
Abstract
We studied the long-term temporal dynamics of the aerobic anoxygenic phototrophic (AAP) bacteria, a relevant functional group in the coastal marine microbial food web, using high-throughput sequencing of the pufM gene coupled with multivariate, time series and co-occurrence analyses at the Blanes Bay Microbial Observatory (NW Mediterranean). Additionally, using metagenomics, we tested whether the used primers captured accurately the seasonality of the most relevant AAP groups. Phylogroup K (Gammaproteobacteria) was the greatest contributor to community structure over all seasons, with phylogroups E and G (Alphaproteobacteria) being prevalent in spring. Diversity indices showed a clear seasonal trend, with maximum values in winter, which was inverse to that of AAP abundance. Multivariate analyses revealed sample clustering by season, with a relevant proportion of the variance explained by day length, temperature, salinity, phototrophic nanoflagellate abundance, chlorophyll a, and silicate concentration. Time series analysis showed robust rhythmic patterns of co-occurrence, but distinct seasonal behaviors within the same phylogroup, and even within different amplicon sequence variants (ASVs) conforming the same operational taxonomic unit (OTU). Altogether, our results picture the AAP assemblage as highly seasonal and recurrent but containing ecotypes showing distinctive temporal niche partitioning, rather than being a cohesive functional group.
Similar content being viewed by others

Introduction
Bacteria are extremely abundant and diverse in the ocean where they drive most biogeochemical cycles. Recent developments in sequencing technologies have allowed studying microbial diversity at unprecedented scales. Mapping microbial communities in hundreds of samples from recent global expeditions has resulted in a comprehensive picture of how they vary across space [1,2,3]. Likewise, long-term microbial observatories are key to understand microbial variation over time, particularly in temperate zones encompassing contrasting meteorological seasons [4, 5]. To date, different seasonal studies conducted in fixed stations in the Atlantic (Bermuda Atlantic Time Series Study, Western English Channel Time Series) and Pacific (Hawaii Ocean Time Series, San Pedro Ocean Time Series (SPOT)) Oceans, and in the Mediterranean Sea (Service d’Observation du Laboratoire Arago Station) concur that plankton turnover is mostly driven by the environment, and that the seasonal patterns are repeatable over time [6,7,8].
Thus far, most seasonal studies have focused on determining the variation of phylogenetic groups based on 16 or 18S rRNA gene sequencing for bacterioplankton and eukaryotic plankton respectively [6, 9,10,11]. However, these phylogenetic units may include different ecotypes given that closely related or even identical rRNA gene-identified species can possess different functional traits [12] as a result of processes such as horizontal gene transfer (HGT) that can disconnect functional from phylogenetic diversity [13]. While a considerable amount of information on the seasonality of bulk microbial communities and of some particular phylogroups exists (i.e. [14]), the seasonality of individual functional groups is barely known.
A functional guild of particular interest is the polyphyletic (i.e., derived from more than one common ancestor through HGT) aerobic anoxygenic phototrophic (AAP) bacteria. These organisms have the ability of photoheterotrophy, that is, they are capable of using both organic matter and light as energy sources [15]. Their discovery challenged previous simplistic views of the structure of ocean microbial food webs [16]. AAP bacteria are relatively common in the euphotic zone of the oceans [17,18,19,20,21], exhibit faster growth rates than other bacterioplankton groups [22, 23] and their cells are in general larger than most marine heterotrophic bacteria [24]. Altogether, these characteristics make them relevant in the ecosystem by processing a large amount of organic matter (see review by [15]).
Phylogenetically, the AAPs belong to the Alphaproteobacteria, Betaproteobacteria and Gammaproteobacteria. However, since these organisms acquired the ability of photoheterotrophy through HGT, the 16S rRNA gene, typically used for identifying prokaryotes, cannot be used as a genetic marker of AAPs in environmental studies. Alternatively, the pufM gene, present in all anoxygenic phototrophs containing type-2 reaction centers, is routinely used in AAP diversity surveys. Based on the phylogeny of this gene and the structure of the puf operon, Yutin et al. [25] defined 12 distinct phylogroups (named from A–L) using metagenomic data from the Global Ocean Survey. Currently, the taxonomic assignation of short environmental sequences of the pufM gene is commonly done using this 12-phylogroup classification. In recent years, several authors have investigated their diversity and community structure in relation to environmental gradients across spatial scales using the variability of this marker gene [18, 20, 25,26,27,28] but much less is known about their temporal dynamics. Two independent studies conducted in the NW Mediterranean [29] and the East coast of Australia [28] examined the variability of AAPs using pufM amplicon sequencing and showed that these assemblages seem to be highly dynamic. These two studies analyzed only 1 year of samples but long-term surveys are necessary to understand their seasonal and interannual patterns of biodiversity, stability, predictability, interactions between species, and responses to environmental changes.
We present here the first long-term exploration of marine AAP assemblages using Illumina sequencing of the amplified pufM gene from monthly samples taken over 10 years at the coastal Blanes Bay Microbial Observatory (BBMO) in the NW Mediterranean Sea. We define the temporal patterns and unveil their recurrence, explore the long-term interactions between the different phylogroups, and identify the main environmental drivers acting upon the observed patterns. Taking advantage of the recent appearance of threshold-free algorithms for amplicon sequence variants (ASVs) analysis, which surpass the clustering of sequences based on similarity cutoffs [30, 31], we have gone one step beyond previous studies and explored the seasonality of ASVs potentially representing different AAP ecotypes at a more fine-grained level. These analyses ultimately allow us to explore the level of ecological consistency within the different phylogenetic clades, i.e. whether the different AAP phylotypes are ecologically cohesive or, contrarily, each phylogroup includes organisms presenting temporal niche partitioning. Additionally, by comparing the sequences recovered through amplicon sequencing to those extracted from metagenomes, we test whether the used primers are adequate to evaluate the seasonality of the dominant AAP groups.
