electron microscopic (TEM) images of cells growing in formate+acetate medium for a 19 days, b 34 days, and c 14 days. d, e Energy-filtered TEM images and corresponding atomic percentages of O, P, S, Ca, and Fe measured in electron-dense structures and cytoplasm. The scale bars are 0.2 μm

Transmission electron microscopic micrographs of ultra-thin sections of the sporulating cells of the strain BYF. a–c Gas vesicles associated with spores. d–f Irregular spore formation by strain BYF. gv gas vesicles, e endospore. The scale bars are 0.2 μm
Physiology and growth conditions
Formate was used as the only electron donor for sulfate reduction for the isolation of strain BYF. It also could grow autotrophically with H2. The strain did not grow if zero-valent iron was excluded from the medium. In attempts to facilitate strain growth, we revealed that addition of acetate (2 mM) to formate-containing medium facilitated growth of the strain and the maximum growth yield reached up to 2 × 105 cells/mL. The use of formate+acetate medium yielded sufficient cell density for taking measurements in liquid culture and to study growth characteristics of the strain. The maximum specific growth rate of 0.027/h with doubling time of 25.8 h was observed when BYF grew in formate+acetate medium at optimum temperature of 55 °C (Supplemental Fig. S2).
Strain BYF could grow at a relatively narrow pH range of 7.0–9.5 with an optimum at 8.0 (Supplemental Fig. S3). The optimal NaCl concentration was 0.1% and growth ceased at concentrations exceeding 1%. The temperature range for BYF growth was 45–60 °С with an optimum at 55 °С. The strain was isolated under anaerobic conditions but it could also grow in cultivation vials with 70% of the volume filled with liquid medium and the remaining headspace with ambient air. Oxygen could be partly reduced by H2S in the headspace, formed from Na2S used as a reducing agent and also produced by sulfate reduction by strain BYF. These conditions slowed the growth and extended the lag phase up to 30–40 days. We did not use anaerobic chambers or jars for strain subcultivation. The cells did not show any changes of morphology or viability when exposed to ambient air for centrifugation.
Physiological tests revealed that strain BYF utilized a broad range of organic electron donors for sulfate reduction, including low-molecular weight carboxylic acids (formate, lactate, pyruvate, fumarate, succinate, propionate, and butyrate), some alcohols (ethanol, isobutanol), and sugars (glucose, sucrose) (Supplemental Table S1). The strain could grow with glucose and sucrose but did not use fructose. BYF did not use as electron donor proteins (peptone, gelatin), cysteine, cellulose, acetate, malate, glycerol, and propanol. We have observed growth when sulfite, thiosulfate, and fumarate were added as electron acceptors to the medium with 7.5 mM formate+2.5 mM acetate. Nitrate, nitrite, elemental sulfur, and Fe(III)NTA did not support growth with formate+acetate as electron donor and carbon source. Strain BYF grew and produced up to 6 and 16 mg/L H2S with SrSO4 and BaSO4, respectively.
Genome comparison of CDA strains BYF and MP104C
The genome of strain BYF was sequenced using a combination of Illumina and Oxford Nanopore techniques. Combined assembly yielded a complete 2,212,498 bp long circular genome in the absence of any extrachromosomal elements (Supplemental Table S2). Although it is shorter than the genome of strain MP104C (2,349,476 bp), the differences were related mostly to the presence/absence of integrated prophages and mobile elements. All genetic determinants of the main metabolic pathways described for strain MP104C [5], particularly the dissimilatory sulfate reduction, glycolysis, autotrophic carbon fixation via the Wood–Ljungdahl pathway, nitrogen fixation, and hydrogen and formate oxidation are present also in the BYF genome consistently with the observed physiological traits. The capability of strain BYF to grow under microaerobic conditions was rather unexpected considering the lack of membrane-bound oxygen reductases, which play important role in oxygen defense [31]. Among the components of oxygen defense system, a Fe-type superoxide dismutase and a rubrerythrin were present while catalase was not found.
