
Abstract
Colonization is a key component of community assembly because it continuously contributes new species that can potentially establish and adds individuals to established populations in local communities. Colonization is determined by the regional species pool, which is typically viewed as stable at ecological time scales. Yet, many natural communities including plants, birds and microbes, are exposed to several distinct and dynamic sources of colonists and how multiple colonist pools interact to shape local communities remains unclear. Using a 16S rRNA amplicon survey, we profiled bacteria within surface, subsurface and burrow sediments and assessed their role as colonist pools for fiddler crab-associated bacteria. We found significant differences in composition among sediment types, driven by halophilic taxa in the surface, and different Desulfobacteraceae taxa in the subsurface and burrow. Bacteria from burrow sediment colonized the crab carapace whereas gut bacterial communities were colonized by burrow and surface sediment bacteria. Despite distinct colonist pools influencing gut bacteria, variation in composition across gut samples did not lead to significant clusters. In contrast, carapace bacterial communities clustered in six distinct groups loosely associated with crab species. Our findings suggest that multiple colonist pools can influence local communities but factors explaining variation in community composition depend on local habitats. Recognizing multiple colonist pools expands our understanding of the interaction between regional and local processes driving community structure and diversity.
Similar content being viewed by others


Introduction
Despite decades of focus on the local scale, ecology now recognizes the influence of regional processes of evolution and dispersal on community diversity and composition [1,2,3]. During all stages of community assembly, colonization can influence community diversity and composition by contributing new species and adding individuals to established populations [4,5,6]. Determining how many and which colonists influence local communities is important to understand community structure because the number and identity of species of colonists can regulate richness [7,8], influence species composition [9], trait distribution [9], and ecosystem function [10] at the local scale. In plant and animal ecology, the traditional concept of the regional species pool hypothesizes that species available for colonization depend on large-scale evolutionary and biogeographical processes [11,12]. Due to the large spatial and long temporal scales of these processes, the regional species pool is often assumed to be stable at ecological time scales [11,13,14] and this view has permeated much of the understanding of local and regional processes [15], interspecific interactions [16], and neutral theory [17]. However, the pool of colonists interacting with local scale habitats is often a subset of this large-regional pool, subject to more proximate habitat factors, particularly in microbial systems [18,19,20].
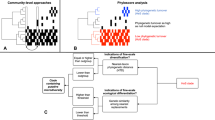
The evolutionary regional species pool can be divided into subsets of colonists that share similar dispersal mechanisms and ecological traits. For example, of all bacteria in alpine glaciers, Rime and collaborators [21] found that microorganisms from glacier sediment, and not from precipitation or streams, colonized newly deglaciated soil. For clarity, we refer to each of these subsets of colonists as a colonist pool, the group of species that arrives to local patches and interacts with local habitats. Previous studies have identified the most important colonist pool among a series of potential pools in some systems (e.g. [20,22,23,24]), but have rarely directly considered the possibility that multiple colonist pools can coexist with several local habitats (but see [25]). For example, although plant communities are often exposed to wind dispersed colonists (seed rain) and colonists from the soil (seed bank), which colonist pool is actually the most influential can depend on soil properties of local habitats [26,27,28]. If a particular local habitat receives colonists from only one colonist pool, comparing the colonist pool with local community composition is informative of the habitat filter [29]. These filters occur when habitat conditions are harsh or stressful for non-tolerant species and local communities become less diverse, less heterogeneous and less similar to the colonist pool [30]. However, there are other possible scenarios when multiple colonist pools influence different habitats (Fig. 1). If neighboring local sites with different habitats receive colonists from the same colonist pool (Fig. 1b), this provides an ideal opportunity to compare their habitat filters. If local sites receive colonists from multiple colonist pools (Fig. 1c), then comparing the local habitat to all contributing colonist pools is necessary to assess the nature of the habitat filters.

a each colonist pool influences one of the habitats; b one colonist pool influences two habitats; and c two colonist pools colonize a local habitat. Small, black arrows indicate the mechanisms that divide the broad regional species pool into colonist pools. Dashed lined circles symbolize each colonist pool and its color indicates species composition. White, thick arrows indicate colonization processes. Solid line circles indicate local communities and their coloration indicates habitat characteristics
Here, we examined the influence of multiple colonist pools of sediment bacteria on fiddler crab-associated bacterial communities and ask whether considering multiple colonist pool influences improves our understanding of this system. When fiddler crabs molt, newly formed chitin surfaces of the carapace and the gut are sterile and receive colonists from sources of bacteria in the surrounding environments [31,32]. Colonists are likely to come from the sediment because fiddler crabs feed on organic matter, microalgae and bacteria that they scrape off the surface sediment [33] and burrow to mate, escape predation and avoid extreme temperatures [34]. Sediment bacteria can be subdivided in three dominant habitats in salt marshes based on physicochemical conditions: sediment surface, subsurface, and crab burrows [35,36,37]. Bacteria in the surface sediment experience high oxygen and light availability [36,38] and should be dominated by aerobic heterotrophs, autotrophs, and nitrogen fixers [39,40,41]. Subsurface bacteria experience low oxygen levels and utilize NO, Fe and SO4 as electron acceptors [35,42] and should thus be dominated by taxa capable of anaerobic metabolism [36]. The sediment on the burrow walls is aerated and mixed as fiddler crabs burrow, developing a diverse bacterial assemblages that differ markedly in composition from surface and subsurface bacteria [43,44,45,46,47]. Although microbial communities in these sediments have been studied separately, their combined influences on the infauna have not been assessed yet. Host-associated microbial communities exposed to colonization from sediment bacteria could reveal alternative multiple colonist pool scenarios (Fig. 1).
