
Abstract
Endosymbiosis is a widespread phenomenon in the microbial world and can be based on diverse interactions between endosymbiont and host cell. The vast majority of the known endosymbiotic interactions involve bacteria that have invaded eukaryotic host cells. However, methanogenic archaea have been found to thrive in anaerobic, hydrogenosome-containing protists and it was suggested that this symbiosis is based on the transfer of hydrogen. Here, we used culture-independent genomics approaches to sequence the genomes of two distantly related methanogenic endosymbionts that have been acquired in two independent events by closely related anaerobic ciliate hosts Nyctotherus ovalis and Metopus contortus, respectively. The sequences obtained were then validated as originating from the ciliate endosymbionts by in situ probing experiments. Comparative analyses of these genomes and their closest free-living counterparts reveal that the genomes of both endosymbionts are in an early stage of adaptation towards endosymbiosis as evidenced by the large number of genes undergoing pseudogenization. For instance, the observed loss of genes involved in amino acid biosynthesis in both endosymbiont genomes indicates that the endosymbionts rely on their hosts for obtaining several essential nutrients. Furthermore, the endosymbionts appear to have gained significant amounts of genes of potentially secreted proteins, providing targets for future studies aiming to elucidate possible mechanisms underpinning host-interactions. Altogether, our results provide the first genomic insights into prokaryotic endosymbioses from the archaeal domain of life.
Similar content being viewed by others


Introduction
Endosymbiosis involves an intimate interaction between a host cell and an endosymbiont. Endosymbiotic interactions involving bacterial partners are plentiful, and are known to have played key roles in several major transitions in the evolution of cellular life on Earth, including the origin of the mitochondria in eukaryotes [1,2,3,4]. Most studies addressing the evolutionary processes that occur during host adaptation have focused on systems that involve bacterial endosymbionts in eukaryotic host cells. One of the few examples outside this realm are the methanogenic archaeal endosymbionts [5, 6] that reside in anaerobic protist hosts [6], including anaerobic amoeba [7], parabasalids and ciliates [8]. These protists generally thrive in oxygen-deprived environments and, as a result, their mitochondria have evolved into anaerobic, hydrogen-producing organelles known as hydrogenosomes [9, 10]. This system is thought to be based on a syntrophic relationship between the host and a methanogenic endosymbiont, which uses the hydrogen produced by the host’s hydrogenosomes [11, 12]. The hosts, particularly large metopid ciliates like Metopus contortus and Nyctotherus ovalis, benefit from the decrease in partial hydrogen pressure in their cells [11, 12], and the endosymbionts benefit from a guaranteed supply of hydrogen, which they can utilize for energy production through methanogenesis. While the endosymbionts may also utilize formate [11], N. ovalis does not seem to produce formate as one of its end products [13], and transcriptomic analysis of both N. ovalis and M. contortus did not provide evidence for genes encoding pyruvate-formate lyase in these species (Lewis et al., manuscript in prep.). Even though these symbiotic systems were discovered in the early 1990’s [5, 14, 15], only little is known about how these archaea have adapted to living inside their ciliate host cells. Here we used cultivation-independent approaches to generate genomic data from the endosymbionts of the anaerobic ciliates N. ovalis and M. contortus. A detailed comparison of these endosymbiont genomes revealed that these are in an early stage of adaptation towards endosymbiosis as evidenced by gene loss and pseudogenization events. Furthermore, both endosymbiont genomes have gained a significant number of genes encoding potentially secreted proteins. These genes represent interesting targets for future studies that aim to reveal potential mechanisms underlying host-interactions. Our study provides the first genomic insights into a prokaryotic endosymbiotic interaction involving an archaeon.
Results
The methanogenic endosymbionts of N. ovalis and M. contortus
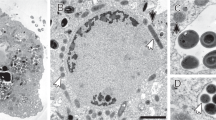
To gain more insights into the intracellular adaptations and evolution of archaeal endosymbiosis, we sequenced the genomes of the methanogenic endosymbionts of the ciliate hosts N. ovalis and M. contortus (Fig. 1a and Suppl. Figure 1). N. ovalis cells were isolated from the hindguts of Blaptica dubia cockroaches, and an anoxic M. contortus enrichment culture was established from marine sediment inoculants sampled near Dorset, UK (see Methods for details). Sequencing of 16S rRNA gene amplicons from lysed cells of both N. ovalis and M. contortus (Suppl. Figure 2) confirmed the presence of Methanobrevibacter-related endosymbionts in N. ovalis (hereafter referred to as Methanobrevibacter sp. NOE, or ‘NOE’ for short), and of Methanocorpusculum-related endosymbionts in M. contortus (hereafter referred to as Methanocorpusculum sp. MCE, or ‘MCE’ for short). Since both endosymbionts are phylogenetically distantly related and emerge from within clades of free-living methanogenic lineages (Fig. 1b, Suppl. Figure 3 and 4), their intracellular lifestyle is inferred to be the result of two recent and independent endosymbiosis events. Transmission electron microscopy (TEM) revealed that NOE cells form close associations with the hydrogenosomes (Fig. 1d and Suppl. Fig. 6) and fluorescence in situ hybridization (FISH) using endosymbiont-specific probes show that the endosymbionts are distributed throughout N. ovalis cells (Fig. 1e, Suppl. Video 1 and Suppl. Fig. 9). The observed adjacent localization and tight interaction between NOE endosymbiont cells and hydrogenosomes is suggestive of a well-established endosymbiotic relationship, in which NOE cells have adapted to an intracellular lifestyle that favours the transfer of hydrogen between NOE and hydrogenosomes. Within M. contortus, we observe a similarly close association of the endosymbiotic methanogen cells and the host hydrogenosomes. However, while the hydrogenosome-methanogen complexes of N. ovalis appear interspersed throughout the cell (Fig. 1c, d, Suppl. Video 1, Suppl. Fig. 5), the hydrogenosomes (Fig. 1d, Suppl. Fig. 6) and methanogens (Fig. 1d, f, Suppl. Video 2, Suppl. Fig. 6) in M. contortus seem positioned more towards the outer membrane. Our identification of hydrogenosomes and methanogens are consistent with previous studies of both M. contortus [15,16,17] and N. ovalis [5] ultrastructure. Although other anaerobic protists exist that contain multiple species of endosymbionts [18,19,20], no additional endosymbiotic organisms could be identified neither in the cells of N. ovalis nor in M. contortus based on FISH experiments using a bacterial-specific probe (negative result not shown), TEM images (Fig. 1c, d) or 16S rRNA amplicon sequencing (Suppl. Figure 2).

