
Abstract
Marine net community production (NCP) tracks uptake of carbon by plankton communities and its potential transport to depth. Relationships between marine microbial community composition and NCP currently remain unclear despite their importance for assessing how different taxa impact carbon export. We conducted 16 and 18S rRNA gene (rDNA) sequencing on samples collected across the Western North Atlantic in parallel with high-resolution O2/Ar-derived NCP measurements. Using an internal standard technique to estimate in-situ prokaryotic and eukaryotic rDNA abundances per liter, we employed statistical approaches to relate patterns of microbial diversity to NCP. Taxonomic abundances calculated using internal standards provided valuable context to traditional relative abundance metrics. A bloom in the Mid-Atlantic Bight featured high eukaryote abundances with low eukaryotic diversity and was associated with the harmful algal bloom-forming Aureococcus anophagefferens, phagotrophic algae, heterotrophic flagellates, and particle-associated bacteria. These results show that coastal Aureococcus blooms host a distinct community associated with regionally significant peaks in NCP. Meanwhile, weak relationships between taxonomy and NCP in less-productive waters suggest that productivity across much of this region is not linked to specific microplankton taxa.
Similar content being viewed by others

Introduction
Uptake of carbon by phytoplankton and its exchange between organisms in the marine environment plays a critical role in the carbon cycle, with primary production in the world’s oceans representing half of global net primary production [1]. A small proportion of surface production [2] is transported to depth via sinking particles, subduction, and other processes, transferring carbon to deep ocean pools with a residence time of millennia or longer [3]. Considerable interest has consequently focused on exploring relationships between surface microbial community structure, marine production [4,5,6], and particulate carbon export [7,8,9].
Net community production (NCP) rates reflect the productivity and metabolic balance of the surface ocean microbial community. Expressed as the difference between gross primary production and community respiration, NCP rates estimate the mixed-layer production of organic carbon available for export [10,11,12,13]. NCP patterns have been well-examined independently, as have patterns of surface ocean community structure. However, direct comparison of relationships between ecology and productivity remains an emerging line of investigation.
The Western North Atlantic is a region of interest for unraveling potential links between community structure and productivity. New production across this region is thought to be driven by a variety of physical and biological processes including nitrogen fixation, mesoscale features, seasonal mixing, and allochtonous nutrient inputs [14,15,16].
A dominant feature of the Western North Atlantic is the Sargasso Sea, an oligotrophic region typical of other subtropical gyre systems [17]. While spring and winter phytoplankton blooms occur following winter mixing of nutrients into the surface layer, the Sargasso Sea in summer exhibits limiting nitrate and phosphate concentrations (N < 50 nmol kg−1, P < 20 nmol kg−1) [18]. Ongoing changes in the biogeochemistry of the Sargasso Sea may impact community composition, carbon export, and nutrient cycling due to increasing stratification [19] and changing nutrient inputs [20]. Records suggest gradual community shifts are underway, with haptophyte populations declining and Synechococcus and dinoflagellate groups increasing in abundance [20, 21]. Globally, oligotrophic subtropical gyres cover some 40% of the planet’s surface [22], and small shifts in microplankton ecology in such regions may have repercussions for biogeochemistry and climate. Large-scale genomics sampling work has suggested that specific key taxa may be important drivers of carbon export in such regions [7].
To the west, the Western North Atlantic is bounded by the North American continental shelf. High rates of production are observed along this coast well into summer [23]. In this region, the shelf, shelf break, shelf slope, and Gulf Stream exert dynamic physical forcings upon resident microplankton, driving variation in community structure and primary production over short transects [24]. Such coastal regions are increasingly being recognized as potentially important carbon sinks [25] and are also predicted to undergo future ecological shifts in response to eutrophication and climate change [26,27,28].
Considering ongoing shifts in microplankton community structure in ecosystems across the Western North Atlantic, evaluating the impact of future community shifts upon primary production and potential carbon export in this region is of great interest. There is thus a need to identify relationships between community composition and NCP. Few regional NCP measurements have been conducted in the Western N. Atlantic to date, with existing NCP data generally coming from time-series measurements [29] or fine-scale studies [30, 31]. Similarly, while community structure at the Bermuda Atlantic Time Series (BATS) has been regularly studied [32, 33], broader rDNA amplicon data surveying the whole region are far sparser.
In this study, we gathered samples for high-throughput 16 and 18S rDNA amplicon sequencing and concurrently conducted high-resolution O2/Ar-based NCP measurements using Equilibrator Inlet Mass Spectrometry (EIMS) [34] along three transects spanning the oligotrophic Sargasso Sea, the Gulf Stream, and the U.S. East Coast. To obtain absolute taxonomic abundances for the sampled communities, we adapted an internal standard approach for 16 and 18S rDNA sequencing to quantitatively characterize community structure [35]. We then assessed trends in whole-community composition and diversity in relation to NCP and evaluated associations between productivity and specific microplankton groups identified in our samples.
Materials and methods
Study Area and collection of O2/Ar and ancillary data
Continuous and discrete measurements were collected over a 3 100 km transect in the western North Atlantic aboard the R/V Atlantic Explorer from 3–12 August 2015. The cruise track progressed west from the BATS Station (32.3°N, −64.6°W) to the North Carolina coast, then northeast to ~50 km south of Long Island, New York before returning to Bermuda (Fig. 1). Fourteen CTD casts were conducted during the cruise at 200–400 km intervals. Underway dissolved O2/Ar measurements were collected alongside discrete sampling for chlorophyll and DNA. O2/Ar was measured continuously from the ship’s underway intake using the EIMS method [34]. Details of O2/Ar-derived NCP calculations and assessment of potential vertical O2/Ar fluxes are described in the Supplementary Methods.

