
Abstract
Microbially mediated anaerobic oxidation of methane (AOM) is a key process in the regulation of methane emissions to the atmosphere. Iron can serve as an electron acceptor for AOM, and it has been suggested that Fe(III)-dependent AOM potentially comprises a major global methane sink. Although it has been proposed that anaerobic methanotrophic (ANME) archaea can facilitate this process, their active metabolic pathways have not been confirmed. Here we report the enrichment and characterisation of a novel archaeon in a laboratory-scale bioreactor fed with Fe(III) oxide (ferrihydrite) and methane. Long-term performance data, in conjunction with the 13C- and 57Fe-labelling batch experiments, demonstrated that AOM was coupled to Fe(III) reduction to Fe(II) in this bioreactor. Metagenomic analysis showed that this archaeon belongs to a novel genus within family Candidatus Methanoperedenaceae, and possesses genes encoding the “reverse methanogenesis” pathway, as well as multi-heme c-type cytochromes which are hypothesised to facilitate dissimilatory Fe(III) reduction. Metatranscriptomic analysis revealed upregulation of these genes, supporting that this archaeon can independently mediate AOM using Fe(III) as the terminal electron acceptor. We propose the name Candidatus “Methanoperedens ferrireducens” for this microorganism. The potential role of “M. ferrireducens” in linking the carbon and iron cycles in environments rich in methane and iron should be investigated in future research.
Similar content being viewed by others

Introduction
Methane is an important greenhouse gas that has relatively stable atmospheric concentration controlled by a balance between sources and sinks [1]. Anaerobic oxidation of methane (AOM) is a major sink estimated to consume >90% of the methane flux from the seafloor before it reaches atmosphere [2]. AOM has been linked to the reduction of sulfate [3], nitrate [4], and nitrite [5] by archaea and bacteria in anaerobic environments. AOM coupled to Fe(III) reduction is also thought to be both thermodynamically favourable and biochemically feasible (equation (1)) [6].
Iron is the fourth most abundant element in the Earth’s crust. Geochemical surveys suggest that Fe(III)-dependent AOM is likely a prevalent process in marine [7,8,9] and freshwater [10,11,12] environments, where large amounts of iron oxides are distributed throughout the sediments [13,14,15,16]. Incubation studies with sediments obtained from anoxic methane-rich environments showed that the addition of Fe(III) oxides can support microbial AOM activity [17, 18].
Previous studies have suggested that anaerobic methanotrophic (ANME) archaea play a role in this process [9, 19, 20]. One study showed that a culture dominated by an archaeon related to Candidatus “Methanoperedens nitroreducens” (referred to as “M. nitroreducens MPEBLZ” [21]) could catalyse Fe(III)-dependent AOM during short-term batch tests [6]. By analysing the published “M. nitroreducens MPEBLZ” genome [21], it was proposed that this archaeon could carry out Fe(III)-dependent AOM independently [6]. Another study showed that ANME archaea related to “M. nitroreducens” were actively involved in AOM in incubations with iron-rich sediment [22].
In this study we aimed to enrich and characterise novel microorganisms that can perform Fe(III)-dependent AOM. Freshwater sediment was incubated with methane and ferrihydrite (Fe(III)). Microbial process was determined through mass- and electron balance and isotopic labelling tests. Microorganism involved in Fe(III)-dependent AOM was identified and characterised through metagenomic and metatranscriptomic analyses.
Materials and methods
Sediment and in-situ water sampling
Sediment samples were taken as inoculum from Gold Creek Reservoir, Queensland, Australia (27° 27′ 37″ S, 152° 52′ 53″ E), in May 2013. The sediment was collected using a core sampler at a water depth of 10 m. Bottom water was collected for use as the initial cultivation medium. Two glass bottles were fully filled with the sediment and the in-situ water, respectively, sealed, and transferred to the laboratory within 2 h of collection. Samples at different depths of the water column and sediment at the sampling location were filtered (0.22 µm filter) for dissolved methane and Fe(II) concentrations measurement (Supplementary Figure 1).
Synthetic growth medium
Synthetic mineral medium was used for the enrichment. The composition of the medium was as follows (per liter): KH2PO4, 0.075 g, MgCl2·7H2O, 0.165 g, CaCl2·2H2O, 0.3 g, alkaline trace element solution, 0.2 mL, and acidic trace element solution, 0.5 mL. The components of the alkaline (10 mM NaOH) trace element solution were (per liter) SeO2, 0.067 g, Na2WO4·2H2O, 0.050 g, and Na2MoO4, 0.242 g, and the components of the acidic (100 mM HCl) trace element solution were (per liter) FeSO4·7H2O, 2.085 g, ZnSO4·7H2O, 0.068 g, CoCl2·6H2O, 0.12 g, MnCl2·4H2O, 0.5 g, CuSO4, 0.32 g, NiCl2·6H2O, 0.095 g, and H3BO3, 0.014 g [23].