Regardless of how many colonist pools influence a site, all propagules must pass through local habitat filters before recruiting to local communities. In the crab’s carapace, bacteria able to attach and compete for space should be able to maintain membership in the community [48,49]. In contrast, to colonize the gut, bacteria must tolerate the physical scrapping and sediment selection of the crab’s oral appendages [50] and the chemical stress of hepatopancreas secretions [51,52]. In addition to local habitat characteristics, host factors like genotype [53,54,55] or species can act as a filter and determine the variation in the composition of associated microbial communities [56,57]. Empirical studies have however found contradictory evidence about these relationships [58,59] vs. [60], which could result from our lack of understanding of the mechanisms through which host genotype or species regulate its habitats. Fiddler crab species have been found to differ in some physiological factors [61] but not others [62] and whether these factors have an effect on their associated microbial communities is unknown. In contrast, the crab’s sex, has been linked with increased richness in male gut bacteria (Cuellar-Gempeler and Munguia [63]). This diversity effect of sex can be mediated by physiological [64,65,66], or behavioral [67,68] differences between fiddler crab males and females. Importantly, to decipher the role of host factors and habitat filters on bacterial community diveristy and composition, we must first investigate the influence of colonization.
The major goal of this study is to investigate the influence of multiple colonist pools on fiddler crab associated bacteria and to compare its effect on composition to that of host-associated factors. Specifically, we asked three questions. First, we ask how different are the colonist pools. We hypothesize that surface, subsurface and burrow bacteria differ based the physicochemical characteristics of each sediment type. Second, we questioned how many and which sediment colonist pool influences the carapace’s and the gut’s bacterial communities. Based on the crab’s interaction with the sediment, we hypothesize that bacteria colonize the gut when the crab feeds on surface sediment and burrow sediment bacteria colonize the carapace as the crab takes refuge. In contrast, we do not expect the subsurface sediment to be an important contributor of colonists. Third, we ask whether host factors like crab sex or species generate variation in community diversity and composition. We hypothesize that sex is more important in determining variation in bacterial composition than host species, based on previous findings that suggest bacterial communities associated with males are more diverse.
Methods
We conducted our study in a 150 × 150 m2 marsh area located on Stedman Island, near Aransas Pass, Texas (27° 53’ 13.56” N, −97° 7’ 0.07 W). The sediment is influenced by inflow from high salinity seawater from Redfish Bay (between 25 and 35 ppt during the summer [69]) and vegetated by scarce black mangrove (Avicenia germinans), saltmarsh cordgrass (Spartina alterniflora) and woody glasswort (Salicornia bigelovii). This salt marsh was chosen because it is co-inhabited by two fiddler crab species (Uca panacea and Uca rapax) that exhibit similar behavior, habitat, and diet [70]. We focused on the bacterial community associated with these crab’s carapace and gut, which represent different habitats for bacteria. While the crab’s carapace is open to colonization [48,49], colonizing the gut requires surpassing physical [50] and chemical filters [51,52].
Sampling procedure
Sample collection was divided evenly in four sampling dates (July 21, 29 and 31, and August 5, 2014). Adult U. panacea and U. rapax (18 males and 18 females) were collected by hand and stored in individual sterile containers. Using sterile spatulas, we collected a total of 10 samples of each sediment type. Approximately 20 g of surface, subsurface and burrow sediment were collected on sterile 50 mL falcon tubes. Replicates of surface, subsurface and burrows were collected approximately 10 m apart from each other at sites with active burrows. Active burrows were identified based on crab activity (leaving and entering) and signs of recent burrowing (freshly disturbed sediment and crab tracks). Surface sediment was scrapped from the top layer (0 to 2 cm deep). For subsurface samples, we mixed sediment from 7 to 15 cm in depth. Although microbial community composition can vary significantly with depth [71], crabs are exposed to all bacteria over this depth range when burrowing. Likewise, burrow samples were obtained from this depth range but along the surface of fiddler crab burrows. We selected only burrows that extended beyond 15 cm in depth and were wider than 2 cm. Crab and sediment samples were transported to the Marine Science Institute from the University of Texas at Austin for further processing.
Sample preparation and DNA extraction
Upon arrival to the lab, samples were prepared for DNA extraction. Each sample from the sediment was homogenized by vortexing at high speed for 30 s and 2 g were separated, in sterile conditions, for DNA extraction. Although this procedure may not completely homogenize a heterogeneous sediment sample, it allowed mixing of the sediment obtained from different depths in subsurface and burrow samples. Surface samples were treated the same for consistency. Crabs were rinsed with sterile deionized water to remove debris and unattached microorganisms. Then, carapaces were swabbed and scraped to profile the surface community. Crabs were sacrificed by freezing and dissected to obtain gut samples. Then, guts were rinsed with sterile deionized water to avoid food bolus interference and to remove unattached bacteria. All samples were kept in MoBio PowerSoil bead tubes at −80 °C and were processed within 2 months of collection. DNA was extracted using the PowerSoil DNA extraction kit (MoBio) as instructed by the manufacturer. We included samples taken of all sterile materials to control for potential contamination. DNA concentration was quantified with Qubit fluorometer (Qubit 2.0, Invitrogen) using high sensitivity assay reagents. Only samples with more than 0.1 ng/μl DNA yield were used for the study to avoid sample bias.
16S rRNA gene library preparation and sequence analysis
Individual samples were prepared for Illumina sequencing using a two-step, gene-specific PCR. To avoid host DNA amplification, we targeted the V4 hypervariable region of bacterial 16S using the 515F/806R primer pair (Ong et al [72], Wang and Qian [73]). We used the MiSeq Illumina platform to obtain pair-end 250 bp nucleotide reads. Library preparation and sequencing was done at the University of Texas Genome Sequencing and Analysis Facility (GSAF). The resulting sequences were processed using custom bash scripts and QIIME [74] with Greengenes as reference databank [75]. OTUs (Operationa Taxonomical Units) were defined at the 97% sequence similarity and were picked with an open frame. Please refer to our Supplementary material for details on library preparation and sequence processing.
Data analysis
The following analyses were conducted in the R environment (version 3.3.2, [76]). We removed OTUs assigned to Archaea or unassigned and those found in less than 3 times in less than 1% of the samples.