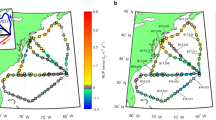
a Map of O2/Ar-derived volumetric NCP measured using EIMS in early August 2015 across the western Sargasso Sea. Positive rates are shown using a warm color scale, while net negative measurements are shown with a cool color scale. Gray values indicate balanced NCP rates. Locations at which samples were collected for community rDNA sequencing are indicated with open circles. b Time series of O2/Ar-derived NCP measurements, with molecular sampling stations indicated by black stars
Microbial community sampling and rDNA amplicon sequencing
Samples for rDNA analysis were obtained from 5 m CTD casts and underway samples (Table S2) pumped from a towfish trailing abeam of the vessel at 3–5 m depth. This custom-built towfish, suspended alongside, is trace metal-clean, using plastic tubing and carrying seawater aboard via an air-driven pump. For each sample, one liter was filtered through a 0.22-μm filter (Millipore, Billerica, MA, USA) using a peristaltic pump, preserved with RNAlater (Thermo Fisher, Waltham, MA, USA), and flash-frozen in liquid nitrogen. At stations with high biomass, the volume of filtrate was reduced to 0.2–0.5 l as filters became clogged.
Internal controls for quantitative sequencing
A quantitative internal standard approach provides information on per-liter abundance of taxa across samples, yielding more meaningful comparisons between taxonomic abundances and biological rate measurements. To quantify rDNA copy numbers l−1, internal genomic standards were added to each sample following [35]. Genomic DNA was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) for Thermus Thermophilus (ATCC #27634D-5), a thermophilic hot springs bacterium, and Schizosaccharomyces pombe (ATCC #24843D-5), a yeast species. The S. pombe genome contains ~110 copies of the 18S V4 rDNA amplicon [35], while the T. thermophilus genome contains two 16S V4 copies [36].
Given the large range in 18S rDNA copy number across eukaryotic genomes, we determined an appropriate spike of control DNA (0.073 ng) by evaluating the average 18S rDNA concentration in our samples using qPCR with 18S V4 primers. To ensure that such a diluted spike would reliably manifest in sequencing output, we conducted a pilot sequencing run on duplicate filters from this study at the Boston University Microarray Core on an Ion Torrent PGM using a 314 chip (Supplementary Methods).
The Ion Torrent test revealed that a 15.2 ng T. thermophilus genomic DNA spike resulted in T. thermophilus reads comprising an average of 5.3% of all reads, while the addition of 0.679 ng of S. pombe gDNA yielded 0.9% S. pombe reads. Based upon these results, we adjusted the spike amounts to quantities expected to constitute <1% of sequenced reads, adding 0.679 ng of the S. pombe standard and 3.04 ng of the T. thermophilus standard to each sample, both in 50 μl volumes. This corresponded to adding c.a. 5 780 000 rDNA copies sample−1 of S. pombe and 2 800 000 rDNA copies sample−1 of T. thermophilus genomic DNA.
DNA extraction for 16S and 18S rDNA sequencing
We conducted DNA extraction using the Qiagen DNeasy Plant Mini Kit (Qiagen, Germantown, MD, USA) following manufacturer instructions with slight modifications [37], with internal gDNA standards added prior to bead-beating [38]. PCR amplification was performed for 30 cycles using custom 16S V4 primers 515F-Y (5′-GTGYCAGCMGCCGCGGTAA-3′) and 805 R (5′-GACTACNVGGGTATCTAAT-3′) and 18S V4 primers F (5′-CCAGCASCYGCGGTAATTCC-3′) and R (5′-ACTTTCGTTCTTGAT-3′), with attached Illumina adapters and barcodes (Supplementary Table 3). These primers are adapted from widely-used universal primers for the amplification of marine prokaryotic [39, 40] and eukaryotic [41] taxa, modified to improve coverage of SAR11 and haptophytes [5, 42]. Primers were each dual-indexed with 6 bp barcodes, using a heterogeneity spacer approach [43, 44]. 16S samples were run at 94 °C for 3 min, 30 cycles at 94 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, followed by a third stage at 72 °C for 10 min. 18S samples were run identically apart from an annealing temperature of 57 °C. Each 16S PCR reaction (25 μl volume) consisted of 2.5 μl 10 × PCR buffer, 0.5 μl dNTP mix (10 μM each), 1 μl 50 mM MgSO4, 0.5 μl each of forward and reverse primer (10 μM), 0.1 μl Platinum Taq Hi-Fidelity Polymerase (Thermo Fisher, Waltham, MA, USA), and 19.4 μl of sterile water. 18S PCR reaction mixtures were identical except polymerase amounts were doubled (0.2 μl per reaction) to address weak amplification, with a compensating water volume decrease to 19.3 μl. PCR products were purified using the Qiagen QIAquick PCR Purification Kit and quantified using a Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA). The samples were then pooled at equimolar concentrations and sequenced using the Illumina MiSeq platform (300 bp PE, V3 chemistry) at the Duke Center for Genomic and Computational Biology.
Analysis pipeline
We obtained 20 450 700 single-end reads from our 25 sequenced samples. Raw single-end reads were trimmed to remove barcodes, assembled, and quality filtered following [43] using pandaseq [45]. 16S amplicon length was 296.7 + / − 2.9 bp (mean + / − sd), while mean 18S amplicon length was 424.1 + / − 4.3 bp. Demultiplexing was performed in QIIME [46]. Five 18S rDNA samples and one 16S rDNA sample contained no reads, the former likely due to a defective forward primer. Primer and other non-biological sequences were subsequently removed using Tagcleaner [47]. We conducted chimera detection and open-reference OTU picking at 97% similarity using the Usearch 6.1 algorithm [48, 49] and Release 123.1 of the SILVA database [50]. OTU clustering was performed using the usearch61 method for de novo OTU picking, and the usearch61_ref method for reference-based OTU picking. Alignment was performed using PyNAST [51] and taxonomy assignment conducted using the RDP classifier 2.2 [52]. Full sequence processing scripts are included in the Supplementary Material. Following taxonomy assignment, internal standard DNA sequences, eukaryotic metazoans, and plastid 16S sequences were filtered out using the QIIME script ‘filter_taxa_from_otu_table.py’. We further discarded one sample due to the low volume of filtrate, leaving 19 eukaryotic and 23 prokaryotic samples.
Sample diversity metrics were calculated for 16S and 18S datasets using the phyloseq package [53] for R 3.4.1 [54]. For alpha diversity analyses only, sample libraries were rarefied to the smallest library size in each set of samples (16S: 98 819; 18S: 33 245). Rarefaction curves begin to level off at the sequencing depths obtained, suggesting that depth was sufficient to represent major patterns of diversity in our samples (Figure S1). Alpha diversity metrics (observed OTUs and Shannon diversity) were calculated using averages from five rarefactions.
