
Abstract
How ecosystem functioning changes with microbial communities remains an open question in natural ecosystems. Both present-day environmental conditions and historical events, such as past differences in dispersal, can have a greater influence over ecosystem function than the diversity or abundance of both taxa and genes. Here, we estimated how individual and interactive effects of microbial community structure defined by diversity and abundance, present-day environmental conditions, and an indicator of historical legacies influenced ecosystem functioning in lake sediments. We studied sediments because they have strong gradients in all three of these ecosystem properties and deliver important functions worldwide. By characterizing bacterial community composition and functional traits at eight sites fed by discrete and contrasting catchments, we found that taxonomic diversity and the normalized abundance of oxidase-encoding genes explained as much variation in CO2 production as present-day gradients of pH and organic matter quantity and quality. Functional gene diversity was not linked to CO2 production rates. Surprisingly, the effects of taxonomic diversity and normalized oxidase abundance in the model predicting CO2 production were attributable to site-level differences in bacterial communities unrelated to the present-day environment, suggesting that colonization history rather than habitat-based filtering indirectly influenced ecosystem functioning. Our findings add to limited evidence that biodiversity and gene abundance explain patterns of microbiome functioning in nature. Yet we highlight among the first time how these relationships depend directly on present-day environmental conditions and indirectly on historical legacies, and so need to be contextualized with these other ecosystem properties.
Similar content being viewed by others


Introduction
Biodiversity-ecosystem functioning (B-EF) relationships are generally expected to be positive because more unique functions are captured as species numbers increase. While this prediction often holds true for macroorganisms [1], it is still contested for microorganisms. Some have found support for positive B-EF relationships in microbial communities [2,3,4,5], as expected if microbes perform a diversity of functions (e.g. litter decomposition, temperature regulation, and nutrient cycling), which increase with numbers of taxa. However, others have found negative and no B-EF relationships [6, 7]. One explanation for this conflicting evidence is that taxonomic diversity may have relatively little influence on functioning in microbial ecosystems that are saturated by thousands of species that overlap in their traits [8]. Empirical evidence to support B-EF theory in microbes has also come from communities where species richness rarely exceeds 100 taxa [9], which is much less than the thousands of taxa found in natural communities, e.g. up to 9,000 prokaryotic taxa in 1 cm3 of soil [10], but see Delgado-Baquerizo et al. [2] and Laforest-Lapointe et al. [3].
Functional information may help resolve the lack of widespread evidence for a positive B-EF relationship in microbial communities by overcoming the limitations of using solely taxonomic information. In particular, taxonomic information may have little value where it does not map onto function, and this may be relatively common in microorganisms because taxa are delineated from classifying closely-related genetic sequences rather than morphological or physiological traits [11]. Horizontal gene transfer can also complicate the use of taxonomic information in microbes because traits might not be vertically transmitted as expected based on phylogeny [12]. Given these concerns, Graham et al. [13] recently found that combining both taxonomic and functional measures of diversity strengthened predictions of ecosystem functioning across 82 microbial systems compared to models including only microbial biomass. While 56% of the variation in functioning was explained by environmental variables, such as pH and temperature, incorporating information about microbial taxonomic diversity explained, on average, 8% of additional variance [13]. Total functional gene abundance equally improved predictions of microbial respiration [13]. These findings underscore the importance of considering abundance and diversity metrics of both function and taxonomy when predicting B-EF relationships.
Past events can also leave a legacy on present-day microbial communities and influence species composition and subsequent ecosystem function as much as the contemporary environment [14, 15]. These events can include past differences in dispersal and environmental conditions that have differentially sorted species composition. Thus, microbial communities can have less gene flow and greater genetic divergence as they become increasingly distant in space irrespective of environmental similarity [16]. For example, recent evidence has shown that historical legacies can result in different microbial communities in similar environmental conditions, partly because microbes are not ubiquitously distributed [17]. Consequently, B-EF relationships have been found to depend on the temporal order of community assembly [18, 19] and to vary over small spatial distances (i.e. 20 m [20]). Despite their potential importance for ecosystem functioning, the effect of historical legacies relative to other ecosystem properties remains unclear.
Here we estimated how three ecosystem properties—microbial diversity and abundance (hereafter “community structure” as defined by Bier et al. [21]), present-day environmental conditions, and historical legacies—influenced ecosystem functioning in lake sediments. Our approach advanced the search for bivariate B-EF relationships by assessing the importance of diversity in the context of other ecosystem properties. Lake sediments are well suited to test the importance of different ecosystem properties because they: (i) share a common microbial species pool from which communities can be differentially assembled according to past events [22], (ii) span large environmental gradients across relatively small distances (i.e. meters), and (iii) carry out functions with widespread importance, such as for carbon (C) cycling [23]. Using next-generation sequencing, we first tested for evidence that historical legacies and environmental conditions influenced microbial community composition in lake sediments. We then tested how much in-situ organic matter mineralization rates—measured as CO2 production under ideal conditions—varied with two diversity and two abundance metrics relative to the influence of historical legacies and the present-day environment. CO2 production is a direct measure of ecosystem function because it is indicative of both food web production and whole-lake C cycling [23]. We predicted that higher levels of diversity, particularly a greater diversity of functional genes, as well as a greater abundance of genes involved in organic matter (OM) decomposition, would increase ecosystem functioning. We also predicted that microbial community structure would be primarily influenced by variation in environmental conditions rather than colonization history, as expected if dispersal was unlimited (‘everything is everywhere, but the environment selects’ hypothesis [24]). Overall, our results add to limited evidence that the diversity and abundance of both taxa and functional genes explain microbiome functioning in nature, and highlight for among the first time how these effects directly depend on local environmental conditions and indirectly on historical legacies.