Material and methods
Location and sample collection
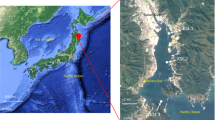
Surface water was collected monthly as described elsewhere [29] from the Blanes Bay Microbial Observatory (41°40′N, 2°48′E), a shallow (~20 m) coastal site about 1 km offshore in the NW Mediterranean coast. A total of 120 samples, from January 2004 to December 2013 were collected and in situ prefiltered through a 200-µm mesh. Several environmental parameters were measured alongside sample collection as described in Supplementary Information 1. The measured variables as well as day length were included in an environmental data table containing a total of 23 biotic and abiotic variables that was used for statistical analysis. The environmental data are shown in Figures S1 and S2. The astronomical seasons (based on equinoxes and solstices) were used for establishing spring, summer, autumn and winter periods. Additionally, the mixing layer depth (MLD) was obtained for the first months of 2004 and from 2008 to 2010 as defined in [32].
DNA extraction, pufM amplification, quantification, sequencing, and sequence processing
About 6 L of 200-µm pre-filtered surface seawater were sequentially filtered through a 20-µm mesh, a 3-µm pore-size polycarbonate filter (Poretics), and a 0.2-µm Sterivex Millipore filter using a peristaltic pump. Sterivex units were filled with 1.8 mL of lysis buffer (50 mM Tris-HCl pH 8.3, 40 mM EDTA pH 8.0, and 0.75 M sucrose), kept at −80 °C and extracted using the phenol-chloroform protocol as in Massana et al. [33]. Note that AAP bacteria attached to particles larger than 3 µm were not the subject of this study.
Partial amplification of the pufM gene (~245 bp fragments) was done in 50 µl reactions using primers pufM forward (5′-TACGGSAACCTGTWCTAC-3′, [34]) and puf_WAW reverse (5′-AYNGCRAACCACCANGCCCA-3′, [35]), each at 0.2 µM final concentration. The final concentration of MgCl2 was 2 mM. PCR conditions were as follows: an initial denaturation step at 95 °C for 5 min and 35 cycles at 95 °C (30s), 58°C (30s), 72 °C (40s) and a final elongation step at 72 °C for 10 min. Sequencing was performed in an Illumina MiSeq sequencer (2 × 250 bp, Research and Testing Laboratory; http://rtlgenomics.com/). Primers and spurious sequences were trimmed using cutadapt v1.14 [36]. DADA2 v1.4 was used to differentiate exact sequence variants and remove chimeras (parameters: maxN = 0, maxEE = c(2,4), trunclen = c(200,200)) [30]. DADA2 resolves ASVs by modeling the errors in Illumina-sequenced amplicon reads. The approach is threshold-free, inferring exact variants up to 1 nucleotide of difference using the quality score distribution in a probability model. For comparison purposes, the ASVs were clustered using UCLUST [37] at 94% of nucleic acid sequence similarity, a threshold typically used for the pufM gene [38]. After filtering for chimeras and spurious sequences with DADA2, 74% of the initial reads (mean 25692, min. 4172, max. 135331) were kept for downstream analyses. Sample BL120313 (13 March 2012) was discarded due to low read counts (836 reads). DADA2 read filtering details can be found in Supplementary Table 1. Moreover, in order to determine whether the primers used in this PCR-based approach captured the seasonality patterns accurately, we used 35 metagenomes generated from the same time-series (samples from years 2011 to 2013; see a detailed explanation in Supplementary Information 2) for comparison. Copy numbers of the marker gene pufM were estimated by quantitative polymerase chain reaction (qPCR) as described in Ferrera et al. [39] (see Supplementary Information 3 for details).
Phylogenetic classification
A custom-made database was generated combining sequences from previous AAP studies [25, 27, 40, 41], variants present in the Integrated Microbial Genomes system [42] and other pufM sequences from the GenBank database. Additionally, the predicted sequences from the BBMO metagenomes were included in this database. The nucleotide sequences were aligned with the guidance of amino acid translations using TranslatorX [43], with a posterior manual curation after filtering the sequences by length (>600 bp). From the alignment, a phylogenetic tree was constructed with RAxML v8.2 [44] (GTRGAMMA model, 1000 bootstraps), and the phylogroups were delimited in the resulting tree using iTOL [45] (Figure S3). Afterwards, the phylogenetic placement of the amplicon nucleotide sequences was performed with the Evolutionary Placement Algorithm v0.2 [46] to establish their phylogroup classification. Finally, to determine potential primer biases, the forward and reverse primers were contrasted against the nucleotide alignment.
Statistical analyses
All analyses were performed using the R language, with phyloseq and vegan packages [47,48,49]. Alphadiversity was analyzed using the Chao1 and Shannon indices [50]. Betadiversity was analyzed using a Bray-Curtis dissimilarity matrix with a previous normalization through rarefying to 4172 reads per sample [51, 52]. We used distance-based redundancy analysis (dbRDA, [53]) to find the environmental predictors (scaled to the mean) that best explained the patterns of community structure and diversity of AAPs over time, with a previous multivariate non-parametric ANOVA for selecting significant variables (p < 0.01). A time-decay analysis of the assemblage was computed excluding rare ASVs as recommended elsewhere [54]. ASVs were considered rare when presenting less than 1% of relative abundance from the rarefied dataset following the Alonso-Sáez et al. [55] criterion.
Time series analysis
Fourier time series analysis was performed to study the AAP assemblage dynamics over a decade. An interpolation of the discarded sample (BL120313) was used to maintain equidistant time points. Values were normalized through the Aitchison log-centered ratio transformation (CLR), adequate for compositional data [56]. A Fisher G-test with the R package GeneCycle was used to determine the significance (p < 0.01) of the periodic components [57]. The time series was decomposed in three components, seasonal periodicity (oscillation inside each period), trend (evolution over time) and residuals, through local regression by the stl function. Additionally, the autocorrelogram was calculated using the acf function.
