
Abstract
The TP53 tumor suppressor is the most frequently altered gene in human cancers, and has been a major focus of oncology research. The p53 protein is a transcription factor that can activate the expression of multiple target genes and plays critical roles in regulating cell cycle, apoptosis, and genomic stability, and is widely regarded as the “guardian of the genome”. Accumulating evidence has shown that p53 also regulates cell metabolism, ferroptosis, tumor microenvironment, autophagy and so on, all of which contribute to tumor suppression. Mutations in TP53 not only impair its tumor suppressor function, but also confer oncogenic properties to p53 mutants. Since p53 is mutated and inactivated in most malignant tumors, it has been a very attractive target for developing new anti-cancer drugs. However, until recently, p53 was considered an “undruggable” target and little progress has been made with p53-targeted therapies. Here, we provide a systematic review of the diverse molecular mechanisms of the p53 signaling pathway and how TP53 mutations impact tumor progression. We also discuss key structural features of the p53 protein and its inactivation by oncogenic mutations. In addition, we review the efforts that have been made in p53-targeted therapies, and discuss the challenges that have been encountered in clinical development.
Similar content being viewed by others


Introduction
The tumor suppressor gene TP53 is the most frequently mutated gene in human tumors.1,2 The process of tumor development is strongly related to the dysfunctions caused by TP53 mutations.3,4 p53 protein functions primarily as a transcription factor, which regulates a wide variety of pathways, such as cell cycle arrest, DNA repair, cell apoptosis, autophagy, and metabolism,1,5,6 and determines whether cells die under stress conditions. Over the years, a growing number of studies have revealed the complexity and connectivity of the p53 pathway and by extension its role in metabolic homeostasis, immune microenvironment, stem cell biology and so on. However, mutant p53 can alter DNA-specific binding, disrupt the spatial conformation of the protein and thermostability of the protein, and result in dysfunction of p53 activity.7,8,9,10
The high frequency of TP53 mutations in tumors and its intrinsic tumor suppressor function make it a highly promising target for tumor therapy. However, the specificity of the p53 structure,11,12 the smooth surface without an ideal drug-binding pocket,13 and the difficulty to restore p53 function, have stalled drug research against p53 for decades. Nevertheless, researchers still believe that this difficult-to-drug target can be tackled, and have made some progress in recent years. In this review, we aim to provide a comprehensive summary of the biological function of p53, p53 signaling pathway, the structural features of p53 protein, as well as the advances in p53-targeted therapies.
The discovery history of p53
The TP53 gene is located on the short arm of chromosome 17 (17p13.1) and encodes a protein with 393 amino acid residues. p53 was initially identified as a host protein that bound to simian virus 40 large T antigen in virally transformed cells,14 and was named p53 in 1979 because its molecular weight was shown to be approximately 53 kilodalton (kDa) in SDS polyacrylamide gel electrophoresis analysis15 (Fig. 1). The actual molecular weight of p53 is 43.7 kDa, due to the large number of proline residues in the protein that slowed down its migration on SDS polyacrylamide gel electrophoresis. TP53 was initially thought to be an oncogene, and high levels of p53 conferred significant tumorigenic potential to rat embryonic fibroblasts.16,17 Subsequent studies have led to a change in the recognition of TP53. The initial p53 cDNA was synthesized using mRNA from tumor cells as a template, and the p53 cDNA subsequently obtained from normal cells did not transform the cells, but rather inhibited tumor cell growth.18 In tumor cells the TP53 gene is often mutated or lost due to chromosome 17 deletion, whereas in normal cells the gene is intact.19,20 When a missense mutation in TP53 occurs, the obtained p53 protein promotes tumorigenic transformation.21,22 TP53 knockout mice have a high probability of developing tumors.23 Overexpression of the wild-type TP53 gene in cells effectively suppressed the transforming effects exerted on cells by those oncogenes, such as the MYC gene and RAS gene.22,24 This series of studies overturned the established paradigm of TP53, which has since become one of the most studied tumor suppressor genes.

Timeline of some major advances in p53 research
The p53 pathway
p53 is a transcription factor that is distributed in the nucleus and cytoplasm, binds specifically to DNA, and regulates a diversity of genes.25,26 Under normal conditions, cellular p53 protein levels are very low owing to strict control by its negative regulators MDM2 and MDMX, which promote p53 degradation through ubiquitination.27,28 When cells are exposed to internal and external stresses, including DNA damage, hypoxia, nutrient deprivation, and cancer cell risk, p53 ubiquitination is inhibited, triggering a rapid increase in intracellular p53 protein levels. Accumulated p53 is activated and stabilized by posttranslational modifications, including phosphorylation, acetylation and methylation.29,30,31,32 Stabilized p53 forms tetramers in the nucleus, binds to target DNA and regulates gene transcription, leading to alterations in downstream signaling pathways.33,34,35,36,37
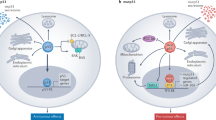
In response to cellular stress, p53 prevents the differentiation of cells with mutated or damaged DNA and terminates cellular processes by transcriptionally activating various genes involved in apoptosis and cell cycle,38,39 which contributes significantly to its tumor suppressor function and is the most studied.33,34,35,36,37,40,41,42,43,44 Meanwhile, a series of studies have shown that p53 also controls a number of “non-classical” pathways (Fig. 2), including metabolic homeostasis, ferroptosis, stem cell differentiation, autophagy, senescence, tumor microenvironment and so on.45,46,47,48,49,50

The p53 pathway. Under normal conditions, p53 protein levels are tightly regulated by MDM2/X, which together ubiquitinate p53, leading to proteasomal degradation of p53. Under stress conditions, p53 is activated and stabilized by post-translational modifications. Stabilized p53 forms tetramers in the nucleus, binds to target DNA, regulates gene transcription, and controls many different biological processes. ALDH4 aldehyde dehydrogenase family 4 member A1, ALOX12 arachidonate-12-lipoxygenase, AMPK 5′-AMP-activated protein kinase, APAF1 apoptotic protease-activating factor 1, Atgs autophagy-related genes, ATM ataxia-telangiectasia mutated proteins, ATR ataxia telangiectasia and Rad3-related, BAX apoptosis regulator BAX, CDC cell division cycle, CDK cyclin-dependent kinase, COX-2 cyclooxygenase-2 (also known as PTGS2), CPT1C carnitine palmitoyltransferase 1C, Cyt C Cytochrome C, DDB2 damage specific DNA binding protein 1, DPP4 dipeptidyl peptidase-4, DRAM damage regulated autophagy modulator 2, FANCC fanconi anemia group C protein, FDXR ferredoxin reductase, GADD45 growth arrest and DNA damage-inducible 45, GLS2 glutaminase 2, GLUT glucose transporter type, GPX glutathione peroxidase, G6PDH glucose‑6‑phosphate (G-6‑P) dehydrogenase, HMGB1 high-mobility group box-1, MCD malonyl-coenzyme A decarboxylase, mTOR mammalian target of rapamycin, NOXA superoxide-generating NADPH oxidase, PAI1 plasminogen activator inhibitor 1, PANK1 pantothenate kinase 1, PDK PDH kinase, PIGs p53-induced gene, PML promyelocytic leukemia protein, PRL3 phosphatase of regenerating liver-3, PTPRV protein tyrosine phosphatase receptor type V, PUMA Bcl-2-binding component 3, ROS reactive oxygen species, RRM2 ribonucleoside-diphosphate reductase subunit M2, SAT1 spermine N1-acetyltransferase 1, SLC7A11 solute carrier family7 member11, TFR1 transferrin receptor 1, TIGAR TP53-induced glycolysis and apoptosis regulator, TP53INP1 tumor protein p53 inducible nuclear protein 1, XPC xeroderma pigmentosum group C protein, YAP1 yes-associated protein 1
The biological function of p53 in cancer
p53 controls a wide range of signaling networks. There is no simple and clear answer to the question of exactly how, when, and what p53 does. Nevertheless, what is clear is that p53 has a very flexible and versatile response, with an integrated response to environmental perturbations determining cell death or maintaining cellular homeostasis. p53 serves as a linkage point for multiple cellular signaling pathways, harmoniously and delicately regulating various biological functions through transcriptional regulation and protein-protein interactions.
Genomic stability
p53 is considered as the guardian of the genome. It plays an important role in maintaining genomic stability. When DNA is damaged, p53 protects the genome by coordinating multiple DNA damage response mechanisms.51 The p53 protein activates the expression of DNA repair proteins DDB2 and XPC.52 The interaction of these proteins with effector proteins may lead to various cell fates, such as apoptosis, senescence or tumorigenesis.53,54
The gene encoding p21 (CDKN1A) is the first transcriptional target identified for p53.55 p21 is a potent inhibitor of binding to cyclin-dependent kinases (CDKs), which inhibits cell cycle proteins and further inhibits phosphorylation of Rb by the cyclin D1-CDK4, cyclin D2-CDK4 and cyclin E-CDK2 complexes.56,57,58 Hypophosphorylated Rb forms a complex with the E2F transcription factor, which inhibits the transcriptional activity of E2F and leads to G1 phase block.40,59 PTPRV, which encodes a transmembrane tyrosine phosphatase, and the phosphatase of regenerating liver-3 are both genes involved in p53-induced G1 phase block.60,61
In addition, p53 represses CDKs and cyclin B, which are required for entry into mitosis and are involved in G2/M phase block.62,63 p53 induces transcription of 14-3-3 sigma and represses the cell cycle protein-dependent kinase Cdc2.64 Gadd45 is a member of the growth arrest and DNA damage gene family. p53 regulates the transcription of Gadd45, disrupts the cyclin B1/Cdc2 complex, and further blocks the G2 phase.63,65 Reprimo is also involved in p53-induced cell cycle arrest in the G2 phase.66 These modulations of p53 reduce the risk of gene mutation and prevent the activation of oncogenes.
On the other hand, p53 can also induce apoptosis in cells with DNA damage.67 p53 causes apoptosis through transcriptional activation of the expression of the pro-apoptotic genes, such as Puma, Bax and Noxa.68,69,70 p53 is also thought to regulate mitochondrial apoptosis through a transcription-independent pathway.33 p53 binds physically to anti-apoptotic proteins (Bcl-2, Bcl-xL and Mcl-1), thereby indirectly inducing apoptosis.71 p53 directly activates the pro-apoptotic protein Bak or disrupts the Mcl-1 and Bak complex, releasing Bak and initiating apoptosis.37,72 In addition, p53-driven miR34a expression may sensitize cells to apoptotic stimuli by decreasing Bcl-2 levels.73,74 In the exogenous apoptotic pathway, p53 induces the expression of the death receptors Fas/Fas ligand and KILLER/DR5 located on the cell membrane, which activates caspase 8 and leads to apoptosis.75,76 p53 also drives the expression of various genes that may have pro-survival or pro-apoptotic functions,77,78 which also contribute to the maintenance of genomic stability.