First, we evaluated differences in diversity and community composition among sediment types. To determine whether these results reflect similar sampling effort across samples, we calculated species accumulation curves. Since the species accumulation curves from sediment reached asymptotes (Fig S1), we considered only raw richness for further analyses. We assessed the difference in richness across sediment type by using a GLM fitted with a negative binomial distribution. Then, we calculated Pielou’s evenness as J = H’/lnS [77] and used an ANOVA to determine differences across sediment types. We assessed normality and homoceidacity of residuals using the Shapiro-Wilks test and the Bartlett test, respectively [78,79]. Evenness was inverse square root transformed to meet parametric assumptions. We calculated distance-to-centroid as a measure of community similarity and evaluated differences in similarity across sediment types. We used an analysis of multivariate homogeneity of group dispersions with 999 permutations. This method is multivariate analogue of Levene’s test for homogeneity of variances where non-euclidean distances between objects and group centroids are reduced to principal coordinates to assess beta diversity [80]. We assessed the role of sampling date by adding it as a random factor in these tests (Table S1) but, since we did not find any effects, we removed it from the statistical models presented here.
We used a Cannonical Correspondance Analysis (CCA) with standardization by row and column weights to assess compositional differences between the three types of sediment bacteria. CCA is a direct ordination technique that can identify correlations between multivariate data on OTU abundances and the sediment type coded as dummy variables. Significance of the differences in composition was assessed with a permutational MANOVA on the CCA scores [81]. We compare the results from the CCA with non metric multidimensional scaling (NMDS) based on Bray-Curtis and a weighted UniFrac (Fig S4). Since we obtained similar results, we felt confident to proceed with the CCA ordination. We chose this specific ordination tool because it can incorporate the function predict, which allows us to find corresponding scores for crab samples in an unbiased way.
Second, we determined the contributions of each sediment pool to each crab-associated community by comparing community composition in each crab sample and the centroid of the various colonist pools. To incorporate the crab sample scores to the sediment CCA, we used the function predict from the package ‘vegan’ (version 2.4.4, [82]). Then, we assigned each crab-associated community to a species pool based on the distance to nearest pool centroid in multivariate CCA space. This approach is useful to estimate similarity between standard and test data without the bias of including all samples in the calculation of the original ordination axes [83]. We assessed the sensitivity of this method by comparing the results from all OTUs with the results from the 100 most abundant taxa (Fig S2) and to the outcome of Source Tracker ([84], Fig S3).
Third, we wanted to determine the effect of host-factors on community diversity and composition. We assessed the effect of host-factors on richness and evenness using a three-way ANOVA including habitat (carapace or gut), sex and species. We tested for normality and homoceidacity as described above for sediment samples. Evenness data was square root transformed to meet parametric assumtions. Then, we used a perMANOVA on CCA scores obtained from predict to determine how host factors, such as the type of habitat, or the crab’s sex or species influence bacterial community diversity and composition. We were also interested in assessing the significance of these groupings independently of the assignment methodology from the predict function. Thus, we used cluster analysis to identify groups of samples based on community composition and asked whether these clusters correspond to host-factors or pool assignments. We used a hierarchical clustering analysis with the “ward” method (hclust function, [85]) and estimated the significant number of clusters using the gap statistic [86]. Lastly, we calculated distance-to-centroid as a measure of community similarity and evaluated whether similarity was determined by the host’s sex or species within each habitat. We used an analysis of multivariate homogeneity of group dispersions as described above and tested the role of habitat type, host sex and species.
We looked at three taxonomical levels to identify the bacteria that contribute to the above diversity patterns. First, we illustrated bacterial phyla that characterize each sediment type and crab habitat these patterns. We built a heatmap detailing the abundance and distribution of bacterial Phyla using heatmap.2 from the package ‘gplots’ (version 3.0.1, [87]). Then, we focused on the family taxonomic level to identify bacterial abundance patterns that underlie colonist pool relationships. To identify specific families contributing to differences and similarities between sediment and crab substrates, we used SIMPER analysis [88]. SIMPER analysis compares normalized relative abundance between samples measuring differences as Bray-Curtis dissimilarity. The contribution of individual OTU i to compositional differences is estimated with the formula
where x represents the abundance of OTU iin simple j and k. Although this method can confound the overall abundance with variation between samples [89], we used it to identify bacterial families explaining 80% of the variation in community composition between carapace, gut, surface, and burrow bacteria. We tested for significant differences in abundance of bacterial families identified by SIMPER across sediment and crab substrates using a permutation test (999 permutations). We selected informative families to further tested differences in abundances among sediment types and crabs. We used ANOVAs unless parametric assumptions were not met after transformations. In these cases Kruskal-Wallis test was used instead. To identify specific differences in abundance across substrates, we used post hoc Tukey or Nemeyeni tests, depending on whether data was parametric or not. To correct for false discoveries over tests for each taxa, we used Bonferroni corrections [90]. We used additional heatmaps to illustrate OTU abundance and occupancy patterns of selected families.
Results
Of the original samples, 65 samples were suitable for analysis. After quality-filtering, we had 21 carapace, 15 gut, and 30 sediment samples with a median of 6605 reads per sample and a range between 3000 and 14346 reads. We identified 517 individual bacterial OTUs distributed across 17 phyla and 80 families.
Colonist pools in the sediment
Sediments contained a total of 459 OTUs. Surface and subsurface had comparable richness levels, while burrow sediments had slightly lower richness, albeit not significantly different (Table 1, χ22, 27 = 4.380, p = 0.996). Evenness followed rather the opposite trend, with burrow having a higher value than subsurface and surface having the lowest value (Table 1, F2, 27=16.83, p < 0.001). Surprisingly, the surface did not contain any unique OTUs, while subsurface and burrow sediments had 14 and 36 unique OTUs, respectively (Table 1). These unique OTUs were rare within each sediment type and did not represent more than 2% of the community (Table 1). The CCA revealed significant differences in bacterial community composition among sediment types (Fig. 2, perMANOVA, F2, 27 = 108.73, R2 = 0.889, p < 0.001). While there were clear differences in composition, sediment types also differed in community similarity, with burrow sediment containing the least similar assemblages (permutation test, F value = 45.898, p < 0.001, Fig. 2)

Crab CCA scores in were calculated with the predict function. Color indicates either sediment type (for sediment samples) or pool assignment (for crab samples). Pool assignment matches the color of the sediment identified as the source of colonists for each crab sample. Polygons in indicate samples from different sediment types (solid lines) and crab habitats (dashed lines)
In the surface sediment, we found high abundance of Firmicutes, and Spirochaetes (Fig. 3). Particularly, surface sediment bacteria were dominated by taxa from Halanaerobacteriaceae and Balneolaceae (relative contribution ranging from 9.2 to 24.8%, Fig. 4, Table S2). Cyanobacteria were consistently found in burrow and surface sediment but never reached high relative abundances (average relative abundance of 0.8% in surface and burrow sediment), and it was extremely rare in subsurface sediment. Subsurface sediments were instead represented by high abundances of OTUs from Caldithrix, and SAR406 (also known as MGA, [91]), while burrow sediments contained high abundance of Acidobacteria and Nitrospirae (Fig. 3). Subsurface and burrow sediments shared high abundances of Rhodobacteriaceae and Desulfobacteriaceae (Fig. 4). Importantly, sediment types differed in OTUs composition within bacterial families such as Desulfobacteriaceae (Fig. 5, Table S3).