The associations between anaerobic metopid ciliates and their methanogenic endosymbionts, and their relationships to other organisms. a A maximum-likelihood tree calculated using IQ-TREE inferred from an alignment of ciliate 18S rRNA genes, showing the relationship of M. contortus and N. ovalis to other ciliates. Host-associated ciliates are marked with pink branches. b A maximum-likelihood tree calculated using IQ-TREE, inferred from an alignment of 57 concatenated single-copy proteins genes from methanogenic archaea, showing the relationship of MCE and NOE to their free-living relatives. The bootstrap supports for both phylogenetic trees (a, b) are represented with unfilled circles <50%, filled circles >50% and no circle >80%. Branch scale bars represent the number of substitutions per site. c, d Transmission electron microscopy (TEM) of N. ovalis (c) and M. contortus (d) cells. The smaller electron dense bodies in M. contortus (d) are likely mucocysts, as identified previously [18]. Examples of hydrogenosomes in the TEM images are marked as ‘H’ and endosymbionts are indicated with red arrows. e, f Confocal maximum intensity projections of Z-stack images of N. ovalis (e) and M. contortus (f) cells, fluorescently labelled with endosymbiont-specific oligonucleotide probes MB and SYM5, respectively
In contrast to species of the Methanocorpusculum genus, which are mainly found in aquatic environments (Suppl. Fig. 4), the known Methanobrevibacter diversity has a well-documented association with mammalian intestinal systems [21,22,23], including with humans [24, 25] (Suppl. Figure 3), resulting in an increased effort to study the Methanobrevibacter genus. As well as prokaryotes, the intestines of ruminants and insects contain a variety of anaerobic ciliates and protists that host intracellular methanogens [26, 27]. Therefore, given that many of the Methanobrevibacter species detected from such environments are only characterised based on their 16S rRNA genes (Suppl. Figure 3), it is possible that some of them could be associated with protists.
Cultivation-independent genomics of methanogenic endosymbionts
We investigated whether we could observe any differences in the degree of host adaptation at the genomic level between the two systems. While isolation and even cultivation of methanogenic endosymbionts has been claimed in previous studies [12, 28,29,30,31], conclusive evidence showing that the isolated organisms are indeed the original endosymbionts, e.g. using in situ methods, has thus far been lacking. Moreover, later studies of the same host organisms identified different species of methanogens as the endosymbionts, and confirmed their findings using in situ oligonucleotide probe hybridization methods [9, 21, 33]. We therefore employed culture-independent approaches to obtain genomic data for NOE and MCE. For NOE, a single-cell genomics approach was used in which we extracted and enriched endosymbiont cells from multiple N. ovalis host cells before sorting individual cells using FACS. Whole-genome amplified (WGA) DNA stemming from individual NOE cells were then identified by PCR-amplification and sequencing of 16S rRNA genes before pooling together for subsequent whole-genome sequencing (see Methods for details). For MCE, we instead used a genome-resolved metagenomics approach whereby multiple M. contortus host cells were lysed and subjected to endosymbiont enrichment prior to bulk WGA and metagenomic sequencing. After metagenomic assembly, contigs belonging to the MCE genome were extracted using a combination of metagenomic binning approaches (see Methods for details). Both cultivation-independent approaches resulted in high quality draft genomes for NOE and MCE. Both genomes were estimated to be over 95% complete (Suppl. Table 1), allowing us to make genomic inferences about the intracellular lifestyle of the respective methanogens and to identify potential host-adaptations at the genomic level.
Methanogenic endosymbiont genomes undergo pseudogenization
Compared to their free-living counterparts, neither the NOE nor the MCE genome showed any significant reduction in genome size (Suppl. Table 1). Whereas the genomes of some bacterial endosymbionts have undergone extensive reduction, especially in the obligate endosymbionts of arthropods, where genome sizes in the range of 100–1000 kb have been observed [33]. In other cases, genome reduction is much less extreme, for example in Polynucleobacter necessarius, the betaproteobacterial endosymbiont of the ciliate Euplotes [34], whose genome is approximately as large as its free-living relatives.
To investigate the genomic events potentially associated with endosymbiosis, we performed an ancestral genome reconstruction of both archaeal endosymbiont genomes. In both genomes we identified an increased number of gene gain, loss, and duplication events in comparison to the free-living congeners of the endosymbionts (Suppl. Table 2). Further investigation revealed that many of these genes were not genuine de novo gene gains and expansions, but rather the remnants of genes undergoing pseudogenization, and thus in the process of being lost. A detailed screen for pseudogenized genes based on the comparison of all predicted endosymbiont genes against closely related sister-species revealed that about 10% (187 genes) of the NOE genes and about 5% (100 genes) of the MCE genes are undergoing pseudogenization. The observed numbers of pseudogenes in these endosymbiont genomes is much higher to those observed in the genomes of their closest free-living relatives (Suppl. Table 1). Accounting for these pseudogenes, the coding density of the NOE genome (63% protein coding content) is significantly lower than that of other free-living members of the Methanobrevibacter genus such as M. arboriphilus DH1 (72%) and M. ruminantium (78%; Fig. 2a, Suppl. Table 1), which already have a markedly lower coding density than other archaea. In fact, the coding density recorded for the Methanobrevibacter sp. NOE genome is lower than any other archaeal genome in the NCBI RefSeq database (5th September 2017; Fig. 2a) that has a unique species taxon ID. The MCE genome also displays a low coding density of 78% protein coding genes, which is lower than its closest free-living relatives Methanocorpusculum labreanum and Methanocorpusculum bavaricum (both 88%; Fig. 2a, Suppl. Table 1). The levels of gene pseudogenization in both methanogenic endosymbionts is characteristic for the early reductive stages of adaptation to an intracellular lifestyle [35]. The ancestral reconstruction was then repeated with all of the pseudogenes removed to recalculate gain and loss of gene families by both endosymbionts (Suppl. Fig. 7 and Suppl. Dataset 1). All inferences regarding gene flow discussed below are drawn from this second, refined ancestral reconstruction.