Retrotransposons are replicated and inserted into new genomic sites by reverse transcription of RNA intermediates, which allows them to increase the copy number or gene mutations in the host genome.79 Disinhibition of retrotransposons has been reported to be closely associated with human tumorigenesis.80 p53 binds to the promoter region of the retrotransposon element LINE1 and prevents the expression of transposon sequences.81 When p53 is absent, cells overexpressing synthetic retrotransposon genes are able to avoid apoptosis.82 Genomic instability caused by deletion or mutation of p53 may accumulate more oncogenes and promote tumorigenesis, proliferation, metastasis and drug resistance. Functional inactivation of p53 not only contributes to genomic mutation and copy number increase, but also maintains the survival of cells carrying faulty genetic information.
Senescence
Senescence is a permanent cell cycle arrest. p53-mediated senescence is closely related to its tumor suppressive effects. DNA damage triggers senescence, a process often referred to as stress-induced premature senescence.83 Various internal or external stressors trigger the DNA damage response pathway, activating the p53 and/or p16INK4A pathways.84 p16INK4A inactivates Cdk4/6, phosphorylates Rb accumulation and inactivates E2F transcription, leading to cell cycle arrest or senescence.85 Alternatively, when UV-induced DNA damage occurs, ATM/ATR activates Chk1/Chk2 kinase, which further activates p53 and p21CIP1, leading to G1 arrest or senescence.86 In addition, p21CIP1 protein levels may lead to inhibition of CDK4/6 activity, resulting in G1 arrest or senescence.87 In addition, p53 directly induces senescence by stabilizing fibrinogen activator inhibitor-1, a marker of senescent cells.88 p53 also activates the transcription of genes that encode promyelocytic leukemia protein, leading to cellular senescence.89
Metabolic homeostasis
Tumor cells require large amounts of biological raw materials and energy to achieve their rapid and sustained growth. The Warburg effect,90 which was first proposed, states that tumor cells metabolize glucose differently than normal cells, as evidenced by enhanced glycolysis and increased lactate production.91 p53 regulation of the glycolytic pathway helps maintain the homeostasis of cellular metabolism and thus acts as a tumor suppressor. p53 can transcribe target genes required for oxidative phosphorylation, such as SCO2,92 or genes that inhibit glycolysis, such as TIGAR and Parkin.93,94 p53 binds to G6PDH, the rate-limiting enzyme of the pentose phosphate pathway, to further inhibit the activated pentose phosphate pathway in tumor cells.95,96 p53 inhibits glucose uptake and glycolysis by suppressing the expression and translocation of glucose transporter proteins such as GLUT1 and GLUT4.97 Glycolysis and gluconeogenesis can be considered reversible processes to some extent. p53 inhibition of glycolysis promotes the process of gluconeogenesis.98 Since tumor cells are highly dependent on glycolysis and the Warburg effect for proliferation and invasion, p53 inhibition of glycolysis tends to impede cancer cell growth.97,99,100
Cancer cells may indeed activate different metabolic pathways under different environmental conditions. Mutant p53 activates the Warburg effect by promoting the translocation of the GLUT1 to the plasma membrane, thereby enhancing tumor metabolism.101 Mutant 53 binds and activates PGC-1α, a major regulator of oxidative phosphorylation, enhancing mitochondrial function and promoting cancer metastasis.102,103 These studies suggest that mutant p53 may confer metabolic plasticity to cancer cells, thereby promote their adaptation to metabolic stress and increase their potential for proliferation and metastasis.
Since tumor cells need to accumulate or synthesize lipids to promote growth and proliferation,104 p53 promotes lipolysis leading to tumor suppression.46 The mevalonate pathway is responsible for the biosynthesis of cholesterol and nonsteroidal isoprenoids, and SREBP2 is a major transcriptional regulator of this pathway.105 p53 blocks the activation of SREBP2 by transcriptionally inducing the ABCA1 cholesterol transporter gene.105 p53 downregulates the expression of USP19 and SOAT1 to inhibit cholesterol esterification.106 p53 also promotes fatty acid oxidation by activating the expression of CPT1C, MCD and PANK1.107
Ammonia is a prevalent product of cellular metabolism. Tumor cells produce large amounts of ammonia during amino acid metabolism, and this ammonia can serve as a nitrogen source for tumor growth.108,109 p53 regulates ammonia content in tumor cells through the urea cycle. p53 regulates ammonia content in tumor cells by inhibiting the expression of three key enzyme genes in the urea cycle of tumor cells, CPS1, OTC and ARG1, which ultimately inhibits tumor growth.110
p53 is also involved in regulating the metabolism of tumor cells along with other metabolic signaling pathways. Increased levels of reactive oxygen species in tumor cells have a dual effect, both by promoting the acquisition of a tumor phenotype and by activating ROS-dependent death signals to kill tumor cells.111,112 The regulation of ROS by p53 also has a dual function. ROS act as an upstream signal to trigger the activation of p53, and p53 transcribes the expression of multiple antioxidant genes, such as GPX1 and manganese superoxide dismutase, to support tumor cell growth or death.113,114,115 ROS can also act as a downstream factor of p53 to drive tumor cell death through apoptosis and ferroptosis.45,116 p53 plays a dual role in inhibiting and promoting the tricarboxylic acid cycle and oxidative phosphorylation.46,117,118 p53 is also involved in regulating cellular redox reactions and mediating cancer cell death.46,119,120 In addition, p53 is involved in the regulation of lipid, amino acid and nucleotide metabolism.46,121,122
Ferroptosis
Ferroptosis is a form of regulated cell death initiated notably by severe lipid peroxidation.123,124,125 p53 was reported to inhibit cystine uptake and promote ferroptosis by transcriptional repression the expression of SLC7A11 which is a key component of the cystine-glutamate antiporter.45,126,127 p53 expression further enhances the ability of GPX4 to antagonize ferroptosis by increasing the biosynthesis of GSH.127 p53 also regulates SLC7A11 expression in a non-transcriptional manner. p53(3KR) is an acetylation-deficient mutant that fails to induce cell cycle arrest, apoptosis and senescence,128 but retains the ability to regulate SLC7A11 expression.45 Another mutant of p53(4KR) lost the ability to regulate SLC7A11 expression.126 This also suggests the importance of acetylation of p53 for the regulation of ferroptosis. H2Bub1 activates SLC7A11 expression. p53 inhibits the level of H2Bub1 by promoting nuclear translocation of the deubiquitinase USP7, which suppresses the expression of SLC7A11 and induces ferroptosis.129
p53 can also activate the expression of SAT1, a rate-limiting enzyme in polyamine catabolism, thereby inducing lipid peroxidation and ferroptosis upon ROS stress.130 SAT1-induced ferroptosis is involved in elevated expression levels of the lipoxygenase ALOX15.130 ALOX12 was also reported to be required for p53-mediated ferroptosis.131 Moreover, p53 promotes ferroptosis through modulation of GLS2, PTGS2, FDXR and noncoding RNAs.132 On the other hand, interaction of p53 with DPP4 promotes nuclear accumulation of DPP4 and blocks plasma-membrane-associated DPP4-dependent lipid peroxidation, thus limiting ferroptosis.133 The p53-p21 axis may contribute to the inhibition of cysteine deprivation-induced ferroptosis,134 whereas it has also been reported that p21 restricts the progression of ferroptosis in a p53-independent way.135
Tumor microenvironment
The status of p53 in tumor cells has a profound impact on the immune microenvironment. p53 regulates the release of cytokines and stimulates macrophage polarization toward the M1 phenotype to suppress tumorigenesis. Macrophages lacking p53 polarize toward M2 and enhance the proliferation of precancerous cells.136 p53 activation stimulates cellular anti-tumor responses,137,138 leading to interferon production, and there is a synergistic effect between cancer immunotherapy.139 Deletion or mutation of p53 in cancer affects the recruitment and activity of T cells, leading to immune evasion of cancer cells.47,140 Restoration of p53 expression enhanced the antitumor effect of anti-PD-1 monoclonal antibody on hepatocellular carcinoma cells and effectively induced reprogramming of the tumor microenvironment.141 TP53 mutations are associated with increased expression of PD-L1, which may be a predictor of response to PD-L1 targeted checkpoint inhibitors.142 Mutant TP53 contributes to the regulation of tumor cells into an immune microenvironment that is conducive to growth.143,144,145,146
Cancer stem cell self-renewal
The Rb/p53 signaling pathway regulates the proliferation and self-renewal process of neuroendocrine cells in the early stages of injury.116 Neuroendocrine cells return to a quiescent state after completing a limited proliferation.147 However, under conditions of Rb/p53 functional deficiency, neuroendocrine cells can acquire a sustained proliferative function and develop into small cell lung cancer.148,149 Activation of p53 in breast,150 prostate,151 epidermis,152 central nervous system153 and hematopoietic stem cells154,155,156 hinders stem cell self-renewal, but the exact mechanism remains to be confirmed.48 p53 has an essential role in regulating normal and malignant stem cell differentiation and self-renewal. Conversely, mutant p53 contributes to the maintenance of cancer stem cells.157 Mutant p53 enhances the proliferation of cancer stem cells by regulating WASP-interacting proteins.158 Mutant p53 also enhances the self-renewal of hematopoietic cells by upregulating FOXH1.159
The epithelial-mesenchymal transition is a form of reprogramming of cancer cells.160 Under pathological conditions, epithelial cells transform into mesenchymal cells, enhancing the invasive metastasis of cancer cells. p53 directly activates miR-200, miR-130 and miR-34 and inhibits the transcription factors Slug, Snail1 and Zeb1, which promote epithelial mesenchymal transformation.161,162,163,164 p53 deletion enhances self-renewal of lung cancer stem cells.165
Autophagy
Autophagy degrades intracellular macromolecules through the lysosomal pathway, thereby enabling intracellular energy supply and organelle renewal.166,167 p53 can directly activate damage-regulated autophagy regulators and induce autophagy.168,169 Autophagy is controlled by autophagy-related genes (Atg).170 Combined ChIP sequencing and RNA sequencing analysis revealed that p53 binds to many autophagy genes, including Atg2, Atg4, Atg7 and Atg10.171 This suggests that p53 induces autophagy in a transcription-dependent manner. Cathepsin D is a lysosomal aspartate protease and overexpression of cathepsin D activates autophagy.172,173 p53 binds to two DNA sites in the cathepsin D promoter region and regulates Cathepsin D expression.174,175 The upregulation of TGM2 expression by p53 enhances autophagy, thereby inhibiting oncogenic transformation and tumor formation in primary human mammary epithelial cells.176
p53 inhibits mTOR and enhances autophagy by activating AMP-responsive protein kinase.177,178,179 The p53 target genes Sestrin1 and Sestrin2 phosphorylate and activate AMPK and promote autophagy.178 p53 induces mitophagy by increasing the level of BNIP3.180 Interaction of beclin1 with Bcl-2 family anti-apoptotic proteins inhibits autophagy.181,182 p53 promotes the expression of BH3-only protein, which competitively binds to Bcl-2 family anti-apoptotic proteins, contributing to the restoration of beclin1 activity and promoting autophagy.181,183
p53 also functions as an inhibitor of autophagy. When p53 with deletion of nuclear localization sequence accumulates in the cytoplasm, it inhibits autophagy.184,185 HMGB1 and p53 regulate autophagy by mutually regulating their distribution in the nucleus and cytoplasm.186,187 In addition, autophagy regulates p53 activity. Atg7 inhibits p53 activation and p53 induced apoptosis.188
Other cellular processes
The signaling pathways involved in the regulation of p53 also include immune responses, non-coding RNAs and so on that exert tumor suppressive effects.49,87,189,190,191,192 In the process of tumor development, p53 suppresses tumor transformation,22 proliferation,193 metastasis194 and drug resistance195 through multifaceted regulation. p53 has an exceptionally flexible biological response that is altered by changes in cell type, cell differentiation status, stress conditions, and different signals in the environment.