Heatmap of bacterial Phyla scaled abundances across sediment and crab samples. Sequence abundances were scaled per sample to obtain relative abundance and colored from white to dark grey, which represent high to low abundance (see color key density plot on the top-left corner). The dendogram on the left side indicates taxa similarities in distributions across samples and was calculated with the hclust function. White vertical lines separate the different substrates

Bacterial relative abundance on crab and sediment samples for selected families. Error bars represent standard error. Letters indicate significant differences in bacterial abundance across substrates

See Fig. 4 for details except for a few changes. White columns indicate samples that did not contain Desulfobacteraceae. Black vertical lines separate the different substrates
Colonist pool influences on crab-associated bacteria
Based on community similarity, most crab-associated communities were assigned to burrow, few to surface sediment, and none to subsurface sediment pool. While all carapace bacterial communities were (100%) assigned to the burrow, 10% gut communities were assigned to the surface and 90% to the burrow (Fig. 2). Crab gut bacterial communities assigned to the surface sediment corresponded to males from both species. A perMANOVA based on predicted CCA scores of crab samples revealed significant differences in bacterial community composition among carapaces and guts, but also showed an effect due to the interaction between host species and sex (Table 2). Crab bacterial communities were characterized by high abundance of Proteobacteria, Bacteroidetes and Actinobacteria and Verrucomicrobia. While most phyla were more abundant in the carapace, Tenericutes and Planctomycetes were more abundant in the gut.
The influences of sediment bacteria were better understood by looking at similarities at the family and OTU taxonomical level (Figs. 4, 5). Carapace and gut bacterial communities held high abundances of Rhodobacteraceae (mostly Paracoccus sp, Rhodobacter sp), Flavobacteriaceae and Saprospiraceae, which are also found, at lower abundance, in burrow and subsurface sediment (Fig. 4). Dominant taxa from the surface (Halanaerobiaceae and Balneolaceae) were absent or present at very low abundances within crab habitats (Fig. 4). Vibrionaceae was an important representative of crab bacterial communities and was found, at lower abundance, in the burrow sediment. At the OTU level, we found that taxa within Desulfobacteraceae found in crab samples reflect those found in burrow sediment (Fig. 5).
Host factors
Habitat within the host was the main explanatory factors for the bacterial communities’ richness and evenness. Carapace bacterial communities had higher richness than gut bacterial communities, with a marginal effect of species (Tables 3, 4). This pattern was probably driven by U. panacea, which had the lowest values of richness in the gut (Table 3). Gut bacterial communities had higher evenness than carapace bacterial communities (Tables 3, 4). There were no significant differences in community similarity between gut and carapace bacterial communities (permutation test, F value = 3.050, p = 0.081, Fig. 2), crab species (F value = 0.616, p = 0.46) or male and female communities (F value = 0.133, p = 0.87).
The cluster analysis showed a contrasting effect of host factors on carapace and gut bacterial communities. Gap analysis indicated that carapace bacterial communities clustered in 6 significant groups (Fig. 6a, b). These groups loosely correspond to host factors (Fig. 6a). For example, the first cluster on the left contains only U. rapax samples and the two clusters on the far right are composed of U. panacea. In contrast, gap analysis indicated only one cluster comprising all gut bacterial communities (Fig. 6c, d).

Cluster analysis a, c and gap statistic calculation results b, d for carapace a,b and gut c, d samples. Height in the y axis represents the distance at which the cluster was formed. Dashed lines show the clustering threshold indicating which sample groups represent significant clusters. Gap statistic plots b,d indicate the gap statistic for different number of clusters (k)
Discussion
Whether a unique regional species pool or multiple sources of colonists influence local communities could have consequences for local community dynamics. Here, we identified three potential colonist pools of bacteria in the salt marsh sediments and their influences in crab-associated bacterial communities. Patterns of similarity in community composition suggested that carapace bacterial communities receive most colonists from burrow sediment (Fig. 2). In contrast, guts received colonists from burrow and, at least in part, from surface bacteria (Fig. 2). This result suggests that multiple colonist pools can influence local patches (Fig. 1b) and individual colonist pool can influence distinct local habitats (Fig. 1c).
We found that the three potential colonist pools within the salt marsh sediment differed in OTU composition (Fig. 2). Our findings coincide with previous studies showing strong physicochemical differences driven sediment depth [47] and bioturbation [37,71,92] and suggest that sediment bacteria are under distinct ecological selection pressures. For example, surface sediment was strongly dominated by salt tolerant taxa (Halanaerobiaceae, Balneolaceae) while other OTUs were rare (Fig. 4, Table S2, [93,94,95]. Even with this strong environmental stress, surface bacteria were more diverse than other sediment types, possibly because oxygen, light availability or immigration from other habitats sustain a long tail of rare OTUs (Fig. 3, Table S2). These results coincide with studies highlighting the strong effects of salinity on autotrophic [40] and nitrogen fixers [41] commonly found in the surface. Interestingly, many of these halophilic taxa are also anaerobic and their abundance in the surface sediment suggests fine scale changes in oxygen availability. Subsurface and burrow communities were dominated by taxa consistent with an environment limited in oxygen but abundant in sulfates and nitrates (Figs. 3, 4, [39,42,96]), but the differences between these two types of sediment were less clear. By looking at the OTU taxonomical level, we found groups of OTU from selected families like Desulfobacteraceae that clearly characterize sediment types (Fig. 5, Table S3). This finding suggests that each group of OTU within this family specialize on each sediment type, partitioning the coastal marsh habitat and reducing competition [97]. Physicochemical differences among sediment type appear to be a stronger factor in driving bacterial composition than physical separation between the sediments driving ecological drift.
