
Abstract
Study design
Propensity score-matched, retrospective cohort study.
Objectives
To determine the risk of developing Alzheimer’s disease (AD) in patients with spinal cord injury (SCI).
Setting
The present study used Taiwan’s National Health Insurance Research Database.
Methods
A total of 9257 patients who had ⩾2 ambulatory visits with a diagnosis of SCI in 2001 were included in the SCI group. The non-SCI group consisted of 37,028 propensity score-matched patients without a diagnosis of SCI. The cumulative incidence of AD was estimated for each of the two patient groups using the Kaplan–Meier method. Stratified Cox proportional hazard regression was then employed to assess the influence of SCI on the risk of AD.
Results
During the follow-up period, 25 subjects in the SCI group and 57 in the non-SCI group developed AD. The cumulative incidence of AD in the SCI group was higher than in the non-SCI group (P = 0.0168); and the hazard ratio of AD for the SCI group, as compared to the non-SCI group, was 1.71 (95% CI 1.06–2.76, P = 0.0273).
Conclusions
This study suggests that patients with SCI have an increased risk of developing AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD), one of the most common neurodegenerative disorders, is characterized by progressive cognitive decline and, eventually, complete functional dependence, causing enormous medical and socioeconomic burdens [1]. The main neuropathological features of AD are extracellular neuritic plaques consisting of beta-amyloid, and intracellular neurofibrillary tangles composed of tau protein. These pathological changes tend to spread gradually throughout the brain as AD progresses [2]. Traumatic brain injury (TBI) has been associated with an increased risk of developing post-traumatic neurodegeneration and chronic traumatic encephalopathy [3, 4]. Spinal cord injury (SCI) is another central nervous system (CNS) injury that can lead to neuronal damage, inflammation, and neurodegeneration [3, 5]. Deposition within the spinal cord of amyloid precursor protein (APP), the precursor of beta-amyloid, has been observed in patients with SCI [6–8]. However, epidemiological data on the association between SCI and AD remain sparse and controversial [9]. Accordingly, we performed this large-scale population-based cohort study to evaluate the risk of AD among patients with SCI in a nationwide population sample.
Methods
Data source
The data used in this study were obtained from Taiwan’s National Health Insurance (NHI) Research Database for the years 2000 through 2003. This data has also been used in our previous studies on the association between SCI and neurodegenerative disorders [10, 11]. The NHI is a compulsory single-payer healthcare system, covering more than 97% of the country’s population. To ensure compliance with the personal privacy regulations in Taiwan, individual data was kept confidential and all personal identification numbers were encrypted before data processing. Because the data were analyzed in a pseudonymized manner for academic research, the present study was exempted from full review by the Research Ethics Committee of the National Taiwan University Hospital, and the individual consent was waived.
Study subjects and design
We used a retrospective cohort study design to investigate the association between SCI and the risk of AD. Propensity-score matching was used to generate two comparable groups of patients, one with and one without SCI, thus minimizing the potential for confounding effects to arise due to imbalances in the baseline characteristics [11, 12]. The advantage of the propensity score matching is that it enables us to investigate the risk of a rare outcome (AD) in SCI patients by simultaneously controlling for many potential confounders. Such study design has also been used in several our previous studies [10, 11, 13].
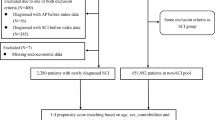
The SCI group consisted of patients from the NHI database who were aged between 40 and 90, and who had a diagnosis of SCI (ICD-9-CM codes 806 or 952) during ambulatory medical-care visits in calendar year 2001. To increase the likelihood that such diagnoses were accurate, only patients who had two or more ambulatory visits in which a diagnosis of SCI was recorded within this 1-year period were included (n = 9321). The first such visit was defined as the index visit. Those who had been diagnosed with AD (ICD-9-CM codes 331.0) prior to the index visit (n = 22) were excluded, leaving a total of 9299 patients in the SCI group at this stage.