Oncogenic effects of wild-type p53
Studies have confirmed that p53 is essential for suppressing cancer in humans. However, a study in 2019 showed that p53 could promote tumor growth by enhancing the metabolism of hepatocellular carcinoma cells117 (Fig. 3). p53 transcription activates Puma expression, which further initiates apoptosis.69 But certain levels of Puma protein interfere with normal mitochondrial function, leading to a shift in mitochondrial energy metabolism from oxidative phosphorylation to glycolysis.117 Another group knocked out MDM2 in hepatocyte-specific KRAS G12D mutant mice and observed p53 accumulation in mouse hepatocytes.196 These p53-activated mice exhibited increased inflammatory responses, hepatocyte apoptosis, and senescence-associated secretory phenotype, leading to the facilitation of a carcinogenic microenvironment196,197 (Fig. 3). Hepatic progenitor cells from p53-accumulating mice were injected into experimental mice growing tumors. The development of hepatocellular carcinoma and other related phenotypes no longer occurred after knockdown of TP53, suggesting that p53-accumulated mice do promote the development of hepatocellular carcinoma.196 Cellular activities regulated by p53 are integrated into tumor suppressive functions, but p53-induced regulation of certain elements may also provide a survival advantage for tumors (Fig. 3).

Pro-tumor effect of p53. In general, p53 is thought to have tumor suppressive effects, but in some cases, p53 promotes tumor growth. In hepatocellular carcinoma cells, p53 transcription activates the expression of Puma, which causes shift in mitochondrial energy metabolism from oxidative phosphorylation to glycolysis, thereby promoting tumorigenesis. In hepatocyte-specific KRASG12D cells knockout of MDM2 to activate p53. Accumulated p53 increased inflammatory responses, hepatocyte apoptosis, and senescence-associated secretory phenotypes that promote carcinogenesis
TP53 mutations in cancer
The TP53 gene is mutated in most tumor cells. Genome sequencing of different human cancer cells showed that 42% of cases carry TP53 mutations.198 The major type of mutation in TP53 is a missense mutation, a single amino acid substitution and the DNA binding domain (DBD) is the most common mutated region199,200 (Fig. 4a, b). p53 mutants are usually classified as structural mutants and DNA contact surface mutants. Structural mutants (R175H, R249S, G245S, Y220C) have reduced protein thermostability, resulting in proteins that do not fold properly at physiological temperatures and lose the ability to bind to DNA. Among them, R175H and C176Y affect the binding of protein to zinc ions. DNA contact surface mutants (R273H/C, R248W) are located in the DNA core binding region with mutations that prevent the binding of protein to DNA. R175, G245, R249, R282, R248 and R273 are the most common mutation sites and are therefore referred to as “hot spot” mutations of TP53201,202,203,204 (Fig. 4b). These mutants not only bind to wild-type p53 to produce dominant-negative (DN) effects but may also be converted to oncogenic proteins via gain-of-function (GOF).203,205,206 Thus, TP53 differs from many “classical” oncogenes, which are usually characterized by nonsense or shift mutations that result in truncated protein inactivation.207,208,209,210

TP53 mutations in cancer. a Frequency of somatic TP53 mutations associated with different types of cancer. b Frequency of missense mutations in TP53 (https://tp53.isb-cgc.org/). c Mutation frequency of TP53 in different tissues and organs
TP53 mutations are prevalent in tumors, but different tissues and organs have different TP53 mutation spectra3 (Fig. 4c). TP53 mutations were commonly found in the ovary (47.27%), colon and rectum (44.55%), lung (40.8%), pancreas (38.53%), stomach (36.78%), urethra (35.01%), liver (29.17%), breast (26.44%), prostate (22.52%), bone (16.19%), thyroid (11.13%), hematopoietic and lymphatic (10.13%) and kidney (8.75%) (https://cancer.sanger.ac.uk/cosmic). TP53 was mutated more frequently in esophageal carcinoma (93.77%), small cell lung cancer (79.06%), ovarian carcinoma (80.46%), colorectal carcinoma (74.45%) and gallbladder carcinoma (57.77%), and less frequently in thyroid carcinoma (3.13%), embryonal tumor (2.08%) and peripheral nervous system (1.25%). Different tumor subtypes of the same tissue and organ also have different TP53 mutation spectra. For example, the frequency of TP53 mutations in non-small cell lung cancer was 57.04%, which was lower than that in small cell lung cancer (79.06%) (https://www.cbioportal.org/). In addition, TP53 mutation spectra differed by race. The frequency of TP53 mutations in breast cancer is 42.9% in Asian and 30–35% in Caucasians.211
Mechanism of action of mutant p53
TP53 acts as a tumor suppressor gene and genome guardian, so cancer cell transformation is unlikely to occur in cells that maintain normal p53 function.212,213 TP53 mutations provide a permissive environment for tumorigenesis.214,215 TP53 mutations are a hallmark of an inherited cancer susceptibility syndrome known as Li-Fraumeni.216,217 The high frequency of TP53 mutations found in tumor cells19,218,219 may be the result of selection pressures that favor tumor cells to escape surveillance and be spared from death.220,221 Many mutant p53 proteins are more stable than wild-type p53 proteins and can accumulate in cells. Some mutant p53 proteins may have completely different functions than wild-type proteins,222 and this effect may be due to altered target gene profiles, mutant p53 secretome or inappropriate protein-protein interactions223 (Fig. 5).

The role of mutant p53 in cancer. Mutant p53 can result in loss-of-function of wild-type p53, dominant-negative repression of wild-type p53 by mutant p53, and gain-of-function with oncogenic properties. Mutant p53 affects various cellular responses, such as genomic instability, metabolic reprogramming, and tumor microenvironment, and promotes cancer cell proliferation, invasion, metastasis and drug resistance. WT wild-type
Loss of p53 transcriptional activity of mutant p53, and to some extent the DN effect are major drivers of the tumor phenotype (Fig. 5). The loss-of-function or DN effect of wild-type p53 enhanced the viability of tumor cells, and this effect was independent of missense mutations in TP53.193 The deletion of TP53 in gastric cells combined with an oncogenic diet confers a selective advantage to cancer cells.224 Analysis of more than 10,000 patient samples from 32 different cancers in The Cancer Genome Atlas revealed that more than 91% of mutant p53 was accompanied by deletion of TP53 alleles.225 Mutations in TP53 are associated with enhanced chromosomal instability,226,227 and are accompanied by amplification of oncogenes and deletion of suppressor genes.227,228 Significant upregulation of proteins associated with cell cycle progression was also observed in p53 mutant cancers, which may be due to loss of control of cell cycle checkpoints by TP53 deletion.225,229,230 Scott W. Lowe’s group used a mutation tracking system to reveal a pattern of TP53 inactivation leading to genomic alterations.231 Although TP53-deficient cells gain the potential for cancer cell transformation, TP53 deletion alone is not sufficient to cause cancer.231 TP53-deficient cells gain additional gene amplification in an ordered way that eventually spirals out of control and develops into cancer.231,232,233,234 Functional, DNA binding and transcriptional analyses against myeloid malignancy cell lines carrying TP53 missense mutations showed loss of p53 function, indicating that DN is the primary choice for TP53 missense mutations in myeloid malignancies.235
The activity of the p53 mutant GOF has been reported to be associated with cellular physiopathology and poor clinical outcomes in cancer patients236,237,238,239,240,241,242,243,244,245,246 (Fig. 5). Mutant p53 binds directly to TBK1, prevents TBK1 from forming a ternary complex with STING and IRF3, and ultimately inhibits the activation of the cGAS-STING pathway.223 Mutant p53 helps tumor tissue evade the killing effect of the immune system by inhibiting the anti-tumor immune response.223,247,248 However, in contrast, the evidence on the effects of p53 mutant GOF is much weaker compared to DN, and it may act modestly or only in certain specific situations.
Mutated TP53 not only lost its normal biological function, but also promoted cancer metastasis. Missense mutations in TP53 were associated with lymph node metastasis in prostate cancer patients.249 TP53 R175H and R273H mutations occurred in more metastatic tumors than in TP53 knockout mice.250,251 In mouse models of pancreatic cancer that specifically express oncogenic KRAS and mutant TP53, more than twice as many metastatic lesions were observed as in TP53 knockout mice.252 Lung adenocarcinoma mice carrying TP53 and KRAS mutations are highly aggressive and metastasize to multiple sites of intrathoracic and extrathoracic in a pattern similar to that of human lung cancer.253 Similarly, TP53 gene deletion induces an increase in systemic neutrophils, which drive systemic inflammation with breast cancer cell metastasis.194
Mutant p53 can lead to treatment resistance in cancer. Multidrug resistance protein 1 (MDR1, also known as P-glycoprotein), encoded by ATP-binding cassette subfamily B member 1, has been shown to be resistant to cytotoxicity and chemotherapy.254 In p53 mutant R248Q-expressing Hep3B cells, expression and activity of the multiple drug resistance gene, P-glycoprotein, are elevated mediating doxorubicin resistance.255 Acetylated mutant p53 interacts with p300 to promote transactivation of ephrin-B2 and enhances ATP-binding cassette subfamily G member 2 levels, thereby promoting chemoresistance.256 In R273H-expressing human squamous cell carcinoma cells, multidrug resistance to doxorubicin, methotrexate and apoptosis-inducing drugs was shown due to downregulation of procaspase-3 levels.257 Wild-type p53 inhibits LRPPRC expression via miR-34a, further reducing MDR1 expression. However, when TP53 is mutated, chemotherapy-induced inactivation of this pathway and the accumulation of LRPPRC and MDR1 promote drug resistance.195 Mutated p53 binds to the miR-223 promoter and reduces its transcriptional activity, and the introduction of exogenous miR-223 makes tumor cells carrying mutated TP53 sensitive to treatment.258
The structures of p53
The comprehensive function of p53 is closely related to its modular structure of typical signaling proteins.201 Human p53 is a multidomain protein that consists of 393 amino acids.259 It contains an N-terminal transactivation domain (TAD, residues 1–61), a proline-rich domain (PRD, residues 64–92), a DNA-binding domain (DBD, residues 96–292) linked to a tetramerization domain (TET, residues 324–356), and a C-terminal regulatory domain (CTD, residues 364–393)260,261 (Fig. 4b). More than 40% of the regions within p53 are intrinsically disordered, including the TAD, CTD, and the linker between DBD and TET.262 These disordered regions allow p53 to interact as a modular protein with a wide range of partner proteins. These structured and unstructured domains individually have unique properties and contribute to the overall functional diversity and complexity of p53. Due to the intrinsic flexibility of the intact protein, the high-resolution structure of full-length p53 remains unclear.