Network construction
We used Local Similarity Analysis (LSA) [58, 59] with a previous CLR transformation for network construction. Briefly, given a time series data and a delay limit, LSA finds the configuration that yields the highest local similarity (LS) score. Only the ASVs present in >5 samples and the environmental variables presenting <5% of missing values were used. The remaining missing values for the variables after filtering were estimated by imputation with the mice package [60]. Only interactions with LS ≥ 0.5, p < 0.001 and 1-month delay were considered. The network was plotted using the ggraph package [61].
Reproducibility
The code for preprocessing and statistical analyses along with package versions is available in the following repository: https://gitlab.com/aauladell/AAP_time_series. Sequence data have been deposited in Genbank under accession number PRJNA449272.
Results and discussion
Patterns of community composition and structure
The amplicon sequences retrieved from 10 years of sampling resulted in a total of 820 ASVs whereas the number of OTUs was 406 (94% similarity cutoff). Of the total ASVs, 276 presented only one nucleotide variation between sequences. In comparison with previous temporal studies (82 OTUs detected in [29], 89 in [28]), our study presents a more complete picture of the pufM diversity and is the largest dataset of AAP diversity reported to date. Estimates of richness were higher during winter (mean 51, max. 126 observed ASVs), decreasing to minimum values in the spring-summer period, i.e. during May–August (mean 35, max. 77) (Fig. 1). The differences between winter and spring/summer were statistically significant (ANOVA, p < 0.05). A similar trend was observed when computing the Shannon index (Fig. 1). Comparing the amplicon with the metagenomic data from 2011 to 2013, we observed that whereas 188 OTUs and 357 ASVs were present in the amplicons for that period, only a total of 84 different pufM sequences were recovered from the metagenomes. However, the Shannon diversity index for the two datasets presented a positive correlation (Pearson R = 0.81, p = 0.001, N = 35) and they followed the same trend of increasing values in winter (Fig. 1).

Alphadiversity distribution of the AAP community for each month colored by season. Richness (number of observed ASVs) and Shannon indexes obtained through amplicon sequencing over a decade (2004–2013) are shown in the top and middle panels, respectively. Each boxplot presents the median and interquartile range of the distribution of 10 data points shown in grey (with the exception of March, with 9 data points). Whiskers represent 1.5 times the interquartile range. The bottom panel shows the Shannon index values obtained from the metagenomic dataset (2011–2013). The colored dots represent the mean monthly values and the bars the standard error of the mean for the 3-year period
A notable negative correlation between day length and the Shannon index was observed (Pearson R = −0.57, p < 0.01, N = 119). That relationship of diversity with day length had previously been observed in long-term bulk bacterioplankton community studies [8], as well as with specific phylogenetic groups such as the SAR11 [62]. A possible explanation is that the deep winter mixing allows the development of high diversity assemblages in contrast to the selection of specific oligotrophic ecotypes occurring during the stratified summer season [62]. In fact, mixed layer depth was a significant predictor of the Shannon diversity index (Spearman R = 0.56, p < 0.001, N = 35, Figure S4). Interestingly, this trend of higher alphadiversity in winter is opposed to that of AAP abundance (Figure S5); higher abundances of the pufM gene during spring and summer were measured by qPCR as compared to winter and fall (p < 0.01). We also found a positive correlation between the qPCR data and the abundance of pufM sequences retrieved in the 3 years of metagenomes (Pearson R = 0.77, p < 0.001, N = 12) (Figure S6), in which higher abundances were found in spring followed by summer. These results support previous observations obtained through various methodological approaches ([29] using microscopy counts, [28] using qPCR and [7] using metagenomics) and confirm that there is a clear inverse relationship between AAP bacterial abundance and diversity.
Regarding community composition across the decadal period (Fig. 2, Figure S7), phylogroup K (Gammaproteobacteria affiliated to the NOR5/OM60 clade) was the most prevalent and dominant over the years (83.8% ± SE 2.3, mean relative abundance), in agreement with previous reports for this station [29] and for other regions such as the Baltic Sea [63]. Yet, a decrease in their contribution was observed during February and March (down to 59.6 and 52% on average respectively). During these months, the contribution of phylogroup E (Rhodobacter-like) to community structure was greater, albeit with a high variation over the decade (±26% SD). The previous 1-year study of AAP diversity conducted by Ferrera et al. [29] reported a similar observation and, moreover, a study of the 16 S rRNA gene diversity from the same location also suggested that Alphaproteobacteria dominate the bacterial assemblages during the local spring bloom [64]. Regarding phylogroups D, F (Rhodobacterales-like), H (uncultured), J (Rhodospirillales-like), I (Betaproteobacteria), Sphingomonas-like and the unclassified ASVs, these presented a mean relative contribution below 1% in the amplicon dataset. These groups displayed occasional peaks (>1% relative abundance) with no clear periodic trend. For example, Sphingomonas-like AAPs showed a contribution of 14% in February 2012 (Figure S7). Overall, these observations are similar to those obtained from the metagenomic distribution (3 years instead of 10) with a good agreement in the relative abundance recovered for the prevailing phylogroups E and K as well as for the less common Sphingomonas-like sequences (correlation values: 0.9, 0.69 and 0.91, respectively; Table S2). Contrarily, phylogroup G seems to be underrepresented in the amplicon dataset likely due to the presence of 3–5 mismatches in the forward primer with the most abundant metagenomic variants.