Enrichment bioreactor operation
A mixture of 0.8 L sediment and 0.8 L in-situ water was incubated in a 2 L bioreactor, leaving a headspace of 0.4 L. The bioreactor was mixed using a magnetic stirrer at 200 rpm, and operated in a temperature controlled laboratory (22 ± 2 °C). The methane partial pressure in the bioreactor headspace was maintained between 0.6 and 1.2 atm by periodically flushing the liquid and headspace with a gas mixture (90% CH4, 5% CO2, and 5% N2). The bioreactor was pressurised to up to 1.3 atm after each flush by manually injecting helium into the headspace to prevent oxygen contamination. Ferrihydrite was pulse-fed to the bioreactor as the electron acceptor for AOM (Fig. 1a). Birnessite (Mn(IV)) was initially added to the bioreactor, along with ferrihydrite, as an alternative electron acceptor for AOM. The synthesis protocols of ferrihydrite and birnessite can be found in Supplementary Information. Addition of birnessite was stopped after ~220 days, when dissolved Fe(II) accumulation was observed. The bioreactor pH was monitored using a pH meter (Oakton, USA), and manually controlled between 6.8 and 7.2 with a 1 M HCl solution. Every 1–3 months, ~100 mL of the bioreactor supernatant was replaced with synthetic growth medium. Methane in gas phase were measured 2–3 times per week and liquid samples were taken via a sampling port in the middle of the vessel for dissolved Fe(II) measurements 2–4 times per month. Total methane was calculated by considering methane in both gas phase (measured) and liquid phase (calculated using Henry’s law). The variation of methane level over time, referred to as methane profile, was used to calculate methane consumption rate of the bioreactor through linear regression. Other potential electron acceptors for AOM, i.e., nitrate, nitrite and sulfate, were also monitored during the enrichment.

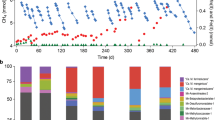
Bioreactor performance data, and mass- and electron balance tests results. a Simultaneous methane consumption and dissolved Fe(II) production. Blue and red arrows represent bioreactor flushing with methane and ferrihydrite addition, respectively. The sharp drop in dissolved Fe(II) following each ferrihydrite addition was determined to be due to abiotic reactions between dissolved Fe(II) and ferrihydrite. b The profiles of Fe(III), Fe(II) and methane during the batch test started on Day 601 as an example. c Average ratios of Fe(III) consumption and Fe(II) production rates, respectively, to methane consumption rate determined in the three batch tests. Error bars represent standard deviations of results using three subsamples from the bioreactor
Mass- and electron balance tests
Subsamples (250 mL) of the bioreactor were used to determine the Fe(III) reduction, Fe(II) production and methane consumption rates on Day 601, 860, and 938. Each batch test was conducted for 15–30 days, and methane in gas phase, and iron in both liquid and solid phases were measured every 3–7 days. The profiles of methane, Fe(III) and Fe(II) were used to determine the consumption rates of methane (rCH4) and Fe(III) (rFe(III)), and the production rate of Fe(II) (rFe(II)) via linear regression. Methane in both gas and liquid phases (calculated using Henry’s law) was considered when calculating methane consumption rates. Mn(II) in liquid and solid phases were also quantified to evaluate its potential contribution to AOM.
Isotopic labelling tests
For each of the duplicate tests, a subsample of bioreactor biomass (250 mL) was transferred anaerobically to a 330 mL glass vessel. The biomass was flushed with a mixed gas (90% CH4, 5% CO2, and 5% N2) for 10 min. Approiximately 10 mL 13C-labelled methane (Sigma-Aldrich, 99 atom % 13C, USA) and ~0.03 g 57Fe-labelled ferrihydrite were added to each vessel on Day 34. Each vessel was pressurised to 1.2 atm through injecting helium. Each test lasted for ~110 days.
During the tests, Fe(III) and Fe(II) in liquid and solid phases were sampled every 3–7 days and measured for their 57Fe(III)/56Fe(III), 54Fe(III)/56Fe(III), 57Fe(II)/56Fe(II) and 54Fe(II)/56Fe(II) ratios. After the 57Fe(III)/56Fe(III) ratio reached a steady state (Supplementary Figure 2), methane in gas phase, CO2 in gas, liquid and solid phases, Fe(III)/Fe(II) in liquid and solid phases were sampled every week and measured for their concentrations and the respective isotopic fractions.
Total methane, Fe(III) and Fe(II) and their consumption/production rates were calculated as described in section “mass- and electron balance tests”. Total CO2 was quantified by considering the amounts in gas, liquid and solid phases, and the profile of CO2 was used to determine its production rate (rCO2) via linear regression. The amounts of methane, CO2, Fe(III) and Fe(II) and their respective isotopic fractions were used to calculate the amounts of 13CH4, 13CO2, 57Fe(III), and 57Fe(II). The profiles of 13CH4, 13CO2, 57Fe(III) and 57Fe(II) were used to determine the consumption rates of 13CH4 (r13CH4) and 57Fe(III) (r57Fe(III)), and the production rates of 13CO2 (r13CO2) and 57Fe(II) (r57Fe(II)) via linear regression.
Iron, manganese, and CO2 extraction
Samples were filtered (0.22 μm) for dissolved Fe(II) and Mn(II) measurement. To quantify the concentrations of Fe(II) and Fe(III) in solid phase, solid was first harvested from 0.1 mL sample by centrifugation (3267 × g, 10 min). A previously described sequential extraction protocol [24] was then used to extract Fe(II) and Fe(III) from different forms of iron compounds: (1) 1 M MgCl2 at pH 7 for 2 h was used to extract adsorbed Fe(II); (2) 1 M sodium acetate at pH 4.5 for 24 h to extract Fe(II) from Fe(II) carbonate; (3) 1 M hydroxylamine-HCl solution in 25% v/v acetic acid for 48 h to extract Fe(III) from ferrihydrite. The same methods were applied for extraction of adsorbed Mn(II) and Mn(II) in Mn(II) carbonate.
CO2 produced exists in liquid phase as dissolved CO2, bicarbonate and carbonate, and in solid phase as carbonate. In order to quantify the produced CO2 in liquid and solid phases, unfiltered samples were injected into evacuated tubes and acidified with 1 M HCl solution immediately. The evacuated tubes were shaken for 72 h to convert dissolved CO2, bicarbonate, and carbonate in liquid and solid phases to CO2 gas in the headspace before measurement.