Consistently with an observation of the gas vesicles, the CDA genome contains a cluster of gvp genes, including gvpA, gvpL/gvpF, gvpK, gvpG, gvpN, and gvpR. The gvp cluster is absent in Candidatus Desulfopertinax cowenii, the closest relative of CDA whose composite genome was obtained from basalt-hosted fluids of the deep subseafloor at Juan de Fuca Ridge [32].
Taking into account the large distance between the South African gold mine and Western Siberian subsurface aquifer and their isolation from the surface, large differences between MP104C and BYF genomes could be expected. However, besides 100% identity of 16S rRNA gene sequences, the ANI of the two genomes was estimated to be 99.95%. Pairwise comparison of MP104C and BYF genomes revealed only 18 regions of structural variations (Fig. 4). Eight of them were related to the presence/absence of mobile elements at particular sites, but strain-specific transposons were not found. The MP104C genome also contains a 132-kb long prophage that is missing in BYF. Other points of divergence are associated with intergenic polymorphic regions and short strain-specific insertions or deletions.

Comparison of the genomes of strains BYF and MP104C. a Distribution of single-nucleotide polymorphisms (SNPs) along the CDA genome. S1–S6 SNP-rich regions. b Mauve alignment of BYF and MP104C genomes. Regions with sequence divergence are shown in intense red color. Strain-specific insertions of mobile elements (IS) are indicated by triangles, the gray box indicates the 132 kb long prophage missing in BYF, S1–S6 denotes six SNP-rich regions shown in a. Coordinates are shown according to the MP104C reference sequence
CRISPR regions implicated in acquired resistance of microorganisms to viruses are one of the most rapidly evolving elements of prokaryotic genomes [33]. These arrays of short repeat sequences separated by unique spacers, acquired from invading viruses, represented a chronological record of viral infections since new spacers are added at the beginning of the array while older ones are located downstream. The MP104C genome harbors two CRISPR regions, containing 51 and 153 spacer-repeat units. The first CRISPR array is intact while the second in interrupted by three insertions of a mobile element and is likely unfunctional. Comparison of the genomes revealed that the first CRISPR locus is identical in BYF and MP104C. In the second locus, there is a single SNP in one spacer sequence (C in BYF at bp 1,767,839 vs T in MP104C), one additional spacer-repeat unit was found in BYF, four such units in MP104C, and in one position different spacers were found in two genomes (Supplemental Figure S4). Notably, these polymorphisms are located not at the beginning of the array, where the novel spacers become added, but are distributed along the array. Probably, the recombination-dependent acquisition and loss are a major mechanism contributing to variability of the second CRISPR locus [34]. The presence of duplicated spacers in this array also supports this proposal.
The search for SNPs between BYF and MP104C genomes revealed 733 such sites (Supplemental Table S3). Most of them (703) are clustered in only 6 regions comprising from 1 (7 SNPs) to 11 (301 SNPs) genes and totalling 48.5 kbp (Table 1). Most of the clustered SNPs are synonymous substitutions (dN/dS = 0.42) pointing not to the adaptive evolution of the corresponding genes but to purifying selection. These regions were probably incorporated into the genomes as a result of horizontal transfer from phylogenetically more distant strains of CDA, followed by recombination replacement. Interestingly, a part of the gvp gene cluster is located within one of SNP-rich regions and could have been acquired laterally. Several SNPs were also found in the fdhD gene encoding the formate dehydrogenase assembly factor. Their presence could be important taking into account that formate is a preferred electron donor for strain BYF. The remaining 30 SNPs were randomly distributed across the genome (Fig. 4) and likely reflect accumulation of mutations in course of evolution of the CDA core genome.
Availability of the metagenome of the groundwater used for isolation of strain BYF enabled genetic comparison between the BYF genome and metagenome-assembled CDA draft genome, BY57 [10]. Both genomes lacked a 132-kb long prophage present in MP104C. The search for SNPs between BYF and BY57 genomes revealed only eight such sites, indicating very low genetic diversity of in situ CDA population in the Siberian aquifer (Supplemental Table S4). Therefore, the observed SNPs between MP104C and BYF were not a consequence of domestication of BYF and its further cultivation in the laboratory but represented actual genetic differences between two natural CDA populations.