Variation in the relative contributions of AAP phylogroup K (top panel), phylogroups G, E (middle panel), phylogroups D, F, I, J, Sphingomonas-like and the unclassified group (bottom panel) for each month over the studied decade (2004–2013). Each boxplot presents the median and the 25 and 75% limits with the distribution of 10 data points in grey (with the exception of March, with 9 data points), and whiskers represent 1.5 times the interquartile range
Metagenomics is often considered the least biased approach for functional gene analysis since the method is PCR-independent and does not suffer from amplification biases that could result in misrepresentation of the relative abundances of certain populations. Nonetheless, for a given time and money investment, metagenomes retrieve less copies of specific marker genes, offering thus less inquiry potential if the main purpose of our study is the barcoding of a particular group of organisms. We found that richness estimates were higher using amplicon data since more variants of the pufM gene were recovered with that approach than from metagenomes, but the seasonal trends in diversity identified by both methodologies were remarkably similar (Fig. 1). Likewise, in terms of community structure there was a good agreement for the most prevailing groups with the exception of phylogroup G. Moreover, the seasonal trends observed at the phylogroup and even at the sequence variant level recovered using these two distinct methodological approaches, presented a close resemblance (Figures S8 and S9, see Seasonality at the fine scale section below).
Noteworthy, the amplicon approach allowed identifying the seasonal tendencies of many more individual ecotypes than what would have been possible through metagenomics, while metagenomics captured some low abundance groups missing in the amplicon dataset. In particular, phylogroups A, B, C, and L, accounting for a total relative abundance of 7.0, 3.1, 3.9, and 0.2%, respectively, were only retrieved through metagenomics. Primer coverage analysis revealed that the forward primer contains between 3 and 8 mismatches with the metagenomic sequences from these phylogroups (details not shown), which could explain their absence in the amplicon dataset and why these groups are rarely reported in AAP surveys based on amplicon sequencing [27, 28]. Exceptionally, Ferrera et al. [29] reported the presence of one single OTU of phylogroup C contributing substantially (13% relative abundance) to the community during winter in Blanes Bay, which differs from the present results. To investigate this discrepancy, we carefully compared the sequence of this OTU to our updated database and found that it had been misclassified and it belongs to phylogroup K while does not show any significant similarity to the new phylogroup C sequences retrieved from the metagenomes. These observations highlight the need to increase the information present in databases to obtain accurate taxonomic assignations. In fact, only a few isolates from phylogroup K exist and none is available for phylogroup C, hampering the classification of these groups as discussed elsewhere [65]. In contrast, phylogroups F, H, I, and J (<1% total relative abundance) were recovered only when using amplicons. Their low relative abundance possibly explains their absence in the metagenomic dataset. Overall, these results remark the need to undertake a revision of the primers typically used for high-throughput sequencing of the pufM gene in order to increase their phylogenetic recovery but, at the same time, demonstrate that PCR-free metagenomics and amplicon-based approaches perform in a comparable fashion in recovering the major AAP groups and, most importantly, that the seasonal patterns observed through amplicon sequencing are robust.
Patterns of betadiversity and recurrence
Non-metric multidimensional scaling (nMDS) using various distance measurements indicated a clear separation of the samples at different temporal scales: by month (Bray-Curtis, PERMANOVA R2 = 0.51, p < 0.001) and by season (Bray-Curtis, PERMANOVA R2 = 0.31, p < 0.001, Figure S10). Spring and winter samples were more dissimilar than those of summer or autumn. The reasons for this pattern are uncertain but could be related to higher date to date environmental variability or to the mixing of the water column that occurs during winter in this station [66].
Community structure was strongly linked to day length, temperature, salinity, phototrophic nanoflagellate abundance, chlorophyll a and silicate concentration, as revealed by distance-based redundancy analysis (dbRDA; Fig. 3, PERMANOVA p < 0.01, Supplementary Information 4), which explained 51.4% of the variation with the first two axis explaining 43.6%. In particular, late spring and early summer samples were mostly influenced by day length and temperature, whereas autumn samples were partially influenced by salinity (Fig. 3). Day length has previously been shown to explain the seasonal variability of the bulk bacterioplankton [8] and AAP community structure [29], but the mechanisms underlying this relationship are unclear. Interestingly, a group of samples from winter and spring appeared to be heavily influenced by the presence of ASVs (ASV8, ASV14, and ASV46) belonging to phylogroup E (Rhodobacter-like, Figure S11), by the abundance of phototrophic nanoflagellate and by the concentration of chlorophyll a (Fig. 3), which could be related to the phytoplankton spring bloom that typically occurs in February–March in Blanes [67]. The summer samples were associated to the high contribution of gammaproteobacterial ASV1 and the fall/early-winter cluster to more diverse communities of other gammaproteobacterial ASVs (Figure S11).

Distance based redundancy analysis of the samples (dots) with the five explanatory variables (arrows) influencing their distribution (PERMANOVA p < 0.01; day length, temperature, salinity, silicate concentration (Si), Chlorophyll a (Chla) concentration and phototrophic nanoflagellate abundance (PNF)). The ordination was performed on the Bray-Curtis dissimilarity of log10 transformed data (with a pseudocount of 1) matrix (after rarefying). Samples are colored by season
Finally, to explore the recurrence of the communities, the Bray-Curtis similarity between samples was plotted against the time lag resulting in the so-called time-decay curve (Fig. 4) [6, 68]. In our study, the assemblage was maintained over time with a median similarity of 0.45, with 6-month oscillations from the yearly maximum (~0.55) to the minimum (~0.39) values. These results indicate that AAP communities are under strong environmental selection that leads to a high seasonal behavior and translates into yearly repeatable communities. To our knowledge, this is the first time that the recurrence of a functional group of planktonic organisms, defined by a marker gene, has been demonstrated. Comparing the results to the 16S rRNA data from the SPOT and the Western Channel time series, we observe that the seasonal turnover at SPOT is less clear than in our location, and an initial decay of similarity is observed reaching a later plateau over time. In the Western Channel, the seasonality is equally marked but the initial decay is even more pronounced than in SPOT (see Fig. 2 in [69]). A possible explanation for these differences is that our comparison accounts only for a highly seasonal sub-community (as these organisms are able to use light, and light varies seasonally) while the overall bacterial/prokaryotic community responds to more variables. Further analyses with other functional genes should help understand whether these patterns are robust for distinct groups.