N-terminal region
The N-terminal region of p53 contains a highly acidic, intrinsically disordered TAD and a proline-rich region.263 TADs, including TAD1 (residues 1–40) and TAD2 (residues 40–61),264 can bind to transcription machinery components and transcriptional coactivators to promote transcriptional initiation and interact with negative regulators to suppress transcriptional activation. TAD can adopt an amphipathic α-helical conformation upon binding of partner proteins,259 which seems to be an essential binding mode, as seen in the structures of TAD bound to the negative regulator MDM2265/MDMX266 (Fig. 6a), transcription coactivator CBP267,268/p300269,270 (Fig. 6b), replication protein A,271 high-mobility group B1, HMGB1,272 Tfb1 subunits of yeast TFIIH273 and metastasis-associated protein S100A4.274 Notably, phosphorylated TAD2 exhibits an acidic string-like conformation when bound to subunit p62 of human TFIIH275 (Fig. 6c). Phosphorylation of TAD has been shown to serve as a switch to rapidly turn p53 function on and off but also as a mechanism for a graded p53 response.276,277 Overall, the conformational plasticity of the TAD, together with posttranslational modifications, makes p53 a highly efficient transcription factor.

TAD binds to partner proteins and regulates transcription. a MDM2/MDMX is the major negative regulator of p53, and the C-terminus of MDM2 has E3 ubiquitin ligase activity (Ub) that promotes p53 degradation. b Under cellular stress conditions, the acetyl group (Ac) is added to the lysine residue of p53-CTD, and p53 binds to the target DNA sequence and interacts with the coactivator CBP or p300 to jointly promote gene transcription. c DNA damage and other stresses induce p53 phosphorylation (P) and binding to TFIIH, which stabilizes p53 and promotes DNA binding and transcription. p53-TAD shown as cartoon. Partner proteins are shown as surfaces
The proline-rich region linking the TAD to DBD is required for p53 to induce cell cycle arrest and apoptosis.278,279 This region plays a role in signal transduction by binding to Src homology 3 domains.280 The PRD is also involved in interactions with focal adhesion kinase (FAK),281 peptidase D,282 ASPP family members,283 and the corepressor protein mSin3a.284 The PRD contains five PXXP motifs, some of which may adopt a polyproline helix-like structure.259 The exact structure and interaction mechanism of PRD are largely unknown.
DNA-binding domain
The structured DBD adopts a basic scaffold of an immunoglobulin-like β-sandwich, a loop-sheet-helix motif (loops L1, S2, and S2’, parts of the extended S10 and H2), and two large loops (L2 and L3) that are stabilized by a tetrahedrally coordinated zinc ion285 (Fig. 7a). The DBD is responsible for binding sequence-specific target DNA, which is central to the biological function of p53 as a transcription factor. In addition, the DBD is also capable of interactions with diverse proteins.

Structures of DBD, TET, CTD and full-length p53. a The structure of DBD in complex with a sequence-specific DNA (PDB: 1TSR). p53-DBD is shown as a cartoon, and the secondary structures are labeled. The interfacial residues are shown as sticks. DNA is shown as sticks and surfaces. b DNA recognition by the p53 tetramer (PDB: 3KMD). c ASPP2 (colored aquamarine). d iASPP (colored pale green). e 53BP1 (colored pink). f NMR structural model of the p53-DBD/BCL-xL complex. g Crystal structure of the p53-DBD/BCL-xL complex. BCL-xL is colored marine. h LTag (colored light blue). i E6/E6AP. E6 is colored light pink. E6AP is colored pale yellow. All p53-DBD molecules are shown as the surface. j Assembly of the p53 tetramerization domain (PDB: 1C26). k CTD sequence. The posttranscriptional modifications are shown as indicated. l A CTD peptide (colored light magenta) became a helical conformation when bound to Ca2+-loaded S100B(ββ) (colored pale cyan and pale green). m A CTD peptide dimethylated at K382 (p53K382me2) binds the tandem Tudor domain (TTD) of 53BP1 (colored yellow) in a U-shape conformation. n A p53 peptide acetylated at K381 and dimethylated at K382 (p53K381acK382me2) forms a helical conformation when interacting with 53BP1-TTD. o A p53K382ac peptide forms β sheet-like contacts with deacetylase Sir2-Af2 (colored light blue). p Structure of the p53/RNA polymerase II assembly. RNA polymerase II assembly is shown as surface. p53 is shown as cartoon. q Full-length p53 structure predicted by AlphaFold2. TAD colored pink, PRD colored green, DBD colored marine, Loop colored yellow, TET colored warm pink, and CTD colored cyan
DNA recognition
In response to a wide variety of stress signals, p53 regulates the transcription of many different genes involved in various pathways. p53 also acts as a pioneer transcription factor, binding to specific DNA sequences in the nucleosome to promote transcriptional activation in chromatin.286,287,288 Most p53 target genes contain a consensus response element (RE) composed of two decametric half-sites of RRRCWWGYYY (R = A, G; W = A, T; Y = C, T) separated by 0–13 base pairs.289,290,291,292 Some REs are cluster sites that have more than two half-sites. p53 also binds a set of noncanonical DNAs, such as cruciforms, left-handed DNA (Z-DNA), quadruplex and triplex DNA.293,294
Various structural studies have demonstrated the molecular mechanism of DNA recognition by p53 (Fig. 7a). Briefly, the residue Arg248 from the large loop L3 interacts with the minor groove.285 The loop-sheet-helix motif docks into the major groove and makes hydrogen bond contacts with bases via key residues Lys120, Cys277 and Arg280.285 Residues Ala276, Ser241 and Arg273 contribute to DNA backbone contacts. p53 binds to a full consensus DNA response element as a tetramer through a highly cooperative self-assembling mechanism (Fig. 7b). Two DBDs bind each decameric half-site as a symmetrical dimer (A-B), and two such dimers (A-B and C-D) constitute a tetramer on a full DNA response element via protein-protein and base stacking interactions.11,70,295,296,297,298,299
Upon DNA binding, the DBD and the DNA helix undergo structural changes. The binding of p53-DBD to the BAX response element causes DNA deformation, which is partially disordered around the spacer region, leading to unwinding and compression of the region to allow protein-protein interactions.70 In some p53/DNA structures, the central A-T doublet of each half-site shows noncanonical Hoogsteen base-pairing geometry instead of standard Watson-Crick base pairs.299,300 Some p53/DNA structures reveal that loop L1 of two p53 subunits adopts a conformational switch from an extended conformation where Lys120 interacts directly with DNA to a recessed conformation where there is no direct DNA contact.301 The conformational switch is related to the DNA-binding specificity of p53. Acetylation of loop L1 residue Lys120 expands the conformational space of loop L1 in the DNA-bound state and promotes sequence-dependent DNA-binding modes for p53.302
DBD-mediated protein-protein interactions
In addition to DNA recognition, the DBD mediates protein-protein interactions with multiple proteins. Interactions with these proteins influence various activities of p53. An increasing number of structures of the DBD/protein complex have been determined, providing fresh perspectives on the mechanisms of p53-DBD binding and functional diversity.
By using a yeast two-hybrid system, 53BP1 and 53BP2 (namely, the C-terminal fragment of ASPP2) were initially identified to bind to p53.303 They interact with p53-DBD through the L3 loop and L2 loop304,305,306 (Fig. 7c, e). 53BP1 plays multiple roles in DNA damage and repair and has been reported to enhance p53-mediated transcriptional activation of ASPP2. The inhibitory member iASPP belongs to the apoptosis-stimulating p53 protein (ASPP) family, which has opposite functions in regulating the apoptotic function of p53.307 Unlike ASPP2, iASPP preferentially binds p53-PRD283 and interacts with the DBD through the L1 loop, helix H2 and N-terminal loop308 (Fig. 7d). The L1 loop of p53 moves away from other DNA-binding modules upon binding iASPP, which disables Lys120 from making contact with a specific base.308 The different binding interfaces with the ASPP family provide insight into the opposing regulatory mechanism of p53.
Cytoplasmic p53 has been reported to regulate the mitochondrial apoptosis pathway by inhibiting antiapoptotic BCL-2 and BCL-xL. In a structural model of BCL-xL/p53 characterized by nuclear magnetic resonance (NMR) spectroscopy and determined using the HADDOCK docking method based on 1:1 stoichiometry, the BCL-xL binding surface of p53-DBD largely overlaps with the DNA-binding surface and encompasses helix H1 and the Zn2+-coordination site309 (Fig. 7f). However, the recently determined crystal structure of the p53/BCL-xL complex indicates that p53 binds BCL-xL with a 2:1 stoichiometry71 (Fig. 7g). Two p53-DBD molecules dimerize through the N-terminal loop and β9-β10 loop of one p53-DBD and the Zn2+-coordination site of the other p53-DBD, involving residues Tyr107 and His178, respectively. The resulting p53-DBD dimer forms a groove and interacts with one BCL-xL to form a ternary complex. The DNA-contacting residues Arg248 and Arg273 form direct hydrogen bonds with BCL-xL. The binding mode is distinct from other p53-DBD binding proteins.
The p53-DBD is also targeted by viral oncoproteins, such as the large T antigen (LTag) of simian virus 40 (SV40)310 (Fig. 7h) and human papillomavirus (HPV) oncoprotein E6311,312 (Fig. 7i). LTag promotes viral replication and cellular transformation. It occupies the entire DNA-binding surface of p53-DBD and interacts with a region of helix H1 that is involved in p53-DBD dimerization. The p53-binding site of HPV E6 substantially overlaps with the binding surface of iASPP, which has been reported to inhibit HPV E6-induced degradation of p53.308
Some of these proteins bind p53 at a surface overlapping with the DNA-binding site, such as 53BP1, ASPP2, Bcl-xL and LTag. Some partner proteins bind at a surface distal from the DNA-binding site, such as iASPP and HPV E6. Notably, the p53 hotspot mutant alleles at residues Arg248 and Arg273 not only make direct contact with DNA but are also involved in direct contact with ASPP, BCL-xL and LTag. Another hotspot residue, Arg282, is involved in iASPP binding. Thus, these hotspot mutants may have multiple hits on p53 activities.