Methods
Study site
We sampled eight littoral sites each located immediately beneath a discrete catchment drained by a single stream in Daisy Lake, Ontario, Canada (46°270 N, 80°520 W; lake area: 36 ha; maximum depth: 14 m, Fig. S1). The sites spanned large gradients in the quantity and quality of terrestrial OM inputs from the surrounding vegetation [25]. These gradients arose from variation in recovery from historical acid and metal contamination, which increased with proximity to a nickel smelter that was closed in 1972 and located 3.5 km northeast of the lake. Following closure of the smelter, lake water chemistry returned to levels characteristic of the broader region: mean ± standard error pH across sites of 6.86 ± 0.02 [26]. However, the surrounding vegetation, primarily comprised of paper birch (Betula papyrifera) and trembling aspen (Populus tremuloides), has been much slower to recover, resulting in a large gradient across sites in terrestrial OM inputs.
Past environmental histories in each site can also result in unique species composition [16]. Thus, we considered that the identity of each site would reflect the legacy of historical events with its own unique soils, geomorphology, and dispersal events and these effects would differ from those of the present-day environment that we could characterize in each site from directly measurable biotic and abiotic variables (described below).
Ecosystem functioning and environmental characterization
We measured ecosystem functioning in 10–15 sediment samples that were collected from each site in waters that ranged from 0.5–1.0 m deep (total n = 97). For each sample, we extruded approximately 35 mL of sediment to a depth of 7.5 cm into 50 mL poly-propylene centrifuge tubes (2.6 cm diameter) using a modified piston corer. Samples were covered with approximately 10 mL lake water from the associated site and transported to the lab on ice.
Ecosystem functioning was measured as total CO2 production per m2 after 20 h. We incubated sediment samples in the dark at 20.5 °C and collected headspace gas at the start and end of the incubation by extracting a 2 mL gas sample with an airtight syringe. Gas samples were analyzed on an infrared CO2 analyzer (Q-S151, Qubit Systems, Kingston, ON, Canada) with a N2 carrier and converted to mass produced per m2 using the ideal gas law and a tube surface area of 5.31 cm2. During sediment sampling, ambient air samples were collected in airtight syringes from each site for subtraction of pre-incubation CO2 from headspace mass. Sediments were then freeze-dried and stored at −20 °C to stabilize the microbial communities and ensure they were representative of CO2 measurements [27].
We initially characterized sediments with 20 environmental variables. To minimize collinearity, we reduced these to four statistically independent predictors that best characterized environmental differences: pH, OM quantity, %terrestrial C, and C:N ratio (see Text S1 and Table S3 for details). Although this reduction in variables somewhat biased our representation of the local environment, we were primarily interested in estimating the importance of the strongest environmental gradients relative to other ecosystem properties. First, we measured pH in porewater of each sediment sample at the time of collection with a handheld pH meter (HI9126/HI1230, Hanna Instruments, Woonsocket, RI, USA). Second, 0.5 g of each sediment core was analyzed after incubation for percent OM content as weight loss on ignition (LOI) for 12 h in a 400 °C muffle furnace, confirming the absence of any visible char [28]. We then used two complementary measurements of OM quality that were averaged at the site-level from a different, unpublished study (Text S1). The first was the percent of terrestrial C in sediment, which characterized the origin of OM. Percent terrestrial C was estimated with a three isotope (δ13C, δ15N, δ34S) mixing model (full details in Text S1). The second measurement was the C:N ratio of sediment, which characterized OM composition. Lower C:N ratios were considered a higher quality to microbial decomposers because they provide more N per mass of sample and are typically associated with more labile materials [29].
Microbial communities
We constructed amplicon sequencing libraries for each sediment sample to characterize microbial community composition. Following careful homogenization, DNA was extracted from 0.25 g of each of the samples that had CO2 measurements using a PowerSoil PowerLyser DNA Isolation Kit (MoBio Laboratories Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions. Insufficient fungal sequences were recovered, so we focused on bacteria as representatives of the microbial community. We targeted the V3–V4 region of the 16S rRNA gene using the bacteria-specific 341F-805R primer pair with a two-stage PCR designed for paired-end sequencing. Amplicons were sequenced on an Illumina MiSeq platform (Illumina, San Diego, CA, USA), quality-filtered to remove low-quality bases and putative chimeras, and clustered into operational taxonomic units (OTUs) at 97% sequence similarity using mothur version 1.39.5 [30]. Any read sequenced fewer than six times was removed from subsequent analyses to minimize the influence of spurious reads [31]. Counts of individual OTUs were then scaled by the total number of reads in each sample to account for sequencing biases using the R package DESeq2 [32]. This measure of “normalized abundance” allows samples with varying read counts to be compared [33]. Such normalization is widely applied for high-throughput count data [34, 35], and all downstream analyses were performed on the DESeq-transformed data to control for these differences in read numbers [33, 36].