Using our non-rarefied sample libraries, calculation of absolute abundances for each OTU was performed following [38]:
where V is the volume filtered and R represents the recovery ratio of internal standards (genomic standards sequenced/molecules of genomic standard added). Output OTU tables are included in the Supplementary Material (Supplementary Tables 5a, 5b)
Further details of downstream statistical analyses including ordination and PLS regression are described in the Supplementary Methods.
Results and Discussion
Patterns of O2/Ar-derived NCP
Underway O2/Ar-derived biological oxygen fluxes within the mixed layer ranged from −2.4 to 17.4 mmol O2 m−3 day−1 (MLD-integrated rates of −25–190 mmol O2 m−2 day−1) (Fig. 1). We observed initial rates below 0.5 mmol O2 m−3 day−1 in the open ocean, increasing to 1 mmol O2 m−3 day−1 within 400 km of the coast. Turning north, fluxes reached 2–4 mmol O2 m−3 day−1 along the Carolina coast. Values were subsequently variable along the coast.
The highest O2/Ar supersaturation occurred at the expedition’s northernmost extent within a productive phytoplankton bloom, with values peaking at 17.4 mmol O2 m−3 day−1 south of Long Island. Passing this bloom, O2/Ar supersaturation declined again to typically below 1 mmol O2 m−3 day−1 during transit back to Bermuda.
We assessed the potential contribution of eddy diffusive and entrainment fluxes to mixed-layer O2/Ar values as minimal (Supplementary Methods). Consequently, we report all biological O2 fluxes as NCP rates henceforth. Except when comparing our data with integrated figures from other literature, we also report rates throughout this manuscript as volumetric values, more suitable for relation to quantitative taxonomic abundances.
Overall, our high-resolution NCP measurements agree well with previously measured patterns, with low NCP rates observed in the open ocean and higher values over the continental shelf along the Mid-Atlantic Bight. The marked peak in productivity at the northern end of the expedition coincided with high measured nitrogen fixation rates [55] and high Chl a. Peak MLD-integrated productivity, reaching 190 mmol O2 m−2 day−1 (136 mmol C m−2 day−1 assuming a photosynthetic quotient of 1.4 [56]), is of a similar magnitude as integrated 14C-derived primary production rates for the Mid-Atlantic Bight spring bloom of up to 158 mmol C m−2 day−1 [57]. Our observed rates are also comparable to summer peak photic-zone primary production of between 145 and 190 mmol C m−2 day−1 modeled for the same area using profile observations [24].
Our low MLD-integrated open-ocean NCP rates, with a mean of 2.2 mmol O2 m−2 day−1, are also consistent with prior Sargasso Sea O2/Ar-based estimates in September/October of 1.1–3.4 mmol O2 m−2 day−1 [30], as well as modeled summer regional NCP values of 3–4 mmol O2 m−2 day−1 [58].
Microbial community quantitative and relative abundance patterns
Analysis of rDNA reads yielded 7 843 eukaryotic and 5 604 prokaryotic OTUs across 19 eukaryotic and 23 prokaryotic samples (Supplementary Table 6). 16S and 18S samples contained at least 98 819 and 33 245 reads per sample.
Our observations of 16S and 18S rDNA abundances per liter were within expected bounds. Bacterial 16S rDNA abundances of 1.78 × 108–5.4 × 109 copies l−1 are consistent with bacterial abundances in the Sargasso Sea and Western North Atlantic of 4.0 × 108–2.3 × 109 cells l−1 [59, 60], assuming a typical 16S copy number of 1–15 [61]. Excluding the three highest NCP stations, where the highest 18S rDNA abundances were observed (range of 1.43 × 108–3.14 × 1010 18S rDNA genes l−1), the median 18S rDNA abundance was 1.4 × 109 sequences l−1.This is high compared with surface ocean eukaryotic cell densities of 1 × 107 protists l−1 and 1 × 106 phytoplankton l−1 [62], but is likely driven by variation in 18S rDNA copy number. Peak 18S rDNA abundances, while high, are also reasonable. Phaeocystis blooms can reach cell counts of 1.5 × 108 cells l−1 [63], and Aureococcus blooms of 6 × 108 cells l−1 have been observed along the Long Island Coast [64].
Absolute abundances of individual taxa are also consistent with previous observations. For example, the median SAR11 16S rDNA abundance in our samples (Fig. 2b) was 6.2 × 108 rDNA genes l−1 (SAR11 contains one 16S gene copy cell−1), compared with previous measurements of 2 × 108 SAR11 cells l−1 in the Sargasso Sea from fluorescence in-situ hybridization counts [65]. Similarly, we observed a median of 1.9 × 108 Prochlorococcus 16S rDNA genes l−1 in our samples (Fig. 2b), consistent with Western North Atlantic observations of 1 × 108 cells l−1 based on qPCR quantification and flow cytometry [66, 67]. Applications of the internal standard approach for samples collected in the lower Amazon River, the Southern Ocean, as well as in soil samples have also demonstrated good correspondence between the standard-derived abundances and complementary abundance data measured using epifluorescence microscopy, photosynthetic pigments, flow cytometry, phospholipid fatty acid analysis, and substrate-induced respiration approaches [35, 36, 68].

rDNA abundances across sampled stations for a Aureococcus anophagefferens b SAR11 and Prochlorococcus
Notably, calculation of absolute taxonomic abundances using internal standards produces patterns distinct from those generated using relative abundance metrics (Fig. 3). This is evident among several abundant 18S and 16S OTUs, including SAR11 clade members, as well as the protist clades Dinoflagellata, Gonyaulacales, Alveolata, and Gymnodiniphycidae. The latter four eukaryotes increase in absolute abundance within the bloom environment, while their relative abundances decrease due to the dominance of Chrysophyceae and Aureococcus anophagefferens within these samples. A similar phenomenon affects SAR11 relative abundances, which are highest between S2–S9 and S20–S25 due to lower 16S rDNA counts for other prokaryotes at those stations. These discrepancies highlight longstanding criticisms of traditionally-used relative abundance metrics [36, 38, 69,70,71,72] and illustrate advantages offered by the internal standard approach. In addition, avoidance of issues caused by compositional community data [73, 74] is valuable when relating taxonomic abundances to microbial or biogeochemical processes like NCP.