Tetramerization domain
Full-length p53 reversibly forms tetramers through the TET. The structural analysis demonstrated that monomeric TET is a V-shaped structure composed of a β-strand and an α-helix linked by hinge residue Gly344313,314,315 (Fig. 7j). The primary dimer relies on eight backbone hydrogen bonds of the β-sheet and is also stabilized by hydrophobic interactions (Phe328, Leu330, Ile332, Phe338 and Phe341) as well as a salt bridge between Arg337 and Asp352.35 The dimer-dimer interactions are primarily mediated by hydrophobic contacts (Met340, Phe341, Leu344, Ala347, Leu348 and Leu350).35 R337C/H/P and L344P affect the formation and transcriptional activity of p53 tetramers and are involved in Li-Fraumeni Syndrome and Li-Fraumeni-Like Syndrome.316 The G334V mutant contributed to a beta-dominated structural transition leading to amyloid formation at physiological temperature.317 A highly conserved leucine-rich nuclear export signal (NES) within the TET is necessary for the subcellular localization of p53.318 Some proteins bind directly to TET, but no complex structure has yet been demonstrated.319
The C-terminal regulatory domain
Similar to the NTD, the CTD is also an intrinsically disordered region. This characteristic endows it with multiple regulatory functions in almost every aspect of p53, including DNA binding, cofactor recruitment, cellular localization, and protein stabilization.320 The CTD possesses many positively charged residues, which are highly conserved in mammals. Extensive posttranslational modifications of the CTD, including acetylation, methylation and phosphorylation, play an essential and interdependent role in its function and stability (Fig. 7k). The CTD adopts different conformations depending on the binding partners and posttranslational modifications201,321 (Fig. 7l–o).
Full-length p53
The highly intrinsic unfolded regions within p53 make it difficultt to determine the high-resolution structure of full-length p53. A 4.6 Å resolution structure of RNA polymerase II (Pol II) with full-length p53,322 has shown that the DBD targets the upstream DNA-binding site within Pol II, and the following TET is exposed on top of the DBD. The TAD distal to the DBD forms helices and binds Pol II’s jaw that contacts downstream DNA (Fig. 7p). This association introduces a conformational change in Pol II, providing insight into the p53-mediated regulation of gene expression.
How an intact p53 tetramer interacts with DNA and proteins is still largely unclear. A combination of NMR, electron microscopy, small-angle X-ray scattering, and FRET techniques indicates that the free p53 tetramer in solution forms an open cross-shaped structure with a pair of loosely coupled DBD.323,324 The oligomers close to form a compact complex upon binding a specific DNA response element. Chemical cross-linking and mass spectrometry methods are also employed to study the structural dynamics of full-length p53.325 A recent study has reported cryo-electron microscopy of p53-DBD and full-length p53 complexed with a nucleosome, in which the DBD binds as a tetramer to the DNA and peels the DNA from the histone surface.288 The N-terminal and C-terminal regions were not observed in the cryo-EM maps, but biochemical analysis suggests that the C-terminus of p53 may contain an additional DNA binding domain.288
The artificial intelligence system AlphaFold has accomplished the great challenge of protein amino acid sequence to structure, bringing great advances to the field of biology, especially structural biology.326,327,328 In 2021, AlphaFold2 released its predicted structure of the full-length p53 protein (https://alphafold.com/) (Fig. 7q).329 The predicted full-length p53 structure is shown as a monomer with the DBD and TET regions resembling the known structures of individual structural domains. Nevertheless, it does not provide more information on how the disordered regions fold and how p53 assembles into a tetramer. In addition, the most important feature of p53 is missense mutations leading to its altered or lost function. AlphaFold2 is currently unable to correctly predict the structural impact of missense mutations.330 Furthermore, the p53 protein functions by binding to various ligands such as DNA, small molecules, metal ions or other proteins. We still lack structural information about this interaction to aid in drug development against p53 and recognition of protein complexes.331,332,333 Following technological revolution, we expect that artificial intelligence will provide more valuable information in the future.
Regardless, structural studies on full-length p53 in complex with different DNA targets, full-length partner proteins, and even higher-order complexes are needed. This structural biology information will not only help to understand the role of p53 in cellular life activities, but these structures may also help to design drugs that target p53.
Structure-based p53 targeting
Structural and biochemical analyses have provided insight into the function of p53. However, many important questions regarding the structure‒function and specific regulation of p53 remain to be determined. In recent years, with the development of NMR, protein crystallography and cryo-electron microscopy techniques, much structural information has been obtained about the binding of p53 to ligands. From this critical information, structure-based design strategies can be applied to optimize the structure and thus improve activity and selectivity.
MDM2/MDMX-p53, a central hub for p53 activation
MDM2/MDMX is the major negative regulator of p53.334 In the MDM2-p53 negative feedback regulatory loop, wild-type p53 activates the transcription of MDM2, while the N-terminal of MDM2 binds to p53-TAD and represses the transcriptional activity of p53.265 In addition, the C-terminus of MDM2 has the enzymatic activity of E3 ubiquitin ligase, which promotes the nuclear export and degradation of p53.335,336 MDMX (or MDM4), a homolog of MDM2, is structurally similar to MDM2, but it lacks ubiquitin ligase activity and can enhance the ubiquitination activity of MDM2 by forming a dimer with MDM2.337,338 Overexpression or activation of MDM2/MDM4 is present in many human tumors, leading to p53 inactivation.334,339,340 Many antitumor drugs targeting MDM2/MDMX-p53 interactions have been developed in recent years (Fig. 8, Table 1).

Structure of MDM2/X with small molecules. a Overlay of the crystal structures of MDM2/p53-TAD (white, sky blue), MDMX/p53-TAD (greencyan, pink) and the three residues of p53-TAD (F19, W23, L26) are shown as sticks. b MDM2 is shown as a surface. c MDMX is shown as surface. d MDM2/RG7112 (PDB: 4IPF). e MDM2/RG7388 (PDB: 4JRG). f MDM2/AMG 232 (PDB: 4OAS). g MDM2/MI-77301 (PDB: 5TRF). h MDM2/NVP-CGM097 (PDB: 4ZYF). Water molecules are red spheres, and hydrogen bonds are black lines. The interacting amino acid residues are shown as sticks (colored gray)
MDM2-p53 inhibitor
The cocrystal structure of p53-TAD with MDM2 shows that Phe19, Trp23 and Leu26 on the α-helix of p53 penetrate deeply into the hydrophobic cleft of MDM2 (Fig. 8a, b), with Leu22 providing additional van der Waals forces.265 These structural features of the p53/MDM2 complex provide a basis for finding inhibitors that block the interaction between these two proteins.
RG7112
Based on the structural features of p53/MDM2, researchers synthesized imidazole-like MDM2 antagonists, among which RG7112 is the first small-molecule inhibitor of MDM2 to enter the clinic. The crystal structure showed that the 4-chlorophenyl ring occupied the Trp23 and Leu26 pockets, while the ethoxy group was prominently located in the Phe19 pocket341 (Fig. 8d). RG7112 had a Kd = 2.9 nM. It effectively blocks p53-MDM2 binding and promotes cancer cell cycle arrest and apoptosis.342 However, the ability of RG7112 to induce apoptosis in cancer cells varies widely. The best responses were observed in MDM2 gene-amplified osteosarcoma cell lines and xenografts.341,343
RG7388
The MDM2 inhibitor RG7388, which was subsequently designed and synthesized, is a class of pyrrolidine derivatives.344 RG7388, Kd = 0.15 nM, induces dose-dependent apoptosis in wild-type p53 cancer cells.342 Structurally, the 4-chlorophenyl ring, 3-chlorophenyl group and neopentyl group mimic p53 occupying the Phe19, Trp23 and Leu26 pockets. In addition, the 3-chlorophenyl group forms a π-π stacking interaction with the His96 residue on MDM2, and the pyrrolidine Cα carbonyl group forms another hydrogen bond with His96344 (Fig. 8e). RG7388 activates p53 to inhibit hematological tumors345 and solid tumors,346,347 but long-term administration may lead to p53 mutations and drug resistance.348,349
AM232
AMG32, Kd = 0.045 nM, is a piperidone analog that potently inhibits the MDM2-p53 interaction.342 Structural analysis revealed that the “cleft” of Gly58 may provide additional assistance for the small molecule to bind MDM2.350 The crystal structure shows that the isopropyl group, C6 aryl group and C5 aryl group occupy the three major binding pockets of p53, namely, Phe19, Trp23 and Leu26, respectively. Meanwhile, the imidazole group of His96 formed π-π stacking interactions with the C5 aryl group and formed hydrogen bonds with carboxylate.342 In addition, the isopropyl group extends into the “cleft” of Gly58, forming CH·O-type interactions with Gly58 and enhancing contacts with nearby hydrophobic residues350 (Fig. 8f). In tumor cells with high MDM2 expression, AMG232 enhances p53 activity and inhibits tumor growth.351 AMG232, in combination with immune checkpoint drugs, also enhances T-cell-mediated tumor killing.352 However, high doses of AMG232 may trigger gastrointestinal side effects, neutropenia and leukopenia, which requires further study.353
SAR405838
SAR405838 (MI77301), Ki = 0.88 nM, is an optimized spiro-oxide compound that blocks the MDM2-p53 interaction and prevents p53 degradation.354 SAR405838 mimics the three key amino acid residues of p53 and forms hydrogen bonds with the His96 residue of MDM2 at different chemical groups, generating π-π stacking, as seen in other MDM2 inhibitors. The structural differences are that SAR405838 mediates the refolding of the MDM2 N-terminus (residues 10–18) and interacts extensively with Val14 and Thr16. Moreover, the hydroxy-cyclohexyl group forms a hydrogen bond with Lys94354 (Fig. 8g). These structural features allow SAR405838 to achieve a tight binding and high specificity for MDM2. SAR405838 activates the p53 pathway, increases the expression of PUMA and P21, and induces complete tumor regression in SJSA-1 osteosarcoma xenograft mice.354 The safety of SAR405838 alone355 or in combination with MEK inhibitors356 has been established, but the drug activity is limited, and TP53 mutations may occur with long-term administration.357
NVP-CGM097
NVP-CGM097, Ki = 1.3 nM, is a novel dihydroisoquinoline-like MDM2 inhibitor obtained by virtual screening and structural optimization.358 The cocrystal structure revealed that in addition to the three key amino acids that mimic the interaction between p53 and MDM2, the isopropyl ether group also forms water-mediated hydrogen bonds with Tyr100, Gln24 and Phe55. NVP-CGM097 induced a conformational change in Phe55; thus, Phe55 formed a π-π stacking interaction with the dihydroisoquinolone core (Fig. 8h). NVP-CGM097 exhibits high selectivity for wild-type p53 and shows potent antiproliferative ability in colorectal cancer and osteosarcoma cells with wild-type p53.358 NVP-CGM097, combined with MEK inhibitors, activates the MAPK signaling pathway and attenuates acute myelogenous leukemia (AML) cell load.359 When combined with BET or Cdk4/6 inhibitors, NVP-CGM097 induces cell death in neuroblastoma or ER-positive breast cancer cells.360,361
MDM2/MDMX-p53 inhibitor
The main features of the MDM2-p53 interaction are preserved in the MDMX-p53 structure, but the central hydrophobic cleft of p53 peptide binding in MDMX is smaller and differently shaped than that of MDM2 (Fig. 8a–c). Therefore, many small molecule drugs targeting MDM2 do not bind MDMX well.