Bray-Curtis similarity between samples plotted against the time lag between each of them (time-decay plot). Mean similarity values for each time lag are plotted in an empty black dot with standard error bars (background grey filled dots show each comparison). A linear regression is plotted, with 95% confidence intervals shown
Patterns of co-occurrence
A co-occurrence network was built with 127 ASVs present in >5 samples and 14 environmental variables, presenting 70 nodes and 142 edges after filtering by local similarity and significance (LS ≥ 0.5, p < 0.001) (Fig. 5). Noteworthy, most of the ASVs retained in the network were seasonal (46 out of 61) (see below). In terms of topology, the network presents one large cluster and other four minor clusters, being the largest one formed by 54 nodes mainly containing ASVs from phylogroups K and E, displaying multiple interactions with various ecosystem variables (temperature, day length, and the abundance of phototrophic picoeukaryotes and nanoflagelates). Temperature was the variable presenting the largest number of the interactions (14), most of them being delayed one month. Out of these, many were of negative nature with Gammaproteobacteria-like ASVs that lower their relative abundance during summer (for example ASV26, 10, 11) while others were positive with ASVs that dominate the AAP community during this season (ASV1). Interestingly, many positive and negative interactions exist between various ASVs of phylogroup K and G and the abundance of phototrophic eukaryotes but none with other phylogroups such as phylogroup E or Sphingomonas-like. Strong biotic relationships between AAP species and phytoplankton have been reported, particularly with dinoflagellates [70, 71] and large fractions of particle-attached AAP bacteria have been observed in various marine environments [72, 73]. Here, we focused on the free-living fraction of AAPs but further interaction network analyses using both free-living and particle-attached AAP bacteria in combination with phototrophic eukaryotic species data would allow to deeper investigate these biotic relationships.

Fast local similarity network showing clusters with ≥3 nodes. Node shape designates the type of variable, with the filling specifying the phylogroup, the size the total relative contribution and the stroke color if the ASV displays a seasonal behavior. Edges can be lagged (discontinuous line) or direct and have negative (i.e., anticorrelation) or positive local scores (LS). The label on the nodes indicates the ASV number. T°: temperature; PNF: abundance of phototrophic nanoflagellates; Peuk1: abundance of picoeukaryotes group I (see Supplementary Information 1 for details)
The majority of correlations occurred within rather than between phylogroups as previously observed [28]; yet, whereas some groups presented mainly positive intergroup interactions (phylogroup E or Sphingomonas-like), phylogroup K showed positive and negative interactions between its ASVs. As an example, a clear negative correlation between ASV1-ASV26 and ASV10-ASV35, all of them being part of phylogroup K, can be observed in Fig. 5. We also noticed that various ASVs of phylogroup E, such as ASV14 and ASV8 (Alphaproteobacteria-like), were positively related among them while presenting negative associations with ASVs from phylogroup K (Gammaproteobacteria-like ASV30 and ASV35). Negative correlations between phylogroup K and phylogroup G (also Alphaproteobacteria-like) had previously been reported [28, 29]. These data thus point towards intergroup competition between members of the Alpha- and Gammaproteobacteria-like AAPs.
Looking at the interactions within closely related ASVs, i.e. those forming the same OTU, we observed multiple connections between them, for example, among ASV1, ASV2, and ASV15, all belonging to OTU1 or ASV14, ASV65 and ASV175, all forming OTU14. Nevertheless, network analysis revealed that sometimes there is a dissociation of these closely related ASVs, as seen for ASV17 and ASV27, belonging to OTU1 which do not present connections with other ASVs from the same OTU. These observations support the idea that ASVs may represent individual AAP ecotypes encompassing distinct ecological patterns and reflects the usefulness of breaking apart sequence clusters into variants in order to dig into the ecology of these organisms.
Seasonality at the fine scale
The seasonality of each ASV was measured by evaluating if their relative abundance distribution presented a significant periodicity (Fisher G-test) through the long-term time series, and if so, by comparing them at different levels of resolution: across closely related sequences (ASVs) and across sequence clusters (OTUs and phylogroups). Seasonal patterns (p < 0.01) were present in 58 out of 127 ASVs analyzed (those ASVs present in >5 samples), affiliated to phylogroups K (44 ASVs), E (9) and G (2), J (1) and the unclassified group (2) (Table 1, Supplementary Table 3). In order to discard that potential amplification artifacts could influence the observed ASV seasonal trends, we mapped representative ASVs from the prevailing phylogroups E, G, and K to the metagenomic sequences and compared the seasonal behavior in both datasets, obtaining a remarkable good concordance (Figure S9).
The seasonal ASVs corresponded to 92% of the total read counts, and 83.4% of the counts corresponded to phylogroup K (Gammaproteobacteria). All periodicities found were of 1 year, with the exception of ASV152 (Gammaproteobacteria), that presented a periodicity of 2 years. Some of these ASVs always presented relative contributions above 1% regardless of season (all from phylogroup K), some presented values above 1% in a specific season (seasonal contributors), and other ASVs peaked (>1%) only occasionally (herein referred as opportunistic; see examples in Figure S12–S15). In fact, most ASVs presented an opportunistic behavior, with low contribution to total community composition during the decade and peaking occasionally. Various studies of the whole bacterioplankton community have observed this variety of strategies coexisting within a given clade [6, 55, 74]. Our results reveal that this trend is maintained for this specific functional assemblage, with a few prevalent ecotypes and a larger pool of specialized ASVs, i.e. appearing within specific environmental conditions, within each phylum.