Based on similarities in OTU relative abundance and occupancy between crab-associated habitats and the sediment pools, burrow and surface sediment bacteria colonized fiddler crabs’ guts and carapaces (Fig. 2). Evidence for influence of the burrow on both crab habitats is better seen at two taxonomic levels. At the family level, Rhodobacteraceae and Flavobacteriaceae were important components of burrow sediment and crab-associated bacterial communities (Fig. 4). These taxa are common to the marine environment and have been previously found in association with crustacean exoskeleton and gut [98,99,100]. At the genus level, different taxa within Desulfobacteriaceae characterized each sediment type, but only burrow taxa matched taxa found associated with crabs (Fig. 5, Table S3). These findings support the idea that both gut and carapace habitats receive colonists from the burrow sediment (Fig. 1b).
In this study, we emphasize the role of the burrow sediment as a source of colonists, yet these results also suggest a two-way colonization feedback between burrow sediments and crab-associated bacteria. There are two reasons why we suggest that burrow to crab colonization is predominant. First, dominant taxa are shared between the subsurface and the burrow, suggesting that these are sediment and not crustacean adapted taxa. Second, Desulfobacteriaceae are known environmental bacteria requiring resources like sulfates and acetates to fulfill metabolic requirements [101]. Desulfobacteriaceae associated with crabs are likely to undergo constant colonization from the burrow, suggesting a source-sink relationship, at least for this group of taxa (also known as mass effects in metacommunity ecology, [102]). However, we want to highlight that the crab’s activity can also have an important effect on the diversity and composition of burrow sediment bacteria. Previous work showed that areas of increased bioturbation are enriched in Vibrio spp. [103,104] and other Proteobacteria [105]. In our study, Vibrionaceae from crab bacterial communities is likely to colonize the burrow sediment. We propose a complex feedback mechanism between the effects of the crab’s activity on burrow sediment physicochemistry, bacterial migration from the crab to the sediment and colonization of crab habitats from burrow sediment bacteria.
We found that crab-associated bacterial communities significantly cluster in groups based on community composition, but only in the carapace (Fig. 6). Contrary to our expectations, the clusters were associated with host species. There may be physiological or behavioral differences between these species that we have not considered. For example, fiddler crab reproductive behavior and physiology varies across species and can influence the associated bacterial communities [65]. In contrast to the carapace bacterial community, there were no significant clusters in gut bacteria (Fig. 6). One possible explanation is that strong filtering in the gut homogenizes these bacterial communities. If this is true, then their composition should remain unchanged and similarity in community composition should be higher [30], yet we failed to find differences in community similarity between gut and carapace bacterial communities. Alternatively, lack of clustering can be explained by considering a gradient in the contribution of multiple colonist pools. On one extreme, guts receive most or all colonists from surface sediment; while towards the other extreme, surface contribution diminishes and burrow contribution increases. Although there are no records of fiddler crab feeding from the burrows, they may be using burrow water when stressed by high temperatures and low water availability [106,107]. In addition, these patterns may be associated with molting stage, or other defining events in the life of each individual crab.
Our results suggest that surface sediment bacteria can influence some gut bacterial communities. While this concurs with our expectations based on crab feeding behavior [67], we found that dominant taxa from the surface sediments were not representative of gut bacterial communities (Figs. 3, 4). Similarly, dominant taxa in the gut were not represented in the surface (Figs. 3, 4). A possible explanation is that rare OTUs from the surface sediment influence the distribution of rare OTUs in the crab’s gut. However, when analyzed the patterns from the 100 most abundant species, we found the same relationship between the gut and the surface sediment bacteria (Fig S2). Influential taxa could thus be intermediate OTUs that are rare within the 100 most abundant taxa. A possible explanation for these patterns is that bacteria able to colonize the crab’s gut survive by remaining inactive and dormant within surface sediment.
The method we use to assign colonist pools to local communities relies on assignments based on similarity to determine species pool influences. This method is limited by the absence of a direct measure of dispersal, which results in two main issues. First, other sources of colonists may influence fiddler crab-associated communities. For example, bacteria in tidal seawater, plants and other crabs could influence fiddler crab associated bacterial communities. However, marsh sediment contains richer and more abundant bacterial assemblages [108] and has a stronger relationship with fiddler crabs based on their activity budgets [67,68,109], when compared to other sources. Second, our supposition that similarity indicates major contribution of a colonist pool assumes that we have identified all the species pools. Even with these limitations, the method used here allows us to identify likely sources of colonists from the host’s surrounding environment with no previous knowledge and it provides results that are comparable to other methods ([84], Fig S3).
Our study highlights colonization from distinct species pools and its consequences for local diversity. Importantly, it suggests that local communities may be influenced by more than one colonist pool and colonist pools can influence more than one local habitat. Grouping species according to their dispersal regimes and large-scale ecological filters (such as sediment type) may result in a more realistic approach to the influence of multiple colonist pools on local communities. This view could aid at bridging our understanding of systems that operate at distinct scales but depend on this balance between colonization and local habitats, such as host-associated bacteria and plant communities. Integrating multiple colonist pools with the traditional evolutionary and biogeographical concept of the species pool may lead to a better understanding of the relationship between colonization, local filters and patterns of diversity across multiple scales.
References
Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecol Lett. 2004;7:601–13.
Ricklefs RE. Community diversity - relative roles of local and regional processes. Science. 1987;235:167–71.
Urban MC, Leibold MA, Amarasekare P, De Meester L, Gomulkiewicz R, Hochberg ME, et al. The evolutionary ecology of metacommunities. Trends in Ecology & Evolution. 2008;23:311–7.
Freestone AL, Osman RW. Latitudinal variation in local interactions and regional enrichment shape patterns of marine community diversity. Ecology. 2011;92:208–17.