Characterization of endosymbiotic genome reduction. a Due to pseudogenization and gene loss both endosymbiotic genomes (marked by red arrows) have the lowest coding densities of their respective genera. b Both endosymbiont genomes show similar functional patterns for genes undergoing pseudogenization. c Overview of presence and absence of genes comprising the biosynthetic pathways for cobalamin, chorismate and aromatic amino acids in the MCE and NOE endosymbiont genomes relative to their closest free-living relatives. Gene presence is indicated with grey circles, while gene absence and pseudogenes are indicated by pink and purple circles, respectively. Please note that gene absence could be the result of genome incompleteness. Genes were classified as pseudogenes if a frameshift or point mutation generated a truncated protein less than 75% of the original protein length, and the gene was no closer than 1000 bp from a contig edge (see Methods for details)
The functional annotation of the genes undergoing pseudogenization in the two endosymbionts revealed that, apart from a large number of genes with unknown functions, many are involved in amino acid transport and metabolism, cell motility, cell defence, and gene regulation (Fig. 2b). While the overall pattern was similar in both NOE and MCE, a larger number of pseudogenes was identified for NOE. These functional patterns of endosymbiont pseudogenes contrast with those observed for the closely related free-living species, which have a more even distribution over all functional categories, with a slight overrepresentation in transposones and prophages (Suppl. Fig. 8). Interestingly, NOE has lost the ability to synthesize cobalamin, as several genes comprising the biosynthesis pathway were found to be missing, or pseudogenized (Fig. 2c and Suppl. Dataset 1). We also noted that in the genome of MCE, the cbiG gene encoding cobalt-precorrin 5A hydrolase was pseudogenized, indicating that cobalamin biosynthesis is likely also impaired in MCE (Fig. 2c). Given that cobalamin (or vitamin B12) is essential in the methanogenesis pathway as part of cobalamin-dependent methyltransferases [36], this indicates that the both endosymbionts possibly rely on an external supply of this vitamin. Furthermore, we found that several genes of the chorismate biosynthesis pathway were missing in the MCE genome, and that several genes of the aromatic amino acid biosynthesis pathways were pseudogenized or missing in both endosymbiont genomes, indicating that these metabolic pathways are likely impaired (Fig. 2c; Suppl. Dataset 1). This idea is further supported by the fact that an inspection of all proteins encoded by un-binned contigs of the M. contortus metagenome did not reveal any proteins that could represent the missing copies of these genes in MCE. Similarly, the draft genomes of the free-living methanogens Methanocorpusculum bavaricum and Methanobrevibacter oralis appear to be missing several aromatic amino acid biosynthesis genes (Fig. 2c). Although we cannot fully exclude the possibility that these genes are missing due to the incompleteness of these draft genomes (Suppl. Table 1), it could also imply that these methanogens are unable to biosynthesize aromatic amino acids. Most Methanobrevibacter species generally thrive in nutrient-rich environments, such as animal digestive tracts. M. oralis [37], which was isolated from the human mouth-cavity in a subgingival plaque [38], has the potential to obtain aromatic amino acids from its local environment. Similarly, living inside the anaerobic ciliate may provide the endosymbiont with access to nutrients and vitamins either from the eukaryotic host, or, in the case of cobalamin, from the bacterial and archaeal cells ingested as food. The loss of genes involved in the synthesis of cobalamin and aromatic amino acids (tryptophan, tyrosine and phenylalanine; Fig. 2c) strongly suggests that both methanogenic endosymbionts are dependent on their intracellular environment for these nutrients. This is similar to insect bacterial endosymbionts, which have also often lost genes for cobalamin biosynthesis [39]. One exception to this trend is the genome of Hodgkinia cicadicola, which has likely has retained its cobalamin synthesis pathway since it relies on the cobalamin-dependent version (MetH) of methionine synthase [39]. Most other insect endosymbionts, and the archaeal endosymbionts presented here, use the cobalamin-independent methionine synthase MetE (Suppl. Dataset 1).
At first glance, it appears that some of the pseudogenized genes from the genomes of NOE and MCE could have potentially disrupted core metabolic functions of these organisms, like the methanogenesis pathway. For example, both the NOE and MCE genomes contain a pseudogenized frhB gene (encoding coenzyme F420-reducing hydrogenase), and the NOE genome contains pseudogenized copies of hdrB and hdrC (encoding the B and C subunits of heterodisulfide reductase). However, both endosymbionts have also retained functional copies of these genes, indicating that only redundant copies of these genes have been pseudogenized (Suppl. Dataset 1).
Apart from the loss of biosynthetic capacity in the NOE and MCE genomes, both these organisms appear to have lost genes involved in adhesion (pilus and flagellin genes), cell surface modification (glycosyl hydrolases and S-layer protein encoding genes) and transcription (transcriptional regulator genes) (Fig. 3 and Suppl. Dataset 1). Furthermore, we found that MCE lost genes involved in mobility (chemotaxis genes) as well as genes encoding the CRISPR-Cas system (see Suppl. Dataset 1 for details). The latter observation contrasts with most other known endosymbiotic bacteria, which seem to have retained at least one copy of an active CRISPR-Cas system [40, 41], possibly to control excessive proliferation of genetic elements. Altogether, in addition to the reduced metabolic capacity, the observed patterns of functional loss can be interpreted as signatures of adaptation towards the more stable environment provided by the host cell [35].

Functional patterns of gene gain and loss in archaeal endosymbiont genomes. Schematic overview of the main functional patterns of gene gain and loss observed in the NOE and MCE genomes. Gained genes encodes many proteins of unknown function and proteins that are putatively secreted and represent possible host-interaction factors. Gene loss and pseudogenization in MCE and NOE were functionally convergent, with observed losses of genes involved in aromatic amino acid metabolism, adhesion, cell surface modification and transcription (see Suppl. Dataset 1 for details). Loss of genes involved in mobility and CRISPR associated genes were specific to MCE, and of genes involved in cobalamin biosynthesis specific to NOE
Potential host-interaction factors
A genetic feature common in some bacterial endosymbionts is the presence of genes encoding protein-protein interaction motifs such as ankyrins, TPR/Sel1 and leucine-rich repeats [42]. A search for the genes encoding these proteins in the genomes of NOE and MCE showed that they were either not enriched or, as in the case of TPR repeats, were actually reduced in number in comparison to their free-living counterparts (Suppl. Dataset 1). Bacterial intracellular parasites are known to utilize diverse strategies to interact with, invade and exploit their host cell including secretion systems [43], proteins with repeat motifs (e.g. ankyrin repeat motifs [44, 45]), and ADP/ATP translocases [46]. We were unable to identify proteins commonly taken to be hallmarks of the bacterial intracellular lifestyle, in the NOE and MCE genomes. Hence, the interaction between these archaeal endosymbionts and their host cells are unlikely to be parasitic in nature, however they could potentially use unidentified strategies or systems to enter and persist inside anaerobic ciliates. This possibility is supported by a large fraction of the proteins, encoded by gained genes in both NOE and MCE genomes, showing either a predicted secretion signal or a trans-membrane helix in the amino-terminus of the protein sequence. We found that 60 (23%) of the MCE gained genes and 77 (18%) of the NOE gained genes encoded proteins that were potentially secreted (Suppl. Table 4). This characteristic seems to represent a common feature of the NOE and MCE genomes that may be related to their shift from a free-living to an endosymbiotic lifestyle. As these endosymbionts most likely employ novel mechanisms for interacting with their host, such as selective retention by the host cell and evasion of the phagocytosis machinery, the enriched sets of putative secreted endosymbiont proteins represent promising targets for studies aiming to elucidate potentially novel mechanisms underlying host-interactions in these organisms.