All of the SCI group’s outpatient and inpatient records in the NHI database from the 12-month period before the index visit were checked for information about pre-existing comorbidities, including diabetes (ICD-9-CM code 250), hypertension (ICD-9-CM codes 401–405), hyperlipidemia (ICD-9-CM code 272), gout (ICD-9-CM code 274), coronary heart disease (ICD-9-CM codes 410–414), atrial fibrillation (ICD-9-CM code 427.31), rheumatic heart disease (ICD-9-CM codes 393–398), stroke (ICD-9-CM codes 430–438), TBI (ICD-9-CM codes 801–804, 850–854), and chronic kidney disease (ICD-9-CM codes 580–587). A particular comorbid condition was deemed present if the patient had ⩾1 hospital discharge or ⩾2 ambulatory visits with the relevant diagnosis code. Socioeconomic variables, including monthly income, geographic region of residence, and place of residence’s level of urbanization, were also used as matching variables because previous studies have shown their impact on the risk of AD [14]. As an indicator of monthly income, we categorized the NHI’s insured payroll-related amounts into four levels (i.e., NT$0, NT$1-15840, NT$15841-25000, and ⩾NT$25001). The first cutoff point of NT$15840 was selected because it was the government-designated minimum monthly salary for full-time employees. The seven-level typology of urbanization in Taiwan devised by the National Health Research Institutes [15] was simplified into a five-level version, with level 1 being “most urbanized”, and level 5 (a combination of the original levels 5, 6, and 7) “least urbanized”. The geographic region of each subject’s residence was classified as Northern, Central, Eastern, or Southern Taiwan. Of the 9299 individuals in the SCI group, 42 were excluded as the information of the geographic location of residence was missing from Taiwan’s household registry, leaving 9257 subjects in the final SCI group.
The non-SCI group was selected from among the remaining patients in the NHI database. For this group, the index visit was defined as the first ambulatory visit in 2001, and only patients who had at least two ambulatory visits in 2001 were included. The exclusion criteria were (1) a diagnosis of SCI (ICD-9-CM codes 806 and 952) prior to the index visit and/or (2) a diagnosis of AD (ICD-9-CM code 331.0) prior to the index visit. Information about comorbid conditions and socioeconomic variables were obtained in the same manner as for the SCI group.
This study used a two-stage approach to carry out propensity-score matching, as described in our previous works [10, 11]. In the first stage, we randomly sampled up to 60 age- and sex-matched non-SCI individuals who meet the above inclusion and exclusion criteria for each patient in the SCI group, yielding a total of 555,390 non-SCI individuals. In the second stage, we fit a logistic regression model that included covariates of age, sex, comorbid conditions, and socioeconomic factors to predict the probability (i.e., propensity score) of SCI for each patient. Then, based on these propensity scores, an eight-to-one digit greedy matching algorithm [16] was employed to select 4 matched control subjects for each SCI patient from the non-SCI pool, resulting in a propensity-score-matched non-SCI group of 37,028 individuals.
Outcomes
From the NHI database, we retrieved the records of each subject from his/her index visit through the end of 2003 (hereafter, the “follow-up period”). Each subject was tracked from the index visit until the earliest of (1) his/her first diagnosis of AD (ICD-9-CM code 331.0, as confirmed by ⩾1 hospital discharge or ⩾2 ambulatory visits); (2) death; or (3) the end of the follow-up period. The primary endpoint of analysis was the date of a given patient’s first occurrence of AD within the follow-up period. The study cohort was linked to Taiwan’s mortality registry to ascertain the patients died during the follow-up period.
Statistical analysis
We used the χ 2 test and Student’s t-test to examine the differences in baseline characteristics between the SCI and non-SCI groups. The covariate balance before and after propensity-score matching was assessed using the standardized-difference method, which is not affected by sample size and is thus preferred over hypothesis-testing methods [17]. An absolute standardized difference of < 0.1 for a given covariate indicates a negligible imbalance. The incidence rate of AD was calculated as the number of AD cases divided by AD-free person-years. Poisson regression was used to estimate the incidence rate ratio of AD between the SCI and non-SCI groups.