Fukami T. Assembly history interacts with ecosystem size to influence species diversity. Ecology. 2004;85:3234–42.
Shipley B, Paine CET, Baraloto C. Quantifying the importance of local niche-based and stochastic processes to tropical tree community assembly. Ecology. 2012;93:760–9.
Karger DN, Tuomisto H, Amoroso VB, Darnaedi D, Hidayat A, Abrahamczyk S, et al. The importance of species pool size for community composition. Ecography. 2015;38:1243–53.
Myers JA, Harms KE. Seed arrival, ecological filters, and plant species richness: a meta-analysis. Ecol Lett. 2009;12:1250–60.
Lessard JP, Belmaker J, Myers JA, Chase JM, Rahbek C. Inferring local ecological processes amid species pool influences. Trends in Ecology & Evolution. 2012;27:600–7.
Belmaker J, Jetz W. Spatial Scaling of Functional Structure in Bird and Mammal Assemblages. American Naturalist. 2013;181:464–78.
Zobel M. The species pool concept as a framework for studying patterns of plant diversity. Journal of Vegetation Science. 2016;27:8–18.
Cornell HV, Harrison SP (2014). What Are Species Pools and When Are They Important? In: Futuyma DJ (ed). Annual Review of Ecology, Evolution, and Systematics, Vol 45. pp 45-67.
Zobel M. Plant species coexistence- the role of historical, evolutionary and ecological factors. Oikos. 1992;65:314–20.
Godfray HCJ, Lawton JH. Scale and species numbers. Trends in Ecology & Evolution. 2001;16:400–4.
Rajaniemi TK, Goldberg DE, Turkington R, Dyer AR. Quantitative partitioning of regional and local processes shaping regional diversity patterns. Ecol Lett. 2006;9:121–8.
Diamond JM. The island dilemma: lessons of modern biogeographic studies for the design of natural reserves. Biol Conserv. 1975;7:129–46.
Hubbell SP (2001) The unified neutral theory of biodiversity and biogeography. Princeton University Press.
da Fonseca-Genevois V, Somerfield PJ, Neves MHB, Coutinho R, Moens T. Colonization and early succession on artificial hard substrata by meiofauna. Marine Biology. 2006;148:1039–50.
Langenheder S, Ragnarsson H. The role of environmental and spatial factors for the composition of aquatic bacterial communities. Ecology. 2007;88:2154–61.
Loudon AH, Woodhams DC, Parfrey LW, Archer H, Knight R, McKenzie V, et al. Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). Isme Journal. 2014;8:830–40.
Rime T, Hartmann M, Frey B. Potential sources of microbial colonizers in an initial soil ecosystem after retreat of an alpine glacier. Isme Journal. 2016;10:1625–1641.
Graves GR, Gotelli NJ. Neotropical land-bridge avifaunas - new approaches to null hypothesis in biogeography. Oikos. 1983;41:322–33.
Holzapfel C, Schmidt W, Shmida A. The role of seed bank and seed rain in the recolonization of disturbed sites along an aridity gradient. Phytocoenologia. 1993;23:561–80.
Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345:1048–52.
Flores GE, Bates ST, Knights D, Lauber CL, Stombaugh J, Knight R, et al. Microbial Biogeography of Public Restroom Surfaces. Plos One. 2011;6:e28132.
Willems JH, Bik LPM. Restoration of high species density in calcareous grassland: the role of seed rain and soil seed bank. Applied Vegetation Science. 1998;1:91–100.
Wolters M, Bakker JP. Soil seed bank and driftline composition along a successional gradient on a temperate salt marsh. Applied Vegetation Science. 2002;5:55–62.
Kalamees R, Zobel M. The role of the seed bank in gap regeneration in a calcareous grassland community. Ecology. 2002;83:1017–25.
Kraft NJB, Adler PB, Godoy O, James EC, Fuller S, Levine JM. Community assembly, coexistence and the environmental filtering metaphor. Functional Ecology. 2015;29:592–9.
Chase JM, Myers JA. Disentangling the importance of ecological niches from stochastic processes across scales. Philosophical Transactions of the Royal Society B-Biological Sciences. 2011;366:2351–63.
Duneau D, Ebert D. The role of moulting in parasite defence. Proceedings of the Royal Society B-Biological Sciences. 2012;279:3049–54.
Gaiser EE, Bachmann RW. The ecology and taxonomy of epizoic diatoms on cladocera. Limnol Oceanogr. 1993;38:628–37.
Dye AH, Lasiak TA. Assimilation efficiencies of fiddler crabs and deposit feeding gastropods from tropical mangrove sediments. Comparative Biochemistry and Physiology a-Physiology. 1987;87:341–4.
Kristensen E. Mangrove crabs as ecosystem engineers; with emphasis on sediment processes. Journal of Sea Research. 2008;59:30–43.
Ferreira TO, Otero XL, Vidal-Torrado P, Macias F. Effects of bioturbation by root and crab activity on iron and sulfur biogeochemistry in mangrove substrate. Geoderma. 2007a;142:36–46.
Kopke B, Wilms R, Engelen B, Cypionka H, Sass H. Microbial diversity in coastal subsurface sediments: a cultivation approach using various electron acceptors and substrate gradients. Appl Environ Microbiol. 2005;71:7819–30.
Papaspyrou S, Gregersen T, Cox RP, Thessalou-Legaki M, Kristensen E. Sediment properties and bacterial community in burrows of the ghost shrimp Pestarella tyrrhena (Decapoda: Thalassinidea). Aquatic Microbial Ecology. 2005;38:181–90.
Moon YJ, Kim SI, Chung YH. Sensing and Responding to UV-A in Cyanobacteria. International Journal of Molecular Sciences. 2012;13:16303–32.
Holguin G, Vazquez P, Bashan Y. The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: an overview. Biology and Fertility of Soils. 2001;33:265–78.
Rejmankova E, Komarkova J. Response of cyanobacterial mats to nutrient and salinity changes. Aquatic Botany. 2005;83:87–107.
Romero IC, Jacobson M, Fuhrman JA, Fogel M, Capone DG. Long-term nitrogen and phosphorus fertilization effects on N-2 fixation rates and nifH gene community patterns in mangrove sediments. Marine Ecology-an Evolutionary Perspective. 2012;33:117–27.
