
Abstract
The methyl-coenzyme M reductase (MCR) complex is a key enzyme in archaeal methane generation and has recently been proposed to also be involved in the oxidation of short-chain hydrocarbons including methane, butane, and potentially propane. The number of archaeal clades encoding the MCR continues to grow, suggesting that this complex was inherited from an ancient ancestor, or has undergone extensive horizontal gene transfer. Expanding the representation of MCR-encoding lineages through metagenomic approaches will help resolve the evolutionary history of this complex. Here, a near-complete Archaeoglobi metagenome-assembled genome (MAG; Ca. Polytropus marinifundus gen. nov. sp. nov.) was recovered from the deep subseafloor along the Juan de Fuca Ridge flank that encodes two divergent McrABG operons similar to those found in Ca. Bathyarchaeota and Ca. Syntrophoarchaeum MAGs. Ca. P. marinifundus is basal to members of the class Archaeoglobi, and encodes the genes for β-oxidation, potentially allowing an alkanotrophic metabolism similar to that proposed for Ca. Syntrophoarchaeum. Ca. P. marinifundus also encodes a respiratory electron transport chain that can potentially utilize nitrate, iron, and sulfur compounds as electron acceptors. Phylogenetic analysis suggests that the Ca. P. marinifundus MCR operons were horizontally transferred, changing our understanding of the evolution and distribution of this complex in the Archaea.
Similar content being viewed by others

Introduction
The methyl-coenzyme M reductase (MCR) complex is a key component of methane metabolism, and until recently had only been found within the Euryarchaeota (Methanococcales, Methanopyrales, Methanobacteriales, Methanomicrobiales, Methanocellales, Methanosarcinales, Methanomassiliicoccales, Methanofastiosales, Methanoflorentales, Methanophagales [1] [ANME-1], and Methanonatronarchaeia). However, recent genome-centric metagenomic studies have led to the discovery of genomes encoding MCR complexes within the phylum Candidatus Bathyarchaeota and the crenarchaeal family Candidatus Methanomethyliaceae (previously Verstraetearchaeota) [2, 3]. Originally, the novel MCR-encoding Ca. Bathyarchaeota and Ca. Methanomethyliaceae were inferred to be capable of hydrogenotrophic and methylotrophic methanogenesis, respectively. Intriguingly, the Ca. Bathyarchaeota also appeared to be capable of producing energy through peptide fermentation and β-oxidation, unusual among MCR-encoding microorganisms. More recently a euryarchaeotal lineage, Candidatus Syntrophoarchaeum, was found to encode Ca. Bathyarchaeota-like MCR homologs and experimentally demonstrated to activate butane for oxidation via modified β-oxidation and Wood-Ljungdahl (WL) pathways [4]. The similarity in the MCR complexes and inferred metabolism of the Ca. Bathyarchaeota and Ca. Syntrophoarchaeum suggest that the Ca. Bathyarchaeota may also oxidize short hydrocarbons. Both organisms are confined to anoxic, hydrocarbon-rich habitats [2,3,4], where abiotically produced short alkanes are abundant and likely to be utilized as carbon and energy sources. The increased number of archaeal lineages encoding the MCR complex and their metabolic flexibility suggests that these microorganisms may have a greater impact on carbon cycling than originally suspected.
The similarity of the MCR complexes encoded by Ca. Bathyarchaeota and Ca. Syntrophoarchaeum is incongruent with their large phylogenetic distance in the genome tree, suggesting that these genes were acquired via horizontal gene transfer (HGT) [1, 5]. Both scenarios indicate that further diversity of divergent MCR-encoding lineages remains to be discovered [5], which has been supported by gene-centric metagenomic analyses of deep-sea and terrestrial hydrothermal environments [6, 7]. Expanding the genomic representation of novel MCR-encoding lineages by targeting these environments using genome-centric metagenomic approaches will help resolve the evolutionary history of the complex and expand the diversity of lineages known to be involved in hydrocarbon cycling.
Archaeoglobi is a class of thermophilic Archaea belonging to the Euryarchaeota that are abundant in subsurface hydrothermal environments, where they likely play a role in carbon and nutrient cycling [8, 9]. The Archaeoglobi are split into three genera: Archaeoglobus, which are all heterotrophic or chemolithotrophic sulfate reducers [10,11,12,13,14,15,16,17,18,19], and Geoglobus and Ferroglobus, which reduce both nitrate and ferric iron [20,21,22]. Pure cultures of Archaeoglobus have been shown to be capable of alkane oxidation [23, 24], and based on their shared metabolic features and close phylogenetic relationship with methanogens [25,26,27,28] are suggested to have an ancestor capable of methanogenesis. However, there are currently no representatives of the Archaeoglobi known to encode the MCR complex, likely a result of poor genomic representation caused by their extreme habitats that are difficult to sample.
Borehole observatories installed on the flank of the Juan de Fuca Ridge in the Pacific Ocean provide pristine fluids from the subseafloor igneous basement aquifer [29]. Previous metagenomic studies on samples collected from these borehole observatories revealed a distinct microbial community, including a number of novel Archaeoglobi [30, 31]. Here, we characterize metagenome assembled genomes (MAGs) from igneous basement fluid samples from the boreholes [31], focusing on a genome encoding two divergent copies of the mcrABG operon. Metabolic reconstruction revealed that the novel Archaeoglobi is potentially capable of hydrocarbon oxidation, amino acid fermentation, and can utilize multiple electron acceptors. Phylogenetic analyses support a horizontal gene transfer hypothesis for the distribution of novel MCR complex among the Archaea, and provides insight into the evolution of both the MCR complex and the Archaeoglobi.