a Comparison of patterns of absolute abundance, calculated using an internal standard approach, to relative abundance for the SAR11 clade of alphaproteobacteria, as well as four groups of eukaryotic protists: b Cryptophyceae, c Retaria, d Gymnodiniphycidae, and e Gonyaulacales. f Scatter plot of 16S and 18S rDNA absolute vs. relative abundance data, with points colored for each prokaryotic (circles) and eukaryotic (diamonds) taxon across all samples, binned at the 5th taxonomic rank
The internal standard approach is nonetheless subject to several assumptions and limitations. A key assumption is that recovery rates of DNA standards are comparable to those of natural sequences within the sample. Particularly given the general implications of primer biases in amplicon work, this premise warrants further investigation. Recovery rate differences due to amplification bias would not alter how the quantitative abundance pattern of a sampled taxon changes across samples, but might result in discrepancies between estimated and actual in-situ abundances. Another important limitation is that quantitative abundance data produced by this method remain sensitive to differences in rDNA copy number across taxa. Although better knowledge of 16S copy number variation across prokaryotes has spurred efforts to correct for copy number differences [75], existing datasets remain limited particularly for eukaryotes, in which rDNA copy number may vary by multiple orders of magnitude. As data collection continues, corrections will likely become more feasible and commonplace.
Among eukaryotes, dinoflagellate lineages dominated all samples except three from the coastal bloom (S14–S16) (Fig. 4a, b). Most of these dinoflagellate sequences corresponded to Syndiniales, alveolate parasites infecting various marine organisms and often detected at high abundances using molecular tools [76,77,78]. While many of these sequences may originate from endosymbionts inside metazoan zooplankton caught on our filters, Syndiniales also infect microzooplankton protists, including ciliates, cercozoa, and other dinoflagellates, and clades targeting both host categories often exhibit a short free-living life stage [76]. Consequently, these sequences may also represent organisms living outside of metazoan hosts, interacting within the marine microbial environment. To a degree, elevated dinoflagellate abundances observed may also reflect high 18S copy numbers, driven by large dinoflagellate genomes [79, 80].

Bar plots of a 18S eukaryotic taxonomy shown at the 5th taxonomic rank using relative abundance and b rDNA gene abundance, and c 16S prokaryotic taxonomy shown at the 4th taxonomic rank using relative abundance and d rDNA abundance
Two samples (S14, S15) associated with the coastal bloom were dominated (>90% relative abundance) by Aureococcus anophagefferens, a pelagophyte that forms coastal “brown tide” harmful algal blooms (HABs) [81], as well as Chrysophyceae (Fig. 4a, b). qPCR surveys have also detected A. anophagefferens at low abundances in pelagic waters, which some suggest indicates an oceanic origin for this nuisance algae [82]. A wide distribution of A. anophagefferens is also supported by our study. We found Aureococcus present in 16 of 19 18S rDNA samples, with a mean of 7.5 × 104 Aureococcus 18S rDNA genes l−1 observed in non-bloom samples. We estimated abundances of 4.4–6.6 × 104 18S rDNA genes l−1 in open-ocean samples (S24, S25) collected near Bermuda. In comparison, estimated Aureococcus 18S rDNA gene abundances ranged between 1.8 × 108 and 2.0 × 1010 rDNA genes l−1 within the observed bloom (Fig. 2a).
Sample 16 featured a high population (~20%) of Prymnesiales, primarily Chrysochromulina and Chrysoculter. Chrysochromulina are another nuisance algae, capable of mixotrophy [83], and forming blooms that can cause fish kills [84]. Other members of Prymnesiales produce harmful hemolytic compounds [85]. Eukaryotic diversity was lower at two bloom stations, S14 and S15, (Supplementary Figure 2) but was similar across our other samples.
Among bacterioplankton, SAR11, SAR86 clade members (appearing as Oceanospirillales in Fig. 4), and Prochlorococcus (Subsection I cyanobacteria) dominated the communities sampled (Fig. 4c, d). The AEGEAN-169 clade of Alphaproteobacteria (Rhodospirillales), as well as MGII Archaea (Thermoplasmatales) also appeared at high proportional abundances. Within the northern bloom, we observed elevated abundances of Planctomycetales, Flavobacteria, Sphingobacteriales, and Order III Cytophagia, with Phycisphaerales appearing at particularly high abundances (>10%) at two stations. Not much is currently known about Phycisphaerales, although they are hypothesized to form associations with macroalgae, with many representatives facultatively anaerobic [86]. In addition, these bloom samples also appear to contain more sequences belonging to less-abundant and “rare” taxa (labeled ‘Other’ in Fig. 4). This phenomenon of elevated abundances of “rare” taxa in bloom events has also been reported elsewhere and may be related to ecological associations with phytoplankton [87, 88]. Bacterial diversity across samples was more uniform than eukaryotic diversity, with prokaryotic Shannon diversity between 4.1–4.5 versus 2.4–5.7 for eukaryotic samples (Supplementary Figure 2).
Relationships between microbial community structure and NCP
At the community level, we observed a negative relationship between measured NCP and eukaryotic Shannon’s H diversity (Pearson: −0.81, Spearman: −0.76, p « 0.01 for both) (Fig. 5), which was strongly driven by low diversity at two highly productive stations. This relationship does not remain significant with those samples excluded (Pearson: −0.56, Spearman: −0.61, p > 0.01). We observed no relationship between prokaryotic diversity and NCP.

a Linear regression of NCP vs. the number of observed 18S eukaryotic OTUs per sample. b Linear regression of NCP vs. 18S Shannon alpha diversity across sample libraries. Dashed lines indicate the resulting regressions when bloom stations (open circles) are excluded. Significance and goodness-of-fit are provided in boldface for the full dataset, and in italics for the dataset with bloom station data excluded
Recent debate over the nature of the relationship between marine microplankton diversity and productivity has been energetic. Any overall relationship between community diversity and productivity would reflect the relative importance of functional diversity, cooperation, competitive exclusion, selective feeding by grazers, and other factors in governing ecosystem production [6, 89, 90]. Earlier research suggests a peak of phytoplankton diversity at locations with moderate production, with decreasing diversity observed for less-productive and highly productive sites [91, 92]. Dominance of a handful of taxa beyond the control of grazers may explain decreased diversity at high productivity. Increased diversity at moderate productivity rates may reflect selective feeding pressures that allow coexistence between a higher diversity of taxa. Within the Western North Atlantic, our data supports the view that the most productive marine communities may exhibit relatively low eukaryotic diversity, a result consistent with meta-analysis and model-based findings that the most productive communities are among the least diverse [6].