The cumulative incidence rate of AD was estimated using the Kaplan–Meier method. Cox proportional-hazards regression stratified by the propensity score matching set was used to estimate the effect of SCI on the subsequent occurrence of AD. An alpha level of 0.05 was considered statistically significant for all analyses, which were performed using SAS 9.4 software (SAS Institute, Cary, NC).
Results
The proportion of SCI patients and controls who had a previous diagnosis of AD before the index visit was 0.24% and 0.14%, respectively. Table 1 presents the socioeconomic and clinical characteristics of the SCI and non-SCI groups at the first stage of matching, with age and sex-matched. As compared to the non-SCI group, the SCI group had a higher prevalence of certain comorbid conditions: i.e., diabetes (P < 0.0001), hypertension (P = 0.0002), coronary heart disease (P = 0.0036), atrial fibrillation (P = 0.0479), stroke (P < 0.0001), TBI (P < 0.0001), and chronic kidney disease (P < 0.0001). There were also significant differences between the SCI and non-SCI groups in terms of monthly income, geographic region, and urbanization level. Before matching, the absolute standardized differences were >0.1 for stroke, TBI, monthly income, urbanization level, geographic region, and propensity score. After propensity-score matching, as shown in Table 2, the matched groups were well balanced in all baseline characteristics, as indicated by the absolute standardized differences being < 0.1 for all covariates.
The number of AD cases and the hazard ratios (HRs) of AD for the two propensity-score-matched groups are presented in Table 3. Of the 9257 patients with SCI, 25 developed AD during 22044.7 person-years of follow-up, representing an incidence rate of 11.34 (95% confidence interval (CI), 7.34–16.74) per 10,000 person-years. Of the 37,028 subjects in the non-SCI group, 57 developed AD during 88374.0 person-years of follow-up: an incidence rate of 6.45 (95% CI, 4.88–8.36) per 10,000 person-years. The incidence ratio of AD for the SCI group was 1.76 (95% CI, 1.10–2.81, P = 0.0186) compared to the non-SCI group. Figure 1 shows the cumulative incidence curves of AD for the two groups. The cumulative incidence of AD during the 3-year follow-up for the SCI group was significantly higher than that for the non-SCI group (P = 0.0168), indicating that the SCI group had a higher risk of developing AD. The stratified Cox regression analysis showed that the SCI group’s HR of AD was 1.71 (95% CI, 1.06–2.76, P = 0.0273) as compared to the non-SCI group.

Cumulative incidence of Alzheimer’s disease (AD) for the spinal cord injury (SCI) group (dotted line) and the non-SCI group (solid line)
Sensitivity analysis
To assess whether our primary results are robust to different case definitions, we conducted the following sensitivity analyses. First, since the SCI subjects had a higher prevalence of TBI, which may confound the association between SCI and AD. We performed sensitivity analysis excluding all patients with a previous diagnosis of TBI. The results show that SCI was still associated with a higher risk of AD (HR 1.92, 95% CI, 1.17–3.17, P = 0.0101). Furthermore, we excluded all subjects with a previous diagnosis of either TBI or post-concussion syndrome (ICD-9-CM code 310.2) from the analysis. The estimated HR was 1.92 (95% CI, 1.16–3.15, P = 0.0106), still suggesting an increased risk of AD in the SCI group. Second, we peformed additonal analysis using dementia (ICD-9-CM codes 290, 294) as the outcome to evaluate the risk of dementia for SCI patients. The results showed that the SCI group had a higher risk of dementia (HR 1.25, 95% CI, 1.12–1.40, P < 0.0001) compared to the non-SCI group.
Discussion
This large-scale longitudinal follow-up study showed that SCI patients had a higher risk of developing AD than non-SCI subjects did. A recent study [9] used one-million beneficiaries sampled from the Taiwan’s NHI database, and found that there was no statistically significant increase in the AD risk for SCI patients (adjusted HR 1.76, 95% CI 0.69–4.48), but SCI patients had a higher risk of other types of dementia [9]. In the present study, we used the large-scale complete NHI database (more than 22 million beneficiaries), and applied propensity score matching to control for all baseline covariates simultaneously. Our study improved the precision of estimation, and showed that SCI patients were at a higher risk of developing AD (HR 1.71, 95% CI, 1.06–2.76, P = 0.0273). Although the exact mechanism(s) by which SCI affects AD risk remains unclear, we can offer the following possible explanations.