Materials and methods
Metagenome generation, assembly, and binning
Two metagenomes from crustal fluids of the JdFR flank were generated as described previously [31]. One sample (SRR3723048) that yielded a novel Archaeoglobi genome was selected for reassembly using metaSPAdes v3.9.0 [32] with default settings, using raw reads as input. Raw reads from SRR3723048 were mapped to the resulting assembly using BWA-MEM [33] v0.7.12. Binning was conducted using MetaBAT v0.32.4 [34] using the --specific setting.
Identification of MCR encoding genomes
Genomes generated by MetaBAT were searched with GraftM v0.11.1 [6] using an McrA-specific GraftM package (gpkg). The McrA gene tree was curated with NCBI taxonomy, with the Bathyarchaeota and Syntrophoarcaheum clade labeled as “divergent”. An evalue of 1e-50 was used to filter for full-length McrA genes.
MAG quality control
The genome encoding the divergent MCR complexes was analysed using RefineM [35] v0.0.23 to identify contigs with divergent tetranucleotide frequencies and GC content. A single 2748 bp contig was removed due to divergent a GC, tetranucleotide and taxon profile (Supplementary Note 1). The remaining contigs were scaffolded with FinishM v0.0.7 roundup using default parameters (github.com/wwood/finishm). The completeness and contamination of the resulting bin was assessed using CheckM v1.0.8 [36] with default settings.
Genome annotation
Genomes were annotated using EnrichM annotate (github.com/geronimp/enrichM). Briefly, EnrichM calls proteins from contigs using Prodigal v2.6.3 [37], and blasts them against UniRef100 using DIAMOND [38] v0.9.22 to obtain KO annotations. Pfam-A [39] (release 32) and TIGRFAM [40] (release 15.0) Hidden Markov Models (HMMs) were run on the proteins using hmmer 3.1b [41] to obtain Pfam and TIGRFAM annotations, respectively. Further manual curation was completed using NCBI BLAST and CD-Search [42].
Genome tree
Using GenomeTreeTK (https://github.com/dparks1134/GenomeTreeTk) v0.0.41, a genome tree of Archaea from NCBI’s RefSeq database (release 80) was created using a concatenated alignment of 122 archaea-specific single marker copy genes. Genomes <50% complete, and with >10% contamination as determined using CheckM were removed from the analysis. After alignment to HMMs constructed for each of the 122 marker genes, alignments were concatenated and genomes with <50% of the alignment were excluded from the analysis. Maximum likelihood trees were constructed using FastTree v2.1.9, and non-parametric bootstrapping was completed using GenomeTreeTK’s bootstrap function.
16S rRNA gene tree
Sequences classified as Archaeoglobi with a pintail score of 100 and an alignment and sequence quality of ≥80 were extracted from the SILVA database [43] (version 132) and used as reference sequences for a 16S rRNA phylogenetic tree. The partial 16S rRNA sequence from the genome encoding the novel MCR complexes was added to the database, sequences were aligned using ssu-align [44] v0.1, and subsequently converted to fasta format using the convert mode from seqmagick v0.6.1 (fhcrc.github.io/seqmagick). Gapped regions in the alignment were removed with trimAl v1.2 using the --gappyout flag [45]. The Maximum likelihood tree was constructed using FastTreeMP [46] with a generalized time-reversible model and --nt flags, bootstrapped with GenomeTreeTK (github.com/dparks1134/GenomeTreeTk), and visualized in ARB [47] v6.0.6.
Protein phylogenies (McrA, McrB, McrG, and RuBisCo)
McrA, McrB, McrG, and RuBisCo sequences were derived from the genomes used in the genome tree. Proteins from all genomes were called using Prodigal v2.6.3, and searched using hmmer v3.1b with Pfam Hidden Markov models (PF02240.15, MCR_gamma; PF02241.17, MCR_beta; PF02249.16, MCR_alpha; PF02745.14, MCR_alpha_N; PF00016.19, RuBisCO_large; PF02788.15, RuBisCO_large_N) with the –cut_tc flag to minimize false positives. For McrA and RuBisCo, both models needed to hit a sequence for it to be included in the analysis. For each gene, sequences were aligned using MAFFT-GINS-i v7.221 and filtered using trimAl with the --gappyout flag. A maximum likelihood tree was constructed using FastTreeMP with default parameters.
Average amino acid identity
Average amino acid identity was calculated with CompareM v0.0.22 (https://github.com/dparks1134/CompareM) using the aai_wf with default parameters.
Network analysis of MHCs
Proteins from the Archaea in NCBI’s RefSeq database (release 80) were searched with the Cytochrome C Pfam HMM (PF00034). Hits were filtered to have at least one of the characteristic cytochrome C CXXCH domains using a custom script (fastacxxch.count.py, github.com/geronimp/HandyScripts/blob/master/99_random/fastacxxch.count.py). After removing duplicate sequences, the closest match for all resulting proteins were identified using DIAMOND with an evalue cutoff of 1e−20. No limit was placed on the number of hits for each protein. The result was visualized in Cytoscape v3.2.0, removing clusters without a Ca. P. marinifundus homolog.