Discussion
CDA strain BYF cultivation
Our attempts to cultivate SRB from the 1-R borehole water were carried out several times. Sulfate reducing Thermodesulfovibrio sp. N1 has been isolated from the sulfidogenic enrichments established in 2013 [12]. Despite the fact that we used the same electron donors, cultivation medium, and techniques as in the present study, no CDA-containing enrichments were obtained from the formate-supplemented media established in 2013. The only difference in cultivation conditions was a pH increase to 8.0 in 2014 as compared to 7.2–7.4 in 2013. Previously, we demonstrated differences in microbial community composition determined in samples collected over several years from another borehole broaching a deep subsurface aquifer in West Siberian Megabasin [35]. The observed differences may relate to the biofilm-like mode of life of microorganisms in the deep subsurface. The changing hydraulic pressure and other random events may influence the microbial community composition from one water sample to another. It is plausible that the successful enrichment of CDA BYF was due to a random event of biofilm occurrence in the water sample. Strain BYF has been detected by the 16S rRNA amplicon sequencing in all 1-R water samples studied in 2013 and 2014. However, it is noteworthy that the water volume used for inoculation of the cultivation vials was 1 mL, as compared to 50 L filtered for sequencing. The pH shift to 8.0 in the 2014 enrichments also might facilitate successful strain BYF cultivation. Another SRB, Thermodesulfovibrio sp. N1, isolated from the same borehole water sample has been shown to have facultatively alkaliphilic growth patterns [12].
Zero-valent iron played a role in the strain BYF cultivation. It could not grow with any of the used electron donors or acceptors if iron wire was excluded from the medium. The iron wire has been successfully used in our previous efforts to facilitate SRB growth [15, 36, 37]. Zero-valent iron generates H2 under anaerobic conditions (Eq. 1):
However, strain BYF required zero-valent iron also when grown in H2-containing media. Sulfide-producing cultures tend to be depleted in dissolved iron, which precipitates in the form of sulfides and becomes unavailable for the cells. Zero-valent iron may provide a microsource of iron in sulfide-containing media. The importance of iron for the bacterium is evidenced by the presence of both Fe(III) ABC transporters of the siderophore/heme/vitamin B12 type and Feo-like Fe(II) transport systems, each encoded in several copies. It is also conceivable that elemental iron scavenges sulfide and reduces its toxicity to the cells. We noticed that addition of cadmium chloride in concentration of 25 mg Cd2+/L increased the BYF cell yield. The possible role of cadmium may be to decrease the dissolved H2S concentration by precipitating it in the form of Cd-sulfide. Further, we cannot exclude the possibility of direct electron uptake from metal iron by BYF, as originally reported for Desulfovibrio by Dinh with co-authors [38] and demonstrated by Deng and Okamoto for Desulfovibrio ferrophilus [39]. Direct electron extraction from extracellular solids has been suggested as an important pathway in the microbial energy acquisition in low-energy subsurface environments [39]. It is possible that steel or cast iron used for the borehole casing may play a role in the CDA BYF enrichment in the 1-R water. Various strings of casing were set in the borehole, ranging from 18” diameter surface guidance pipe and 14” conductor casing, down to a final string of 5” diameter casing in the productive horizons.
Strain BYF morphology and physiology
The striking morphological feature of the strain BYF was gas vesicles, which occurred in sporulating cells only. The gas-filled proteinaceous gas vesicles are known to provide cells with buoyancy [40]. Thus CDA spores associated with gas vesicles may represent a mechanism, which allows the strain to spread to large distances, making it a “bold traveler,” the English translation of the Latin “audax-viator.” One of the early studies of a Desulfotomaculum, the closest relative of Desulforudis, reported spores associated with gas vesicles [41]. Subsequent research showed that Desulfotomaculum spores could survive harsh temperature conditions and spread to the cold marine sediments from crustal hot seeps [42].