First, studies of humans [6–8] and animals [18, 19] have shown that after SCI occurs, APP is deposited throughout the spinal cord, and can persist for a prolonged period – sometimes decades – after the traumatic event [6–8]. Post-SCI accumulation of APP can lead to increased beta-amyloid production in the spinal cord, as has been observed in animal models [18, 19]. Beta-amyloid is not only the key component of neuritic plaques, the histopathological hallmark of AD, but also an active player in the pathogenesis of the disease. Beta-amyloid accumulation, in turn, can lead to excessive oxidative stress, increased inflammation, and mitochondrial dysfunction, resulting in neuronal degeneration and death [20]. Beta-amyloid can be taken up by axons, transported retrogradely to the cell body, and then transmitted to various regions of the brain via neuronal connections [21]. This transmission process may be essential to the progression of AD within the brain [21]. We therefore suggest that increased accumulation of beta-amyloid and APP in the CNS may predispose patients with SCI to a higher risk of AD.
Second, several human [22, 23] and animal [24] studies have found elevated concentrations of tau protein in the cerebrospinal fluid (CSF) after SCI. Tau protein is a key element in AD pathogenesis, and tau accumulation has been found to progress in a caudal to rostral direction in AD [2]. While it has been suggested that the transmission of tau follows a prion-like neuron-to-neuron pattern [2, 25, 26], some evidence indicates another possible route for its propagation: through the CSF [25, 26]. In an animal model, tau protein released by damaged neurons was shown to reach the CSF and spread to distant regions; to be taken up by neurons and glia cells [25]; and then to spread further via neuron-to-neuron transmission [26]. The same transmission mechanism for tau may also occur in the human CNS [25]. Tau exerts its toxicity by impairing axonal transport, leading to cytoskeletal collapse and eventually, neuronal dysfunction and death [20]. We therefore speculate that elevated tau concentrations in the CSF after SCI, and the subsequent spread of tau, may also contribute to SCI patients’ higher risk of developing AD.
Third, SCI patients exhibit chronic systemic inflammation, as demonstrated by their persistently elevated concentrations of proinflammatory markers such as serum TNF-α [27, 28], IL-6 [27], and C-reactive protein (CRP) [29]. Systemic inflammation can be an important driving force for neuroinflammation [30, 31], which in turn plays an important role in the pathogenesis of AD [30–32]. Animal studies have shown that peripheral inflammation exaggerates both CNS inflammation [33, 34] and beta-amyloid deposition [33]; and human studies have demonstrated that elevated levels of IL-6 [35], TNF-α [36], and CRP [35] are associated with an increased risk of AD [35, 36]. We therefore hypothesize that the association between SCI and AD may be mediated by SCI-related systemic inflammation, which by inducing neuroinflammation could lead to a higher risk of AD.
While previous histopathological studies based on animal and human models have suggested neurodegeneration in CNS may occur after SCI, epidemiological evidence for the association between SCI and neurodegenerative disorders is limited. The present cohort study demonstrated a temporal sequence between SCI and AD, which suggests a temporal association between SCI and AD, and provides epidemiological evidence supporting the hypothesis that SCI may contribute to a higher risk of neurodegenerative disorders in CNS. Moreover, in the sensitivity analysis, the positive association between SCI and AD was not attenuated by excluding all subjects with previous diagnosis of TBI or post-concussion syndrome, suggesting that SCI is independently associated with an increased risk of AD.