KO analysis
Proteins from the genomes were searched using DIAMOND blastp against UniRef100 with an evalue cutoff of 1e-05. For each protein, the KO annotations were derived from the top hit. The presence/absence of each KO in each genome was used as input to a principal component analysis (PCA) using the prcomp function in R.
Sliding window GC and tetranucleotide frequencies
A custom script (https://github.com/geronimp/window_sequence) was written to fragment contigs into short sequences, in a sliding window. For each fragment, the percent GC and 4mer frequency was calculated using seqstat in the biosquid package v1.9 g (packages.debian.org/sid/biosquid) and Jellyfish [48] v2.2.6.
Data visualization
Figures were generated in R [49] v3.0.1 using ggplot [50] v1.0.0 and refined using Inkscape v0.91 (inkscape.org/en).
Results and discussion
To investigate the microbial diversity within Juan de Fuca Ridge flank boreholes, a metagenome (45.4 Gbp total raw reads) was generated, assembled, and binned. One of the 98 MAGs (Supplementary Note 1; Supplementary Figure 1) was found to encode two divergent McrAs that were most similar to Ca. Syntrophoarchaeum caldarius (52% AAI) and Ca. Syntrophoarchaeum butanivorans (56% AAI). Based on 228 Euryarchaeota-specific marker genes, this MAG was estimated to be nearly complete (99.84%) with low contamination (1.96%), and a genome size of ~2.13 Mbp. Annotation of the 2305 proteins this MAG encoded revealed all subunits of the MCR complex arranged in operons (mcrABG), including two copies of the mcrC subunit and an ancillary mcrD subunit, all of which have highest sequence similarity to homologs within Ca. Syntrophoarchaeum.
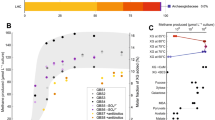
Phylogenetic analysis of the two MCR complexes encoded by the MAG revealed that they branched with high support with divergent MCRs from Ca. Syntrophoarchaeum and Ca. Bathyarchaeota (Fig. 1a; Supplementary Figure 2–6). Notably, the average branch length within the divergent McrA clade was double (1.05 ± 0.24 substitutions per site) that of traditional hydrogenotrophic, acetoclastic and H2-dependent methylotrophic methanogens (0.46 ± 0.10 substitutions per site), suggesting an accelerated rate of evolution following duplication or HGT [51], the latter being more likely due the fewer evolutionary events required. To determine the taxonomy of the MAG, a genome tree was constructed from a concatenated alignment of 122 archaeal single copy marker genes. In both FastTree and IQ-TREE phylogenies, the MAG was positioned basal to other members within the class Archaeoglobi with strong bootstrap support (100%; Fig. 1b; Supplementary Figure 7,8), including other Archaeoglobi previously recovered from the Juan de Fuca Ridge [31]. Phylogenetic analysis of the partial 16S rRNA gene (904 bp) confirmed the position of the MAG within the Archaeoglobi (Supplementary Figure 9). The average AAI between the MAG and other Archaeoglobi recovered from the Juan de Fuca Ridge (54.2% ± 0.7 AAI; Supplementary Figure 10) and relative evolutionary divergence [52, 53], suggest it is the first representative of a novel family within the Archaeoglobi [54]. We propose the name Candidatus ‘Polytropus marinifundus’ gen. nov., sp. nov. for this MAG, as the first representative of a new family within the Archaeoglobi, Candidatus ‘Polytropaceae fam. nov. The incongruencies between the genome tree and MCR phylogenies for Ca. P. marinifundus, Ca. Syntrophoarchaeum, and the Ca. Bathyarchaeota are most parsimoniously explained by HGT of the MCR.

McrA gene phylogeny and genome tree phylogeny. Maximum-likelihood trees of a McrA proteins, and b a concatenated alignment of 122 single copy archaeal marker genes from high quality archaeal RefSeq genomes (release 80). Hydrogenotrophic and acetoclastic methanogens are shaded blue, H2-dependent methylotrophic methanogens are shaded green, known/putative methane oxidizers are shaded yellow, and lineages encoding divergent McrAs are shaded red. Bootstrap support was generated from 100 replicates, and white, gray and black nodes represent ≥50%, ≥75%, and ≥90% support, respectively
Metabolic reconstruction of the Ca. P. marinifundus MAG highlighted the potential for diverse metabolic capabilities, including amino acid fermentation and short chain alkane oxidation using a wide variety of electron acceptors (Fig. 2). Ca. P. marinifundus encodes a complete Wood-Ljungdahl pathway, and consistent with the Ca. Bathyarchaeota and Ca. Methanomethyliaceae, encodes five copies of the H subunit of the methyltetrahydromethanopterin (H4MPT): coenzyme M methyltransferase complex (mtrH), each co-located with predicted di-methylamine and tri-methylamine corrinoid proteins (Supplementary Table 1). This suggests that Ca. P. marinifundus encodes a diverse range of methyltransferases, but is unlikely to conserve energy via methane oxidation or hydrogenotrophic methanogenesis [26]. The