We also constructed shotgun sequencing libraries for 22 of the 97 samples in order to characterize functional genes present in each site (n = 2−3 samples per site). Sequencing libraries were prepared with 1 ng of genomic DNA per sample using the Nextera XT DNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions and sequenced on an Illumina NextSeq platform. Raw sequences were processed following the EMBL-EBI pipeline version 3.0 [37] and summarized using Gene Ontology (GO) terms. Sequences were deposited in EBI under project number ERP01606 (full details in Text S1).
Using the microbial sequencing data, we calculated two diversity and two abundance metrics. First, we calculated normalized bacterial abundance by summing the total number of OTUs per sample. The number of OTUs were DESeq-transformed counts rather than relative abundances, so their sum was not equal to 1 and represented differences in normalized abundances between samples [33, 36]. Second, we calculated taxonomic diversity as Shannon’s H’ for each sample at both the OTU- and the family-level. As both measures were strongly correlated (ρ = 0.92, p < 0.0001), we used the family-level Shannon’s H’ in our analyses to limit the number of unclassified taxa whilst retaining as much information as possible about taxonomic diversity (42% of reads were classified to this rank). Shannon’s H’ is considered a robust estimator of diversity for microbial communities as it accounts for both abundance and evenness [38], and has been widely used, thereby allowing for comparison with other studies [2, 39]. Similarly, we calculated functional diversity on the data obtained from shotgun sequencing using Shannon’s H’. Finally, we defined four subsets of functional genes that were involved in different aspects of terrestrial OM decomposition and consequently CO2 production (after Refs [40,41,42,43]). We summed the DESeq-transformed abundance of these genes, which were broadly associated with: (1) hydrolase enzymes that break down cellulose, hemicellulose and xylan, (2) oxidases that break down a range of compounds and/or are involved in assimilatory and dissimilatory P and N transformations, (3) intracellular-level carbohydrate metabolism, and (4) aromatic compound catabolism (see Table S4 for full list of GO categories).
Is there evidence of legacy and environmental effects on community composition?
We assessed similarity between microbial communities as geographic and environmental distances increased to test if they were associated with historical legacies and present-day conditions. We calculated the Morisita–Horn similarity index for all pair-wise combinations of normalized microbial abundance, Euclidean distances for geographic distance, and Mahalanobis distances for environmental similarity (i.e. standardized Euclidean distances accommodating for different measurement units and covariance structure among pH, OM quantity, %terrestrial C, C:N ratio). These indexes were calculated using the 97 samples rather than the 22 sample functional gene subset, and significance of the associations between distance matrices was assessed using partial Mantel tests with Pearson’s correlation coefficient and 999 permutations constrained within sites. Partial Mantel tests are commonly used to disentangle the effects of present-day environmental conditions on community composition from those of historical legacies, especially when continuous habitat variables and geographic distances are available [16]. Here they allowed us to assess the relationship between microbial community and geographic distance while controlling for environmental similarity and vice versa.
We performed a canonical correspondence analysis (CCA) to explore further how bacterial community composition varied in relation to historical legacies and present-day environmental conditions, and in particular, to identify the environmental variables that most explained differences among sites. The CCA was constrained by site, pH, OM quantity, %terrestrial C, and C:N ratio. To test whether community composition varied more with present-day environmental conditions or historical legacies, which we interpreted as being associated with the variation among sites that was unexplained by pH, OM quantity, %terrestrial C, and C:N ratio, we ran a permutational multivariate analysis of variance (PERMANOVA) using the ‘adonis2’ function in the ‘vegan’ R package. Significance of marginal effects was assessed with 999 permutations of the community data constrained within sites, with the environmental variables and site identity as predictors. To achieve normality, C:N ratio was log-transformed while OM quantity (%LOI) and %terrestrial C were logit-transformed.
What is the relative importance of different properties for ecosystem functioning?
We developed a conceptual model to test our hypotheses about the relative importance of community structure, present-day environmental conditions, and historical legacies for ecosystem functioning. This model considered four different pathways by which the different properties could influence ecosystem functioning. Firstly, the model let ecosystem functioning vary with the direct effects of the four environment variables and four measures of community structure (Fig. 1). By considering genomic data that could be linked to specific functions, the model also incorporated a trait-based approach that offered more insight into ecosystem functioning than solely based on community-level diversity measures [9, 44]. With the trait-based approach in mind, the normalized abundances of the functional gene sets and measures of OM quality could also interact (dashed arrows in Fig. 1), as expected because the efficacy of traits involved in decomposition generally depend on OM quality rather than quantity [39, 45]. Thirdly, the model also accounted for the indirect effects that historical legacies, associated with each of the eight sites, and present-day environmental conditions could have an ecosystem functioning by influencing the four measures of community structure. Finally, we estimated the effects of these four measures on one another to test how the relationship between taxonomy and function indirectly affected ecosystem functioning (small arrows in Fig. 1).