Isolation in pure culture and physiological experiments with strain BYF confirmed provisions made from the CDA MP104C genome analysis [5]. Strain BYF is a thermophilic chemolithoautotroph capable of sulfate reduction and hydrogenotrophy. Additionally, we have demonstrated that BYF uses a broad range of electron donors for sulfate reduction. Thus CDA is well adapted for switching the “fuel” source and can not only grow autotrophically when H2 is available but can also consume organic acids, alcohols, or sugars. In general, both ecosystems harboring CDA, the Mponeng mine fracture water and the aquifer broached by the 1-R borehole, have similar origin (a mixture of relict and meteoric water) and hydrochemical characteristics. The thermal (40–60 °C) water reported for both ecosystems is reduced, alkaline, saline, and contains H2S and methane. The difference is the sulfate concentration, which varies in the Mponeng fracture water from 50.8 to 178 mg/L [43] but does not exceed the detection limit of 5 mg/L in the 1-R borehole water [10]. The low sulfate concentration in the Siberian aquifer raises a question about the abundant sulfate-reducing consortium revealed in the aquifer by metagenomic study, which included Desulfobacca and Desulfotomaculum along with cultivated strains CDA BYF and Thermodesulfovibrio sp. N1 [10, 12]. Interestingly, we could cultivate and isolate in pure culture not only CDA BYF and Thermodesulfovibrio sp. N1 but also a pure culture of close relative of Desulfotomaculum profundi and members of the Desulfobacca genus in the enrichment culture (Frank et al., unpublished). All these cultures were isolated with sulfate as electron acceptor. Several explanations for the occurrence of abundant sulfate-reducing consortium in the low-sulfate aquifer are possible. The bacteria can use electron acceptors other than sulfate. CDA BYF could use fumarate, but not Fe, the concentration of which is relatively high in 1-R water. SRB can use solid-phase electron acceptors, such as gypsum, hannebachite, anglesite, or barite [44]. Our experiments showed that strain BYF could grow with barite or celestine. The barite occurrence in the Bazhenov Formation rocks has been documented [45]. The barium concentration in the 1-R water is not sufficiently high to speculate that barite fuels sulfate reduction in the aquifer. However, the elevated strontium concentrations from 0.946 to 1.011 mg/L were detected in the 1-R water samples. High strontium content up to 500 mg/L is a well-known characteristic of the West Siberian Megabasin [46]. It is also conceivable that concentration of sulfate could be higher in pore water of the rock matrix due to oxidation of such minerals as pyrite, the high content of which is recorded in the Bazhenov Formation [45]. Sulfate may also remain undetected in the relict marine water localized in spatially isolated sulfate-rich parts of the aquifer system.
Global dispersion and evolution of CDA
The determination of the complete genome sequence of strain BYF allowed us to compare it with the genome of strain MP104C. A surprisingly high similarity was discovered both at the level of the genome structure and the point nucleotide sequence variations. The finding of near identical CRISPR loci, known as one of the most rapidly evolving elements of prokaryotic genomes [33], in two strains was also unexpected.
The presence of only 30 SNPs dispersed in a 2-Mbp long core genome is an exceptional feature for а microorganism that is not spread globally but found only in a few spatially distant subsurface ecosystems on different continents. Equally unusual is the high similarity of CRISPR loci, which reflect the history of the interaction of a microorganism with viruses expected to be specific for a given habitat. Estimating the rate of accumulation of mutations in natural populations of microorganisms always presents a problem, but as a rough estimate one can use the data on the genome evolution of seven strains of the thermophilic archaeal species Sulfolobus islandicus isolated from three locations separated during different times beginning 910,000 years ago [47]. Similar to what we found for CDA, genome evolution of S. islandicus involved strain-specific integration of mobile elements, gene loss, and gain of genetic material from related species and accumulation of SNPs in the core genomes [47]. The average rate of nucleotide substitution of 4.66 × 10–9 (± 6.76 × 10–10) substitutions per site per year were reported for S. islandicus [47], similar to the universal rates estimated from comparisons of other species [48, 49]. Using these values, it is possible to estimate the divergence time between conservative regions of the genomes of strains MP104C and BYF (30 SNPs in 2.3 Mb) at about 2800 years and between variable regions (703 SNP in 48.5 kb) at about 3.1 million years. Both these values are orders of magnitude smaller than the time of a split of an ancient supercontinent Pangea approximately 175 million years ago leading to the formation of Africa, Eurasia, and America. However, the rates of nucleotide substitutions per site per year could be much lower in the slow-growing microbes. For example, mutation rates from 10−4 to 10−3 per genome per generation were inferred for bacterial populations in subseafloor sediments, where the generation times reached dozens of years [50]. Generalization of these values to a bacterium dividing once in 1000 years would estimate the mutation rate in about 1 SNP per 1–10 million years. However, contribution of replication-independent factors (e.g., radiation-induced DNA damage) could be more important for accumulation of mutations at large timescales.