ALRN-6924
Currently, there are no inhibitors that act on MDMX alone, but one stapled peptide, ALRN-6924, binds both MDM2 (Kd = 10.9 nM) and MDMX (Kd = 57 nM).362 In AML cells, ALRN-6924 induces cell cycle arrest and apoptosis and significantly prolongs the survival of AML model mice.362 ALRN-6924 is well tolerated in phase I clinical trials in patients with solid tumors and lymphomas carrying wild-type p53.363 However, molecular simulations reveal that ATSP-7041, an analog of ALRN-6924, may bind to the p53 coactivator p300 and isolate free p300, thus reducing the transcriptional activity of p53.364 This mechanism needs further investigation.
Other MDM2 inhibitors
Other MDM2 inhibitors currently in clinical trials include HDM201,365,366 MK-8242,367,368,369 BI-907828,370 APG-115,371,372,373 and milademetan (DS-3032b)374 (Table 1). Although the specific interaction sites are unknown, they all induce cell death in a variety of tumor cells, particularly in patients with MDM2 amplification and intact p53 expression. APG-115 was the first MDM2 inhibitor to enter the clinic in China and was granted Fast Track Designation (FTD) by the U.S. Food and Drug Administration. APG-115 interrupts the p53-MDM2 interaction, increases the abundance of MDM2 in T cells, and plays a critical biological role in maintaining T-cell stability, survival, and antitumor immunity.371
In addition, researchers designed Proteolysis-targeting chimera (PROTAC) degraders based on MDM2 inhibitors.375 WB156, consisting of a nutlin derivative linked to the CRBN ligand lenalidomide, effectively depleted MDM2 and activated wild-type p53, thereby inducing apoptosis.376,377 The rapid degradation of MDM2 by MD-224 resulted in complete tumor regression in leukemia cells carrying wild-type p53.378
Overall, the design and development of drugs targeting MDM2/MDMX-p53 is a hot spot and priority in the field of oncology drug research worldwide. However, the specificity of MDM2/MDMX-p53 protein interactions poses a great difficulty in the development of small molecule inhibitors. Although some MDM2 inhibitors have entered clinical trials, there are currently no marketed drugs, and it is hoped that structure-based guidance will lead to new breakthroughs for MDM2-p53 inhibitors.
Tackling the p53 mutation
Under normal conditions, wild-type p53 suppresses tumor development through transcriptional regulation and protein-protein interactions. However, in many situations, missense mutant p53 is expressed at high levels in tumor cells, partly due to the inability of mutant p53 to induce gene expression of MDM2,379 which supports the reactivation of mutant p53 as a therapeutic option.5 Most TP53 mutations are missense mutations located in the DBD.3,4,203,205,380 p53 mutants mainly affect the thermostability of p53 protein, structural stability (structural mutants such as 175, 220, 245 and 249) or p53-DNA contact (DNA contact mutants such as 248 and 273). Therefore, a number of small molecule compounds, peptide or antibody drugs targeting p53 mutants have been developed to recover the native conformation or normal function of the p53 protein.381,382,383,384,385
Broad-spectrum mutant p53 rescue compounds
Employing library screens, structure-based design and other methods, drugs or compounds were discovered to have effects on the thermostability, specific DNA binding capacity or transcriptional activity of p53 mutant proteins. Some of them were initially identified as p53 mutant activators, but as research progressed, it was discovered that they could also exert antitumor activity independent of p53 status (Tables 2–3).
PRIMA-1 and APR-246
PRIMA-1 and APR-246 are prodrugs that are converted into the bioactive compound methylene quinuclidinone (MQ). PRIMA-1 was obtained by screening a library of compounds.386 PRIMA-1 restores the wild-type conformation and DNA contact of p53 mutants, and induces transcription of the downstream target genes BAX, P21 and PUMA.386,387,388 After PRIMA-1 treatment of SKOV-His-175 cells, a 46% increase in folded p53 protein was observed using the p53 conformation-specific monoclonal antibody PAb1620.386 Further studies revealed that APR-246 (PRIMA-1Met, eprenetapopt), a methylated derivative of PRIMA-1,389 has stronger antitumor activity and fewer toxic side effects than PRIMA-1, and inhibits the growth of p53 mutant tumors of various origins in combination with other anticancer drugs.390,391,392,393,394,395,396 Tumor cell lines carrying TP53, TP53 mutations, or TP53 deletions responded to APR-246 treatment.397,398,399,400 However, growing evidence suggests that APR-246 exerts its effects through a multitude of pathways independent of p53,401,402,403,404 such as downregulating glutathione concentrations in tumor cells,405,406 regulating oxidation-reduction homeostasis407,408,409 in tumor cells to trigger ferroptosis,410,411,412 and inducing endoplasmic reticulum stress413 or unfolded protein responses.414
Clinical and preclinical data showed that the combination of APR-246 and azacytidine (AZA) showed synergistic activity in patients with myelodysplastic syndromes (MDS), AML and solid tumors carrying TP53 mutations with an acceptable safety and tolerability profile.415,416,417,418,419,420 Food and Drug Administration and European Medicines Agency have granted APR-246 orphan drug status and FTD for the treatment of MDS carrying the TP53 mutation. Aprea Therapeutics recently announced preliminary data from its Phase III clinical trial. In a cohort of 154 intention-to-treat patients, the complete remission rate for APR-246 in combination with AZA was 33.3%, compared to 22.4% in the AZA alone group. Although the complete remission rate was higher with the combination, it did not reach statistical significance (https://www.aprea.com). Data from another clinical study could explain this phenomenon. Decitabine alone (a hypomethylating agent, similar to AZA) was able to produce a very high response rate (100%) in patients with AML or MDS carrying a TP53 mutation.421 This suggests that the therapeutic effect of the hypomethylating agent may be dominant in myeloid malignancies carrying TP53 mutations.
MQ can covalently bind to cysteine to restore the function and conformation of mutant p53. MQ is a very active Michael receptor that preferentially and reversibly binds to the soft nucleophile cysteine thiol of p53.422 Theoretically, all cysteines exposed on the surface of p53 are potential modification sites of Michael addition reaction285,423 (Fig. 9a). Computational docking indicated that Cys124, located in the center of the L1/S3 pocket of the p53 core region, may be the site of the MQ modification.424 In 2021, Degtjarik and colleagues investigated the mechanism of MQ reactivation of mutant p53 based on structure.425 Based on the DNA contact surface mutant R273H/R273C and the structural mutant R282W, six cysteines, Cys124, Cys182, Cys229, Cys273, Cys275 and Cys277, were identified to bind to MQ. Among them, the formation of new hydrogen bonds between MQ-Cys277 and DNA stabilizes the protein‒DNA interface, while MQ-Cys124 and MQ-Cys229 stabilize the local conformation and support the p53 dimer interface (Fig. 9b). MQ-Cys182, MQ-Cys275 and MQ-Cys273 are only observed in the structures absent from DNA, and seem to be incompatible with DNA binding. These conjugates form intramolecular interactions or intermolecular interactions with neighboring p53 molecules, stabilizing a p53 dimer different from that in p53-DNA tetrameric complexes. Therefore, MQ shows great diversity in reacting with p53 cysteines, while it worth considering whether each conjugate is beneficial for p53 rescue. Theoretically, MQ can bind to any exposed cysteine and GSH can also bind covalently to MQ,411 which may be one of the reasons for the antitumor activity of MQ.

Compounds targeting cysteine in p53-DBD. a The amino acid sequence and structure of p53-DBD, cysteine is highlighted and labeled, p53 tetramers are labeled as (a–d), respectively and cysteines are shown as sticks. b MQ bound to C124, C229 and C277 in p53-R282W-DNA tetramers. The MQ conjugates are in stick representation (green). c Structure of p53-bound arsenic and zinc ions. d Structure of p53-bound antimony and zinc ions. The interacting amino acid residues are shown as sticks
ATO
Arsenic trioxide (ATO) is a traditional Chinese medicine that leads to complete remission in patients with acute promyelocytic leukemia (APL).426 The specific fusion protein PML-RARα is present in more than 98% of APL patients. One of the mechanisms by which ATO treats APL is its ability to bind directly to the cysteine in the zinc finger structure of the fusion protein, mediating the ubiquitinated degradation of PML-RARα.427 Subsequently, a series of reports showed that ATO induced apoptosis in tumor cells carrying TP53 mutations.428,429,430
Lu Min’s team found that ATO could help stabilize the structural mutation of p53 (R175H) and restore the cancer suppressive activity of p53.431 The crystal structure showed that the DNA-binding domains of Cys124, Cys135, Cys141 and Met133 formed an As-binding pocket, with direct interaction of arsenic with Cys124, Cys135 and Cys141 and van der Waals forces between the side chain of Met133 and the arsenic atom (Fig. 9c). Most structural p53 mutants were rescued to varying degrees by ATO, but mutations in the DNA contact surface (R248Q, R273H) were not observed. ATO increased the thermostability of structural mutant p53 by 0.9–6.5 °C, restored the protein folding state close to wild-type, and increased transcriptional activity by several fold to 44-fold. Studies on hematologic tumors showed that ATO significantly prolonged the median survival time in mice; and studies on solid tumors showed that ATO-treated mice had only 10–20% of the tumor volume of the control group.431
Potassium antimony tartrate (PAT)
Subsequently, the Lu Min’s team screened compounds for temperature-sensitive p53 mutant proteins, using thermostability as a criterion for rescue. Potassium antimony tartrate (PAT) was identified as binding to the p53 V272M mutant (Kd = 9.09 μM) in a non-covalent manner, increasing its thermostability by 3.6 °C and remarkably restoring protein folding and transcriptional activity.432 PAT is an antiparasitic agent that has been used as a treatment for leishmaniasis,433,434 and has since been found to have antitumor activity.435,436 PAT is similar to ATO in that both can induce apoptosis in APL cells.437,438 PAT and ATO bind to mutant p53 at similar sites (Fig. 9d), but differ in two aspects, with PAT non-covalently binding mutant p53 and preferentially rescuing temperature-sensitive mutants (a subtype of structural mutants), whereas ATO covalently binding mutant p53 and apparently rescuing more structural p53 mutants.432 Notably, the ATO and PAT studies comprehensively and experimentally compared the set of representative mutant p53-rescuing compounds. PAT exhibited specific p53 V272M antitumor activity in both cellular and xenograft mouse models. Further testing identified 65 of p53 mutants that could be rescued by PAT, but all were non-hotspot mutations.432 A clinical trial of antimony for the treatment of MDS/AML with TP53 mutations is currently underway.