Trait-based conceptual model of pathways by which different ecosystem properties (community structure, present-day environment, and historical legacies) affect ecosystem functioning. Measured variables associated with different ecosystem properties are in solid white boxes, with the normalized functional gene abundance subsets in dashed white boxes. Solid arrows represent potential pathways between ecosystem properties and dashed arrows represent potential interactions between ecosystem properties
We used path analysis to estimate the strength and direction of presumed direct and indirect causal linkages that described our conceptual model (Fig. 1). In this analysis, we only used the 22 samples for which we had both taxonomic and functional information. The model formula for log-transformed CO2 production as a response in R pseudo-code was:
where environment included pH, OM quantity, %terrestrial C, and C:N ratio; community structure was each of normalized bacterial abundance, taxonomic diversity, functional diversity, and normalized functional gene abundance; the interaction was between normalized functional gene abundance and each measure of OM quality (%terrestrial C and C:N ratio); and historical legacies were represented by a site-level random effect. We acknowledge that this random effect can also incorporate other present-day environmental variables that systematically varied across sites, but these are unlikely to be more important or uncorrelated with the 20 variables that we actually measured (Table S1). In total, we fitted the model separately with each of the four normalized functional gene abundance subsets. As gene counts were DESeq-transformed, there was no dependency of one gene on another across samples, and genes could therefore be summed into independent subsets. All of the models also estimated residual (i.e. random) error for each of the focal responses.
We also fitted four separate models to estimate each measure of community structure as a response of the environment, historical legacies, and the other measures of community structure (small arrows in Fig. 1). For functional gene abundance, we only modeled the subset(s) of genes identified as significant in the model with CO2 production as a response. Allowing the measures of community structure to be both dependent variables and independent predictors of ecosystem functioning is consistent with treating them as endogenous variables in a path analysis that teases apart direct and indirect correlations [46, 47].
Each model described above was fitted with linear mixed models using Bayesian inference by calling the ‘blme’ function in the R package ‘blmer’ [48]. All measured variables were standardized to a common scale with a mean of 0 and an SD of 1, so that we could compare the relative importance of different linkages. To infer effects, we calculated posterior means and 95% confidence intervals (CI) for each parameter by bootstrapping model parameter estimates 800 times with the ‘boot’ package in R. Effects were considered significant when 95% CI around estimated effect sizes excluded zero. To assess the overall goodness-of-fit of models, marginal R2 values were calculated.
As all our four models with different functional gene subsets were within 2 small sample Akaike information criterion (AICc) units of each other ([49], Table S5), we averaged parameter estimates across the model set. The posterior means of each model were multiplied by their respective AICc weight and summed to determine the average parameter estimates and 95% CI. For functional gene abundance and its interaction with OM quality, no averaging was performed across the model set. We instead reported the effects associated with each of the four unique subsets of functional genes.
Results
Bacterial community composition
Overall, we found considerable bacterial biodiversity. We obtained about 25,000 OTUs that corresponded with about 540 families in each of the 97 and 22 sample datasets (Table S6). The most common OTUs were in the Koribacteraceae, Hyphomicrobiaceae and Solibacteraceae, each accounting for 0.5% of all normalized abundances per sample. The 22 samples with functional data that we considered in our path analyses subsequently also showed relatively high taxonomic and functional diversity, with H’ values exceeding 5 (Table S7). The highest number of genes involved in CO2 production (i.e. normalized functional gene abundance) came from oxidase genes, followed by hydrolases, carbohydrate catabolism, and aromatic compound catabolism (Table S7).
Biogeographical patterning of the study sites
The eight sites each showed evidence of having unique bacterial communities that reflected both past events and present-day environmental conditions. We specifically found that communities became less similar as they were increasingly distant in space when controlling for the effects of the environment (partial Mantel test: r = −0.15, p = 0.003, Fig. 2a), suggesting that isolation by distance may maintain differences in biotic assemblages that arose from past events such as differential establishment and persistence of species through time. These differences could not be attributed to dispersal-limitation as most taxa were ‘everywhere’. 369 of the 551 bacterial families occurred at all eight sites, with another 66 present at seven sites (Fig. S2), evidence that abundances rather than presence varied across space. We also found that community similarity increased with environmental similarity when controlling for geographical distances (partial Mantel tests: r = 0.43, p = 0.005, Fig. 2b), suggesting that different present-day environments also influenced microbial communities within our study.

Bacterial communities differ across geographic and environmental space. Community similarity (Horn–Morisita index) at the family level between all pair-wise combinations of 97 sediment samples (a) decreased with geographic distance and (b) increased with environmental similarity (Mahalanobis distance) according to partial Mantel tests
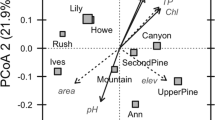
We found further evidence of past and present-day influences over bacterial communities when clustering compositional differences among sites (Table S8a). Distinct communities were observed across sites (F = 3.92, p = 0.01), even after constraining composition by present-day environmental variables (Fig. 3). Both measures of OM quality significantly differentiated communities (F = 5.64, p = 0.001 for %terrestrial C and F = 4.61, p = 0.001 for C:N ratio), with no effect of either pH or OM quantity (Table S8b). We reached a similar conclusion when using a partial redundancy analysis to compare the effects of site identity and environmental variables on community composition (Table S8c).