Discussion
Overall, we observed a relatively low level of potential genomic adaptation towards an intracellular lifestyle in the NOE and MCE genomes. This could be expected if the endosymbionts were acquired relatively recently, and there is evidence available to suggest that this could be the case. Firstly, phylogenetic studies have provided evidence that endosymbiotic methanogens and their anaerobic ciliate hosts do not co-speciate [32, 47], suggesting that the interaction between them is facultative in nature and that the endosymbionts can regularly be replaced [11, 12]. In support of this, several ciliates, including M. contortus, have been shown to survive when their endosymbionts were removed by treatment with the methanogenesis inhibitor 2-bromoethanesulphonate (BES), with little or no negative affect on their growth rate and yield [16, 49]. In their natural environments however, there could be a number of possible mechanisms or causes to explain why ciliates lose their methanogenic endosymbionts. One such explanation is that, once acquired by the host cell, the methanogens are subjected to the effect of Muller’s ratchet, due to their small effective population size and limited opportunity for genomic recombination [48]. As a result, the endosymbionts would accumulate deleterious mutations, causing frequent pseudogenization events and eventually resulting in extinction. Alternatively, since the ciliates are not dependent on their endosymbionts, it is possible that they could, in some cases, relinquish them as a mechanism to adapt to changing environmental conditions.
Despite evidence for possible cyclic loss and reacquisition of different methanogenic endosymbionts, there is data to suggest that, at least for limited periods of evolution, the associations with their ciliate hosts are stable. For example, the endosymbionts of the M. contortus strain from the present study, which was isolated in Dorset (UK), were identified as being the same species of Methanocorpusculum as those from another M. contortus strain [14] isolated more than 20 years earlier from a spatially distant site (>1000 km) in Denmark [11]. Based on this, along with similar observations from another anaerobic ciliate species [50], it is likely that methanogenic endosymbionts in anaerobic ciliates will exhibit genomic adaptations to their intracellular lifestyles. In support of this, analysis of the first genomes sequenced from methanogenic archaeal endosymbionts in the present study revealed an overall loss of several biosynthetic capabilities, as well as the loss of cellular functions, which are likely to be the direct result of adaptation towards a more stable intracellular environment provided by the ciliate hosts (Fig. 3). Furthermore, we identified a pool of gained genes encoding proteins that are likely to be secreted, and some of which might potentially represent novel host-interaction factors. Future studies of these factors might reveal details about the mechanisms underlying selective retention in the ciliate host cell. Ultimately, such studies might reveal why so few archaeal lineages have established endosymbiotic interactions with eukaryotes as compared to the plethora of such interactions observed in the bacterial realm.
Methods
Ciliate isolation
Nyctotherus ovalis ciliates were isolated from the hindgut of Blaptica dubia cockroaches via electromigration as previously described [51]. The cockroaches were obtained from Cricket Express (Bohus, Sweden; http://www.cricketexpress.se), kept in plastic boxes, and fed ad libitum. Ciliates from the species Metopus contortus were sampled in 2013 from a brackish lake in Poole Park, Dorset (UK) (decimal degrees: 50.716155, −1.972559) and cultured using N75S media (https://www.ccap.ac.uk/media/documents/N75S.pdf), to which wheat grains were added, in sealed vials flushed with N2 to maintain anoxic conditions. Cells were isolated from the cultures by hand picking using a drawn-out Pasteur pipette.
Electron microscopy
Isolated ciliates were fixed in ice cold 0.15 M HEPES (Sigma-Aldrich, CAS 7365-45-9), pH 7.4, with 2.5% glutaraldehyde (Sigma-Aldrich, CAS 111-30-8). Imaging was performed by the Microscopy Imaging Center (University of Bern, Bern, Switzerland).
Fluorescent in situ hybridization (FISH)
Endosymbiont species were identified by FISH using the following oligonucleotide probes designed to specifically bind to regions of 16S rRNA: Methanocorpusculum-specific probe (SYM5) (5′-CTGCATCGACAGGCACT) [14] and Methanobrevibacter-specific probe (MB) (5′-CCGTTAAGGATGGCACT) (this study). An archaea-specific positive-control probe (ARCH915) (5′-GTGCTCCCCCGCCAATTCCT) [52] (Suppl. Fig. 9) and a negative-control probe (NONEUB/NON338) (5′-ACTCCTACGGGAGGCAGC) [53] were also used in FISH experiments. All probes were synthesised by biomers.net GmbH and double-labelled with either 6-Fam, Cy3 or Cy5 fluorescent dyes. Due to auto-fluorescence emitted from the sample at emission spectra similar to 6-Fam and Cy3, only Cy5 could be used to visualise endosymbionts of N. ovalis. Ciliate cells were fixed in 4% paraformaldehyde at 4 °C and washed in PBS. Cells of N. ovalis and M. contortus were attached to gelatine or poly-l-lysine coated slides, respectively. Sample dehydration, probe hybridisation and washing were the same as described previously [54], except that formamide was removed from the hybridisation buffer as its presence caused non-specific binding of probes to samples. Therefore instead of using formamide, the stringency of the hybridisation reactions was ensured by increasing the hybridisation temperature according to the estimated probe dissociation temperatures (Td), which were calculated as described by [52]. The Td of probe SYM5 was calculated as 56.6 °C and the Td of probe MB was calculated as 54.2 °C. Probes were hybridised for 2 h at 2 °C lower than their respective Td to ensure stringency. After washing, dried samples were mounted with ProLong Diamond antifade mountant. Z-sections were imaged using a confocal microscope (A1R, Nikon) with a ×60/1.4 objective lens. Vertical z-stacks were deconvolved using Huygens deconvolution software (Scientific Volume Imaging) with empirically measured point spread functions. Image projections were reconstructed using the program Fiji (ImageJ).
16S rRNA amplicon sequencing
Amplicons from washed and lysed M. contortus cells and ciliate-free culture media, as well as washed and lysed N. ovalis cells and hindgut extract filtered through a 5 µm Minisart filter (Sartorius), were generated using universal A519F [55] (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNTACAGCMGCCGCGGTAA-3′) and U1391R [56] (5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTATACGGGCGGTGWGTRC-3′) primers with a 5′ overhang containing Illumina sequencing primers. An initial denaturation step was run at 94 °C for 2 min, followed by 20 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 45 s and extension at 72 °C for 80 s. A final extension was performed at 72 °C for 10 min. A second PCR reaction using forward (5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACG-3′) and reverse (5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′) primers containing barcodes (marked as ‘X’) and Illumina MiSeq adapters was carried out using an initial denaturation at 95 °C for 15 min, followed by 10 cycles of denaturation at 95 °C for 20 s, annealing at 61 °C for 30 s and extension at 72 °C for 80 s. A final extension was performed at 72 °C for 7 min.