Nevertheless, several limitations of this research should be noted. First, the diagnoses of SCI, AD, and comorbid conditions were entirely determined using the ICD codes from the NHI database, which may raise concerns about diagnostic accuracy. However, the Bureau of the NHI samples claim data from every hospital in the NHI program, and reviews charts regularly to verify diagnostic validity as well as the quality of care being provided. For these reasons, the NHI database is widely accepted as an appropriate database for biomedical research [11, 13]. Second, due to the inherent limitation of the NHI database, the information about certain lifestyle factors such as smoking, alcohol consumption, physical inactivity, and obesity is lacking, which may influence the interpretation of our results. Third, since the present study used NHI database from 2000 to 2003, the median follow-up time was only 31 months and therefore the long-term effects of SCI on the risk of developing AD cannot be evaluated. And finally, most inhabitants of Taiwan are of Chinese ethnicity, and it is unclear whether the findings from the present study can be generalized to other ethnic populations.
In conclusion, the present longitudinal follow-up study of a nationwide population-based cohort shows that patients with SCI are at an increased risk of developing AD. Further studies are required to elucidate the mechanism(s) that underlie this association.
References
Cummings JL, Isaacson RS, Schmitt FA, Velting DM. A practical algorithm for managing Alzheimer’s disease: what, when, and why? Ann Clin Transl Neurol. 2015;2:307–23.
Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–20
Daneshvar DH, Goldstein LE, Kiernan PT, Stein TD, McKee AC. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci. 2015;66:81–90.
Collins JM, King AE, Woodhouse A, Kirkcaldie MT, Vickers JC. The effect of focal brain injury on beta-amyloid plaque deposition, inflammation and synapses in the APP/PS1 mouse model of Alzheimer’s disease. Exp Neurol. 2015;267:219–29.
Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–74.
Cornish R, Blumbergs PC, Manavis J, Scott G, Jones NR, Reilly PL. Topography and severity of axonal injury in human spinal cord trauma using amyloid precursor protein as a marker of axonal injury. Spine (Phila Pa 1976). 2000;25:1227–33.
Ahlgren S, Li GL, Olsson Y. Accumulation of beta-amyloid precursor protein and ubiquitin in axons after spinal cord trauma in humans: immunohistochemical observations on autopsy material. Acta Neuropathol. 1996;92:49–55.
Olsson Y, Ahlgren S, Farooque M, Holtz A, Li GL, Yu WR. Pathophysiology of spinal cord trauma: observations on vasogenic oedema and axonal injuries in human and experimental material. Neuropathol Appl Neurobiol. 1996;22:518–20.
Huang SW, Wang WT, Chou LC, Liou TH, Lin HW. Risk of dementia in patients with spinal cord injury: a nationwide population-based cohort study. J Neurotrauma. 2017;34:615–22.
Lin CW, Huang YP, Pan SL. Spinal cord injury is related to an increased risk of multiple sclerosis: a population-based, propensity score-matched, longitudinal follow-up study. J Neurotrauma. 2015;32:655–9.
Yeh TS, Huang YP, Wang HI, Pan SL. Spinal cord injury and Parkinson/‘s disease: a population-based, propensity score-matched, longitudinal follow-up study. Spinal Cord. 2016;54:1215–9.
D’Agostino RB Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat Med. 1998;17:2265–81.
Huang YP, Chen LS, Yen MF, Fann CY, Chiu YH, Chen HH, et al. Parkinson’s disease is related to an increased risk of ischemic stroke-a population-based propensity score-matched follow-up study. PLoS ONE. 2013;8:e68314.
Fox M, Knapp LA, Andrews PW, Fincher CL. Hygiene and the world distribution of Alzheimer’s disease: epidemiological evidence for a relationship between microbial environment and age-adjusted disease burden. Evol Med Public Health. 2013;2013:173–86.
Liu C, Hung Y, Chuang Y, Chen Y, Weng W, Liu J, et al. Incorporating development stratification of Taiwan townships into sampling design of large scale health interview survey (in Chinese). J Health Manag. 2006;4:1–22.
Parsons L. Performing a 1:N case-control match on propensity score. In: Proceedings of the Twenty-ninth Annual SAS Users Group International Conference: 9-12 May 2004; Montreal, Canada. North Carolina: SAS Institute Inc.