WL pathway may be used for oxidation of acetyl-CoA as previously observed in heterotrophic Archaeoglobales isolates [12]. Ca. Syntrophoarchaeum caldarius and Ca. Syntrophoarchaeum butanivorans have been inferred to oxidize alkanes activated by the MCR complex, putatively via the β-oxidation and WL pathways [4]. Ca. P. marinifundus encodes β-oxidation and methyltransferase enzymes that would allow short alkane oxidation (Fig. 2), and the two copies of mcrA share catalytic residues with Ca. Syntrophoarchaeum homologs (Supplementary Figure 11). However, unlike Ca. Syntrophoarchaeum, the presence of a short chain acyl-CoA and butyryl-CoA dehydrogenase (acd and bcd, respectively), and a long-chain acyl-CoA synthetase (fadD) may allow Ca. P. marinifundus to oxidize long chain fatty acids (Fig. 2). The energy for hydrocarbon activation may be produced via either a soluble or membrane bound heterodisulfide reductase (hdrABC, hdrDE, respectively), both of which are encoded by the Ca. P. marinifundus genome. While mvhAG was not encoded by Ca. P. marinifundus, the C-terminal of hdrA is fused to the mvhD subunit as previously observed in Methanosarcina acetivorans [55], suggesting it plays a similar role in disulfide reduction. A further seven putative hdrD subunits co-located with flavin adenine dinucleotide-containing dehydrogenases (glcD) potentially oxidize coenzyme M (CoM-SH) and coenzyme B (CoB-SH) as proposed for the Ca. Methanomethyliaceae and Ca. Bathyarchaeota (Fig. 2) [2, 3]. While common in the Archaeoglobi, the membrane bound HdrDE has not been observed in Ca. Bathyarchaeota and Ca. Syntrophoarchaeum. The functional redundancy of hdrABC/DE has also been observed in Archaeoglobus profundus [56], where they were suggested to play a role in sulfur metabolism. However, without the dissimilatory sulfate reductase (dsrAB) gene, their role in Ca. P. marinifundus remains unclear.

Metabolic reconstruction of the Ca. P. marinifundus genome. Metabolic reconstruction of the Ca. P. marinifundus genome. a Respiratory and fermentative pathways are shaded blue and red, respectively. b Proposed CoM-S-S-CoB disulfide regeneration and membrane energetics of Ca. P. marinifundus are shown. Genes associated with the pathways shown can be found in Supplementary Table 1
Alkane oxidation can be energetically favorable when coupled to an electron accepting process such as sulfate [57], nitrate [58], nitrite [59], and metal oxide [60] reduction, or transfer to a syntrophic partner via direct interspecies electron transfer (DIET) [61]. Similar to the iron metabolizing Geoglobus [22, 62, 63] and Ferroglobus [64] within the Archaeoglobi, Ca. P. marinifundus does not encode dsrAB, but was found to encode 10 multi-haem c-type cytochromes (MHCs) with 4–31 haem binding motifs that may facilitate iron reduction [8, 62, 63, 65] or DIET as proposed in ANME-2 [61]. To compare the multi-haem cytochrome profile of Ca. P. marinifundus with other Bacteria and Archaea, a network analysis was conducted on genomes from NCBI’s RefSeq database. Each MHC encoded by Ca. P. marinifundus share high sequence similarity to homologs encoded by archaeal (e.g., Ferroglobus placidus, Geoglobus acetivorans) and bacterial (e.g., Ferrimonas and Shewanella) iron reducers, Ca. Methanoperedens nitroreducens, and Ca. Syntrophoarchaeum (Fig. 3). Four multi-haem cytochromes similar to Methanoperedens, Ferroglobus and Geoglobus homologs are organized into three contiguous operons (Fig. 3d) encoding membrane-bound, redox-active complexes, including a bc1-like complex with a Rieske iron sulfur protein and cytochrome b that may generate a proton gradient with a Q-cycle, and two complexes associated with the transfer of electrons to the membrane (Fig. 3d). One complex includes an enzyme with two haem binding domains that is conserved among iron metabolizing Geoglobus and Ferroglobus (ORF 212). Intriguingly, Ca. P. marinifundus encodes an operon of three MHCs, two of which are specific to Archaeoglobi, Methanoperedenaceae, and Ca. Syntrophoarchaeum, suggesting these MHCs play a specific role in alkane oxidizers (Fig. 3e). The final gene in this operon is homologous to a large C-type cytochrome also found in Geoglobus acetivorans SBH6T that is a possible genomic determinant of iron reduction [21]. Ca. P. marinifundus also encodes a dissimilatory nitrate reductase (narGHJIK; Fig. 3a), which may allow alkane oxidation coupled to nitrate reduction [58]. A further two operons encode sulfur reductase-like complexes that were previously only found in the hyperthermophile Aquifex aeolicus [66], and shown to allow tetrathionate, polysulfide, and elemental sulfur to be used as terminal electron acceptors (Fig. 3b, c). The potential to use partially oxidized forms of sulfur, nitrogen, and iron as electron acceptors suggests that Ca. P. marinifundus can use different electron sinks depending on environmental conditions [67].