Principal coordinate analysis (PCoA) of both prokaryotic and eukaryotic samples demonstrated distinctions between coastal bloom and other samples (Fig. 6), indicating community dissimilarities. Linear regressions of environmental parameters against the first principal component revealed significant correlations between NCP, temperature, latitude, Chlorophyll, and PC1 for both our 18 and 16S datasets (Supplementary Table 1), suggesting associations between these parameters and community structure. None of these trends remained significant once data from bloom stations S14, S15, and S16 were excluded, however, indicating that these relationships were driven largely by these samples, which possess distinctive community structure, high Chl and NCP, and low water temperatures compared to all other stations.

Principal Coordinates Analysis of a eukaryotic 18S and b bacterial/archaeal 16S samples, ordinated by Bray-Curtis dissimilarity. Samples are color coded based on general region of origin, as visually determined from OSCAR ocean current data for the date of sampling (https://earth.nullschool.net/), with green indicating stations located within the Gulf Stream and with red and blue samples originating from waters inshore and offshore of the current, respectively
Relationships between NCP and specific microplankton taxa
Partial Least Squares (PLS) regression analysis revealed groups of prokaryotic and eukaryotic taxa associated with high volumetric NCP rates (Supplementary Tables 4a-4f), with these relationships again strongly driven by the bloom community. Eukaryotic taxa associated with NCP included Ochrophyta, Aureococcus anophagefferens, picozoa, cryptophytes, prymnesiophytes, and stramenopiles, such as several uncultured MArine STramenopile (MAST) clades (Fig. 7b).

Heatmaps showing strength of correlations, determined using Partial Least Squares regression analysis between a prokaryotic and b eukaryotic taxa and environmental parameters, including NCP. Plots were produced at the fourth taxonomic rank for prokaryotes and eukaryotes. Strength of correlations is denoted by the color scale, with negative and positive correlations in blue and red tones, respectively. Rows bounded by black rectangles indicate taxa with a correlation with NCP greater than 0.2. (Full tables of correlation coefficients included with supplementary material). Dendrograms link taxa with similar relationships to the set of variables, and vice versa
Many of these protists are commonly associated with phytoplankton bloom conditions. Aureococcus anophagefferens possesses a large genome optimized for uptake of ambient dissolved organic carbon and nitrogen and is adapted for fast growth under turbid, low-light conditions [81, 93]. Members of Chrysophyceae also form blooms and practice phagotrophy, engulfing, and processing particulate matter [94]. The high abundance of these two taxa within the bloom implies an environment favoring opportunistic uptake of available particulate and dissolved organic material.
Other eukaryotes strongly associated with high NCP include groups of heterotrophic protists: radiolarians, centrohelids, Labyrinthulomycetes, Ciliophora, as well as flagellates such as Kathablepharidae, Choanomonada, and uncultured marine stramenopiles. Many of these taxa feed upon algae, bacteria, detritus, and other particles. The associations between these taxa and NCP may indicate flourishing of heterotrophs within an environment with enhanced food and prey concentrations.
The bacterial taxa most correlated with NCP corroborate this picture of a productive bloom ecosystem driven by high phytoplankton productivity. Groups of Bacteriodetes, a class of heterotrophic bacteria generally observed to thrive in particle-rich bloom environments [95], are strongly associated with NCP. Other bacterial groups primarily exhibiting surface or particle-associated lifestyles, including Verrucomicrobia and Planctomycetes, also display high correlations with NCP. Numerous Gammaproteobacteria taxa, including the fast-growing Vibrionales clade, are also strongly associated with productivity (Fig. 7a).
We acknowledge that our community sampling represents a snapshot of this bloom and cannot capture successional dynamics. 8-day MODIS satellite chlorophyll data measured before and after our cruise suggest that the bloom first appeared in late July one to two weeks before sampling. Our expedition likely encountered the bloom at its temporal midpoint, with the bloom then fading by late August. We further note that taxa associated with this event may not be characteristic of other blooms that might occur throughout the region. Although satellite imagery indicates that a large bloom often recurs annually in the Mid-Atlantic Bight in late summer, additional sampling is required to confirm whether the observed community structure also recurs.
Interestingly, when PLS regression analyses were repeated while excluding bloom stations S14–S16, only a handful of bacterial taxa and eukaryotic taxa remained associated with NCP rates, and the overall strength of associations weakened. Outside of the observed bloom, moderate correlations with productivity were displayed by just several groups of cryptophytes and bacterioplankton (Fig. 8a, b). These results might indicate that relationships between specific groups of eukaryotic and prokaryotic taxa and NCP in less-productive locations are either undetected by our study or hidden within the uncertainties of the measurements conducted. At the same time, such a finding may suggest that links between productivity and community structure in this region are complex, with the abundance of any given taxa not strongly associated with measured productivity.

Heatmaps of PLS correlations for a prokaryotic and b eukaryotic taxa at the 4th taxonomic rank, following removal of data from bloom stations S14 to S16. Strength of correlations is denoted by the color scale, with negative and positive correlations in blue and red tones, respectively. Asterisks mark taxa with a correlation with NCP greater than 0.2. Dendrograms link taxa with similar relationships to the set of variables, and vice versa
The relationships we have detailed between productivity and selected microplankton taxa exhibit interesting discrepancies with findings from similar work conducted in other regions of the global ocean. A TARA Oceans study of associations between bacterial, eukaryotic, and viral taxa, NPP, and particulate carbon export linked some of the same microplankton groups to primary production and to particle export that were productivity-associated within our full dataset, including Vibrio and Alteromonadales among bacteria, as well as dinoflagellates, Labyrinthula, Cercozoa, Picozoa, prymnesiophytes, MAST-3, and Radiolaria [7].