Chemical, protein and isotopic analyses
Gaseous methane and CO2 were measured with a Gas Chromatograph (GC) equipped with a Porapak Q column and a thermal conductivity detector (Shimadzu GC-8A, Japan), and liquid phase nitrite and nitrate concentrations were measured using a Flow Injection Analyzer (FIA) (Lachat QuickChem8000, USA), as described previously [25]. Dissolved methane was measured using a GC equipped with a flame ionization detector (Agilent 7890 A, USA) according to the method described previously [26]. Dissolved and extracted iron and manganese were measured using Inductively Coupled Plasma-Optical Emission Spectrophotometer (ICP-OES) (PerkinElmer Optima 7300DV, USA). Total protein was determined using the bicinchoninic acid assay kit (Pierce, USA) with bovine serum albumin as the standard protein [27]. The ratios of 57Fe(III)/56Fe(III), 54Fe(III)/56Fe(III), 57Fe(II)/56Fe(II) and 54Fe(II)/56Fe(II) were measured on an Agilent 7900 Inductively Coupled Plasma-Mass Spectrometer (ICP-MS) in the Radiogenic Isotope Facility, The University of Queensland (UQ). A helium collision cell gas was used for the removal of ArO+ isobaric interference on iron isotopes, resulting in accurate measurements of the above isotopic ratios to precisions typically <1% (dependent on the count rates of measured isotopes in each sub-sample solution). The ratios of 13CH4/12CH4 and 13CO2/12CO2 were measured with Gas Chromatograph-Combustion-Isotope Ratio Mass Spectrometer (GC-C-IRMS) (Isoprime, UK) in the Stable Isotope Geochemistry Laboratory at UQ. Sulfate was measured using Ion Chromatograph (IC) with an AD25 absorbance and a DS6 heated conductivity detector (Dionex ICS-2000, USA).
DNA extraction
DNA from biomass samples (~2 mL) was extracted using the FastDNA SPIN kit for soil (MPBio, USA) according to the manufacturer’s protocol.
PCR amplification and 16S rRNA amplicon sequencing and analysis
Primers 926F (5′-AAACTYAAAKGAATTGACGG-3′) and 1392R (5′-ACGGGCGGTGTRC-3′) were used for 16S rRNA PCR [28]. DNA concentrations were assessed by gel electrophoresis (1% agarose), and a Qubit fluorometer with Quant-iT dsDNA HS Assay kits (Invitrogen, USA). 16S rRNA gene amplicons were sequenced at the Australian Centre for Ecogenomics (ACE) and analysed using the ACE pyrotag pipeline (http://wiki.ecogenomic.org/doku.php?id=amplicon_pipeline_readme). Operational taxonomic units (OTU) tables were generated from the pipeline.
DNA library preparation
A paired-end library was prepared using the TruSeq Nano DNA library preparation kit (Illumina, USA) for DNA extracted on Day 624 (for the initial metagenome). A paired-end library was prepared using the Nextera XT DNA library preparation kit (Illumina, USA) for DNA extracted on Day 1106 (for the shallow metagenome). The libraries were sequenced on a NextSeq500 (Illumina, USA) platform generating 2 × 150 bp paired-end reads with an average insert length of 300 bp. In total, 126,367,422 and 8,500,985 paired-end reads for the initial and shallow metagenome were generated from the library, respectively.
The shallow metagenome sequencing was performed during metatranscriptomic sequencing to confirm the microbial composition of the bioreactor remained consistent with the initial metagenome assessment.
Quality control and assembly of sequence data
Paired-end reads for the initial metagenome were trimmed using Trimmomatic [29] and BBMerge (http://sourceforge.net/projects/bbmap/) using default settings. Reads were assembled using the de novo assembly method in CLC Genomics Workbench v7.5 (CLCBio, Denmark) using default settings. Scaffolding of the assembled contigs was also performed using CLC Genomics Workbench. Ambiguous bases from scaffolding were resolved using abyss-sealer v1.9.0 as part of the ABySS software suite [30]. This reduced the number of ambiguous bases by 35.83% (45,581 of 127,232). The resulting assembly consisted of 363,468 scaffolds (437,445 contigs) ≥500 bp with an N50 of 1930 bp, a median length of 832 bp, and a total of ~553 Mbp of sequence data.
Recovery and assessment of population genomes
Population genomes were recovered from the assembled contigs using MetaBat [31]. Scaffolds within the genomes with divergent GC-content or tetranucleotide frequencies were removed using the outlier method in RefineM v0.0.7 (https://github.com/dparks1134/RefineM). Additional scaffolding and resolving of ambiguous bases of the “M. ferrireducens” genome was performed using the “roundup” mode of FinishM v0.0.7 (https://github.com/wwood/finishm). Completeness and contamination rates of the population bins were assessed using CheckM v1.05 [32] with the “lineage wf” command.
Functional annotation
Assembled contigs of “M. ferrireducens” were annotated using AnnotateM (https://github.com/fauziharoon/annotateM) which uses Prodigal [33] to call ORFs and annotations from several databases: Prokka v.1.11 [34], IMG (Release: May 2015) [35], UniRef90 (Release: November 2015) [36], clusters of orthologous groups (COG) [37], Pfam 28 [38] and TIGRfam (Release: January 2014) [39]. Genes of interest were further verified using NCBI’s conserved domain search to identify conserved motif(s) present within the gene [40]. Psortb v3.0 [41] was used to predict subcellular localisation of the putative proteins. Pred-Tat was used to predict putative signal peptides [42]. Assembled contigs of the metagenome were annotated by searching against Uniref100 (accessed October 2016) using the “blastp verysensitive” setting in Diamond v0.8.23 (https://github.com/bbuchfink/diamond.git). The top hit for each gene with an e-value < 1e−3 was mapped to the KO database [43] using the Uniprot ID mapping files. KO annotations linked to microbial metabolism of interest are summarised with TPM (Transcripts per million) values (Supplementary Figure 3 and Supplementary Dataset 1).