Bacterial communities differed across sites and environments. The CCA plot shows associations between bacterial community composition at the family level when constrained by site and environmental variables (n = 97). Each color is a distinct site with ellipses representing the standard error around the centroid. Arrows show vector fitting of the constrained environmental variables. *** associated with a variable at p < 0.001 in the PERMANOVA
Linking biogeography and community composition to ecosystem functioning
We found that community structure explained as much variation in ecosystem functioning [median (95% CI): 26% (16–33%)] as the present-day environment [20% (13–29%)], revealing that other properties in addition to those of microbial communities make relatively large contributions to ecosystem functioning (Fig. 4). Half of the variation in community structure was attributable to taxonomic diversity (Fig. 4). Subsequently, a 1 standard deviation (SD) increase in taxonomic diversity above its mean H’ of 4.4 was sufficient to increase CO2 production by 77 (95% CI: 36–127) mg m−2 from an average of 118 (102–135) to 195 (134–285) mg m−2 (Fig. 5a). The other half of the variation in ecosystem functioning explained by community structure came from functional genes encoding for oxidases, which were the only gene subset with a statistically significant effect (Tables S5 and S9). For example, a 1 SD increase in normalized oxidase abundance above its DESeq-normalized mean of 56.1 increased CO2 production by 93 (2–317) mg m−2 from an average of 139 (99–190) to 232 (101–507) mg m−2 (Fig. 5b).

Individual and interactive effects of the environment and microbial communities largely explain ecosystem functioning. Predictors were averaged across the model set except for normalized functional gene abundance, where oxidases were the best supported subset (Table S5). Boxes are shown only for variables with a direct or indirect effect on ecosystem function with 95% CI that exclude zero. Numbers accompanying each arrow are median (95% CI) percentage of variance in the associated response explained by a focal effect, with arrow width proportional to these values. Dashed lines represent interactions

Environment and community structure influence ecosystem functioning of littoral lake sediments. CO2 production increases with (a) taxonomic diversity, (b) normalized functional gene abundance (oxidases) as %terrestrial C also increases, and (c) percent OM quantity measured as loss on ignition (%LOI). (d) CO2 production decreases with pH. Lines are mean model fit at mean values of the other variables. Polygons are 95% confidence intervals. In (b), we show the statistical interaction between normalized functional gene abundance and %terrestrial C by plotting lines at the mean of the latter ± 1 standard deviation (σ)
An additional 12% (median, 95% CI: 2–18%) of variation in ecosystem functioning was explained by the interaction between community structure and the environment (Fig. 4). We specifically found that oxidases further increased CO2 production when terrestrial C was relatively abundant in sediment, highlighting the dependency of some functional genes on specific environmental conditions for influencing ecosystem functioning. For example, a 1 SD increase in terrestrial C at the mean oxidase abundance increased CO2 production from the average of 139 mg m−2 by 88 (95% CI: 46–154) mg m−2 (Fig. 5b).
Finally, the averaged model predicting CO2 production showed that both OM quantity and pH were the strongest environmental correlates of ecosystem functioning in our lake sediments (Fig. 4, Table S9). For example, if OM quantity doubled above its mean value of 13% across our sediment cores, CO2 production increased on average (95% CI) by 83 (35–167) mg m−2 from its mean of 118 (Fig. 5c). By contrast, a 1 SD increase in pH above its mean of 5.9 decreased CO2 production by 23 (9–31) mg m−2 (Fig. 5d).
Indirect effects of historical legacies and present-day environment on ecosystem functioning
The path analysis suggested that the effect of taxonomic diversity in predicting CO2 production was associated with differences in bacterial communities among sites, indicative of historical legacies, rather than filtering by the measured environmental variables. A median of 12% (95% CI: 6–28%) of the variation in taxonomic diversity was directly explained by site, with an additional 34% (95% CI: 15–48%), on average, explained by normalized bacterial abundance, which itself was 20% (95% CI: 7–42%) dependent on site (Fig. 4). Taxonomic diversity also had an indirect effect on CO2 production by explaining a median of 24% of the variation in the normalized abundance of oxidase genes (95% CI: 11–32%). This relationship was negative, suggesting that less taxonomically diverse communities were more likely to be dominated by taxa that relied on oxidizing OM as opposed to higher diversity communities where more functions were present (Table S10). Overall, however, the indirect effects of historical legacies on ecosystem functioning mediated by community structure were relatively small. Historical legacies explained <20% of the variation in each of the measures of community structure, none of which individually or interactively explained more than 13% of variation in CO2 production (Fig. 4). Thus, even if our site-level random effect included unmeasured present-day environmental variables, these effects were minimal. By contrast, no environmental variables influenced community structure, consequently having no indirect effect on ecosystem functioning (Table S10). While functional diversity depended on taxonomic diversity, it did not directly influence CO2 production (Table S10). We also verified that there were no missing linkages in our model, namely from CO2 production to community structure, which could feedback onto the latter (Table S11).
Discussion
Our study is the first, to our knowledge, that estimates the relative importance of individual and interactive effects of three fundamental properties – community structure, present-day environmental conditions, and historical legacies – on ecosystem functioning. We found that the present-day environment and community structure explained roughly the same amount of variation in ecosystem function, adding to limited evidence of a positive B-EF relationship in natural microbial communities [2, 3]. Importantly, we found that ecosystem processes were predicted better by also considering other characteristics of community structure, like functional gene abundances, and particularly their interactive effects with the environment. These findings, along with the evidence that microbial diversity and abundance varied more with site identity than with the four present-day environmental variables that most differed among sites, highlight the large influence that ecosystem properties other than biodiversity have upon ecosystem functioning.