Comparing among the seasonal ASVs, we distinguished different behaviors. For example, ASVs divergent enough to form distinct OTUs but belonging to the same phylogroup did not always follow the same distribution (Fig. 6a); e.g. for phylogroup K, the annual maxima of ASV1 occurred during June and July with a minimum in February/March, whereas ASV10 presents the opposite distribution. Contrarily, most ASVs belonging to phylogroups G and E followed a similar trend among them, with their maxima in March, being ASV86 an exception presenting a maximum in September (not shown). Looking at a further level of resolution, i.e., comparing the seasonality of closely related ASVs (that would form the same OTU), we observed that these generally displayed similar temporal patterns although some notable exceptions existed. An example is represented in Fig. 6b in which the seasonal periodicities of five closely related ASVs, all corresponding to OTU1, are plotted together. In this figure, a slight succession of the summer maxima can be observed (ASV2 peaking before ASVs 33 and 1, with ASV57 afterwards), being all these only 1 nucleotide different among them. Yet, ASV128 (presenting a distance of 4 nucleotides to OTU1) displays a different distribution peaking during winter. The existence of divergent distributions of ASVs composing the same OTU demonstrates the need to break apart the clusters of related sequences, since these can hide distinct ecological patterns. Furthermore, while the previous AAP temporal studies provided insights of the inter-annual community structure, this is the first study that identifies the long-term tendencies of individual ecotypes.

Seasonal component of the relative abundance distribution (log10 + 1 transformed) for some remarked ASVs fitted with a polynomial function. a Various ASVs with distant nucleotide similarity, colored by phylogroup assignation. b Various ASVs belonging to OTU1 (dashed line corresponds to OTU1). The patterns were defined based on the relative abundance dynamics of 10 years by time series analysis
At the other end, when we explored the seasonality at the phylogroup level, we found that phylogroup K as a whole did not present a statistically significant seasonal pattern (p > 0.01) (Figure S16-S17). The disparity of distributions of the various sequences within may be the reason of the loss of a significant signal when computing seasonality at the group level. Contrarily, the autocorrelograms showed phylogroup E presenting a high value (max. 0.34 over a year), followed by phylogroup J (Figure S16B). These results could indicate a higher degree of ecotype differentiation in gammaproteobacterial phylogroup K as compared to alphaproteobacterial phylogroups E and G. A possible explanation is that phylogroup K is phylogenetically broader (based on the 16S rRNA gene sequences) as compared to phylogroups E and G, resulting in more variable tendencies within it. Further analyses including the genomic context with the assignation of sequence variants to metagenome assembled genomes (MAGs) or genome sequencing of new isolates of phylogroup K, would help splitting this phylogroup into smaller phylogenetic clusters, perhaps showing ecological coherence. Lehours et al. [27] recently tested the ecological consistency of the AAP bacteria across different oceanic regions and, interestingly, identified clades with good ecological and phylogenetic coherence. Our temporal analyses add a new level of complexity by showing that, despite a certain degree of consistency exists, highly similar ASVs can present very different seasonal distributions that could translate into different ecology.
Concluding remarks
This work shows that the AAPs present a peak of diversity during winter, contrary to their abundance, and that gammaproteobacterial AAPs are the prevalent members of the community in the Mediterranean Sea year-round. Our results also evidence that the AAP assemblages show seasonal patterns repeatable over long periods of time. This study also demonstrates that PCR-free metagenomics and amplicon-based approaches perform in a comparable fashion in recovering major AAP groups and that the seasonal patterns observed through amplicon sequencing are robust. Interestingly, distinct seasonal behaviors were observed within the same phylogroup and even within different ASVs conforming the same OTU. In contrast to the recent spatial study of Lehours et al. [27], in which they reported ecological cohesiveness when comparing contrasting biomes, we found that the different AAP phylotypes do not appear as coherent when studying their seasonal behavior and seem to be rather composed of different ecotypes with distinctive temporal niche partitioning. Overall, these results show that the analysis of long time series allows exploring in-depth patterns of a highly dynamic microbial group and provides a framework for modeling their ecological role in relation to seasonality of marine carbon cycling.
References
Salazar G, Cornejo-Castillo FM, Benítez-Barrios V, Fraile-Nuez E, Álvarez-Salgado XA, Duarte CM, et al. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 2015;10:1–13.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science. 2015;348:1261359.
Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, et al. The Sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:0432–66.
Buttigieg PL, Fadeev E, Bienhold C, Hehemann L, Offre P, Boetius A. Marine microbes in 4D—using time series observation to assess the dynamics of the ocean microbiome and its links to ocean health. Curr Opin Microbiol. 2018;43:169–85.
Kane MD. Microbial observatories: exploring and discovering microbial diversity in the 21st century. Microb Ecol. 2004;48:447–8.
Fuhrman JA, Cram JA, Needham DM. Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol. 2015;13:133–46.
Galand PE, Pereira O, Hochart C, Auguet JC, Debroas D. A strong link between marine microbial community composition and function challenges the idea of functional redundancy. ISME J. 2018;12:2470–8.
Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B, et al. Defining seasonal marine microbial community dynamics. ISME J. 2012;6:298–308.
Giner CR, Balagué V, Krabberød AK, Ferrera I, Reñé A, Garcés E, et al. (2018) Quantifying long-term recurrence in planktonic microbial eukaryotes. Mol. Ecol. https://doi.org/10.1111/mec.14929
Kim DY, Countway PD, Jones AC, Schnetzer A, Yamashita W, Tung C, et al. Monthly to interannual variability of microbial eukaryote assemblages at four depths in the eastern North Pacific. ISME J. 2014;8:515–30.
Martin-Platero AM, Cleary B, Kauffman K, Preheim SP, McGillicuddy DJ, Alm EJ, et al. High resolution time series reveals cohesive but short-lived communities in coastal plankton. Nat Commun. 2018;9:266.
Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013;7:830–8.
Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272 LP–7.
Galand PE, Gutiérrez-Provecho C, Massana R, Gasol JM, Casamayor EO. Inter-annual recurrence of archaeal assemblages in the coastal NW Mediterranean Sea (Blanes Bay Microbial Observatory). Limnol Oceanogr. 2010;55:2117–25.