Identification of c-type cytochromes
Putative multi-heme c-type cytochromes (MHCs) were identified by ORFs possessing ≥3 CXXCH motifs in the draft genomes and the metagenome. Putative MHCs were subsequently searched for cytochrome c-type protein domains using hmmsearch (HMMER v.3.1) [44] with PfamA [45].
Construction of genome trees
The archaeal and bacterial genome trees were constructed using the Genome Tree Database (GTDB v1.1.0, https://github.com/Ecogenomics/GTDBNCBI) with a concatenated set of 122 archaeal-specific and 120 bacterial-specific conserved marker genes inferred from genomes available in NCBI (accessed June 2016) using GenomeTreeTK (https://github.com/dparks1134/GenomeTreeTK). Marker genes were identified from the “M. ferrireducens” genome and 354 NCBI archaeal genomes using 122 single-copy marker genes (Supplementary Dataset 2). Marker genes were identified from the other 14 population genomes and 12,400 bacterial genomes using 120 single-copy marker genes (Supplementary Dataset 2). These marker genes were identified and aligned in each genome using HMMER v.3.1 [44], concatenated, and trees were constructed using FastTree V.2.1.8 [46] with the WAG+GAMMA models. The tree was decorated with taxonomic information using Phylorank v.0.0.29 (https://github.com/dparks1134/PhyloRank). This is done through determining the placement of each taxa that results in the highest F-measure [47], and support values were determined using 100 nonparametric bootstrapping with GenomeTreeTK. The trees were visualised using ARB [48] and modified using Adobe illustrator (Adobe, USA).
Construction of 16S rRNA gene tree
The Methanoperedeneceae 16S rRNA gene was identified in the genome and used to infer taxonomic assignment of the population genome. Sequences were compared against the SILVA 16S rRNA database (Version 123.1). Sequences were aligned with 330 16S rRNA sequences retrieved from the SILVA database using SSU-align v0.1 [49]. Sequences were imported into ARB (version 6.0.2). The phylogenetic tree was constructed using FastTree V2.1.8 [46] using the Generalized Time-Reversible and GAMMA models. Support values were determined using 100 nonparametric bootstrapping. The trees were visualised using ARB [48] and modified using Adobe illustrator.
RNA extraction and library preparation
A subsample of the bioreactor biomass was collected and stored in Lifeguard Soil Preservation Solution (MoBio, USA). The sample was then centrifuged and the sediment (~200 mg) was transferred into a 2 mL Lysing Matrix E tube (MP Biomedicals, USA). Lysis solution (1.2 mL) containing sodium phosphate buffer (750 mM, pH 7), NaCl (500 mM), cethyltrimethyl ammonium bromide (CTAB, 0.5%), and EDTA (50 mM) was added.
The tube was then homogenised using PowerLyzer Homogenizer (MoBio, USA) for 30 s twice, and cooled on ice for 60 s in between homogenisations at 4500 rpm at room temperature (25 °C). The tube was centrifuged at 16,000 × g for 5 min at 10 °C. The supernatant was subjected to phenol/chlorofoam/isomyl alcohol extraction (pH 6.5), followed by two chloroform extractions. The aqueous phase was purified using RNeasy MinElute cleanup kit (Qiagen, Germany). Turbo DNA-free kit (Thermo Fisher Scientific, USA) was used to remove genomic DNA, followed by concentration using a RNA Clean & Concentrator-5 Kit (Zymo Research, USA) following the manufacturer’s protocol. Removal of rRNA was performed using the Ribo-Zero Magenetic kit (Epicentre, USA) as per manufacturer’s protocol. RNA was prepared for sequencing using the ScriptSeq stranded mRNA library prep kit (Illumina, USA) following the manufacturer’s protocol. The library was sequenced on a NextSeq500 (Illumina, USA) platform generating 2 × 150 bp paired-end reads with an average insert length of 300 bp.
Read mapping of raw mRNA reads to ORFs
Metatranscriptomic paired-end reads were processed using the TranscriptM program with a few modifications (https://github.com/wwood/transcriptM). Briefly, reads underwent quality trimming, PhiX (bacteriophage) contaminant reads removal (used as a control during illumina sequencing), rRNA, tRNA, and tmRNA removal [50]. The processed reads were mapped against all metagenomic ORFs using 0.97 as the minimum percentage identity of mapped read and 0.95 as minimum percentage alignment for a mapped read. Modifications include quality trimming and Illumina adapter sequence removal with SeqPrep (https://github.com/jstjohn/SeqPrep) and calculation of TPM values using the tpm_table.py script (https://github.com/EnvGen/metagenomics-workshop/blob/master/in-house/tpm_table.py) with the –nonunique = all option to include mapped reads that overlaps two genes and count them as 1 for each gene. TPM values were calculated as follows:
where rg is reads mapped to gene g, rl is read length, cl is coding sequence (CDS) length, and T is sum of rg×rl/cl for all genes.
Data availability
All data supporting the findings of this study are available in this paper and the Supplementary Information. Sequencing data are deposited at the NCBI Short Read Archive under accession numbers SAMN06544930, SAMN06544931, and SAMN06544932. All draft genome nucleotide sequences have been deposited under the NCBI Biosample accession SAMN08364719- SAMN08364733.