Our results suggested that, despite their relatively small effects, historical legacies were more important than present-day environmental filtering in explaining the diversity and abundance of microbial communities and thus indirectly influencing ecosystem function. This finding is consistent with others that have shown historical legacies to be important for microbial-mediated ecosystem functions [14, 50]. In our study, legacies were likely the result of random differences in colonization history, such as arrival order and timing [18, 19], rather than past geographic events or dispersal limitation, as sites were all located within the same lake and most OTUs occurred at all sites. While measures of OM quality influenced microbial community composition, consistent with others [39, 51], there was no direct effect of these present-day environmental conditions on diversity and abundance, and subsequently ecosystem function. Thus, the biogeographical patterning across sites that we interpreted as being associated with past events adds to growing evidence that, while ‘everything’ may be ‘everywhere’ [24], not everything flourishes everywhere. One caveat is that there were still many OTUs unidentifiable at the family level.
Contrary to our prediction, we found that environmental conditions directly explained variation in CO2 production rather than doing so indirectly by changing microbial communities. For example, more acidic samples released more CO2 due to less inorganic carbon speciation [52]. We also found that higher OM quantities increased CO2 production, potentially because of non-microbial processes, such as extracellular oxidative metabolism and inorganic chemical reactions [53]. Photo-oxidation or thermal degradation of our samples was unlikely as they were incubated in the dark at controlled temperatures. Alternatively, the increase in CO2 production could be due to an increase in microbial biomass or in total abundance, but we only estimated normalized microbial abundance in our study. These findings are also consistent with a recent meta-analysis of 58 studies that found microbial biodiversity was not consistently associated with soil environmental variables, but rather more influenced by climate, ecological legacies and evolutionary history [50]. Microbial communities may in part be controlled by processes occurring at finer spatiotemporal scales than routinely measured, e.g. millimeters and minutes [54].
We found that increasing taxonomic diversity promoted ecosystem functioning more than functional gene diversity, which had no effect on CO2 production. These results suggest that multiple taxa may perform the same tasks associated with as broad a function as C utilization [2]. Given this convergence of function, increasing numbers of taxa may be sufficient to increase ecosystem functioning irrespective of their specific traits, resulting in high levels of complementarity [3, 5]. Our results therefore support the need to focus on the identity of traits associated with a response of interest rather than solely on functional diversity [9, 55, 56]. Other metrics, such as phylogenetic diversity, may also enhance predictions of ecosystem functioning by capturing additional axes of trait variation to those directly measured by functional data [57]. However, phylogenetic diversity may only be a useful proxy where the associated functions are evolutionarily conserved and not widely dispersed across lineages, as may be the case for a universal function like C utilization.
Oxidase-encoding genes were the only functional gene subset that we found to be associated with ecosystem functioning. Oxidases break down complex and recalcitrant organic polymers that come from terrestrial OM, such as lignin and humic acids [45], and which would have varied considerably across sites given the surrounding forest gradient [25]. The increasing association between oxidases and CO2 production as terrestrial C inputs to sediments increased was also unsurprising as aquatic microbial communities are adapted to utilize complex organic polymers derived from litterfall [58, 59]. We also found more oxidase genes in less taxonomically diverse assemblages, which may have arisen if a few taxa containing a higher proportion of oxidase genes became disproportionally active with increasing inputs of terrestrial C [60]. The other subsets of genes may have not explained much variation in CO2 production because they affected simpler molecules that were less associated with our specific terrestrial C gradient [61]. Additionally, shotgun metagenomics can only ascertain the presence of genes, not their expression, so some gene sets may have had nonsignificant effects because they were inactive.
Our results also suggest that future increases of OM inputs may promote benthic respiration and reduce the large C sink capacity of many northern lakes [62]. Northern lakes are burying increasingly more terrestrial OM into their sediments, primarily due to human activities [63]. Across our sites, a difference in sediment OM of 2 vs 55%, associated with a doubling of surrounding forest cover from 36 to 64% [26], was sufficient to increase CO2 production by an average (95% CI) of 5-times (3–7 times). Thus, our results also show how models that integrate biodiversity and trait-based approaches can better predict the outcomes of future changes to lake C cycles. More broadly, predictions of how ecosystem functioning varies with biodiversity will be improved if placed in the context of other ecosystem properties, such as past and present-day environments. Future studies should consider generalizing the importance of these other properties relative to the taxonomic and functional aspects of biodiversity in different spatial and temporal contexts. Another next step from our study would be to disentangle past and present-day influences more directly, such as by manipulating colonization dynamics in different environmental contexts and measuring how ecosystem function responds (e.g. [64]).
References
Tilman D, Isbell F, Cowles JM. Biodiversity and ecosystem functioning. Annu Rev. Ecol Evol S. 2014;45:471–93.
Delgado-Baquerizo M, Giaramida L, Reich PB, Khachane AN, Hamonts K, Edwards C, et al. Lack of functional redundancy in the relationship between microbial diversity and ecosystem functioning. J Ecol. 2016;104:936–46.
Laforest-Lapointe I, Paquette A, Messier C, Kembel SW. Leaf bacterial diversity mediates plant diversity and ecosystem function relationships. Nature. 2017;546:145–7.
Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK. The contribution of species richness and composition to bacterial services. Nature. 2005;436:1157–60.
Venail PA, Vives MJ. Positive effects of bacterial diversity on ecosystem functioning driven by complementarity effects in a bioremediation context. PLoS ONE. 2013;8:e72561.