Segarra KEA, Comerford C, Slaughter J, Joye SB. Impact of electron acceptor availability on the anaerobic oxidation of methane in coastal freshwater and brackish wetland sediments. Geochim Cosmochim Acta. 2013;115:15–30.
Fanjul E, Grela MA, Iribarne O. Effects of the dominant SW Atlantic intertidal burrowing crab Chasmagnathus granulatus on sediment chemistry and nutrient distribution. Mar Ecol Prog Ser. 2007;341:177–90.
Fanjul E, Escapa M, Montemayor D, Addino M, Alvarez MF, Grela MA, et al. Effect of crab bioturbation on organic matter processing in South West Atlantic intertidal sediments. Journal of Sea Research. 2015;95:206–16.
Hunting ER, Whatley MH, van der Geest HG, Mulder C, Kraak MHS, Breure AM, et al. Invertebrate footprints on detritus processing, bacterial community structure, and spatiotemporal redox profiles. Freshwater Science. 2012;31:724–32.
Marinelli RL, Lovell CR, Wakeham SG, Ringelberg DB, White DC. Experimental investigation of the control of bacterial community composition in macrofaunal burrows. Mar Ecol Prog Ser. 2002;235:1–13.
Wang JQ, Zhang XD, Jiang LF, Bertness MD, Fang CM, Chen JK, et al. Bioturbation of Burrowing Crabs Promotes Sediment Turnover and Carbon and Nitrogen Movements in an Estuarine Salt Marsh. Ecosystems. 2010;13:586–99.
Harris JM. The presence, nature and role of gut microflora in aquatic invertebrates - a synthesis. Microb Ecol. 1993;25:195–231.
Robinson CJ, Bohannan BJM, Young VB. From structure to function: the ecology of host-associated microbial communities. Microbiology and Molecular Biology Reviews. 2010;74:453–76.
Brosing A. Recent developments on the morphology of the brachyuran foregut ossicles and gastric teeth. Zootaxa. 2010;44:33–41.
Vogt G, Stocker W, Storch V, Zwilling R. Biosynthesis of Astacus protease, a digestive enzyme from crayfish. Histochemistry. 1989;91:373–81.
Wang W, Wu XG, Liu ZJ, Zheng HJ, Cheng YX. Insights into hepatopancreatic functions for nutrition metabolism and ovarian development in the crab Portunus trituberculatus: gene discovery in the comparative transcriptome of different hepatopancreas stages. Plos One. 2014;9:e84921.
Benson AK, Kelly SA, Legge R, Ma FR, Low SJ, Kim J, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18933–8.
Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–33.
Smith CCR, Snowberg LK, Caporaso JG, Knight R, Bolnick DI. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. Isme Journal. 2015;9:2515–26.
Dillon RJ, Dillon VM. The gut bacteria of insects: Nonpathogenic interactions. Annu Rev Entomol. 2004;49:71–92.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science. 2011;332:970–4.
Brucker RM, Bordenstein SR. The Hologenomic Basis of Speciation: Gut Bacteria Cause Hybrid Lethality in the Genus Nasonia. Science. 2013;341:667–9.
Fraune S, Bosch TCG. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13146–51.
Martinson VG, Douglas AE, Jaenike J. Community structure of the gut microbiota in sympatric species of wild Drosophila. Ecol Lett. 2017;20:629–39.
Munguia P, Levinton JS, Silbiger NJ. Latitudinal differences in thermoregulatory color change in Uca pugilator. J Exp Mar Bio Ecol. 2013;440:8–14.
Levinton J, Lord S, Higeshide Y. Are crabs stressed for water on a hot sand flat? Water loss and field water state of two species of intertidal fiddler crabs. J Exp Mar Bio Ecol. 2015;469:57–62.
Cuellar-Gempeler C, Munguia P. Fiddler crabs (Uca thayeri, Brachyura: Ocypodidae) affect bacterial assemblages in mangrove forest sediments. Community Ecology. 2013;14:59–66.
Darnell MZ, Fowler KK, Munguia P. Sex-specific thermal constraints on fiddler crab behavior. Behavioral Ecology. 2013;24:997–1003.
Hartnoll RG. Reproductive investment in Brachyura. Hydrobiologia. 2006;557:31–40.
Pennoyer KE, Himes AR, Frederich M (2016). Effects of sex and color phase on ion regulation in the invasive European green crab, Carcinus maenas. Marine Biology 163.
Caravello HE, Cameron GN. Foraging time allocation in relation to sex by the Gulf Coast fiddler crab (Uca panacea). Oecologia. 1987;72:123–6.
Caravello HE, Cameron GN. Time activity budgets of the Gulf-coast fiddler-crab (Uca panacea). American Midland Naturalist. 1991;126:403–7.
Bittler K. Salinity gradients in the Mission-Aransas National Estuarine Research REserve. Port Aransas TX: The Marine Science Institute from the University of Texas at Austin; 2011.
Thurman CL. Fiddler-crabs (Genus Uca) of Eastern Mexico (Decapoda, Brachyura, Ocypodidae). Crustaceana. 1987;53:94–105.
Bertics VJ, Ziebis W. Biodiversity of benthic microbial communities in bioturbated coastal sediments is controlled by geochemical microniches. Isme Journal. 2009;3:1269–85.
Ong SH, Kukkillaya VU, Wilm A, Lay C, Ho EXP, Low L et al. (2013). Species Identification and Profiling of Complex Microbial Communities Using Shotgun Illumina Sequencing of 16S rRNA Amplicon Sequences. Plos One 8
Wang Y, Qian PY (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. Plos One 4.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010b;7:335–6.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72:5069–72.
Team Rdc (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria.
Pielou EC. The measurement of diversity in different types of biological collections. J Theor Biol. 1966;13:131–44.
Bartlett MS. Properties of sufficiency and statistical tests. Proceedings of the Royal Society of London Series a-Mathematical and Physical Sciences. 1937;160:0268–82.
Shapiro SS, Wilk MB. An analysis of variance test for normality (complete series). Biometrika. 1965;52:591–&.
Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–53.