Operons encoding redox active complexes within Ca. P. marinifundus. White arrows represent hypothetical proteins. Networks are of MHCs and represent clusters of related proteins and their organism of origin
Ca. P. marinifundus putatively generates ATP via anaerobic respiration of short chain alkanes. A proton motive force is generated by the F420H2:quinone oxidoreductase, and ATP is generated by an archaeal type ATP synthase (Fig. 2B). However, Ca. P. marinifundus is predicted to be a facultative fermenter, with the ability to ferment via a number of pathways (Fig. 2). While no glucose transporters could be identified, glucose can be fermented via glycolysis, producing acetyl-CoA via pyruvate-ferredoxin oxidoreductase (por) or 2-oxoglutarate ferredoxin oxidoreductase (kor). Ca. P. marinifundus also encodes genes to ferment organic acids such as lactate via lactate dehydrogenase (ldh) and succinate, fumarate and malate via a partial citric acid cycle (Fig. 2). A type II/III ribulose 1,5-bisphosphate carboxylase/oxygenase homologous to Ca. Bathyarchaeota, Ca. Methanomethyliaceae, and Altiarchaeales may play a role in nucleotide catabolism [2, 68] (Archaeal type RuBisCO; Supplementary Figure 8 and 9). The RuBisCo encoded by Ca. P. marinifundus contained all catalytic residues but one (K177 [69]), a feature shared with the subsurface dwelling Altiarchaeales (Supplementary File 16). A number of amino acid transporters and peptidases (tetrahedral aminopeptidase, peptidase family M50, Xaa-Pro dipeptidase, methionine aminopeptidase) indicate that Ca. P. marinifundus can also ferment peptides (Fig. 2). Pathways for the fermentation of glutamate, glutamine, alanine, cysteine, aspartate and asparagine exist, as well as a number of aminotransferases (aspB, hisC, cobD) and two copies of por, which is involved in the fermentation of aromatic amino acids in other hyperthermophiles [70]. Ca. P. marinifundus also encodes a benzoyl-CoA reductase complex (bcrABCD) indicating the potential to degrade aromatic compounds. The acetyl-CoA generated via glycolysis and via fermentation of organic and amino acids may be used via a reversed Wood-Ljungdahl pathway, or substrate level-phosphorylation using acetyl-CoA synthetase (acs) or acetate-CoA ligase (acd). The various fermentative strategies used by Ca. P. marinifundus suggest that it is adapted to a fluctuating availability of organic compounds.
To compare the metabolic capabilities of Ca. P. marinifundus with publically available archaeal genomes from RefSeq and GenBank, a global analyses of KEGG Orthologous (KO) genes was conducted. The KO profile of Ca. P. marinifundus was most similar to other members of the Archaeoglobi, Methanomassiliicoccales, Methanophagales [1] (ANME-1), Ca. Syntrophoarchaeales and Methanonatronarchaeia sp., but distant from the Ca. Bathyarchaeota and Ca. Methanomethyliaceae (Fig. 4a, b). To further examine the shared genomic content of novel MCR encoding lineages, orthologous clusters (OCs) were generated using proteinortho [71]. Within the Archaeoglobi, 134 OCs were unique to Ca. P. marinifundus, mapping to 71 KOs primarily associated with carbon metabolism (Supplementary Table 1). The few clusters that were unique to the Ca. P. marinifundus, Ca. Bathyarchaeota and Ca. Syntrophoarchaeum (seven OCs) were limited to subunits from the MCR complex, a putative methanogenesis marker (TIGR03275), and a gene associated with cobalamin biosynthesis (cob(I)alamin adenosyltransferase; cobA; Supplementary Table 1). A further 25 OCs were specific only to Ca. P. marinifundus and Ca. Syntrophoarchaeum, including four further methanogenesis markers (TIGR03271, TIGR03291, TIGR03268, TIGR03282), a sugar-specific transcriptional regulator, and a class II fumarate hydratase. Many OCs shared between Ca. P. marinifundus and Ca. Syntrophoarchaeum were annotated as “hypothetical protein”, suggesting much of the metabolic similarities of novel hydrocarbon metabolisers have yet to be functionally characterized.

Comparative genomics of the Ca. P. marinifundus genome. a PCA of the presence/absence of KEGG Orthologous (KO) genes in all archaea, b within the Euryarchaeota, with the exception of the Haloarchaea, and c within the Archaeoglobi
To explore the evolutionary history of the MCR complex, the gene phylogeny of the McrA subunit was compared with the archaeal genome phylogeny (Supplementary Figure 14). Traditional euryarchaeal methanogens are largely congruent with the branching order of the genome tree (Supplementary Figure 14), suggesting that the evolutionary history of the McrA largely follows vertical inheritance. However, the H2-dependent methylotroph and Ca. Bathyarchaeota/Ca. Syntrophoarchaeum/Ca. P. marinifundus clades are not monophyletic in the genome tree (Supplementary Figure 14). Ca. Bathyarchaeota and Ca. P. marinifundus McrA cluster with different homologs of the Ca. Syntrophoarchaeum (Fig. 1a), a phylogenetic pattern most parsimoniously explained by HGT (Supplementary Figure 14). The basal phylogenetic position of Ca. P. marinifundus and divergence to other Ca. Syntrophoarcheum MCRcrA would suggest either the donor is unknown or this event did not occur in recent evolutionary history, supported by the lack of a divergent GC or kmer profile surrounding the gene context of the MCRs encoded by Ca. P. marinifundus [72] (Supplementary Figure 15 and 16). Potential HGT to an ancestor of Ca. P. marinifundus contradicts the traditional vertical inheritance pattern of the MCR complex within the Archaea. Syntrophoarchaeum and Ca. P. marinifundus encode multiple copies of the mcrABG operon, suggesting they may be expressed under different environmental conditions as observed for the mcr and mrt of the Methanococci and Methanobacteria [73], or are used for the oxidation of different length alkanes, as is the hypothesized for the Ca. Syntrophoarchaeum [4].