Intriguingly, however, many of these abovementioned associations vanish from our analysis when our dataset is limited to non-bloom station data, whereas Guidi et al. suggest that these same relationships are strong within the oligotrophic ocean. It is also worth noting that several taxa implicated in carbon export by Guidi et al. show no or even negative correlations with NCP in our analysis, such as Synechococcus (Subsection I Cyanobacteria) and Oceanospirillales. Dissimilarities may be attributable to differences in abundance metrics, molecular methods, and the distinctions between in-situ O2/Ar-derived NCP, modeled NPP, and optically-determined particle export (i.e., not all NCP is exported). Further, ecological dynamics encompassed by our regional study may not be extrapolatable to global open-ocean data. Yet our work nevertheless spans a considerable area and range of marine biomes. Rather, our results suggest that outside of the observed bloom, productivity across a relatively wide region is not strongly associated with specific microbial taxa. Such questions warrant further investigation.
Conclusions
Our results document a dramatic bloom in Mid-Atlantic Bight coastal waters, where the harmful algal bloom-forming taxon Aureococcus, Chrysophyceae, heterotrophic protists, and particle-associated bacterioplankton were strongly associated with this productivity peak. This result emphasizes the potential significance of large coastal blooms to productivity patterns in the Western North Atlantic, and highlights HAB-forming Aureococcus as a taxon of particular interest. We also find few associations between taxonomy and NCP across a wide range of less-productive waters, suggesting that specific microplankton taxa may not be responsible for driving broader patterns of production across much of this region.
Our quantitative amplicon sequencing approach serves as a useful tool in investigating the ocean microbiome and its influence on the marine environment, providing important additional context beyond relative abundance metrics. Coupled with the ever-increasing resolution and capabilities of in-situ biogeochemical methods, adoption of similar study designs can enable more nuanced examination of the role of the microplankton community across diverse ocean environments.
Supplementary information is available at the ISME Journal’s website. Sequences and metadata are available from the NCBI Sequence Read Archive under accession number SRP126177.
References
Field CB, Behrenfeld MJ, Randerson JT, Falkowski P. Primary production of the biosphere: integrating terrestrial and oceanic components. Science. 1998;281:237–40.
Buesseler KO. The decoupling of production and particulate export in the surface ocean. Glob Biogeochem Cycles. 1998;12:297–310.
Williams R, Follows M. Ocean dynamics and the carbon cycle: principles and mechanisms. Cambridge, United Kingdom: Cambridge University Press; 2011.
Cassar N, Wright SW, Thomson PG, Trull TW, Westwood KJ, de Salas M, et al. The relation of mixed-layer net community production to phytoplankton community composition in the Southern Ocean. Glob Biogeochem Cycles. 2015;29:446–62.
Lin YJ, Cassar N, Marchetti A, Moreno C, Ducklow H, Li ZC. Specific eukaryotic plankton are good predictors of net community production in the Western Antarctic Peninsula. Scientific Reports 2017;7.
Vallina SM, Follows MJ, Dutkiewicz S, Montoya JM, Cermeno P, Loreau M. Global relationship between phytoplankton diversity and productivity in the ocean. Nat Commun. 2014;5:4299.
Guidi L, Chaffron S, Bittner L, Eveillard D, Larhlimi A, Roux S, et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature. 2016;532:465.
Boyd P, Newton P. Evidence of the potential influence of planktonic community structure on the interannual variability of particulate organic-carbon flux. Deep-Sea Res Part I-Oceanogr Res Pap. 1995;42:619–39.
Richardson TL, Jackson GA. Small phytoplankton and carbon export from the surface ocean. Science. 2007;315:838–840.
Dugdale RC, Goering JJ. Uptake of new and regenerated forms of nitrogen in primary productivity. Limnology Oceanography 1967;12:196.
Li ZC, Cassar N. A mechanistic model of an upper bound on oceanic carbon export as a function of mixed layer depth and temperature. Biogeosciences. 2017;14:5015–27.
Berger WH, Wefer G. Export production: seasonality and intermittency, and paleoceanographic implications. Palaeogeogr, Palaeoclimatol, Palaeoecol. 1990;89:245–54.
Williams, et al. Group report: Export productivity from the photic zone. In: Berger WH, Smetacek V, Wefer G, (eds). Productivity of the ocean: present and past. Hoboken, N.J: John Wiley and Sons; 1989. p. 99–115. PJL
Fawcett SE, Lomas MW, Ward BB, Sigman DM. The counterintuitive effect of summer-to-fall mixed layer deepening on eukaryotic new production in the Sargasso Sea. Glob Biogeochem Cycles. 2014;28:86–102.
Lipschultz F, Bates NR, Carlson CA, Hansell DA. New production in the Sargasso Sea: history and current status. Glob Biogeochem Cycles. 2002;16:1-1–7.
McGillicuddy DJ, Robinson AR, Siegel DA, Jannasch HW, Johnson R, Dickeys T, et al. Influence of mesoscale eddies on new production in the Sargasso Sea. Nature. 1998;394:263–6.
Steinberg DK, Carlson CA, Bates NR, Johnson RJ, Michaels AF, Knap AH. Overview of the US JGOFS Bermuda Atlantic Time-series Study (BATS): a decade-scale look at ocean biology and biogeochemistry. Deep-Sea Res Pt Ii. 2001;48:1405–47.
Lomas MW, Bates NR, Johnson RJ, Knap AH, Steinberg DK, Carlson CA. Two decades and counting: 24-years of sustained open ocean biogeochemical measurements in the Sargasso Sea. Deep-Sea Res Pt Ii. 2013;93:16–32.
Krause JW,Lomas MW,Nelson DM, Biogenic silica at the Bermuda Atlantic Time-series Study site in the Sargasso Sea: Temporal changes and their inferred controls based on a 15-year record. Glob Biogeochem Cycles. 2009;23.
Lomas MW, Steinberg DK, Dickey T, Carlson CA, Nelson NB, Condon RH, et al. Increased ocean carbon export in the Sargasso Sea linked to climate variability is countered by its enhanced mesopelagic attenuation. Biogeosciences. 2010;7:57–70.
Bates NR, Best MHP, Neely K, Garley R, Dickson AG, Johnson RJ. Detecting anthropogenic carbon dioxide uptake and ocean acidification in the North Atlantic Ocean. Biogeosciences. 2012;9:2509–22.