Anderson MJ, Walsh DCI. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol Monogr. 2013;83:557–74.
Oksanen J, Blanchet G, Kindt R, Legendre P, Minchin P, O’Hara GL et al (2015). vegan: Community ecology package, 2.2-1 edn. p R package.
Petraitis PS, Methratta ET, Rhile EC, Vidargas NA, Dudgeon SR. Experimental confirmation of multiple community states in a marine ecosystem. Oecologia. 2009;161:139–48.
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Meth. 2011;8:761–3.
Ward JH. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;58:236–44.
Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2001;63:411–23.
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A et al (2016). gplots: Various R programming tools for plotting data, 3.0.1. edn.
Clarke KR. Nonparametric multivariate analyses of changes in community structure. Australian Journal of Ecology. 1993;18:117–43.
Warton DI, Wright ST, Wang Y. Distance-based multivariate analyses confound location and dispersion effects. Methods in Ecology and Evolution. 2012;3:89–101.
Dunn OJ. Multiple comparison among means. J Am Stat Assoc. 1961;56:52–64.
Wright JJ, Mewis K, Hanson NW, Konwar KM, Maas KR, Hallam SJ. Genomic properties of Marine Group A bacteria indicate a role in the marine sulfur cycle. ISME J. 2014;8:455–68.
Plante CJ, Wilde SB. Biotic disturbance, recolonization, and early succession of bacterial assemblages in intertidal sediments. Microb Ecol. 2004;48:154–66.
Abdeljabbar H, Cayol JL, Ben Hania W, Boudabous A, Sadfi N, Fardeau ML. Halanaerobium sehlinense sp nov., an extremely halophilic, fermentative, strictly anaerobic bacterium from sediments of the hypersaline lake Sehline Sebkha. Int J Syst Evol Microbiol. 2013;63:2069–74.
Bardavid RE, Oren A. The amino acid composition of proteins from anaerobic halophilic bacteria of the order Halanaerobiales. Extremophiles. 2012;16:567–72.
Oren A. Life at high salt concentrations, intracellular KCl concentrations, and acidic proteomes. Frontiers in Microbiology. 2013;4:315.
Ferreira TO, Otero XL, Vidal-Torrado P, Macias F. Redox processes in mangrove soils under Rhizophora mangle in relation to different environmental conditions. Soil Science Society of America Journal. 2007b;71:484–91.
Oakley BB, Carbonero F, van der Gast C, Hawkins RJ, Purdy KJ. Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J. 2010;4:488–97.
Dang HY, Lovell CR. Numerical dominance and phylotype diversity of marine Rhodobacter species during early colonization of submerged surfaces in coastal marine waters as determined by 16S ribosomal DNA sequence analysis and fluorescence in situ hybridization. Appl Environ Microbiol. 2002;66:467–75.
Kirchman DL. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100.
Wang W. Bacterial diseases of crabs: a review. J Invertebr Pathol. 2011;106:18–26.
Miletto M, Williams KH, N’Guessan AL, Lovley DR. Molecular analysis of the metabolic rates of discrete subsurface populations of sulfate reducers. Appl Environ Microbiol. 2011;77:6502–9.
Shmida A, Wilson MV (1985). Biological determinants of species diversity. Journal of biogeography : 1-20.
Gamble MD, Lovell CR. Infaunal Burrows Are Enrichment Zones for Vibrio parahaemolyticus. Appl Environ Microbiol. 2011;77:3703–14.
Pruzzo C, Vezzulli L, Colwell RR. Global impact of Vibrio cholerae interactions with chitin. Environ Microbiol. 2008;10:1400–10.
Laverock B, Smith CJ, Tait K, Osborn AM, Widdicombe S, Gilbert JA. Bioturbating shrimp alter the structure and diversity of bacterial communities in coastal marine sediments. Isme Journal. 2010;4:1531–44.
Thompson WE, Molinaro PJ, Greco TM, Tedeschi JB, Holliday CW. Regulation of hemolymph volume by uptake of sand capilary water in desiccated fiddler crabs, Uca pugialtor and Uca pugnax. Comparative Biochemistry and Physiology a-Physiology. 1989;94:531–8.
Thurman CL. Osmoregulation in six sympatric fiddler crabs (genus Uca) from the northwestern Gulf of Mexico. Marine Ecology-Pubblicazioni Della Stazione Zoologica Di Napoli I. 2002;23:269–84.
Lasher C, Dyszynski G, Everett K, Edmonds J, Ye WY, Sheldon W, et al. The Diverse Bacterial Community in Intertidal, Anaerobic Sediments at Sapelo Island, Georgia. Microb Ecol. 2009;58:244–61.
Weissburg M. Functional analysis of fiddler crab foraging sex-specific mechanics and constraints in Uca pugnax (Smith). J Exp Mar Bio Ecol. 1992;156:105–24.
Acknowledgements
For financial support, we thank the Graduate Doctoral Dissertation Improvement Grant from the Ecology Evolution and Behavior program at the University of Texas at Austin for funding. We would like to thank Deana Erdner for useful comments guiding our molecular work and for her generous sharing of laboratory space. Three anonymous reviewers provided remarkable advice that improved the manuscript considerably. Lastly, we thank undergraduate and graduate students as well as postdocs and friends that contributed to field sampling.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
:The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Cuellar-Gempeler, C., Leibold, M.A. Multiple colonist pools shape fiddler crab-associated bacterial communities. ISME J 12, 825–837 (2018). https://doi.org/10.1038/s41396-017-0014-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-017-0014-8
This article is cited by
-
Profile of the gut microbiota of Pacific white shrimp under industrial indoor farming system
Applied Microbiology and Biotechnology (2024)
-
Partitioning benthic nitrogen cycle processes among three common macrofauna holobionts
Biogeochemistry (2022)
-
A high abundance of Firmicutes in the intestine of chinese mitten crabs (Eriocheir sinensis) cultured in an alkaline region
AMB Express (2021)
-
Environment and host-related factors modulate gut and carapace bacterial diversity of the invasive red swamp crayfish (Procambarus clarkii)
Hydrobiologia (2021)
-
N2 fixation dominates nitrogen cycling in a mangrove fiddler crab holobiont
Scientific Reports (2020)