Koblížek M. Ecology of aerobic anoxygenic phototrophs in aquatic environments. FEMS Microbiol Rev. 2015;39:854–70.
Fenchel T. Marine bugs and carbon flow. Science. 2001;292:2444 LP–5.
Cottrell MT, Kirchman DL. Photoheterotrophic microbes in the arctic ocean in summer and winter. Appl Environ Microbiol. 2009;75:4958–66.
Jiao N, Zhang Y, Zeng Y, Hong N, Liu R, Chen F, et al. Distinct distribution pattern of abundance and diversity of aerobic anoxygenic phototrophic bacteria in the global ocean. Environ Microbiol. 2007;9:3091–9.
Lami R, Cottrell MT, Ras J, Ulloa O, Obernosterer I, Claustre H, et al. High abundances of aerobic anoxygenic photosynthetic bacteria in the South Pacific Ocean. Appl Environ Microbiol. 2007;73:4198–205.
Ritchie AE, Johnson ZI. Abundance and genetic diversity of aerobic anoxygenic phototrophic bacteria of coastal regions of the pacific ocean. Appl Environ Microbiol. 2012;78:2858–66.
Schwalbach MS, Fuhrman JA. Wide-ranging abundances of aerobic anoxygenic phototrophic bacteria in the world ocean revealed by epifluorescence microscopy and quantitative PCR. Limnol Oceanogr. 2005;50:620–8.
Ferrera I, Gasol JM, Sebastián M, Hojerová E, Koblížek M. Comparison of growth rates of aerobic anoxygenic phototrophic bacteria and other bacterioplankton groups in coastal mediterranean waters. Appl Environ Microbiol. 2011;77:7451–8.
Ferrera I, Sánchez O, Kolářová E, Koblížek M, Gasol JM. Light enhances the growth rates of natural populations of aerobic anoxygenic phototrophic bacteria. ISME J. 2017a;11:2391–3.
Sieracki ME, Gilg IC, Thier EC, Poulton NJ, Goericke R. Distribution of planktonic aerobic anoxygenic photoheterotrophic bacteria in the northwest Atlantic. Limnol Oceanogr. 2006;51:38–46.
Yutin N, Suzuki MT, Teeling H, Weber M, Venter JC, Rusch DB, et al. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean sampling expedition metagenomes. Environ Microbiol. 2007;9:1464–75.
Boeuf D, Cottrell MT, Kirchman DL, Lebaron P, Jeanthon C. Summer community structure of aerobic anoxygenic phototrophic bacteria in the western Arctic Ocean. FEMS Microbiol Ecol. 2013;85:417–32.
Lehours A-C, Enault F, Boeuf D, Jeanthon C. Biogeographic patterns of aerobic anoxygenic phototrophic bacteria reveal an ecological consistency of phylogenetic clades in different oceanic biomes. Sci Rep. 2018;8:4105.
Bibiloni-Isaksson J, Seymour JR, Ingleton T, van de Kamp J, Bodrossy L, Brown MV. Spatial and temporal variability of aerobic anoxygenic photoheterotrophic bacteria along the east coast of Australia. Environ Microbiol. 2016;18:4485–500.
Ferrera I, Borrego CM, Salazar G, Gasol JM. Marked seasonality of aerobic anoxygenic phototrophic bacteria in the coastal NW Mediterranean Sea as revealed by cell abundance, pigment concentration and pyrosequencing of pufM gene. Environ Microbiol. 2014;16:2953–65.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581.
Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 2015;9:968–79.
Galí M, Simó R, Vila-Costa M, Ruiz-González C, Gasol JM, Matrai P. Diel patterns of oceanic dimethylsulfide (DMS) cycling: microbial and physical drivers. Glob Biogeochem Cycles. 2013;27:620–36.
Massana R, Murray AE, Preston CM, Delong EF. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl Environ Microbiol. 1997;63:50–56.
Béjà O, Suzuki MT, Heidelberg JF, Nelson WC, Preston CM, Hamada T, et al. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature. 2002;415:630–3.
Yutin N, Suzuki MT, Béjà O. Novel Primers reveal wider diversity among marine aerobic anoxygenic phototrophs. Appl Environ Microbiol. 2005;71:8958–62.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Zeng YH, Chen XH, Jiao NZ. Genetic diversity assessment of anoxygenic photosynthetic bacteria by distance-based grouping analysis of pufM sequences. Lett Appl Microbiol. 2007;45:639–45.
Ferrera I, Sarmento H, Priscu J, Chiuchiolo A, González JM, Grossart H-P. Diversity and distribution of freshwater aerobic anoxygenic phototrophic bacteria across a wide latitudinal gradient. Front Microbiol. 2017b;8:175.
Cuadrat RRC, Ferrera I, Grossart H-P, Dávila AMR. Picoplankton bloom in Global South? A high fraction of aerobic anoxygenic phototrophic bacteria in metagenomes from a coastal bay (Arraial do Cabo—Brazil). Omi A J Integr Biol. 2016a;20:76–87.
Graham ED, Heidelberg JF, Tully BJ. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 2018;12:1861–6.
Markowitz VM, Korzeniewski F, Palaniappan K, Szeto E, Werner G, Padki A, et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 2006;34:D344–8.
Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–13.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Letunic I, Bork P. Interactive tree of life (iTOL)v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Barbera P, Kozlov AM, Czech L, Morel B, Darriba D, Flouri T, et al. EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst Biol. 2018;68:syy054–syy054.
McMurdie PJ, Holmes S. Phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
R Core Team R: a language and environment for statistical computing. 2014
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. 2018
Smith B, Wilson JB. A consumer’ s guide to evenness indices. Oikos. 1996;76:70–82.
Faith DP, Minchin PR, Belbin L. Compositional dissimilarity as a robust measure of ecological distance. Vegetatio. 1987;69:57–68.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27.
Legendre P, Legendre L. Numerical Ecology. Dev Environ Model. 1988;24:870.