Karl DM, Church MJ. Microbial oceanography and the Hawaii Ocean Time-series programme. Nat Rev Microbiol. 2014;12:699–713.
O’Reilly J, Busch D. Phytoplankton primary production on the northwestern Atlantic shelf. Rapp PV Reun Cons Int Explor Mer. 1984;183:255–68.
Mouw CB, Yoder JA. Primary production calculations in the Mid-Atlantic Bight, including effects of phytoplankton community size structure. Limnol Oceanogr. 2005;50:1232–43.
Bauer JE, Cai W-J, Raymond PA, Bianchi TS, Hopkinson CS, Regnier PAG. The changing carbon cycle of the coastal ocean. Nature. 2013;504:61–70.
Cai W-J. Estuarine and coastal ocean carbon paradox: CO2 sinks or sites of terrestrial carbon incineration? Annu Rev Mar Sci. 2011;3:123–45.
Doney SC. The growing human footprint on coastal and open-ocean biogeochemistry. Science. 2010;328:1512–6.
Lomas MW, Glibert PM, Shiah FK, Smith EM. Microbial processes and temperature in Chesapeake Bay: current relationships and potential impacts of regional warming. Glob Change Biol. 2002;8:51–70.
Brix H, Gruber N, Karl DM, Bates NR. On the relationships between primary, net community, and export production in subtropical gyres. Deep-Sea Res Pt Ii. 2006;53:698–717.
Estapa ML, Siegel DA, Buesseler KO, Stanley RHR, Lomas MW, Nelson NB. Decoupling of net community and export production on submesoscales. Glob Biogeochem Cycles. 2015;29:1266–82.
Mourino-Carballido B, McGillicuddy DJ. Mesoscale variability in the metabolic balance of the Sargasso Sea. Limnol Oceanogr. 2006;51:2675–89.
Treusch AH, Demir-Hilton E, Vergin KL, Worden AZ, Carlson CA, Donatz MG, et al. Phytoplankton distribution patterns in the northwestern Sargasso Sea revealed by small subunit rRNA genes from plastids. Isme J. 2012;6:481–92.
Vergin KL, Done B, Carlson CA, Giovannoni SJ. Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat Microb Ecol. 2013;71:1–U129.
Cassar N, Barnett B, Bender M, Kaiser J, Hamme R, Tilbrook B. Continuous high-frequency dissolved O-2/Ar measurements by equilibrator inlet mass spectrometry. Anal Chem. 2009;81:1855–64.
Lin Y, Gifford S, Ducklow H, Schofield O, Cassar N (submitted). Towards quantitative marine microbiome community profiling using internal standards.
Smets W, Leff JW, Bradford MA, McCulley RL, Lebeer S, Fierer N. A method for simultaneous measurement of soil bacterial abundances and community composition via 16S rRNA gene sequencing. Soil Biol Biochem. 2016;96:145–51.
Moisander PH, Beinart RA, Voss M, Zehr JP. Diversity and abundance of diazotrophic microorganisms in the South China Sea during intermonsoon. Isme J. 2008;2:954–67.
Satinsky BM, Gifford SM, Crump BC, Moran MA. Use of internal standards for quantitative metatranscriptome and metagenome analysis. Microb Metagenomics, Metatranscriptomics, Metaproteomics. 2013;531:237–50.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–22.
Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, Knight R. PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics. 2011;27:1159–61.
Stoeck T, Bass D, Nebel M, Christen R, Jones MDM, Breiner HW, et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol. 2010;19:21–31.
Bradley IM, Pinto AJ, Guest JS. Design and evaluation of illumina MiSeq-compatible, 18S rRNA gene-specific primers for improved characterization of mixed phototrophic communities. Appl Environ Microbiol. 2016;82:5878–91.
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:6.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: PAired-eND assembler for illumina sequences. BMC Bioinformatics 2012;13.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Schmieder R, Lim YW, Rohwer F, Edwards R. TagCleaner: identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinformatics 2010;11.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–7.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
McMurdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 2013; 8.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria; 2017
Tang W, Wang S, Batista D, Dehairs F, Gifford S, Gonzalez A et al (submitted). Coastal oceans broaden the biogeography of marine N2 fixation.
Laws EA. Photosynthetic quotients, new production and net community production in the open ocean. Deep-Sea Res Part a-Oceanogr Res Pap. 1991;38:143–67.
Falkowski PG, Flagg CN, Rowe GT, Smith SL, Whitledge TE, Wirick CD. The fate of a spring phytoplankton bloom—export or oxidation. Cont Shelf Res. 1988;8:457–84.
Letscher RT, Moore JK. Modest net autotrophy in the oligotrophic ocean. Glob Biogeochem Cycles. 2017;31:699–708.
Malmstrom RR, Kiene RP, Cottrell MT, Kirchman DL. Contribution of SAR11 bacteria to dissolved dimethylsulfoniopropionate and amino acid uptake in the North Atlantic ocean. Appl Environ Microbiol. 2004;70:4129–35.
Rowe JM, DeBruyn JM, Poorvin L, LeCleir GR, Johnson ZI, Zinser ER, et al. Viral and bacterial abundance and production in the Western Pacific Ocean and the relation to other oceanic realms. Fems Microbiol Ecol. 2012;79:359–70.
Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE. 2013;8:e57923.
Kirchman DL. Processes in microbial ecology. New York: Oxford University Press; 2012.
Rutten TPA, Sandee B, Hofman ART. Phytoplankton monitoring by high performance flow cytometry: a successful approach? Cytom Part A. 2005;64A:16–26.
Gobler CJ, Renaghan MJ, Buck NJ. Impacts of nutrients and grazing mortality on the abundance of Aureococcus anophagefferens during a New York brown tide bloom. Limnol Oceanogr. 2002;47:129–41.
Morris RM, Rappe MS, Connon SA, Vergin KL, Siebold WA, Carlson CA, et al. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–10.
Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science. 2006;311:1737–40.
Zinser ER, Coe A, Johnson ZI, Martiny AC, Fuller NJ, Scanlan DJ, et al. Prochlorococcus ecotype abundances in the North Atlantic Ocean as revealed by an improved quantitative PCR method. Appl Environ Microbiol. 2006;72:723–32.