16S rRNA genetic diversity analysis
All sequences in the SILVA release 128 database classified as belonging to the Methanocorpusculum or Methanobrevibacter genera were downloaded. Sequences shorter than 600 bp were removed, and remaining sequences were clustered at 96% sequence similarity using VSEARCH [57]. Multiple sequence alignments were made using mafft-linsi 7.309 [58] and phylogenetic trees were constructed using IQ-TREE with the following settings:
-bb 1000 -seed 123456 -m TESTNEW -mset GTR
The model selected for the Methanocorpusculum dataset was GTR + R3 and for the Methanobrevibacter dataset GTR + R5.
Endosymbiont enrichment
Isolated ciliates were cleaned from contaminating environmental prokaryotes by seven cycles of centrifugation at 400×g for 1 min and replacement of electromigration buffer [59] with filter-sterilized electromigration buffer (0.2 µm Minisart filter, Sartorius). Purified ciliates were then left at room temperature overnight to allow for digestion of phagocytosed prokaryotes. Ciliates were mechanically lysed using a sterile pestle. Enrichment for endosymbionts and removal of ciliate nuclear DNA was performed by spinning down lysed ciliates for 5 min at 400×g, thereby pelleting nuclei and large cell debris. The supernatant was then transferred to a new Eppendorf tube and centrifuged at 12,000×g for 5 min, and the supernatant was removed. The pellet was re-suspended in 200 µl sterile PBS and centrifuged at 400×g for 5 min to remove any remaining nuclei or cell debris. The supernatant was consecutively filtered through 5 µm Minisart filters (Sartorius) and 2.7 µm glass microfiber GF/D filters (Whatman) and finally centrifuged at 12,000×g for 15 min to pellet the endosymbionts [60].
Genome sequencing
The enriched endosymbionts from N. ovalis were sorted with fluorescent activated cell sorting (FACS) and their DNA amplified using multiple displacement amplification (MDA) at the Single Cell Genomics Centre at Bigelow Laboratory for Ocean Sciences (East Boothbay, ME, USA). A set of 19 single amplified genomes (SAGs), positively identified as the endosymbiotic archaea using PCR with endosymbiotic specific primers (Forward: 5′-GGATGGGTCTGCGGCCGATT-3′, Reverse: 5′-CCCAGGCGGCGGACTTAACA-3′), were selected and pooled together. Enriched endosymbionts from M. contortus were MDA-amplified using a REPLI-g Mini Kit (Qiagen) according to the manufacturer’s instructions.
Illumina sequencing libraries were generated and sequenced at the National Genomics Infrastructure sequencing platforms at the Science for Life Laboratory at Uppsala University. The library created from amplified SAGs from N. ovalis endosymbionts were sequenced with 2 × 100 bp paired-end sequencing using the Illumina HiSeq, and the M. contortus enriched endosymbiont library was sequenced as 2 × 250 bp paired-end sequences on the Illumina MiSeq. Sequencing primers were removed from read pairs using SeqPrep [61] and low quality data was filtered out using Trimmomatic [62], with the TRAILING:20 and MINLEN:(80 for N. ovalis and 200 for M. contortus) trimmers. A final screen for any remaining adapter sequences was performed by aligning all reads to the NCBI UniVec database using BLAST [63].
Genome assembly
The N. ovalis sequence data was assembled using SPAdes 3.5 [64] with k-mers 21,33,55,75, and the M. contortus data using SPAdes 3.0 with k-mer values of 33,55,75,101. Both assemblies were done using the parameters: –sc and –careful. Assemblies were screened for any human or phiX spike-in sequences by aligning the contigs against NCBI’s nt database using BLASTn [63]. Contigs smaller than 1000 bp were removed. Homologues for all proteins from each contig were identified using BLAST (using the NCBI nr database and an e-value of 1 × 10−4. All contigs contained at least 1 protein with best blast hit against Methanobrevibacter indicating that they are not contaminating contigs from other bacteria or archaea. All genome metrics were calculated using Quast 2.3 [65] and completeness was calculated with the in-house micomplete script [66], using a custom database where marker genes not found in genomes from the same genus (Methanobrevibacter and Methanocorpusculum, respectively) were removed.
Metagenomic binning
All contigs from the M. contortus dataset were aligned with BLAST against the genome of Methanocorpusculum labreanum Z, and any contigs that showed homology to M. labreanum Z with an E-value of 0 were used as a training dataset in PhymmBL v4.0 [67] in order to identify any remaining contigs. The endosymbiotic bin was verified by using ESOM (Emergent Self-Organizing Maps) 1.1 [68] using a minimum contig size of 2000 bp and maximum of 5000 bp. The output was overlaid with the bin from PhymmBL and both analyses showed a full overlap. Homologues for all proteins from each contig were identified using BLAST (using the NCBI nr database and an e-value cut-off of 1 × 10-4). All contigs contained at least 1 protein with best blast hit against Methanocorpusculum indicating that they are not contaminating contigs from other bacteria or archaea.
Standard information for SAG and MAG quality
The standard information suggested by [69] that is required for reporting SAG and MAG quality, as well as additional metadata, is presented in Supplementary Table 5.
Genome annotation
Protein coding genes were predicted using Prodigal 2.5 [70]. Genes encoding ribosomal RNA were predicted using RNAmmer 1.2 [71]. All predicted proteins were queried against the non-redundant database of NCBI with an E-value threshold of 10−4 using BLAST [63], and protein domains were predicted using InterProScan 5.16.55 [72]. Each protein was assigned to either an existing arCOG or to a newly created one, as described previously [73].
Absence of genes
Absent genes from key biosynthetic pathways were screened for in the original M. contortus metagenome by psi-blast of the relevant arCOGs against all proteins predicted in the metagenome. All hits were concatenated together with the known arCOG sequences and aligned using mafft-linsi [58] and trees were created using IQ-TREE [74] with the following parameters: -seed 123456 -fast -alrt 1000 -m TESTONLY. The resulting trees were then examined for any monophyletic groups between the predicted proteins of the metagenome and the species of the same genus as the endosymbiont (Methanocorpusculum).
Pseudogene identification in endosymbiont genomes
Pseudogenes were identified in several steps. Genes closer than 1000 bp from a contig edge were excluded and never annotated as true pseudogenes. A custom Perl script was used to identify candidates, genes located next to each other, where both shared their common best BLAST hit in the closest relative genome (M. arboriphilus for NOE and Methanocorpusculum labreanum in MCE). The NOE and MCE contigs were aligned with BLAST (tblastx) to the closest relative genome. Candidate pseudogenes were visually inspected in ACT [75] and annotated as pseudogenes if they aligned to consecutive sections of the matching gene. All genes which had a homologue in the closest genome were also visually inspected and those which were shorter than 75% of their homologue were annotated as pseudogenes. Genes that had no homologue in the closest relative genome were aligned to all proteins from genomes in the same genus and marked as pseudogene if their top hit had an expected value less than 1e−4 but were shorter than 75% of the top hit, and manually verified. For verifying that any stop codons in the annotated pseudogenes were not assembly artefacts, all reads of the assembly were mapped using bowtie2 [76] and inspected to verify that nonsense and frameshift mutations were involved. To compare with numbers of pseudogenes reported for the genomes of free-living relatives, pseudogenes were also predicted using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP), and any predicted pseudogenes within 1000 bp of a contig edge were ignored (also see below).