There are probably two not mutually exclusive explanations for the high genetic similarity between the Siberian and South African CDA strains. The first is a slow rate of evolution of microorganisms of the deep subsurface biosphere, thriving under energy-limiting conditions. The generation time in such environments can reach hundreds to thousands of years [51,52,53]. However, unlike the South African gold mine, the deep subsurface aquifer in the Western Siberia is not an organic-poor environment and harbors a diverse microbial community with a significant proportion of heterotrophs [10]. The doubling time of the strain BYF under optimal growth conditions was about 29 h, suggesting the possibility of its fast growth also in the natural environment when nutrients are available in excess.
The second explanation could be relatively fast global dispersion of CDA. Its spores enabled by gas vesicles with improved buoyancy may represent a mechanism that allows CDA to spread over long distances and colonize subsurface environments where it can outcompete other prokaryotes due to its specific nutritional style, as observed in the South African mines. CDA may originate from the deep terrestrial subsurface and can be delivered to the surface with the discharge of groundwater into surface environments. On the surface, microorganisms can spread over long distances, for example, with atmospheric aerosols [54]. The colonization of new terrestrial subsurface environments by CDA might happen via meteoric water discharge to the deeper horizons.
The present mosaic structure of MP104C and BYF genomes, consisting of a highly conserved core and a few more diverged regions, could result from the genetic interaction between the quickly distributed CDA “strain” and genetically more distant, site-specific strains or species related to CDA. Such more distant lineages could make different contributions to CDA genomes, as illustrated by the discovery in South African ecosystems of both SAGs, genetically very similar to MP104C and BYF, and more distant lineages having an average nucleotide identity <90% [2]. The analysis of the CDA genomes from other geographic regions will clarify the mechanisms of the global spread of this unique microorganism.
Deep subsurface environments have been considered as ecosystems harboring slow-growing prokaryotes due to extreme energy limitation. Our study showed that the typical subsurface organism can be adapted to relatively fast growth patterns under nutrient-rich conditions. The extremely low metabolic rates and generation time ascribed to CDA represent only “one side of medal” and the organism is rather flexible and can switch to different energy sources and growth patterns.
Data availability
The annotated genome sequence of CDA strain BYF has been deposited in the NCBI GenBank database under the accession number CP034260. The Supplementary Information includes Supplementary Figures 1–4 and Supplementary Tables 1–4. Other data that support the findings of this study are available from the corresponding author upon request.