Satinsky BM, Fortunato CS, Doherty M, Smith CB, Sharma S, Ward ND et al. Metagenomic and metatranscriptomic inventories of the lower Amazon River, May 2011. Microbiome 2015;3.
Farrelly V, Rainey FA, Stackebrandt E. Effect of genome size and rrn gene copy number on pcr amplification of 16s ribosomal-RNA genes from a mixture of bacterial species. Appl Environ Microbiol. 1995;61:2798–801.
Barber RT, Hiscock MR. A rising tide lifts all phytoplankton: growth response of other phytoplankton taxa in diatom-dominated blooms. Global Biogeochem Cycles 2006;20.
Gifford SM, Sharma S, Rinta-Kanto JM, Moran MA. Quantitative analysis of a deeply sequenced marine microbial metatranscriptome. Isme J. 2011;5:461–72.
Moran MA, Satinsky B, Gifford SM, Luo H, Rivers A, Chan L-K, et al. Sizing up metatranscriptomics. ISME J. 2013;7:237–43.
Aitchison J. A new approach to null correlations of proportions. J Int Assoc Math Geol. 1981;13:175–89.
Aitchison J. The statistical-analysis of compositional data. J R Stat Soc Ser B-Methodol. 1982;44:139–77.
Kembel SW, Wu M, Eisen JA, Green JL. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PloS Comput Biol 2012;8.
Guillou L, Viprey M, Chambouvet A, Welsh RM, Kirkham AR, Massana R, et al. Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environ Microbiol. 2008;10:3349–65.
Not F, Gausling R, Azam F, Heidelberg JF, Worden AZ. Vertical distribution of picoeukaryotic diversity in the Sargasso Sea. Environ Microbiol. 2007;9:1233–52.
Romari K, Vaulot D. Composition and temporal variability of picoeukaryote communities at a coastal site of the English Channel from 18S rDNA sequences. Limnol Oceanogr. 2004;49:784–98.
Figueroa RI, Cuadrado A, Stuken A, Rodriguez F, Fraga S. Ribosomal DNA organization patterns within the dinoflagellate genus alexandrium as revealed by fish: life cycle and evolutionary implications. Protist. 2014;165:343–63.
Prokopowich CD, Gregory TR, Crease TJ. The correlation between rDNA copy number and genome size in eukaryotes. Genome. 2003;46:48–50.
Gobler CJ, Lonsdale DJ, Boyer GL. A review of the causes, effects, and potential management of harmful brown tide blooms caused by Aureococcus anophagefferens (Hargraves et Sieburth). Estuaries. 2005;28:726–49.
Popels LC, Cary SC, Hutchins DA, Forbes R, Pustizzi F, Gobler CJ, et al. The use of quantitative polymerase chain reaction for the detection and enumeration of the harmful alga Aureococcus anophagefferens in environmental samples along the United States East Coast. Limnol Oceanogr-Methods. 2003;1:92–102.
Jones HLJ, Leadbeater BSC, Green JC. Mixotrophy in marine species of chrysochromulina (prymnesiophyceae)– ingestion and digestion of a small green flagellate. J Mar Biol Assoc U K. 1993;73:283–96.
Richardson K. Harmful or exceptional phytoplankton blooms in the marine ecosystem. Adv Mar Biol. 1997;31:301–85.
Graneli E, Edvardsen B, Roelke DL, Hagstrom JA. The ecophysiology and bloom dynamics of Prymnesium spp. Harmful Algae. 2012;14:260–70.
Lage OM, Bondoso J. Planctomycetes and macroalgae, a striking association. Front Microbiol 2014;5.
Gilbert JA, Steele JA, Caporaso JG, Steinbruck L, Reeder J, Temperton B, et al. Defining seasonal marine microbial community dynamics. ISME J. 2012;6:298–308.
Hatosy SM, Martiny JBH, Sachdeva R, Steele J, Fuhrman JA, Martiny AC. Beta diversity of marine bacteria depends on temporal scale. Ecology. 2013;94:1898–1904.
Cermeño P, Rodríguez-Ramos T, Dornelas M, Figueiras FG, Marañón E, Teixeira IG, et al. Species richness in marine phytoplankton communities is not correlated to ecosystem productivity. Mar Ecol Progress Ser. 2013;488:1–9.
Goebel NL, Edwards CA, Zehr JP, Follows MJ, Morgan SG. Modeled phytoplankton diversity and productivity in the California Current System. Ecol Model. 2013;264:37–47.
Irigoien X, Huisman J, Harris RP. Global biodiversity patterns of marine phytoplankton and zooplankton. Nature. 2004;429:863–7.
Li WKW. Macroecological patterns of phytoplankton in the northwestern North Atlantic Ocean. Nature. 2002;419:154–7.
Gobler CJ, Berry DL, Dyhrman ST, Wilhelm SW, Salamov A, Lobanov AV, et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc Natl Acad Sci USA. 2011;108:4352–7.
Caron DA, Alexander H, Allen AE, Archibald JM, Armbrust EV, Bachy C, et al. Probing the evolution, ecology and physiology of marine protists using transcriptomics. Nat Rev Microbiol. 2017;15:6–20.
Buchan A, LeCleir GR, Gulvik CA, Gonzalez JM. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol. 2014;12:686–98.
Acknowledgements
This research was supported by an NSF-CAREER grant awarded to NC (#1350710) and a Chateaubri and Fellowship awarded to SW. NC was also supported by the "Laboratoire d'Excellence" LabexMER (ANR-10-LABX-19) and co-funded by a grant from the French government under the program "Investissements d'Avenir". RE was supported by an NSF GRFP award (1106401). We thank the staff of the Bermuda Institute of Ocean Sciences as well as the crew and technicians of the R/V Atlantic Explorer for their valuable assistance in organizing and conducting our field study. We are also thankful to Rod Johnson, Bruce Williams, and Natasha McDonald at BIOS for their help with sample shipping and analysis. We are additionally grateful to Karoline Faust for her input on network approaches and to Geoffrey Smith for his help and for our use of his towfish equipment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Subject Category: Geomicrobiology and microbial contributions to geochemical cycles
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wang, S., Lin, Y., Gifford, S. et al. Linking patterns of net community production and marine microbial community structure in the western North Atlantic. ISME J 12, 2582–2595 (2018). https://doi.org/10.1038/s41396-018-0163-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0163-4