Faust K, Lahti L, Gonze D, de Vos WM, Raes J. Metagenomics meets time series analysis: Unraveling microbial community dynamics. Curr Opin Microbiol. 2015;25:56–66.
Alonso-Sáez L, Díaz-Pérez L, Morán XAG. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ Microbiol. 2015;17:3766–80.
Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:1–6.
Ahdesmaki M, Fokianos K, Strimmer K. Package ‘GeneCycle’. (2015)
Durno WE, Hanson NW, Konwar KM, Hallam SJ. Expanding the boundaries of local similarity analysis. BMC Genom. 2013;14:1–14.
Xia LC, Steele JA, Cram JA, Cardon ZG, Simmons SL, Vallino JJ, et al. Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Syst Biol. 2011;5:S15.
Azur MJ, Stuart EA, Frangakis C, Leaf PJ. Multiple imputation by chained equations: what is it and how does it work? Int J Methods Psychiatr Res. 2012;20:40–49.
Pedersen TL. ggraph: An implementation of grammar of graphics for graphs and networks. 2017
Salter I, Galand PE, Fagervold SK, Lebaron P, Obernosterer I, Oliver MJ, et al. Seasonal dynamics of active SAR11 ecotypes in the oligotrophic Northwest Mediterranean Sea. Isme J. 2015;9:347–60.
Mašín M, Zdun A, Stoń-Egiert J, Nausch M, Labrenz M, Moulisová V, et al. Seasonal changes and diversity of aerobic anoxygenic phototrophs in the Baltic Sea. Aquat Microb Ecol. 2006;45:247–54.
Alonso-Sáez L, Balagué V, Sà EL, Sánchez O, González JM, Pinhassi J, et al. Seasonality in bacterial diversity in north-west Mediterranean coastal waters: assessment through clone libraries, fingerprinting and FISH. FEMS Microbiol Ecol. 2007;60:98–12.
Caliz J, Casamayor EO. Environmental controls and composition of anoxygenic photoheterotrophs in ultraoligotrophic high-altitude lakes (Central Pyrenees). Environ Microbiol Rep. 2014;6:145–51.
Gasol JM, Cardelús C, Morán XAG, Balagué V, Forn I, Marrasé C, et al. Seasonal patterns in phytoplankton photosynthetic parameters and primary production at a coastal NW Mediterranean site. Sci Mar. 2016;80S1:63–77.
Nunes S, Latasa M, Gasol JM, Estrada M. Seasonal and interannual variability of phytoplankton community structure in a Mediterranean coastal site. Mar Ecol Prog Ser. 2017;592:57–75.
Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J. 2013;754:1493–506.
Hatosy SM, Martiny JBH, Sachdeva R, STeele J, Fuhrman JA. Beta diversity of marine bacteria depends on temporal scale. Ecology. 2013;94:1898–904.
Yang Q, Jiang Z-W, Huang C-H, Zhang R-N, Li L-Z, Yang G, et al. Hoeflea prorocentri sp. nov., isolated from a culture of the marine dinoflagellate Prorocentrum mexicanum PM01. Antonie Van Leeuwenhoek. 2018;111:1845–53.
Biebl H, Allgaier M, Tindall BJ, Koblížek M, Lünsdorf H, Pukall R, et al. Dinoroseobacter shibae gen. nov., sp. nov., a new aerobic phototrophic bacterium isolated from dinoflagellates. Int J Syst Evol Microbiol. 2005;55:1089–96.
Lami R, Ras J, Lebaron P, Koblížek M. Distribution of free-living and particle-attached aerobic anoxygenic phototrophic bacteria in marine environments. Aquat Microb Ecol. 2009;55:31–8.
Waidner La, Kirchman DL. Aerobic anoxygenic phototrophic bacteria attached to particles in turbid waters of the Delaware and Chesapeake estuaries. Appl Environ Microbiol. 2007;73:3936–44.
Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, et al. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio. 2014;5:1–9.
Acknowledgements
We thank the people involved in maintaining the BBMO and those taking care of sampling, particularly Clara Cardelús and Vanessa Balagué. We would also like to thank Ramon Massana and Irene Forn for providing microscopy counts. We thank the Marbits bioinformatics platform at ICM-CSIC, particularly Ramiro Logares and Célio Dias for providing the metagenomic data and Anders Kristian Krabberød (University of Oslo) for help with network analyses. Access to the MareNostrum Supercomputer was granted to Ramiro Logares under agreement BCV-2017-3-0001. qPCR analyses was done at the Institute of Evolutionary Biology (Barcelona) thanks to José Luís Maestro. This research was funded by grant REMEI (CTM2015-70340-R) from the Spanish Ministry of Economy, Industry and Competitiveness and Grant 2017SGR/1568 from CIRIT-Generalitat de Catalunya to Consolidated Research Groups.
Author information
Authors and Affiliations
Contributions
IF and JMG conceived the study; IF and OS designed and performed laboratory analyses; AA, PS, OS, and IF analyzed the data; AA and IF wrote the paper and all authors commented and revised it.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Auladell, A., Sánchez, P., Sánchez, O. et al. Long-term seasonal and interannual variability of marine aerobic anoxygenic photoheterotrophic bacteria. ISME J 13, 1975–1987 (2019). https://doi.org/10.1038/s41396-019-0401-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0401-4
This article is cited by
-
Phenology and ecological role of aerobic anoxygenic phototrophs in freshwaters
Microbiome (2024)
-
A Metagenomic and Amplicon Sequencing Combined Approach Reveals the Best Primers to Study Marine Aerobic Anoxygenic Phototrophs
Microbial Ecology (2023)
-
Long-term patterns of an interconnected core marine microbiota
Environmental Microbiome (2022)
-
Influence of short and long term processes on SAR11 communities in open ocean and coastal systems
ISME Communications (2022)
-
Seasonal niche differentiation among closely related marine bacteria
The ISME Journal (2022)