References
Whitman WB, Coleman DC, Wiebe JW. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–83. https://doi.org/10.1073/pnas.95.12.6578
Labonté JM, Field EK, Lau M, Chivian D, Van Heerden E, Wommack KE, et al. Single cell genomics indicates horizontal gene transfer and viral infections in a deep subsurface Firmicutes population. Front Microbiol. 2015;22:349. https://doi.org/10.3389/fmicb.2015.00349
Magnabosco C, Lin LH, Dong H, Bomberg M, Ghiorse W, Stan-Lotter H, et al. The biomass and biodiversity of the continental subsurface. Nat Geosci. 2018;11:707–17. https://doi.org/10.1038/s41561-018-0221-6
Inagaki F, Hinrichs K-U, Kubo Y, Bowles MW, Heuer VB, HongInagaki W-L, et al. Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor. Science. 2015;349:420–4. https://doi.org/10.1126/science.aaa6882
Chivian D, Brodie EL, Alm EJ, Culley DE, Dehal PS, DeSantis TZ, et al. Environmental genomics revealed single-species ecosystem deep within Earth. Science. 2008;322:275–8. https://doi.org/10.1126/science.1155495
Davidson MM, Silver BJ, Onstott TC, Moser DP, Gihring TM, Pratt LM, et al. Capture of planktonic microbial diversity in fractures by long-term monitoring of flowing boreholes, Evander Basin, South Africa. Geomicrobiol J. 2011;28:275–300. https://doi.org/10.1080/01490451.2010.499928
Magnabosco C, Tekere M, Lau MCY, Linage B, Kuloyo O, Erasmus M, et al. Comparisons of the composition and biogeographic distribution of the bacterial communities occupying South African thermal springs with those inhabiting deep subsurface fracture water. Front Microbiol. 2014;5:679. https://doi.org/10.3389/fmicb.2014.00679
Tiago I, Veríssimo A. Microbial and functional diversity of a subterrestrial high pH groundwater associated to serpentinization. Environ Microbiol. 2013;15:1687–706. https://doi.org/10.1111/1462-2920.12034
Kjeldsen KU, Kjellerup BV, Egli K, Frølund B, Nielsen PH, Ingvorsen K. Phylogenetic and functional diversity of bacteria in biofilms from metal surfaces of an alkaline district heating system. FEMS Microbiol Ecol. 2007;61:384–97. https://doi.org/10.1111/j.1574-6941.2006.00255.x
Kadnikov VV, Mardanov AV, Beletsky AV, Banks D, Pimenov NV, Frank Y, et al. A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition. FEMS Microbiol Ecol. 2018;94:fiy152. https://doi.org/10.1093/femsec/fiy152
Banks D, Frank YA, Kadnikov VV, Karnachuk OV, Watts M, Boyce A, et al. Hydrochemical data report from sampling of two deep abandoned hydrocarbon exploration wells: Byelii Yar and Parabel’, Tomsk Oblast’, Western Siberia, Russian Federation. NGU Report, 2014.034. Trondheim: Geological Survey of Norway; 2014.
Frank YA, Kadnikov VV, Lukina AP, Banks D, Beletsky AV, Mardanov AV, et al. Characterization and genome analysis of the first facultatively alkaliphilic Thermodesulfovibrio isolated from the deep terrestrial subsurface. Front Microbiol. 2016;7:2000. https://doi.org/10.3389/fmicb.2016.02000
Cline JD. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr. 1969;14:454–8. https://doi.org/10.4319/lo.1969.14.3.0454
Widdel FF, Bak R. Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Truper HG, Dworkin M, Harder W, Schleifer KH, editors. The Prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications. 2nd ed. Berlin: Springer; 1992. p. 3352–78. https://doi.org/10.1007/978-1-4757-2191-1_21.
Karnachuk OV, Pimenov NV, Yusupov SK, Frank YA, Puhakka JA, Ivanov MV. Distribution, diversity, and activity of sulfate-reducing bacteria in the water column in Gek-Gel lake, Azerbaijan. Mikrobiologiia. 2006;75:101–9. https://doi.org/10.1134/S0026261706010152
Frank YA, Kadnikov VV, Gavrilov SN, Banks D, Gerasimchuk AL, Podosokorskaya OA, et al. Stable and variable parts of microbial community in Siberian deep subsurface thermal aquifer system revealed in a long-term monitoring study. Front Microbiol. 2016;7:2101. https://doi.org/10.3389/fmicb.2016.02101
Ikkert OP, Gerasimchuk AL, Bukhtiyarova PA, Tuovinen OH, Karnachuk OV. Characterization of precipitates formed by H2S-producing, Cu-resistant Firmicute isolates of Tissierella from human gut and Desulfosporosinus from mine waste. Antonie Van Leeuwenhoek. 2013;103:1221–34. https://doi.org/10.1007/s10482-013-9900-x
Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2001;56:2.4. 1–2.4.5. https://doi.org/10.1002/0471142727.mb0204s56
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2. https://doi.org/10.14806/ej.17.1.200
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. https://doi.org/10.1089/cmb.2012.0021
Cao MD, Nguyen SH, Ganesamoorthy D, Elliott AG, Cooper MA, Coin LJ. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat Commun. 2017;8:14515. https://doi.org/10.1038/ncomms14515
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. https://doi.org/10.1093/bioinformatics/btp698
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/nmeth.1923
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. https://doi.org/10.1038/nbt.1754
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. https://doi.org/10.1371/journal.pone.0112963
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365. https://doi.org/10.1038/srep08365
Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. https://doi.org/10.1093/nar/gkm360
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907 [q-bio.GN]; 2012.