Selection of genome data for comparative analyses
For the comparative analyses, we decided to exclude partial genes at contig ends that had been automatically annotated as pseudogenes (Suppl. Table 1). Furthermore, genomes previously sequenced using 454 and IonTorrent sequencing platforms, which also appeared enriched for pseudogenes, were discarded from any comparison following an investigation into this issue (see below for details). Finally, we also excluded the published genome from a supposed archaeal endosymbiont from our analysis, which was cultured and deposited in a culture collection, Methanobacterium formicicum [77] and was isolated from its host Pelomyxa palustris. Since previous studies have failed to confirm that this is the actual endosymbiont of this species [8, 20, 78] based on FISH, it seems more likely that this isolate was a contaminating free-living methanogen. The fact that the 16S rRNA gene sequence of M. formicicum was identical to another cultured methanogen [8], which was supposedly isolated from the ciliate Metopus striatus, and that both these hosts happened to be isolated from the same aquarium [12, 79], also lends credibility to this idea.
Homopolymer detection in free-living methanogenic relatives
For all genomes from free-living members of Methanobrevibacter and Methanocorpusculum sequenced using technologies known to have issues with homopolymer sequences (≥3 identical nucleotides in a row; 454 and IonTorrent [80]). 20 ORFs annotated as pseudogenes were selected and aligned using mafft 7.017 [58] to the closest homologues from other free-living archaea (based on BLAST hits). Sites causing the pseudogenization were manually inspected to verify whether these were homopolymer sites. Genomes where a significant (>80%) of apparent pseudogenization events corresponded to homopolymeric sites were discarded from the analysis (as indicated in Suppl. Table 1).
Identification of pseudogenes in free-living methanogenic relatives
Genomes included in NCBI’s Refseq database have pseudogenes automatically annotated by PGAP (Prokaryotic Genome Annotation Pipeline). PGAP searches for truncated proteins, but does not take contig edges into account, which artificially inflates the number of pseudogenes. The number of pseudogenes thus correlates with the number of contigs in an assembly. In order to correct for this behaviour, we discarded pseudogenes within 1000 bp from a contig edge, as was done for the endosymbiont genomes. The pseudogenes were automatically annotated by identifying homologues in species of the same taxonomic order via BLAST (blastx), using a minimum query coverage of 50%, sequence identity of 50% and e-value of 1 × 10−4.
Furthermore, to assess potential bias in the manual pseudogenes detection in the endosymbiont genomes (see above) compared to the automated detection in the genomes of their free-living relatives, we also employed the automated PGAP method for the endosymbiont genomes. This analysis revealed no strong deviation, and retrieved about 90% of the pseudogenes detected using the manual methodology (data not shown).
Coding density
Gene coding density was calculated by dividing the total length of all non-overlapping protein coding genes with the total genome size. Pseudogenes were not considered coding sequences either for the endosymbionts or the free-living species. The species selected for the analysis consisted of all archaeal genomes in the NCBI RefSeq database (as of 5th September 2017). A single genome of each unique species taxonomy ID was selected in order to ensure redundant information was not included. All genomes sequenced using 454 or IonTorrent technology were removed, as discussed above. Genomes of the genera Methanobrevibacter and Methanocorpusculum missing from the RefSeq database were also included in the analysis. These include the genomes with the following assembly accessions: GCF_000320505.1, GCF_001639295.1, GCF_001639285.1, GCF_001639265.1 GCF_900109595.1, GCF_002208625.1, GCF_001729385.1, GCF_001729455.1 GCF_000621965.1, GCF_000430905.1 and GCF_002072215.
Trans-membrane and secretion signal domain identification
Signal peptides in the gained genes were identified in two steps. An initial prediction was made using PRED-SIGNAL [81]. Remaining proteins were again scanned using TMHMM 2.0c [82] and any predicted single trans-membrane helix located in the first 60 amino acids of the protein was scored as a signal peptide [82]. The remaining proteins with predicted trans-membrane helices were annotated as trans-membrane proteins.
Phylogenetic analysis
All multiple sequence alignments were performed with mafft-linsi 7.037b [58]. Positions missing in more than 50% of the sequences were removed with trimAl 1.4 [83]. Phylogenies from concatenated proteins were based on 57 single-copy orthologs listed in Supplementary Table 2. Phylogenetic trees of 18S rRNA genes and the 57 concatenated proteins were inferred with IQ-TREE 1.5.0a using the built-in modeltest, preselecting the GTR model for the rRNA and the LG matrix for the concatenated protein one. The concatenated protein dataset was analysed as follows:
-bb 1000 –mset LG –m TEST –madd LG + C10, LG +
C20, LG + C30, LG + C40, LG + C50, LG + C60.
The LG + C60 model was selected for the concatenated protein dataset [74].
Ancestral reconstruction
Clusters of orthologous groups were generated as previously described [66] and used to create a presence/absence matrix for all taxa in the euryarchaeal tree based on 57 marker genes (as described above). Reconstruction of gene flux was analysed using count [84] with the rates optimized using the gain-loss-duplication model with a Poisson distribution at the root. All parameters were set to default values. The family history was calculated using posterior probabilities. Threshold for gained and lost genes was set to 0.90. A second ancestral reconstruction was performed in the same way but with all identified pseudogenes removed prior to clustering of the orthologous groups.
Data availability
Sequence data for 16S rRNA amplicons are deposited to the NCBI Sequence Read Archive under GenBank BioProject PRJNA380999. Sequence data for Methanobrevibacter sp. NOE and Methanocorpusculum sp. MCE are available under DDBJ/EMBL/GenBank Biosamples SAMN06660699 and SAMN06660698 respectively.
References
Embley TM, Martin W. Eukaryotic evolution, changes and challenges. Nature. 2006;440:623–30.
Ettema TJ. Evolution: mitochondria in the second act. Nature. 2016;531:39–40.
Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–81.
Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol. 2017;27:R1177–92.
Gijzen HJ, Broers C, Barughare M, Stumm CK. Methanogenic bacteria as endosymbionts of the ciliate Nyctotherus ovalis in the cockroach hindgut. Appl Environ Microbiol. 1991;57:1630–4.