Given the metabolic similarities between the Archaeoglobi and methanogens [26], it has been hypothesized that the last common ancestor (LCA) of the Archaeoglobi encoded the MCR complex, which was subsequently lost following HGT of the dsrAB gene from the Bacteria [74, 75]. While it is unclear whether the LCA of the Archaeoglobi encoded a conventional or divergent MCR complex, four scenarios explain the current distribution of dsrAB and the MCR complex in this lineage: (i) HGT of dsrAB into the LCA of the Archaeoglobi, followed by loss of this metabolism after acquisition of the divergent MCR complex in Ca. P. marinifundus (Supplementary Figure 17A), (ii) HGT of the divergent MCR complex into the LCA of the Archaeoglobi, followed by loss of the complex after the acquisition of dsrAB in the Archaeoglobaceae (Supplementary Figure 17B) (iii) the separate acquisition of the divergent MCR complex and dsrAB by Ca. P. marinifundus and the Archaeoglobaceae (Supplementary Figure 17C), or (iv) differential loss of the dsrAB gene and the divergent MCR complex from an ancestor to the Archaeoglobi that encoded both operons (Supplementary Figure 17D). Greater genomic representation and comparative genomics of the Archaeoglobi will clarify the evolutionary story, particularly if lineages are found within the Archaeoglobi that encode the MCR complex. The evolutionary history of the MCR is highly complex, with evidence for both vertical inheritance and HGT (Supplementary Figure 17). It is likely that archaeal lineages encoding the divergent MCR complex will continue to be discovered, and with the genomic representation they add to public databases, their evolutionary history and metabolic role in the hydrothermal subsurface biosphere will become increasingly clear.
Description of candidatus “Polytropus marinifundus”
Candidatus Polytropus (Po.ly.tro’pus. N.L. masc. n. Polytropus (from Gr. masc. adj. polytropus) one that turns many ways, versatile). Candidatus Polytropus marinifundi (ma.ri.ni.fun’di. L. adj. marinus of the sea, marine; L. masc. n. fundus the bottom; N.L. gen. n. marinifundi of/from the sea floor). “Polytropaceae” (Po.ly.tro’pa.ce.a.e N.L. n. “Polytropaceae” -entis, type genus of the family; suff. -aceae, ending to denote a family; N.L. masc. pl. n. Polytropaceae, the family of the genus “Polytropus”).
Code availability
All unpublished software used in this publication are available on github; sequence_windower (https://github.com/geronimp/window_sequence); EnrichM (github.com/geronimp/enrichM); GenomeTreeTK (github.com/dparks1134/GenomeTreeTk); CompareM v0.0.22 (https://github.com/dparks1134/CompareM); fastacxxch.count.py, (github.com/geronimp/HandyScripts/blob/master/99_random/fastacxxch.count.py); FinishM (v0.0.7 github.com/wwood/finishm). GTDB-Tk (github.com/Ecogenomics/GTDBTk).
Data availability
References
Adam PS, Borrel G, Brochier-Armanet C, Gribaldo S. The growing tree of Archaea: new perspectives on their diversity, evolution and ecology. ISME J. 2017;11:2407–25.
Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science. 2015;350:434–8.
Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:16170.
Laso-Pérez R, Wegener G, Knittel K, Widdel F, Harding KJ, Krukenberg V, et al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature. 2016;539:396–401.
Evans P, Boyd J, Leu A, Donovan WBP, Tyson G. Phylogenetic diversity and metabolic capacity of mcr and mcr-like containing archaeal lineages. Submitted.
Boyd JA, Woodcroft BJ, Tyson GW. GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res. 2018;46:e59–e59.
McKay LJ, Hatzenpichler R, Inskeep WP, Fields MW. Occurrence and expression of novel methyl-coenzyme M reductase gene (mcrA) variants in hot spring sediments. Sci Rep. 2017;7:7252.
Brileya K, Reysenbach A-L. The class Archaeoglobi. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes. Berlin, Heidelberg: Springer; 2014. p. 15–23.
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ. Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev. 2011;75:361–422.
Birkeland N-K, Schönheit P, Poghosyan L, Fiebig A, Klenk H-P. Complete genome sequence analysis of Archaeoglobus fulgidus strain 7324 (DSM 8774), a hyperthermophilic archaeal sulfate reducer from a North Sea oil field. Stand Genom Sci. 2017;12:79.
Stetter KO. Archaeoglobus fulgidus gen. nov., sp. nov.: a new taxon of extremely thermophilic archaebacteria. Appl Environ Microbiol. 1988;10:172–3.
Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–70.
Burggraf S, Jannasch HW, Nicolaus B, Stetter KO. Archaeoglobus profundus sp. nov., Represents a New Species within the Sulfate-reducing Archaebacteria. Syst Appl Microbiol. 1990;13:24–28.
von Jan M, Lapidus A, Del Rio TG, Copeland A, Tice H, Cheng J-F, et al. Complete genome sequence of Archaeoglobus profundus type strain (AV18). Stand Genom Sci. 2010;2:327–46.
Huber H, Jannasch H, Rachel R, Fuchs T, Stetter KO. Archaeoglobus veneficus sp. nov., a Novel Facultative Chemolithoautotrophic Hyperthermophilic Sulfite Reducer, Isolated from Abyssal Black Smokers. Syst Appl Microbiol. 1997;20:374–80.