Austin PC. A critical appraisal of propensity-score matching in the medical literature between 1996 and 2003. Stat Med. 2008;27:2037–49.
Pajoohesh-Ganji A, Burns MP, Pal-Ghosh S, Tadvalkar G, Hokenbury NG, Stepp MA, et al. Inhibition of amyloid precursor protein secretases reduces recovery after spinal cord injury. Brain Res. 2014;1560:73–82.
Kobayashi S, Sasaki T, Katayama T, Hasegawa T, Nagano A, Sato K. Temporal-spatial expression of presenilin 1 and the production of amyloid-beta after acute spinal cord injury in adult rat. Neurochem Int. 2010;56:387–93.
De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer’s disease. Subcell Biochem. 2012;65:329–52.
Song HL, Shim S, Kim DH, Won SH, Joo S, Kim S, et al. beta-Amyloid is transmitted via neuronal connections along axonal membranes. Ann Neurol. 2014;75:88–97.
Pouw MH, Kwon BK, Verbeek MM, Vos PE, van Kampen A, Fisher CG, et al. Structural biomarkers in the cerebrospinal fluid within 24 h after a traumatic spinal cord injury: a descriptive analysis of 16 subjects. Spinal Cord. 2014;52:428–33.
Kwon BK, Stammers AM, Belanger LM, Bernardo A, Chan D, Bishop CM, et al. Cerebrospinal fluid inflammatory cytokines and biomarkers of injury severity in acute human spinal cord injury. J Neurotrauma. 2010;27:669–82.
Roerig A, Carlson R, Tipold A, Stein VM. Cerebrospinal fluid tau protein as a biomarker for severity of spinal cord injury in dogs with intervertebral disc herniation. Vet J. 2013;197:253–8.
Le MN, Kim W, Lee S, McKee AC, Hall GF. Multiple mechanisms of extracellular tau spreading in a non-transgenic tauopathy model. Am J Neurodegener Dis. 2012;1:316–33.
Hall GF, Patuto BA. Is tau ready for admission to the prion club? Prion. 2012;6:223–33.
Davies AL, Hayes KC, Dekaban GA. Clinical correlates of elevated serum concentrations of cytokines and autoantibodies in patients with spinal cord injury. Arch Phys Med Rehabil. 2007;88:1384–93.
Hayes KC, Hull TC, Delaney GA, Potter PJ, Sequeira KA, Campbell K, et al. Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J Neurotrauma. 2002;19:753–61.
Manns PJ, McCubbin JA, Williams DP. Fitness, inflammation, and the metabolic syndrome in men with paraplegia. Arch Phys Med Rehabil. 2005;86:1176–81.
Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405.
Pasqualetti G, Brooks DJ, Edison P. The role of neuroinflammation in dementias. Curr Neurol Neurosci Rep. 2015;15:531.
Fischer R, Maier O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxid Med Cell Longev. 2015;2015:610813.
Kyrkanides S, Tallents RH, Miller JN, Olschowka ME, Johnson R, Yang M, et al. Osteoarthritis accelerates and exacerbates Alzheimer’s disease pathology in mice. J Neuroinflammation. 2011;8:112.
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–84.
Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–72.
Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68:1902–8.
Acknowledgements
This work was supported by the Department of Health, Executive Yuan, Republic of China [DOH93-TD-M-113-030, DOH94-TD-M-113-004, and DOH95-TD-M-113-002].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Funding
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional information
Tian-Shin Yeh and Yu-Chun Ho contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yeh, TS., Ho, YC., Hsu, CL. et al. Spinal cord injury and Alzheimer’s disease risk: a population-based, retrospective cohort study. Spinal Cord 56, 151–157 (2018). https://doi.org/10.1038/s41393-017-0009-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41393-017-0009-3
This article is cited by
-
Acute Transcriptomic and Epigenetic Alterations at T12 After Rat T10 Spinal Cord Contusive Injury
Molecular Neurobiology (2023)
-
Lentivirus-mediated downregulation of α-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury
Journal of Neuroinflammation (2019)