Hackstein JH. (Endo)symbiotic methanogenic archaea. Berlin: Springer Science & Business Media; 2010.
van Bruggen JJ, Stumm CK, Zwart KB, Vogels GD. Endosymbiotic methanogenic bacteria of the sapropelic amoeba Mastigella. FEMS Microbiol Lett. 1985;31:187–92.
Embley TM, Finlay B. Systematic and morphological diversity of endosymbiotic methanogens in anaerobic ciliates. Antonie Van Leeuwenhoek. 1993;64:261–71.
Akhmanova A, Voncken F, van Alen T, van Hoek A, Boxma B, Vogels G, et al. A hydrogenosome with a genome. Nature. 1998;396:527–8.
Embley TM, Finlay BJ, Dyal PL, Hirt RP, Wilkinson M, Williams AG. Multiple origins of anaerobic ciliates with hydrogenosomes within the radiation of aerobic ciliates. Proc R Soc Lond B. 1995;262:87–93.
Fenchel T, Finlay B. Production of methane and hydrogen by anaerobic ciliates containing symbiotic methanogens. Arch Microbiol. 1992;157:475–80.
van Bruggen JJ, Zwart KB, van Assema RM, Stumm CK, Vogels GG. Methanobacterium formicicum, an endosymbiont of the anaerobic ciliate Metopus striatus McMurrich. Arch Microbiol. 1984;139:1–7.
Hackstein J, de Graaf R, van Hellemond J, Tielens A. Hydrogenosomes of anaerobic ciliates. In: Tachezy J, editor. Hydrogenosomes and mitosomes: mitochondria of anaerobic eukaryotes. Berlin: Springer-Verlag; 2008. pp. 97–112.
Embley TM, Finlay BJ, Brown S. RNA sequence analysis shows that the symbionts in the ciliate Metopus contortus are polymorphs of a single methanogen species. FEMS Microbiol Lett. 1992;97:57–61.
Finlay B, Fenchel T. Polymorphic bacterial symbionts in the anaerobic ciliated protozoon Metopus. FEMS Microbiol Lett. 1991;79:187–90.
Esteban G, Fenchel T, Finlay B. Diversity of free-living morphospecies in the ciliate genus Metopus. Arch für Protistenkd. 1995;146:137–64.
Finlay B, Fenchel T. Hydrogenosomes in some anaerobic protozoa resemble mitochondria. FEMS Microbiol Lett. 1989;65:311–4.
Esteban G, Guhl BE, Clarke KJ, Embley TM, Finlay BJ. Cyclidium porcatum n. sp.: a free-living anaerobic scuticociliate containing a stable complex of hydrogenosomes, eubacteria and archaeobacteria. Eur J Protistol. 1993;29:262–70.
Goosen NK, Wagener S, Stumm CK. A comparison of two strains of the anaerobic ciliate Trimyema compressum. Arch Microbiol. 1990;153:187–92.
Gutiérrez G, Chistyakova LV, Villalobo E, Kostygov AY, Frolov AO. Identification of Pelomyxa palustris Endosymbionts. Protist. 2017;168:408–24.
Attwood G, Kelly W, Altermann E, Leahy S. Analysis of the Methanobrevibacter ruminantium draft genome: understanding methanogen biology to inhibit their action in the rumen. Aust J Exp Agric. 2008;48:83–8.
Rea S, Bowman JP, Popovski S, Pimm C, Wright A-DG. Methanobrevibacter millerae sp. nov. and Methanobrevibacter olleyae sp. nov., methanogens from the ovine and bovine rumen that can utilize formate for growth. Int J Syst Evolut Microbiol. 2007;57:450–6.
Wright A-DG, Ma X, Obispo NE. Methanobrevibacter phylotypes are the dominant methanogens in sheep from Venezuela. Microb Ecol. 2008;56:390–4.
Miller TL, Wolin M, de Macario EC, Macario A. Isolation of Methanobrevibacter smithii from human feces. Appl Environ Microbiol. 1982;43:227–32.
Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci USA. 2007;104:10643–8.
Finlay B, Esteban G, Clarke K, Williams A. Some rumen ciliates have endosymbiotic methanogens. FEMS Microbiol Lett. 1994;117:157–61.
Hackstein J, Stumm C. Methane production in terrestrial arthropods. Proc Natl Acad Sci USA. 1994;91:5441–5.
Broers CA, Meijers HH, Symens JC, Stumm CK, Vogels GD. Symbiotic association of Psalteriomonas vulgaris n. sp. with Methanobacterium formicicum. Eur J Protistol. 1993;29:98–105.
Goosen NK, Horemans AM, Hillebrand SJ, Stumm CK, Vogels GD. Cultivation of the sapropelic ciliate Plagiopyla nasuta Stein and isolation of the endosymbiont Methanobacterium formicicum. Arch Microbiol. 1988;150:165–70.
Tokura M, Tajima K, Ushida K. Isolation of Methanobrevibacter sp. as a ciliate-associated ruminal methanogen. J General Appl Microbiol. 1999;45:43–7.
Van Bruggen J, Zwart K, Hermans J, Van Hove E, Stumm CK, Vogels GD. Isolation and characterization of Methanoplanus endosymbiosus sp. nov., an endosymbiont of the marine sapropelic ciliate Metopus contortus Quennerstedt. Arch Microbiol. 1986;144:367–74.
Embley TM, Finlay BJ. The use of small subunit rRNA sequences to unravel the relationships between anaerobic ciliates and their methanogen endosymbionts. Microbiology. 1994;140:225–35.
McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26.
Boscaro V, Felletti M, Vannini C, Ackerman MS, Chain PSG, Malfatti S, et al. Polynucleobacter necessarius, a model for genome reduction in both free-living and symbiotic bacteria. Proc Natl Acad Sci USA. 2013;110:18590–5.
Toft C, Andersson SG. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 2010;11:465–75.
Matthews RG. Cobalamin-dependent methyltransferases. Acc Chem Res. 2001;34:681–9.
Khelaifia S, Garibal M, Robert C, Raoult D, Drancourt M. Draft genome sequencing of Methanobrevibacter oralis strain JMR01, isolated from the human intestinal microbiota. Genome Announc. 2014;2:e00073-14.
Ferrari A, Brusa T, Rutili A, Canzi E, Biavati B. Isolation and characterization of Methanobrevibacter oralis sp. nov. Curr Microbiol. 1994;29:7–12.
McCutcheon JP, McDonald BR, Moran NA. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci USA. 2009;106:15394–9.
Izawa K, Kuwahara H, Kihara K, Yuki M, Lo N, Itoh T, et al. Comparison of intracellular “Ca. Endomicrobium Trichonymphae” genomovars illuminates the requirement and decay of defense systems against foreign DNA. Genome Biol Evol. 2016;8:3099–107.