Jiang L. Negative selection effects suppress relationships between bacterial diversity and ecosystem functioning. Ecology. 2007;88:1075–85.
Becker J, Eisenhauer N, Scheu S, Jousset A. Increasing antagonistic interactions cause bacterial communities to collapse at high diversity. Ecol Lett. 2012;15:468–74.
Nielsen UN, Ayres E, Walln DH, Bardgett RD. Soil biodiversity and carbon cycling: a review and synthesis of studies examining diversity-function relationships. Eur J Soil Sci. 2011;62:105–16.
Krause S, Le Roux X, Niklaus PA, Van Bodegom PM, Lennon JT, Bertilsson S, et al. Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front Microbiol. 2014;5:251.
Bardgett RD, van der Putten WH. Belowground biodiversity and ecosystem functioning. Nature. 2014;515:505–11.
Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013;7:830–8.
Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–9.
Graham EB, Knelman JE, Schindlbacher A, Siciliano S, Breulmann M, Yannarell A, et al. Microbes as engines of ecosystem function: when does community structure enhance predictions of ecosystem processes? Front Microbiol. 2016;7:214.
Martiny JB, Martiny AC, Weihe C, Lu Y, Berlemont R, Brodie EL, et al. Microbial legacies alter decomposition in response to simulated global change. ISME J. 2017;11:490–9.
Vass M, Langenheder S. The legacy of the past: effects of historical processes on microbial metacommunities. Aquat Microb Ecol. 2017;79:13–19.
Martiny JB, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–12.
Friedline CJ, Franklin RB, McCallister SL, Rivera MC. Bacterial assemblages of the eastern Atlantic Ocean reveal both vertical and latitudinal biogeographic signatures. Biogeosciences. 2012;9:2177–93.
Fukami T, Morin PJ. Productivity–biodiversity relationships depend on the history of community assembly. Nature. 2003;424:423–6.
Fukami T, Dickie IA, Paula Wilkie J, Paulus BC, Park D, Roberts A, et al. Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol Lett. 2010;13:675–84.
Lear G, Bellamy J, Case BS, Lee JE, Buckley HL. Fine-scale spatial patterns in bacterial community composition and function within freshwater ponds. ISME J. 2014;8:1715–26.
Bier RL, Bernhardt ES, Boot CM, Graham EB, Hall EK, Lennon JT, et al. Linking microbial community structure and microbial processes: an empirical and conceptual overview. FEMS Microbiol Ecol. 2015;91:fiv113.
Niño-García JP, Ruiz-Gonzalez C, del Giorgio PA. Landscape-scale spatial abundance distributions discriminate core from random components of boreal lake bacterioplankton. Ecol Lett. 2016;19:1506–15.
Tranvik LJ, Downing JA, Cotner JB, Loiselle SA, Striegl RG, Ballatore TJ, et al. Lakes and reservoirs as regulators of carbon cycling and climate. Limnol Oceanogr. 2009;54:2298–314.
Baas-Becking LGM. Geobiologie of inleiding tot de milieukunde. The Hague, Netherlands: WP Van Stockum & Zoon; 1934.
Tanentzap AJ, Szkokan-Emilson EJ, Kielstra BW, Arts MT, Yan ND, Gunn JM. Forests fuel fish growth in freshwater deltas. Nat Commun. 2014;5:4077.
Szkokan-Emilson EJ, Wesolek BE, Gunn JM. Terrestrial organic matter as subsidies that aid in the recovery of macroinvertebrates in industrially damaged lakes. Ecol Appl. 2011;21:2082–93.
Miller DN, Bryant JE, Madsen EL, Ghiorse WC. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microb. 1999;65:4715–24.
Ball DF. Loss-on-ignition as an estimate of organic matter and organic carbon in non-calcareous soils. J Soil Sci. 1964;15:84–92.
Taylor BR, Parkinson D, Parsons WFJ. Nitrogen and lignin content as predictors of litter decay rates: a microcosm test. Ecology. 1989;70:97–104.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb. 2009;75:7537–41.
Curd EE, Martiny JB, Li H, Smith TN. Bacterial diversity is positively correlated with soil heterogeneity. Ecosphere. 2018;9:e02079.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16:410–22.
Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013;14:671–83.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27.
Mitchell A, Bucchini F, Cochrane G, Denise H, ten Hoopen P, Fraser M, et al. EBI metagenomics in 2016 - an expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Res. 2016;44:595–603.
Haegeman B, Hamelin J, Moriarty J, Neal P, Dushoff J, Weitz JS. Robust estimation of microbial diversity in theory and in practice. ISME J. 2013;7:1092–101.
Ruiz-González C, Niño‐García JP, del Giorgio PA. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol Lett. 2015;18:1198–206.
Kirk TK, Farrell RL. “Enzymatic combustion”: the microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–501.
Sinsabaugh RL, Moorhead DL. Resource allocation to extracellular enzyme production: a model for nitrogen and phosphorus control of litter decomposition. Soil Biol Biochem. 1994;26:1305–11.
Golchin A, Oades JM, Skjemstad JO, Clarke P. Soil structure and carbon cycling. Soil Res. 1994;32:1043–68.