Results and discussion
The laboratory-scale bioreactor was inoculated with sediment collected from a freshwater reservoir exhibiting high dissolved methane and Fe(II) concentrations (Supplementary Figure 1), and fed with methane and Fe(III) oxide in the form of ferrihydrite. After ~220 days of operation, the accumulation of dissolved Fe(II), accompanied by methane consumption, suggested that Fe(III)-dependent AOM was likely occurring in the bioreactor (Fig. 1a and Supplementary Figure 4).
Mass- and electron balance tests were conducted on Days 601, 860, and 938 using subsamples from the bioreactor. Since dissolved Fe(II) only accounted for a small fraction of Fe(II) produced in the system, extraction of Fe(II) and Fe(III) from the solid phase was required in order to confirm the reduction of Fe(III) to Fe(II). The methane- and Fe(III) consumption rates, and Fe(II) production rate were determined for each test (Fig. 1b and Supplementary Table 1). The average rate ratio between Fe(III) to methane consumption was close to its predicted stoichiometry of 8:1 (equation (1)), as was the rate ratio between Fe(II) production to methane consumption (Fig. 1c). The involvement of sulfate [3], nitrate [4], nitrite [5], and manganese [9] as electron acceptors for AOM was not observed (Supplementary Figure 5).
To further verify that this enrichment culture was able to couple AOM to Fe(III) reduction, isotopic labelling experiments using 13C-labelled methane and 57Fe-labelled ferrihydrite were conducted with subsamples from the bioreactor (Fig. 2 and Supplementary Figure 2). The 13CO2 and 57Fe(II) production rates were concordant with the 13CH4 and 57Fe(III) consumption rates (Fig. 2a, b), as were the total CO2 and total Fe(II) production rates to the total methane and total Fe(III) consumption rates, respectively (Fig. 2c, d). These results indicated full methane oxidation to CO2, and Fe(III) reduction to Fe(II). The methane oxidation to Fe(III) reduction ratio was 1:8.2, strongly suggesting that all electrons generated from methane oxidation were used for Fe(III) reduction, consistent with equation (1). The specific AOM rate of the culture was 69 μmol mg(protein)−1 h−1, which is one order of magnitude higher than the rate reported previously for the “M. nitroreducens MPEBLZ”-dominated culture when fed with ferrihydrite [6].

AOM coupled to Fe(III) reduction by the enrichment culture in one of the duplicate isotopic labelling tests (grey area in Supplementary Figure 2a). a–d Conversion of 13CH4 to 13CO2 (a), 57Fe(III) to 57Fe(II) (b), CH4 to CO2 (c), and Fe(III) to Fe(II) (d)
16S rRNA gene amplicon and metagenomic sequencing of bioreactor samples between Day 300 and 1100 indicated that the bioreactor community was dominated by an archaeon belonging to the recently described Methanoperedenaceae [4] (17.0–53.6% relative abundance; Supplementary Table 2 and Supplementary Dataset 3). No 16S rRNA genes of this archaeon were detected in the initial inoculum sample indicating strong selective growth for this microorganism in our bioreactor. Metagenomic data generated from a sample collected on Day 624 (15.8 Gb of 150 bp, paired-end Illumina) was assembled and binned into population genomes, leading to the recovery of 15 substantially completed genomes (≥70% completeness and ≤10% contamination rates [32]; Supplementary Figure 6 and Supplementary Table 3). This included the genome of the dominant archaeon in the bioreactor (representing 28.1% relative abundance; Supplementary Table 2), which we propose the name Candidatus “Methanoperedens ferrireducens” based on its proposed metabolism (see below).
A genome tree constructed using 122 conserved single copy archaeal marker genes confirmed the placement of the “M. ferrireducens” genome within the Methanoperedenaceae (Fig. 3a). A 16S rRNA gene tree constructed with a near full-length 16S rRNA gene (1470 bp) recovered from the “M. ferrireducens” genome supported this phylogenetic placement (Fig. 3b). Comparative genomics analysis of the “M. ferrireducens” genome against the two publicly available Methanoperedenaceae genomes recovered from nitrate-reducing bioreactors (“M. nitroreducens” [4] and “M. nitroreducens sp. BLZ1” [21] revealed an average amino acid identity (AAI) of 72.5% and 81.9%, respectively (Supplementary Table 4), indicating that each of these genomes likely represents a separate genus [51].