ZMC1
Zn2+ is crucial for DNA recognition by p53. Zn2+ can help stabilize the interaction between p53 and DNA by tetrahedral coordination with the imidazole group of H179 and the thiol groups of Cys238, Cys242 and Cys176298 (Fig. 9c, d). Conversely, the lack of Zn2+ affects the correct folding and DNA recognition of the p53-DBD protein.439 Therefore, helping zinc ions to bind in the correct position may be an effective strategy to target p53.
R175H is one of the p53 hotspot mutations and is classified as a Zn2+-binding mutation. The binding affinity of DBD-R175H to Zn2+ was significantly reduced compared to that of wild type.440,441 On the basis of this hotspot mutation, NSC319726, also known as ZMC1, was identified from 48129 compounds.441 ZMC1 helped coordinate zinc ions at appropriate sites to restore the structure and function of the R175 mutant442 and significantly inhibited tumor growth in mice carrying the R175H mutation.441 ZMCI treatment increased the folded R175 mutant protein by twofold.442 ZMC1 acts as an ionophore,443 transferring zinc ions from the extracellular space to the cytoplasm, maintaining the cytoplasmic zinc ion concentration within an appropriate range and thus reactivating the p53 mutant.444,445,446 In addition, ZMC1 can chelate redox-active copper,447 increase intracellular reactive oxygen levels and decrease glutathione concentrations to exert antitumor effects.441,444
ZMC1 acts as an ionophore,443 transferring zinc ions from the extracellular space to the cytoplasm, maintaining the cytoplasmic zinc ion concentration within an appropriate range and thus reactivating the p53 mutant.444,445,446 In addition, ZMC1 can chelate redox-active copper,447 increase intracellular reactive oxygen levels and decrease glutathione concentrations to exert antitumor effects.441,444
Other compounds
Salim et al. identified a thiosemicarbazideb compound COTI-2 that showed inhibitory activity in a variety of tumors by using a computational platform.448 COTI-2 inhibits tumor growth in a p53-dependent and p53-independent manner, ultimately leading to cell cycle arrest and apoptosis.448,449,450 COTI-2 was reported to restore DNA-binding properties to the p53-mutant protein,449,451,452,453 whereas it is obscure whether COTI-2 can physically bind p53. COTI-2 is currently in phase I clinical trials for the treatment of gynecologic malignancies or head and neck squamous cell carcinomas.
There are also small molecule drugs that have not been structurally studied but may interact with the cysteine residues of p53-DBD and restore the wild-type conformational and transcriptional function of p53. CP-31398 was screened from a library of >100,000 compounds originally identified to rescue the mutant p53.454,455,456 However, subsequent studies have shown that CP-31398 does not interact with p53-DBD or full-length p53, but rather acts as an intercalator and may interact with p53 during biosynthesis. CP-31398 inhibited ubiquitination and degradation of p53457 and activated BAX independently of p53 to promote apoptosis of tumor cells.455 Molecular docking structures revealed that STIMA-1,458 MIRA-1459,460,461 stictic acid424 and UCI-LC0023462 could bind in the L1/S3 pocket of p53-DBD, probably to the thiol group of Cys124 in this transiently opened pocket.424
Some of these broad-spectrum mutant p53 rescue compounds have been reported to bind p53-DBD cysteine, helping restore the correct folding and thermostability of mutant p53. Complex structures show that MQ can bind to six of the ten cysteines,425 while ATO431 and PAT432 bind at a pocket formed by Cys124, Cys135, Cys141 and Met133. p53 has ten cysteines in its DBD (Fig. 9a). Theoretically, these cysteines are all potential sites for modification. Amino acids exposed on the surface of p53-DBD, Cys182, Cys229 and Cys277, are the most susceptible sites for modification. Cys182 and Cys229 are located on the p53 dimer contact surface and modification of these two sites may cause non-functional artificial dimers.463 Cys277 is located on the DNA contact surface and modification of this site may result in spatial site blocking that inhibits p53 binding to DNA.463 Cys176, Cys238 and Cys242 are relatively unlikely to be modified, as they involve the coordination of a zinc atom.298 Besides, Cys124, Cys135, Cys141 and Cys275 are located in the p53-DBD core region, which is exposed when the protein is not fully folded or conformationally altered, and may also be the site of modification. Specific targeting of these sites may be more helpful in restoring the structure and function of mutant p53.
Allele-specific mutant p53 rescue compounds
Y220C
The Y220C mutation in p53 is a relatively specific mutation. Y220 is far from the site where p53 binds to DNA, while the tyrosine mutation between S7/S8 becomes cysteine, forming a hydrophobic cavity on the p53 surface.464,465 Therefore, targeting the hydrophobic pocket of Y220C becomes an ideal drug target, helping p53 to restore normal folding without interfering with p53 binding to DNA.466 Several small molecules have been shown to bind to the hydrophobic pocket of Y220C and restore p53 function467,468,469,470,471,472,473,474,475,476,477,478 (Tables 2–3, Fig. 10a–i).

Structure of the p53 mutant Y220C with small molecules. a p53-WT surface (PDB: 3KMD). b p53-Y220C surface (PDB: 2VUK). c p53-Y220C/Phikan083 (PDB: 2VUK). d p53-Y220C/Phikan5196 (PDB: 4AGQ). e p53-Y220C/PK7242 (PDB: 3ZME). f p53-Y220C/Compound9 (PDB: 5AOJ). g p53-Y220C/Compound6 (PDB: 5G4O). h p53-Y220C/MB710 (PDB: 5O1I). i p53-Y220C/PK9318 (PDB: 6GGB). Water molecules are red spheres, and hydrogen bonds are black lines. The interacting amino acid residues are shown as sticks (colored greencyan)
C14586 is the first orally bioavailable mutant p53 protein-selective reactivator that selectively binds to the cleft produced by the p53 Y220C mutant protein, thereby restoring wild-type p53 protein structure and tumor suppressor function.479 Preclinical trials have shown that continuous oral administration leads to complete tumor regression in 80% of mice.479 An ongoing clinical phase 1/2 study will evaluate the safety, tolerability and antitumor activity of C14586 in adult patients with advanced or metastatic solid tumors with the Y220C mutation.480
The hot spot mutation Y220C is a particularly suitable target site for structure-based drug design. The mutant p53 temperature sensitivity can be repaired by small molecule drugs, and the cavity that appears due to the mutation is also a good target for small molecules. Structure-based drug design and optimization may lead to new strategies for patients with tumors carrying the p53-Y220C mutation.
Targeting p53 aggregation
p53 is a temperature-sensitive protein. Once mutated, its structural stability is compromised, exposing adhesion sequences encased in the hydrophobic core of p53, which drives the formation of p53 aggregates,481 thereby depriving p53 of its DNA recognition function and proapoptotic capacity482,483 (Tables 2–3, Fig. 11). Amyloid aggregates caused by TP53 mutations have been found in biopsy specimens from ovarian and breast cancers.484,485

Targeting p53 aggregation. p53-DBD has an amyloid-forming segment, LTIITLE, that forms Mutant p53 aggregates. ReAcp53, ADH-6 and LI inhibit p53 aggregation and restore the conformation and function of p53
ReACp53
The formation of p53 aggregates is considered to be an amyloid lesion, where two β-sheet layers stack in parallel to form a tight complementary spatial zipper, forming amyloid fibrils mainly through hydrogen bonding interactions between the main and side chains of the β-sheet layer.486,487 Several research groups have reported similar sequence positions as highly adherent fragments of p53 (251–258, ILTIITLE) (Fig. 11). p53 mutations result in reduced p53 stability and expose the highly adherent fragment that drives p53 aggregation, which is effectively inhibited by the I254R mutation.457,488,489 Soragni et al. designed a cell-penetrating peptide (named ReACp53) that inhibits amyloid aggregation of the mutant p53 protein and restores the transcriptional function of p53 and its ability to induce mitochondrial apoptosis.490 ReACp53 treatment increased the nuclear localization of mutant p53 by 70–100% and approximately doubled the transcriptional activity.490
Molecular dynamics simulations were used to study the binding characteristics of the ReACp53 peptide to the R175H mutant p53-DBD.491 The results show that ReACp53 has hydrophobic interactions with residues Leu188 and Leu201 and forms salt bridges or hydrogen bonds with residues Asp186, Glu198, Asp204, Glu 221 and Glu 224, which prevent the aggregation-prone region (residues 182–213) from being exposed. In addition, the complex formed by the ReACp53 peptide and the R175H mutant p53-DBD has the same positive net charge as wild-type p53. The aggregation of p53 was inhibited by electrostatic repulsion.491 ReACp53 also inhibits the growth of various tumors characterized by p53 mutations.490,492,493,494
ReACp53 inhibits the aggregation of mutant p53 to rescue p53 function, but it is clear that its non-p53 targets are also present in cancer cells. In p53-silenced cells, the response of ReACp53 was not abolished, and addition of this peptide to p53-silenced two-dimensional cultured cancer cells resulted in rapid apoptosis.495 Thus, although ReACp53 was designed as a mutant p53 rescue compound, its exact mechanism of action and targeting in tumors and whether it acts primarily by targeting p53 need to be further elucidated.
ADH-6
ADH-6, Kd = 366 nM, is a cationic tripyridylamide obtained by screening the oligopyridylamide library.496 Previous studies have reported that these α-helix-like mimics can mimic the secondary structure of proteins and effectively regulate amyloid aggregation497,498 (Fig. 11). Using NMR spectroscopy, ADH-6 was shown to bind not only p53-prone aggregation sites but also multiple regions of p53-DBD, including sheet 1, 3, 4, 6, 7 and helix 2. Subsequently, researchers also found that ADH-6 dissociated p53 mutant aggregates and selectively induced apoptosis in multiple p53 aggregation-prone mutated cancer cells (R248W, R248Q, R175H, R273H, Y220C and R280K). In addition, the unfolded state of mutant p53 was reduced by 24–50% after ADH-6 treatment of cells, and the expression of downstream target genes p21, Noxa and BAX was significantly increased.496
LI/LH
Miller et al. designed two bifunctional ligands (LI/LH) based on other compounds that bind amyloid proteins.499 LI and LH have the same structural features except for an iodine substituent at the ortho position. The iodine atoms may form halogen bonds that bind to exposed hydrophobic amino acid residues and regulate the aggregation of p53 (Fig. 11). LI and LH acted as metallochaperones binding to zinc ions, which increased the intracellular zinc ion level and improved the binding ability of zinc ions to mutant p53, thus regulating mutant p53 aggregation. LI and LH also increased the expression of Noxa and p21 by 1.5 to 4.3-fold. Meanwhile, transmission electron microscopy observation confirmed that mutant p53 could significantly inhibit the formation of aggregates when cocultured with LI.499
HDAC6/Hsp90 inhibitors
Mutant p53 aggregates not only induced wild-type p53 aggregates but also coaggregated with p63 and p73495,500 (Fig. 11). The p53 aggregates led to upregulation of Hsp70 and Hsp9098 (Fig. 11). Hsp70 inhibited MDM2-mediated degradation of mutant p53 ubiquitination and led to transient exposure of p53 adhesion sequences, increasing the formation of p53 aggregates.501 The interaction between Hsp90 and mutant p53 prevented the ubiquitinated degradation of mutant p53 protein.502 Thus, disruption of the HDAC6/Hsp90 complex by HDAC inhibitors503 or Hsp90 inhibitors504 induces degradation of the p53 mutant.