Mori K, Maruyama A, Urabe T, Suzuki K-I, Hanada S. Archaeoglobus infectus sp. nov., a novel thermophilic, chemolithoheterotrophic archaeon isolated from a deep-sea rock collected at Suiyo Seamount, Izu-Bonin Arc, western Pacific Ocean. Int J Syst Evolut Microbiol. 2008;58:810–6.
Steinsbu BO, Thorseth IH, Nakagawa S, Inagaki F, Lever MA, Engelen B, et al. Archaeoglobus sulfaticallidus sp. nov., a thermophilic and facultatively lithoautotrophic sulfate-reducer isolated from black rust exposed to hot ridge flank crustal fluids. Int J Syst Evolut Microbiol. 2010;60:2745–52.
Stokke R, Hocking WP, Steinsbu BO, Steen IH. Complete genome sequence of the thermophilic and facultatively chemolithoautotrophic sulfate reducer Archaeoglobus sulfaticallidus strain PM70-1T. Genome Announc 2013; 1.
Stetter KO, Huber R, Blöchl E, Kurr M, Eden RD, Fielder M, et al. Hyperthermophilic archaea are thriving in deep North Sea and Alaskan oil reservoirs. Nature. 1993;365:743.
Anderson I, Risso C, Holmes D, Lucas S, Copeland A, Lapidus A, et al. Complete genome sequence of Ferroglobus placidus AEDII12DO. Stand Genom Sci. 2011;5:50–60.
Mardanov A, Slododkina G, Slobodkin A, Beletsky A, Gavrilov S, Kublanov I, et al. The Geoglobus acetivorans genome: Fe(III) reduction, acetate utilization, autotrophic growth, and degradation of aromatic compounds in a hyperthermophilic archaeon. Appl Environ Microbiol. 2015;81:1003–12.
Manzella MP, Holmes D, Rocheleau J, Chung A, Reguera G, Kashefi K. The complete genome sequence and emendation of the hyperthermophilic, obligate iron-reducing archaeon ‘Geoglobus ahangari’ strain 234 T. Stand Genom Sci 2015; 10.
Khelifi N, Amin Ali O, Roche P, Grossi V, Brochier-Armanet C, Valette O, et al. Anaerobic oxidation of long-chain n-alkanes by the hyperthermophilic sulfate-reducing archaeon, Archaeoglobus fulgidus. ISME J. 2014;8:2153–66.
Khelifi N, Grossi V, Hamdi M, Dolla A, Tholozan J-L, Ollivier B, et al. Anaerobic oxidation of fatty acids and alkenes by the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus. Appl Environ Microbiol. 2010;76:3057–60.
Ney B, Ahmed FH, Carere CR, Biswas A, Warden AC, Morales SE, et al. The methanogenic redox cofactor F420 is widely synthesized by aerobic soil bacteria. ISME J. 2017;11:125–37.
Borrel G, Adam PS, Gribaldo S. Methanogenesis and the Wood–Ljungdahl pathway: an ancient, versatile, and fragile association. Genome Biol Evol. 2016;8:1706–1711.
Vornolt J, Kunow J, Stetter KO, Thauer RK. Enzymes and coenzymes of the carbon monoxide dehydrogenase pathway for autotrophic CO2 fixation in Archaeoglobus lithotrophicus and the lack of carbon monoxide dehydrogenase in the heterotrophic A. profundus. Arch Microbiol. 1995;163:112–8.
Bapteste E, Brochier C, Boucher Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea. 2005;1:353–63.
Lin H-T, Cowen JP, Olson EJ, Amend JP, Lilley MD. Inorganic chemistry, gas compositions and dissolved organic carbon in fluids from sedimented young basaltic crust on the Juan de Fuca Ridge flanks. Geochim Et Cosmochim Acta. 2012;85:213–27.
Jungbluth SP, Bowers RM, Lin H-T, Cowen JP, Rappé MS. Novel microbial assemblages inhabiting crustal fluids within mid-ocean ridge flank subsurface basalt. ISME J. 2016;10:2033–47.
Jungbluth SP, Amend JP, Rappé MS. Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids. Sci Data. 2017;4:170037.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165
Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–42.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2014;12:59–60.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2015;44:D279–85.
Selengut JD, Haft DH, Davidsen T, Ganapathy A, Gwinn-Giglio M, Nelson WC, et al. TIGRFAMs and Genome Properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007;35:D260–4.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–31.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Nawrocki EP, Eddy SR. ssu-align: a tool for structural alignment of SSU rRNA sequences. 2010. http://selab.janelia.org/software.html.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Price M, Dehal P, Arkin A. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70.
Team RC, Others. R: a language and environment for statistical computing 2013.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2009.
Petitjean C, Makarova K, Wolf Y, Koonin E. Extreme deviations from expected evolutionary rates in archaeal protein families. Genome Biol Evol. 2017;9:2791–2811.
Chaumeil P-A, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. in preparation 2018.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018:36;996–1004
Konstantinidis KT, Tiedje JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258–6264.
Yan Z, Wang M, Ferry JG. A ferredoxin- and F420H2-dependent, electron-bifurcating, heterodisulfide reductase with homologs in the domains bacteria and archaea. MBio 2017;8.
Mander GJ, Pierik AJ, Huber H, Hedderich R. Two distinct heterodisulfide reductase-like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. FEBS J. 2004;271:1106–16.
Milucka J, Ferdelman TG, Polerecky L, Franzke D, Wegener G, Schmid M, et al. Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature. 2012;491:541–6.
Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature. 2013;500:567–70.
Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM, et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature. 2010;464:543–8.
Ettwig KF, Zhu B, Speth D, Keltjens JT, Jetten MSM, Kartal B. Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc Natl Acad Sci USA. 2016;113:12792–6.
McGlynn SE, Chadwick GL, Kempes CP, Orphan VJ. Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature. 2015;526:531–5.
Slobodkina G, Kolganova T, Querellou J, Bonch-Osmolovskaya E, Slobodkin A. Geoglobus acetivorans sp. nov., an iron (III)-reducing archaeon from a deep-sea hydrothermal vent. Int J Syst Evolut Microbiol. 2009;59:2880–3.
Kashefi K, Tor JM, Holmes DE, Gaw Van Praagh CV, Reysenbach A-L, Lovley DR. Geoglobus ahangari gen. nov., sp. nov., a novel hyperthermophilic archaeon capable of oxidizing organic acids and growing autotrophically on hydrogen with Fe(III) serving as the sole electron acceptor. Int J Syst Evolut Microbiol. 2002;52:719–28.
Hafenbradl D, Keller M, Dirmeier R, Rachel R, Roßnagel P, Burggraf S, et al. Ferroglobus placidus gen. nov., sp. nov., a novel hyperthermophilic archaeum that oxidizes Fe2+ at neutral pH under anoxic conditions. Arch Microbiol. 1996;166:308–14.
Tor JM, Lovley DR. Anaerobic degradation of aromatic compounds coupled to Fe(III) reduction by Ferroglobus placidus. Environ Microbiol. 2001;3:281–7.
Guiral M, Tron P, Aubert C, Gloter A, Iobbi-Nivol C, Giudici-Orticoni M-T. A membrane-bound multienzyme, hydrogen-oxidizing, and sulfur-reducing complex from the hyperthermophilic bacterium Aquifex aeolicus. J Biol Chem. 2005;280:42004–15.
Wankel SD, Adams MM, Johnston DT, Hansel CM, Joye SB, Girguis PR. Anaerobic methane oxidation in metalliferous hydrothermal sediments: influence on carbon flux and decoupling from sulfate reduction. Environ Microbiol. 2012;14:2726–40.
Wrighton KC, Castelle CJ, Wilkins MJ, Hug LA, Sharon I, Thomas BC, et al. Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J. 2014;8:1452–63.
Cleland WW, Andrews TJ, Gutteridge S, Hartman FC, Lorimer GH. Mechanism of rubisco: the carbamate as general base. Chem Rev. 1998;98:549–62.
Mai X, Adams MW. Indolepyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. A new enzyme involved in peptide fermentation. J Biol Chem. 1994;269:16726–32.
Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011;12:124.
Ravenhall M, Škunca N, Lassalle F, Dessimoz C. Inferring horizontal gene transfer. PLoS Comput Biol. 2015;11:e1004095.
Reeve JN, Nölling J, Morgan RM, Smith DR. Methanogenesis: genes, genomes, and who’s on first? J Bacteriol. 1997;179:5975–86.
Klein M, Friedrich M, Roger AJ, Hugenholtz P, Fishbain S, Abicht H, et al. Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. J Bacteriol. 2001;183:6028–35.
Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2014;9:1152.
Acknowledgements
We thank the captain and crew, A. Fisher, K. Becker, C. G. Wheat, and other members of the science teams on board R/V Atlantis cruise AT18-07. We also thank the pilots and crew of remote-operated vehicle Jason II. This research was supported by two grants from the National Science Foundation: Microbial Observatories (MCB06-04014 to MSR), and the Science and Technology Center for Dark Energy Biosphere Investigations (C-DEBI; OCE-0939564 to JPA). This study used samples and data provided by the Integrated Ocean Drilling Program. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research program under Award Number. Genomic Science Program of the United States Department of Energy Office of Biological and Environmental Research (DE-SC0016469, DE-SC0010580, DE-SC0016440); Australian Research Council (ARC) Future Fellowship (FT170100070 to G.W.T.); ARC Postgraduate Award (to JAB); ARC Discovery Early Career Researcher Award (DECRA; DE-160100248 to BJW); ARC DECRA (DE-170100428 to PNE).
Author contributions
JAB and SPJ contributed equally to this work. MSR, JPA, and GWT designed the overall study and procured funding. JAB, SPJ, MSR, and GWT designed and carried out experiments and analyses around specific microbial hypotheses. JAB, SPJ, and GWT wrote the manuscript. All authors edited, reviewed and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boyd, J.A., Jungbluth, S.P., Leu, A.O. et al. Divergent methyl-coenzyme M reductase genes in a deep-subseafloor Archaeoglobi. ISME J 13, 1269–1279 (2019). https://doi.org/10.1038/s41396-018-0343-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0343-2
This article is cited by
-
A compendium of viruses from methanogenic archaea reveals their diversity and adaptations to the gut environment
Nature Microbiology (2023)
-
Mcr-dependent methanogenesis in Archaeoglobaceae enriched from a terrestrial hot spring
The ISME Journal (2023)
-
Diversity and function of methyl-coenzyme M reductase-encoding archaea in Yellowstone hot springs revealed by metagenomics and mesocosm experiments
ISME Communications (2023)
-
Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits
Microbiome (2022)
-
Conserved and lineage-specific hypothetical proteins may have played a central role in the rise and diversification of major archaeal groups
BMC Biology (2022)