Phylogenetic placement of the dominant “M. ferrireducens” population. a, b Genome tree (a) and 16 S rRNA gene tree (b) showing the phylogenetic placement of the “M. ferrireducens” genome inferred by maximum-likelihood. In both trees, the Methanoperedenaceae genomes and 16 S rRNA gene sequences are highlighted in red with “M. ferrireducens” indicated with an asterisk. Black and white dots indicate >90% and >70% bootstrap values, respectively. The scale bars in a and b represent amino acids and nucleotide changes, respectively
Consistent with the other Methanoperedenaceae genomes, metabolic reconstruction of the “M. ferrireducens” genome revealed the presence of genes encoding a complete “reverse methanogenesis” pathway [4, 52], supporting its role in methane oxidation (Fig. 4). Biosynthetic genes for menaquinone (MK) were also identified in the genome (Supplementary Dataset 1), suggesting its use as a membrane soluble electron carrier [21]. The conversion of methane to CO2 generates reducing equivalents which are likely used to reduce the menaquinone pool through two membrane-bound enzyme complexes, F420H2 (Fpo) dehydrogenase and heterodisulfide reductase (HdrDE) [21, 53]. The “M. ferrireducens” genome encoded 46 putative MHCs (Supplementary Dataset 4), consistent with the high numbers of MHCs identified in other members of Methanoperedenaceae and the ANME-2a/b genome [53,54,55,56]. Extracellular MHCs have been implicated in direct interspecies electron transfer (DIET) between ANME archaea and sulfate-reducing bacteria [53, 56], and dissimilatory metal reduction in numerous metal-reducing microorganisms [57]. Several of the MHCs were found adjacent to gene clusters encoding menaquinone:cytochrome c oxidoreductases, including the bc-like complex, non-canonical Rieske/cytb complex, and NrfD-like complexes encoding a NrfD protein and an ferredoxin iron-sulfur (FeS) protein [21, 53]. These complexes likely transport electrons from the cytoplasmic membrane to MHCs outside the cytoplasmic membrane, which subsequently interacts with other MHCs for Fe(III) reduction (Fig. 4). One 22-heme MHC contained a S-layer domain, and is hypothesised to extend from the archaeal S-layer [53]. This MHC may be involved as the terminal reductase for extracellular Fe(III). The nitrate reductase genes (narGH) found in other members of the Methanoperedenaceae were not identified in the “M. ferrireducens” genome or in the unassembled metagenomic reads, indicating that nitrate is not used as an electron acceptor by this microorganism. No NiFe hydrogenases were identified in this genome, excluding hydrogen as a putative electron donor source as seen in other Methanoperedeneceae genomes [58, 59].

Metabolic construction of the putative pathway for AOM coupled to Fe(III) reduction in “M. ferrireducens“. Electrons from methane are generated through the “reverse methanogenesis” pathway and transferred to the menaquinone pool (MK/MKH2) via the Fpo and Hdr complexes, which oxidise F420H2 and CoM-SH+CoB-SH, respectively. CoM-SH and CoB-SH can be oxidised by the HdrDE complex [53] or the cytoplasmic HdrABC–FrhB complex that also reoxidise ferredoxin and reduce two F420 [21]. Reducing equivalents are transferred via the menaquinone:cytochrome c oxidoreductases to MHCs located outside the cytoplasm to reduce the Fe(III) oxides. Abbreviations for enzymes and co-factors: H4MPT, tetrahydromethanopterin; MFR, methanofuran; Fwd, formyl-methanofuran dehydrogenase; Ftr, formylmethanofuran/H4MPT formyltransferase; Mch, methenyl-H4MPT cyclohydrolase; Mtd, F420-dependent methylene H4MPT dehydrogenase; Mer, F420-dependent methylene-H4MPT reductase; Mtr, Na+-translocating methyl-H4MPT:coenzyme M methyltransferase; Mcr, methyl-coenzyme M reductase; Fpo, F420H2 dehydrogenase; MK, menaquinone; CoB-SH, coenzyme B; CoM-SH, coenzyme M; Fd, ferredoxin; Hdr, heterodisulfide reductase; FrhB, F420-reducing hydrogenase subunit B; Cytb, b-type cytochrome; NrfD, polysulfide reductase subunit D; FeS, ferredoxin iron sulfur protein. The MHCs are denoted in as blue and the number of hemes are indicated as red diamonds. Normalised gene expression is indicated as TPM. More information on the metabolism in ANME archaea can be found in the previous reviews [61, 62]
Metatranscriptomic sequencing was performed (Day 1106) to examine the metabolically active members of the community during AOM coupled to Fe(III) reduction. Shallow metagenomic sequencing was also performed simultaneously to ensure the microbial community profile was consistent with the initial metagenome assembly used for the analysis (Supplementary Table 2). The majority of the reads from the metatranscriptome mapped to the “M. ferrireducens” genome (41.1% of total mapped transcriptome reads), suggesting that this microorganism represented the most dominant population transcriptionally in the bioreactor community at the time of sampling.
All genes involved in the “reverse methanogenesis” and putative extracellular electron transfer pathways were highly expressed in “M. ferrireducens”, indicating their potential role in active methane oxidation and metal respiration (Fig. 4; Supplementary Figure 3). Two menaquinone:cytochrome c oxidoreductase gene clusters (NrfD-like and bc-like) appeared to be highly expressed, and are hypothesised to interact with MHCs located outside the cytoplasmic membrane. Out of all the MHC genes, 14 were found to be highly expressed (Supplementary Dataset 4), including a 4-heme MHC and 8-heme MHC that were amongst the most expressed genes in the genome. Intriguingly, the 8-heme MHC gene is located adjacent to a highly expressed peptidase gene, which encodes a S8/S53 peptidase conserved domain and a N-terminal signal peptide for translocation across the membrane. This putative peptidase is highly similar to a serine peptidase identified in the Fe(III) reducer, “Shewanella oneidensis”, which has been implicated in the removal of extracellular polymeric substances for increased connection between the MHCs and Fe(III) oxide surfaces [43]. The high gene expression of these MHCs indicate their potential involvement in Fe(III) reduction (Fig. 4). Further studies will be necessary to identify the location of the highly expressed MHCs involved with Fe(III) oxides. The S-layer domain containing MHC was lowly expressed, and hence may not be involved in metal reduction. One hypothesis for this low expression may be their utilisation under different conditions such as electron transfer to syntrophic bacteria or other extracellular electron acceptors [53].
The metabolic capabilities of the other community members including aerobic methanotrophs, nitrate reducers and iron reducers were also analysed (Supplementary Dataset 1), however their relative transcriptome expression levels were low, strongly indicating “M. ferrireducens” as the main contributor of Fe(III)-dependent AOM (Supplementary Figure 3).