Darling AE, Mau B, Perna NT. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. https://doi.org/10.1371/journal.pone.0011147
Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J Prepr. 2016;4:e1900v1. https://doi.org/10.7287/peerj.preprints.1900v1
Ramel F, Amrani A, Pieulle L, Lamrabet O, Voordouw G, Seddiki N, et al. Membrane-bound oxygen reductases of the anaerobic sulfate-reducing Desulfovibrio vulgaris Hildenborough: roles in oxygen defence and electron link with periplasmic hydrogen oxidation. Microbiology. 2013;159:2663–73. https://doi.org/10.1099/mic.0.071282-0
Jungbluth SP, Glavina del Rio T, Tringe SG, Stepanauskas R, Rappé MS. Genomic comparisons of a bacterial lineage that inhabits both marine and terrestrial deep subsurface systems. PeerJ. 2017;5:e3134. https://doi.org/10.7717/peerj.3134
Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol. 2008;10:200–7. https://doi.org/10.1111/j.1462-2920.2007.01444.x
Kupczok A, Landan G, Dagan T. The contribution of genetic recombination to CRISPR array evolution. Genome Biol Evol. 2015;7:1925–39. https://doi.org/10.1093/gbe/evv113
Frank Y, Banks D, Avakian M, Antsiferov D, Kadychagov P, Karnachuk O. Firmicutes is an important component of microbial communities in water-injected and pristine oil reservoirs, Western Siberia, Russia. Geomicrobiol J. 2016;33:387–400. https://doi.org/10.1080/01490451.2015.1045635
Karnachuk OV, Sasaki K, Gerasimchuk AL, Sukhanova O, Ivasenko DA, Kaksonen AH, et al. Precipitation of Cu-sulfides by copper-tolerant Desulfovibrio isolates. Geomicrobiol J. 2008;25:219–27. https://doi.org/10.1080/01490450802153124
Mardanov AV, Panova IA, Beletsky AV, Avakyan MR, Kadnikov VV, Antsiferov DV, et al. Genomic insights into a new acidophilic, copper-resistant Desulfosporosinus isolate from the oxidized tailings area of an abandoned gold mine. FEMS Microbiol Ecol. 2016;92:fiw111. https://doi.org/10.1093/femsec/fiw111
Dinh HT, Kuever J, Mussmann M, Hassel AW, Stratmann M, Widdel F. Iron corrosion by novel anaerobic microorganisms. Nature. 2004;427:829–32. https://doi.org/10.1038/nature02321
Deng X, Okamoto A. Electrode potential dependency of single-cell activity identifies the energetics of slow microbial electron uptake process. Front Microbiol. 2018;9:2744. https://doi.org/10.3389/fmicb.2018.02744
Pfeifer F. Distribution, formation and regulation of gas vesicles. Nat Rev Microbiol. 2012;10:705–15. https://doi.org/10.1038/nrmicro2834
Widdel F, Pfennig N. Sporulation and further nutritional characteristics of Desulfotomaculum acetoxidans. Arch Microbiol. 1981;129:401–2. https://doi.org/10.1007/BF00406471
O’Sullivan LA, Roussel EG, Weightman AJ, Webster G, Hubert CRJ, Bell E, et al. Survival of Desulfotomaculum spores from estuarine sediments after serial autoclaving and high-temperature exposure. ISME J. 2015;9:922–33. https://doi.org/10.1038/ismej.2014.190
Lin L-H, Wang P-L, Rumble D, Lippmann-Pipke J, Boice E, Pratt LM, et al. Long-term sustainability of a high-energy, low-diversity crustal biome. Science. 2006;314:479 https://doi.org/10.1126/science.1127376
Karnachuk OV, Kurochkina SY, Tuovinen OH. Growth of sulfate-reducing bacteria with solid-phase electron acceptors. Appl Microbiol Biotechnol. 2002;58:482–6. https://doi.org/10.1007/s00253-001-0914-3
Kontorovich AE, Yan PA, Zamirailova AG, Kostyreva EA, Eder VG. Classification of rocks of the Bazhenov Formation. Russ Geol Geophys. 2016;57:1606–12. https://doi.org/10.1016/j.rgg.2016.10.006