Yang JC, Madupu R, Durkin AS, Ekborg NA, Pedamallu CS, Hostetler JB, et al. The complete genome of Teredinibacter turnerae T7901: an intracellular endosymbiont of marine wood-boring bivalves (Shipworms). PLoS ONE. 2009;4:e6085.
Schmitz-Esser S, Tischler P, Arnold R, Montanaro J, Wagner M, Rattei T, et al. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J Bacteriol. 2010;192:1045–57.
Dale C, Plague GR, Wang B, Ochman H, Moran NA. Type III secretion systems and the evolution of mutualistic endosymbiosis. Proc Natl Acad Sci USA. 2002;99:12397–402.
Al-Khodor S, Price CT, Kalia A, Kwaik YA. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 2010;18:132–9.
Jernigan KK, Bordenstein SR. Ankyrin domains across the Tree of Life. PeerJ. 2014;2:e264.
Schmitz-Esser S, Linka N, Collingro A, Beier CL, Neuhaus HE, Wagner M, et al. ATP/ADP translocases: a common feature of obligate intracellular amoebal symbionts related to Chlamydiae and Rickettsiae. J Bacteriol. 2004;186:683–91.
van Hoek AHAM, van Alen TA, Sprakel VSI, Leunissen JAM, Brigge T, Vogels GD, et al. Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates. Mol Biol Evol. 2000;17:251–8.
Moran NA. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc Natl Acad Sci USA. 1996;93:2873–8.
Fenchel T, Finlay BJ. The biology of free-living anaerobic ciliates. Eur J Protistol. 1991b;26:201–15.
Lewis WH, Sendra KM, Embley TM, Esteban GF. Morphology and phylogeny of a new species of anaerobic ciliate, Trimyema finlayi n. sp., with endosymbiotic methanogens. Front Microbiol. 2018;9:140.
Hoek AHAMV, Sprakel VSI, Alen TAV, Theuvenet APR, Vogels GD, Hackstein JHP. Voltage-dependent reversal of anodic galvanotaxis in Nyctotherus ovalis. J Eukaryot Microbiol. 1999;46:427–33.
Stahl A, Amann R. Development and application of nucleic acid probes. Nucleic acid techniques in bacterial systematics. Chichester: John Wiley & Sons;1991. pp. 205–48.
Wallner G, Amann R, Beisker W. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry. 1993;14:136–43.
Daims H, Stoecker K, Wagner M. Fluorescence in situ hybridization for the detection of prokaryotes. Mol Microb Ecol. 2005;213:239.
Ovreås L, Forney L, Daae FL, Torsvik V. Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol. 1997;63:3367–73.
Jorgensen SL, Hannisdal B, Lanzén A, Baumberger T, Flesland K, Fonseca R, et al. Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic Mid-Ocean Ridge. Proc Natl Acad Sci USA. 2012;109:E2846–55.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
van Hoek AH, van Alen TA, Sprakel VS, Hackstein JH, Vogels GD. Evolution of anaerobic ciliates from the gastrointestinal tract: phylogenetic analysis of the ribosomal repeat from Nyctotherus ovalis and its relatives. Mol Biol Evol. 1998;15:1195–206.
Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SGE. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 2013;9:e1003381.
St. John, J. SeqPrep. 2011. https://github.com/jstjohn/SeqPrep.
Bolger AM, Lohse M, Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov A, S., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5.
Spang A, Saw JH, Jørgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015;521:173–9.
Brady A, Salzberg SL. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Methods. 2009;6:673–6.
Ultsch A, Mörchen F. 2005. ESOM-Maps: tools for clustering, visualization, and classification with Emergent SOM. Marburg: University of Marburg.
Bowers RM, Kyrpides CN, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–32.
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2011;40:gkr948.
Saw JH, Spang A, Zaremba-Niedzwiedzka K, Juzokaite L, Dodsworth JA, Murugapiran SK, et al. Exploring microbial dark matter to resolve the deep archaeal ancestry of eukaryotes. Philos Trans R Soc B. 2015;370:20140328.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis comparison tool. Bioinformatics. 2005;21:3422–3.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Gutiérrez G. Draft genome sequence of Methanobacterium formicicum DSM 3637, an archaebacterium isolated from the methane producer amoeba Pelomyxa palustris. J Bacteriol. 2012;194:6967–8.
Fenchel T, Finlay BJ. Ecology and evolution in anoxic worlds. Oxford: Oxford University Press; 1995.
van Bruggen JJ, Stumm CK, Vogels GD. Symbiosis of methanogenic bacteria and sapropelic protozoa. Arch Microbiol. 1983;136:89–95.
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–9.
Bagos P, Tsirigos K, Plessas S, Liakopoulos T, Hamodrakas S. Prediction of signal peptides in archaea. Protein Eng Des Sel. 2008;22:27–35.
Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Csűös M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–2.
Acknowledgements
We thank Prof. Genoveva Esteban for help in isolating and identifying cultures of M. contortus, and we are grateful to Dr. Nicole Poulton for the initial evaluation of N. ovalis lysates. We would also like to thank Roel van Eijk and Lina Juzokaite for their help with sequencing library construction and Daniel Lundblad for his help in establishing cockroach cultivation. All sequencing was performed by the National Genomics Infrastructure sequencing platforms at the Science for Life Laboratory at Uppsala University, a national infrastructure supported by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation. We thank the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) at Uppsala University and the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High-Performance Computing for providing computational resources. This work was supported by grants of the Swedish Research Council (VR grant 621-2009-4813), the European Research Council (ERC Starting grant 310039-PUZZLE_CELL) and the Swedish Foundation for Strategic Research (SSF-FFL5) to TJGE and to TME (ERC Advanced grant 20100317-EUKORIGINMIT).
Author contributions
TJGE conceived and supervised the study. AEL isolated the endosymbionts and performed genomic sequence assemblies and analysis. AEL performed comparative genomics and phylogenomics analyses with occasional support from LG. WHL and TME isolated and cultured M. contortus samples and performed the fluorescent in situ hybridization experiments, as well as microscopic imaging. AS performed the metabolic analysis. AEL and TJGE wrote the manuscript, which was edited and approved by all authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Lind, A.E., Lewis, W.H., Spang, A. et al. Genomes of two archaeal endosymbionts show convergent adaptations to an intracellular lifestyle. ISME J 12, 2655–2667 (2018). https://doi.org/10.1038/s41396-018-0207-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0207-9
This article is cited by
-
Symbionts of Ciliates and Ciliates as Symbionts
Indian Journal of Microbiology (2024)
-
The energy metabolism of Balantidium polyvacuolum inhabiting the hindgut of Xenocypris davidi
BMC Genomics (2023)
-
Comparative genomic analysis of Methanimicrococcus blatticola provides insights into host adaptation in archaea and the evolution of methanogenesis
ISME Communications (2021)
-
The host-associated archaeome
Nature Reviews Microbiology (2020)