Previous studies have suggested that ANME archaea may be able to couple AOM to Fe(III) reduction [6, 9]. In this study we demonstrated that a new member of Methanoperedenaceae is capable of coupling AOM to Fe(III) reduction independently through the “reverse methanogenesis” pathway and a putative extracellular electron transfer pathway. Environments rich in methane and iron are abundant on Earth [7, 9, 11, 60], possibly providing suitable habitats for “M. ferrireducens”. The ubiquity of MHCs in Methanoperedenaceae [4, 6] suggest this lineage is metabolically versatile and has the potential to use a wide range of different electron acceptors, which could have broad implication to the global methane cycle. The potential role of “M. ferrireducens” in the global carbon and iron cycles requires further investigation.
References
Conrad R. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep. 2009;1:285–92.
Knittel K, Boetius A. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol. 2009;63:311–34.
Hoehler TM, Alperin MJ, Albert DB, Martens CS. Field and laboratory studies of methane oxidation in an anoxic marine sediment: evidence for a methanogen-sulfate reducer consortium. Glob Biogeochem Cycles. 1994;8:451–63.
Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature. 2013;500:567–70.
Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM, et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature. 2010;464:543–8.
Ettwig KF, Zhu B, Speth D, Keltjens JT, Jetten MSM, Kartal B. Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc Natl Acad Sci USA. 2016;113:12792–6.
Egger M, Rasigraf O, Sapart CJ, Jilbert T, Jetten MSM, Röckmann T, et al. Iron-mediated anaerobic oxidation of methane in brackish coastal sediments. Environ Sci Technol. 2015;49:277–83.
Riedinger N, Formolo MJ, Lyons TW, Henkel S, Beck A, Kasten S. An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments. Geobiology. 2014;12:172–81.
Beal EJ, House CH, Orphan VJ. Manganese and iron-dependent marine methane oxidation. Science. 2009;325:184–7.
Kà Norði, Thamdrup B, Schubert CJ. Anaerobic oxidation of methane in an iron-rich Danish freshwater lake sediment. Limnol Oceanogr. 2013;58:546–54.
Sivan O, Adler M, Pearson A, Gelman F, Bar-Or I, John SG, et al. Geochemical evidence for iron-mediated anaerobic oxidation of methane. Limnol Oceanogr. 2011;56:1536–44.
Crowe S, Katsev S, Leslie K, Sturm A, Magen C, Nomosatryo S, et al. The methane cycle in ferruginous Lake Matano. Geobiology. 2011;9:61–78.
Lovley DR, Phillips EJ. Availability of ferric iron for microbial reduction in bottom sediments of the freshwater tidal Potomac River. Appl Environ Microbiol. 1986;52:751–7.
Martin J-M, Meybeck M. Elemental mass-balance of material carried by major world rivers. Mar Chem. 1979;7:173–206.
Wersin P, Höhener P, Giovanoli R, Stumm W. Early diagenetic influences on iron transformations in a freshwater lake sediment. Chem Geol. 1991;90:233–52.
Widerlund A, Ingri J. Early diagenesis of arsenic in sediments of the Kalix River estuary, northern Sweden. Chem Geol. 1995;125:185–96.
Segarra KEA, Comerford C, Slaughter J, Joye SB. Impact of electron acceptor availability on the anaerobic oxidation of methane in coastal freshwater and brackish wetland sediments. Geochim Cosmochim Acta. 2013;115:15–30.
Sivan O, Antler G, Turchyn AV, Marlow JJ, Orphan VJ. Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps. Proc Natl Acad Sci USA. 2014;111:E4139–47.
House CH, Beal EJ, Orphan VJ. The apparent involvement of ANMEs in mineral dependent methane oxidation, as an analog for possible Martian methanotrophy. Life. 2011;1:19–33.
Scheller S, Yu H, Chadwick GL, McGlynn SE, Orphan VJ. Artificial electron acceptors decouple archaeal methane oxidation from sulfate reduction. Science. 2016;351:703–7.
Arshad A, Speth DR, de Graaf RM, den Camp HJO, Jetten MSM, Welte CU. A metagenomics-based metabolic model of nitrate-dependent anaerobic oxidation of methane by Methanoperedens-like archaea. Front Microbiol. 2015;6:1423.
Weber HS, Habicht KS, Thamdrup B. Anaerobic methanotrophic archaea of the ANME-2d cluster are active in a low-sulfate, iron-rich freshwater sediment. Front Microbiol. 2017;8:619.
Ettwig KF, van Alen T, van de Pas-Schoonen KT, Jetten MSM, Strous M. Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl Environ Microbiol. 2009;75:3656–62.
Poulton SW, Canfield DE. Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem Geol. 2005;214:209–21.
Hu S, Zeng RJ, Burow LC, Lant P, Keller J, Yuan Z. Enrichment of denitrifying anaerobic methane oxidizing microorganisms. Environ Microbiol Rep. 2009;1:377–84.
Guisasola A, de Haas D, Keller J, Yuan Z. Methane formation in sewer systems. Water Res. 2008;42:1421–30.
Ettwig KF, Shima S, van de Pas-Schoonen KT, Kahnt J, Medema MH, Op den Camp HJM, et al. Denitrifying bacteria anaerobically oxidize methane in the absence of Archaea. Environ Microbiol. 2008;10:3164–73.
Amann RI, Ludwig W, Schleifer K-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–69.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;11:119.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–22.
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–8.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinf. 2003;4:41.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–85.
Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41:D387–95.
Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–31.
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–15.
Bagos PG, Nikolaou EP, Liakopoulos TD, Tsirigos KD. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics. 2010;26:2811–7.
Burns JL, Ginn BR, Bates DJ, Dublin SN, Taylor JV, Apkarian RP, et al. Outer membrane-associated serine protease involved in adhesion of Shewanella oneidensis to Fe (III) oxides. Environ Sci Technol. 2009;44:68–73.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths‐Jones S, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–41.
Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Buchner A, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–7.
Mukherjee S, Huntemann M, Ivanova N, Kyrpides NC, Pati A. Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand Genom Sci. 2015;10:18.
Konstantinidis KT, Tiedje JM. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol. 2007;10:504–9.
Hallam SJ, Putnam N, Preston CM, Detter JC, Rokhsar D, Richardson PM, et al. Reverse methanogenesis: testing the hypothesis with environmental genomics. Science. 2004;305:1457–62.
McGlynn SE, Chadwick GL, Kempes CP, Orphan VJ. Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature. 2015;526:531–5.
Kletzin A, Heimerl T, Flechsler J, van Niftrik L, Rachel R, Klingl A. Cytochromes c in Archaea: distribution, maturation, cell architecture, and the special case of Ignicoccus hospitalis. Front Microbiol. 2015;6:439.
Wang F-P, Zhang Y, Chen Y, He Y, Qi J, Hinrichs K-U, et al. Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J. 2014;8:1069–78.
Wegener G, Krukenberg V, Riedel D, Tegetmeyer HE, Boetius A. Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature. 2015;526:587–90.
Shi L, Dong H, Reguera G, Beyenal H, Lu A, Liu J, et al. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat Rev Microbiol. 2016;14:651–62.
Hernsdorf AW, Amano Y, Miyakawa K, Ise K, Suzuki Y, Anantharaman K, et al. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017;11:1915.
Ino K, Hernsdorf AW, Konno U, Kouduka M, Yanagawa K, Kato S, et al. Ecological and genomic profiling of anaerobic methane-oxidizing archaea in a deep granitic environment. ISME J. 2017;12:31.
Wankel SD, Adams MM, Johnston DT, Hansel CM, Joye SB, Girguis PR. Anaerobic methane oxidation in metalliferous hydrothermal sediments: influence on carbon flux and decoupling from sulfate reduction. Environ Microbiol. 2012;14:2726–40.
McGlynn SE. Energy metabolism during anaerobic methane oxidation in ANME archaea. Microbes Environ. 2017;32:5–13.
Timmers PHA, Welte CU, Koehorst JJ, Plugge CM, Jetten MSM, Stams AJM. Reverse methanogenesis and respiration in methanotrophic Archaea. Archaea. 2017;2017:22.
Acknowledgements
We thank Kim Baublys, Sue Golding, Faye Liu, and Ai Nguyen for their help with isotopic analyses, Alistair Grinham and Lu Fan for helping with inoculum collection and in-situ methane and Fe(II) profiling, and Jiaoyang Pu for helping with bioreactor maintenance. We also thank Serene Low and Nicola Angel for library preparation and Illumina sequencing. We are grateful to Eloise Larsen for reviewing and editing the manuscript. This work is supported by the Australian Research Council (ARC) through projects DP120100163 and DP170104038 and the U.S. Department of Energy’s Office of Biological Environmental Research (DE-SC0010574). C.C. is supported by The University of Queensland International Scholarship and China Scholarship Council Scholarship. A.O.L. is supported by the Australian Postgraduate Award. G.X. is supported by The University of Queensland Postdoctoral Fellowship. J.G. is supported by the ARC Discovery Early Career Researcher Award. Y.F. and J.Z. acknowledge funding support from the ARC and The University of Queensland to the Radiogenic Isotope Facility. G.W.T. is supported by The University of Queensland Vice-Chancellor’s Research Focused Fellowship. S.H. is supported by an Advanced Queensland Research Fellowship.
Author contributions
Z.Y. and S.H. designed the study. C.C. enriched the microorganisms and conducted mass and electron balance and isotopic labelling batch tests. C.C., J.G., Z.Y., and S.H. analysed the process data. A.O.L. performed the sampling, preservation and nucleic acid extractions. A.O.L. prepared samples for metagenomic and metatranscriptomic sequencing. A.O.L. and G.W.T. performed the microbial community analysis and the bioinformatics analysis. G.X. verified the extraction methods. Y.F. and J.Z. performed the isotopic iron measurement. C.C., A.O.L., G.W.T., Z.Y., and S.H. wrote the manuscript in consultation with all other authors.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Candidatus“Methanoperedens ferrireducens” gen. et sp. nov.
Etymology. Methanoperedens. methano (new Latin): pertaining to methane; peredens (Latin): consuming, devouring; ferrireducens. ferri (Latin): pertaining to Fe(III) oxide; reducens (Latin): leading back, bringing back and in chemistry converting to a different oxidation state. The name implies an organism capable of consuming methane and also reducing iron-related compounds.
Locality. Enriched from sediment collected from a freshwater reservoir in Brisbane, Australia.
Rights and permissions
About this article

Cite this article
Cai, C., Leu, A.O., Xie, GJ. et al. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J 12, 1929–1939 (2018). https://doi.org/10.1038/s41396-018-0109-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0109-x
This article is cited by
-
Mechanisms of extracellular electron transfer in anaerobic methanotrophic archaea
Nature Communications (2024)
-
Effects of pockmark activity on iron cycling and mineral composition in continental shelf sediments (southern Baltic Sea)
Biogeochemistry (2024)
-
Characterization and environmental applications of soil biofilms: a review
Environmental Chemistry Letters (2024)
-
Vertical distribution of Candidatus Methylomirabilis and Methanoperedens in agricultural soils
Applied Microbiology and Biotechnology (2024)
-
Anaerobic methanotroph ‘Candidatus Methanoperedens nitroreducens’ has a pleomorphic life cycle
Nature Microbiology (2023)