Novikov DA, Shvartsev SL. Hydrogeological conditions of the Pre-Enisei petroleum subprovince. Russ Geol Geophys. 2009;50:873–83. https://doi.org/10.1016/j.rgg.2009.09.005
Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci USA. 2009;106:8605–10. https://doi.org/10.1073/pnas.0808945106
Ochman H, Elwyn S, Moran NA. Calibrating bacterial evolution. Proc Natl Acad Sci USA. 1999;96:12638–43. https://doi.org/10.1073/pnas.96.22.12638
Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–17. https://doi.org/10.1073/pnas.95.16.9413
Starnawski P, Bataillon T, Ettema TJG, Jochum LM, Schreiber L, Chen X, et al. Microbial community assembly and evolution in subseafloor sediment. Proc Natl Acad Sci USA. 2017;114:2940–5. https://doi.org/10.1073/pnas.1614190114
Lomstein BA, Langerhuus AT, D’Hondt S, Jørgensen BB, Spivack AJ. Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature. 2012;484:101–14. https://doi.org/10.1038/nature10905
Jørgensen BB, D’Hondt S. A starving majority deep beneath the seafloor. Science. 2006;314:932–34. https://doi.org/10.1126/science.1133796
Phelps TJ, Murphy EM, Pfiffner SM, White DC. Comparison between geochemical and biological estimates of subsurface microbial activities. Microb Ecol. 1994;28:335–49. https://doi.org/10.1007/BF00662027
Smith DJ, Timonen HJ, Jaffe DA, Griffin DW, Birmele MN, Perry KD, et al. Intercontinental dispersal of bacteria and archaea by transpacific winds. Appl Environ Microbiol. 2013;79:1134–9. https://doi.org/10.1128/AEM.03029-12
Acknowledgements
Strain BYF isolation was supported by the Russian Foundation for Basic Research (grant # 18–04–00181) to O.V.K. group. Studies of strain BYF morphology and physiology were supported by the Russian Science Foundation (grant # 18-14-00130) to O.V.K. group. The work of N.V.R. group on sequencing and analysis of CDA genome was supported by the Russian Science Foundation (grant 14–14–01016) and the Ministry of Science and Higher Education of the Russian Federation. We thank Andrei Miller for his excellent assistance with TEM. We appreciate the three anonymous reviewers’ suggestions that helped to improve the manuscript.
Author information
Authors and Affiliations
Contributions
O.V.K. and N.V.R. designed the study and wrote the manuscript. Y.A.F. and A.P.L. performed the research work on CDA isolation, cultivation, morphology, and physiology. A.V.M. and V.V.K. sequenced the genome of strain BYF and performed comparative genomics analysis. A.V.B. performed SNP analysis and genome comparisons. All authors commented on and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karnachuk, O.V., Frank, Y.A., Lukina, A.P. et al. Domestication of previously uncultivated Candidatus Desulforudis audaxviator from a deep aquifer in Siberia sheds light on its physiology and evolution. ISME J 13, 1947–1959 (2019). https://doi.org/10.1038/s41396-019-0402-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0402-3
This article is cited by
-
Unique H2-utilizing lithotrophy in serpentinite-hosted systems
The ISME Journal (2023)
-
Hydrogen and dark oxygen drive microbial productivity in diverse groundwater ecosystems
Nature Communications (2023)
-
DNA uptake from a laboratory environment drives unexpected adaptation of a thermophile to a minor medium component
ISME Communications (2023)
-
Coexistence of Psychrophilic, Mesophilic, and Thermophilic Sulfate-Reducing Bacteria in a Deep Subsurface Aquifer Associated with Coal-Bed Methane Production
Microbial Ecology (2023)
-
Water and soil contaminated by arsenic: the use of microorganisms and plants in bioremediation
Environmental Science and Pollution Research (2022)