Zhang Y, Zhang X, Liu X, Xiao Y, Qu L, Wu L, et al. Microarray-based analysis of changes in diversity of microbial genes involved in organic carbon decomposition following land use/cover changes. FEMS Microbiol Lett. 2006;266:144–51.
Martiny JB, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: a phylogenetic perspective. Science. 2015;350:aac9323.
Sinsabaugh RL. Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biol Biochem. 2010;42:391–404.
Grace JB, Schoolmaster DR, Guntenspergen GR, Little AM, Mitchell BR, Miller KM, et al. Guidelines for a graph‐theoretic implementation of structural equation modeling. Ecosphere. 2012;3:1–44.
Shipley B. Cause and correlation in biology: a user’s guide to path analysis, structural equations and causal inference with R. Cambridge, UK: Cambridge University Press; 2016.
Chung Y, Rabe-Hesketh S, Dorie V, Gelman A, Liu J. A nondegenerate penalized likelihood estimator for variance parameters in multilevel models. Psychometrika. 2013;78:685–709.
Burnham KP, Anderson DR. Model Selection and Multimodel Inference. A Practical Information-Theoretical Approach. Heidelberg, Germany: Springer; 2002.
Hendershot JN, Read QD, Henning JA, Sanders NJ, Classen AT. Consistently inconsistent drivers of microbial diversity and abundance at macroecological scales. Ecology. 2017;98:1757–63.
Schallenberg M, Kalff J. The ecology of sediment bacteria in lakes and comparisons with other aquatic ecosystems. Ecology. 1993;74:919–34.
Morel FM, Hering JG. Principles and applications of aquatic chemistry. NY, USA: John Wiley & Sons; 1993.
Wang B, Lerdau M, He Y. Widespread production of nonmicrobial greenhouse gases in soils. Glob Change Biol. 2017;23:4472–82.
Grundmann GL, Debouzie D. Geostatistical analysis of the distribution of NH4 + and NO2 − oxidizing bacteria and serotypes at the millimeter scale along a soil transect. FEMS Microbiol Ecol. 2000;34:57–62.
Roger F, Bertilsson S, Langenheder S, Osman OA, Gamfeldt L. Effects of multiple dimensions of bacterial diversity on functioning, stability and multifunctionality. Ecology. 2016;97:2716–28.
Trivedi P, Delgado-Baquerizo M, Trivedi C, Hu H, Anderson JC, Jeffries TC, et al. Microbial regulation of the soil carbon cycle: evidence from gene–enzyme relationships. ISME J. 2016;10:2593–604.
Flynn DF, Mirotchnick N, Jain M, Palmer MI, Naeem S. Functional and phylogenetic diversity as predictors of biodiversity–ecosystem‐function relationships. Ecology. 2011;92:1573–81.
Emilson CE, Kreutzweiser DP, Gunn JM, Mykytczuk N. Leaf-litter microbial communities in boreal streams linked to forest and wetland sources of dissolved organic carbon. Ecosphere. 2017;8:e01678.
Judd KE, Crump BC, Kling GW. Bacterial responses in activity and community composition to photo-oxidation of dissolved organic matter from soil and surface waters. Aquat Sci. 2007;69:96–107.
Muscarella ME, Jones SE, Lennon JT. Species sorting along a subsidy gradient alters bacterial community stability. Ecology. 2016;97:2034–43.
Rocca JD, Hall EK, Lennon JT, Evans SE, Waldrop MP, Cotner JB, et al. Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 2015;9:1693–9.
Gudasz C, Ruppenthal M, Kalbitz K, Cerli C, Fiedler S, Oelmann Y, et al. Contributions of terrestrial organic carbon to northern lake sediments. Limnol Oceanogr. 2017;2:218–27.
Heathcote AJ, Anderson NJ, Prairie YT, Engstrom DR, del Giorgio PA. Large increases in carbon burial in northern lakes during the Anthropocene. Nat Commun. 2015;6:10016.
Reed HE, Martiny JB. Microbial composition affects the functioning of estuarine sediments. ISME J. 2013;7:868–79.
Acknowledgements
We thank J. Wolstenholme for fieldwork, D. Baulcombe and I. Henderson for offering lab space, G. Jones and M. Meyer for assisting with sample processing, S. Dickens and P. Marriot for help with sequencing, and three anonymous reviewers for comments that improved an earlier draft. Support for this work came from NERC Standard Grant NE/L006561/1 and Gatsby Fellowship GAT2962 to A.J.T.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Orland, C., Emilson, E.J.S., Basiliko, N. et al. Microbiome functioning depends on individual and interactive effects of the environment and community structure. ISME J 13, 1–11 (2019). https://doi.org/10.1038/s41396-018-0230-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0230-x
This article is cited by
-
Exercise and the gut microbiome: implications for supportive care in cancer
Supportive Care in Cancer (2023)
-
Spatial and Seasonal Patterns of Sediment Bacterial Communities in Large River Cascade Reservoirs: Drivers, Assembly Processes, and Co-occurrence Relationship
Microbial Ecology (2023)
-
Will free-living microbial community composition drive biogeochemical responses to global change?
Biogeochemistry (2023)
-
Plastic pollution fosters more microbial growth in lakes than natural organic matter
Nature Communications (2022)
-
The effects of g-C3N4/biochar and g-C3N4 on bacterial community in riverbed sediment
Environmental Science and Pollution Research (2022)