Peptide and antibody drugs targeting protein interactions
Peptide drugs and antibody drugs are characterized by high specificity and good safety profiles. With the maturation of biotechnology, an increasing number of peptide and antibody therapeutics are beginning to emerge that can bind to mutated p53 and restore the function of the p53 mutant (Tables 2–3). However, although peptide drugs can enter cells to restore p53 function, it is difficult to maintain stability and function after entering the human body.505,506
CDB3
A nine amino acid peptide, CDB3, was designed based on the p53 binding protein 53BP2 region (490–498).507 The NMR structure showed that CDB3 interacts with multiple sites in the DBD region, concentrated between loop 1, helix 2 and sheet 8, located at the edge of the DNA-binding site and partially overlapping with the DNA-binding site.507 Further experiments revealed that CDB3 could bind to G245S and R249S structural mutants,508 restore conformational folding,509 improve the affinity of the β-sheet I195T mutant to DNA, and restore the transcriptional activation function of p53 (R175H, R237H). Upon binding of CDB3 to mutant p53, the p53 protein is activated, and target genes compete for the position of binding CDB3, which in turn is released back into the cell and continues to target other mutants.507,510
pCAPs
A series of p53 conformation-activating peptides, or pCAPs, were identified using phage display technology. Despite the absence of clear structural information, pCAPs were shown to bind to misfolded p53 mutants, forcing p53 conformational recovery and activating transcription of p53 downstream target genes. Significantly reduced mouse tumor size in mouse xenograft models of breast, ovarian and colon cancer.511
p28
The amphipathic penetrating peptide p28 contains twenty-eight amino acids from Pseudomonas aeruginosa.512,513,514 It preferentially enters cancer cells and inhibits their proliferation by stabilizing the expression of p53.515 p28 forms a complex with the DBD of p53, and structural simulations show that the main binding sites of p28 are in the nonmutagenic loop L1 (amino acids 112–124) and the mutagenic loop L7/L8 regions (Y220C, P223L).516 p28 inhibits the interaction between the E3 ligase COP1 and p53,517 improves the posttranslational stability of p53, and increases p53 protein expression in various p53 wild-type or mutated tumor cell lines.518,519 These results provide foundational evidence that p28 performs well in phase 1 clinical trials in patients with various p53 wild-type or mutant tumors.520,521,522
Nb3 and Nb139
Nanobodies containing only a heavy chain variable region (VHH) but still retain antigen specificity and high affinity523 have proven their value in cancer therapy and diagnosis in recent years.524,525,526 Two p53-binding nanobodies, Nb3 and Nb139, were obtained by immunization and panning procedures.527 Nb3 binds to the “structural mutations” R175H and R282W, and Nb139 binds to both wild-type and mutant p53, inhibiting the transcriptional capacity of p53. The cocrystal structure shows that the three complementary determining regions (CDRs) of Nb139 interact with p53-DBD by forming backbone or side chain hydrogen bonds and van der Waals forces (Fig. 12a). Compared with the wild-type p53 structure, the Nb139/p53-DBD complex maintains the structural and DNA-binding properties of p53, which provides an innovative approach to study p53.527

Antibody drugs against p53. a Structure of the Nb139/p53-DBD complex. p53-DBD shown as cartoon/surface (colored pink). Nb139 shown as cartoon (colored marine). b Mechanism of action and structure of the bispecific antibody H2-scDb. H2-scDb is a bispecific antibody that recognizes the p53R175H mutant peptide presented on the cell surface by HLA-A and binds to the T-cell receptor, activating T cells and releasing cytokines to kill tumor cells. The interacting amino acid residues are shown as sticks. Hydrogen bonds as black lines
H2-scDb
Mutant p53 is hydrolyzed intracellularly to produce a peptide known as a mutation-associated neoantigen that can be presented on the cell surface after forming a complex (pHLA) with human leukocyte antigen (HLA) proteins.528,529 In two patients with metastatic epithelial carcinoma carrying the p53R175H mutation, Malekzadeh et al. identified the neoantigen as the HMTEVVRHC peptide, which can be recognized and formed into a complex by HLA*02:01530 (Fig. 12b). To exploit this tumor-specific surface antigen, the researchers screened the antibody fragment library and optimized the design to identify a TCR-mimetic antibody in the form of a single-chain double antibody (scDb) with Kd = 86 nM.529 The structure revealed that the p53R175H peptide occupies the binding cleft between α1 and α2 of HLA-A*02:01. The His175 amino acid residue in the p53R175H peptide fragment plays a significant role in direct contact with H2-Fab. The imidazole side chain of His175 forms hydrogen bonds with Asp54 (CDR-H2) and Tyr94 (CDR-L3) and forms a π-π stacking interaction with Tyr52 (CDR-H2) (Fig. 12b). Functionally, H2-scDb bound specifically to the p53R175H/HLA complex and efficiently induced T-cell immune responses. In multiple myeloma xenograft mice carrying p53R175H, H2-scDb effectively regressed tumors.529
Second-site suppressor mutation
Mutant p53 usually affects the binding of p53 to DNA or the conformation of p53. Second site suppressor mutations could help correct the conformation of mutant p53, improve the stability of p53-DNA binding, and provide a theoretical basis for the reactivation of mutant p53 (Table 4).531,532,533,534,535,536,537,538,539,540,541,542,543,544,545
Crystal structures show that the second-site suppressor mutations usually cause only small local structural changes, whereas do not alter the overall structure of the p53 protein.534,536,545,546 These second-site suppressor mutations act through two types of mechanisms: supplying a new DNA-contacting amino acids when rescuing DNA-contacting mutants, or increasing thermostability when rescue structural mutants.537,547 These mutants provide the basis for designing drugs that restore the transcriptional inactivation and instability of p53 caused by mutations.
Other therapies
The TET and CTD have essential roles in the proper folding and functional regulation of p53. There are no drugs that specifically target these two structural domains. A synthetic peptide (361–382) extracted from the p53-CTD region (361–382, peptide 46) can interact with the DBD and CTD regions of p53,548 and the addition of different concentrations of peptide 46 restores the DNA binding ability of p53 mutants (R273H, R248W, R175H, R249S, V143A).549 The fusion of the p53 protein with the N-terminal spider silk domain contributes to the stability of the p53 protein.550 The therapeutic effects and specific mechanisms targeting these two structural domains need further exploration.
Researchers have also developed innovative therapies that target mRNA for protein degradation. For example, a specific deoxyribozyme (DZ-249A) has been designed to target the mutation site of TP53 to degrade the mRNA of mutant TP53, thereby reducing the expression of the mutant p53 protein.551,552 Combination therapy of multiple mechanisms is also a promising strategy; for instance, the combination of MDM2 and BCL-2 inhibitors can effectively induce apoptosis.553,554 Moreover, gene therapy and immunotherapy are also new options for targeting p53.
Therapeutic strategy for truncated p53
Nonsense mutations cause proteins to terminate or end translation earlier than expected, generating truncated proteins. A total of 8.19% of TP53 mutant tumors were detected to carry nonsense mutations and were cleared by the nonsense-mediated mRNA decay (NMD) machinery during transcription555,556 (Fig. 1a). Currently, there are two approaches to rescue truncated p53.557 One is to restore production of full-length p53 protein by drug-induced read-through of the premature termination codon. For instance, G418 and NB124 can stably promote the expression of full-length p53 protein.558,559 The other is the inhibition of NMD.560,561 However, NMD is a potent cellular surveillance mechanism562 and non-specific inhibition of NMD may do more harm than good.
Therapeutic strategy for p53 isoforms
Fourteen isoforms of p53 have been identified: p53, p53β, p53γ, Δ40p53, Δ40p53β, Δ40p53γ, Δ133p53, Δ133p53β, Δ133p53γ, Δ160p53, Δ160p53β, Δ160p53γ, Δp53, and p53ψ.563,564,565,566,567,568 It was shown that Δ133p53 activates the anti-apoptotic BCL-2 protein and antagonizes p53-induced apoptosis.563 p53Ψ cannot bind to DNA or transcriptionally activate p53 target genes. However, it induces the expression of epithelial-mesenchymal transition markers and enhances the viability and invasiveness of cancer cells.565 Overexpression of Δ133p53α facilitates the long-term propagation of primary epithelial cells in vitro.569 These evidences suggest that some p53 isoforms are in marked contradiction to the function of wild-type full-length p53, exhibit oncogenic effects, and support cancer cell proliferation and invasion. Therefore, rescue therapy may not be appropriate for these p53 isoforms, and targeted degradation may be a better strategy to remove their oncogenic effects, such as PROTAC,570 ATTEC,571 AUTAC,572 and LYTAC.573
Conclusion
The TP53 gene is the most frequently mutated gene in humans. In the 40 years since the discovery of p53, new insights have been made into the gene regulatory mechanisms and tumor suppression pathways of p53. We have learned about an elaborate and complex tumor suppression network, but have not yet discovered its accurate and complete picture. Based on the relationship between the structure and function of p53 and cancer development, many anticancer drugs targeting p53 have been developed. However, there are no approved drugs to date.
Summary points
Among the molecules developed to target p53, two main mechanistic approaches have been implemented. One is to target wild-type p53 and inhibit the p53/MDM2 complex to prevent its degradation. To date, eleven MDM2 inhibitors are in clinical trials. However, long-term administration of an MDM2 inhibitor may induce acquired resistance related to TP53 mutations and increased MDMX expression.574,575 There is still a lack of MDM2/MDMX dual-target or specific MDMX inhibitors. Moreover, the accumulation of p53 in normal tissues may produce greater toxic effects.353
The second is to target p53 mutants. Many therapeutic approaches aim to rescue the function of mutated p53 through small molecule compounds. The development of p53 mutant-rescuing drugs has been hampered by false trials that have proven not to be reproducible in other labs or to function by p53-unrelated pathways. Some drugs were initially identified as p53 mutant activators, but it remains puzzling whether they actually rely on p53 for their action. For example, CP-31398 was initially thought to maintain the wild-type conformation of the mutant p53,454 but was shown to be unable to bind p53 in a different lab.455 Dr. Soragni states on her website that “we don’t really know what the mechanism is for ReACp53 to kill cancer cells” (http://alice.mbi.ucla.edu/reacp53.html). This may be because most experiments to rescue mutant p53 drugs are performed in cells, and the lack of information on the direct interaction of compounds with proteins leads to our incomplete understanding of the mechanism of drug action. Most of the currently reported drugs that activate p53 mutants have p53-independent antitumor effects. It is undeniable that these drugs do act to rescue mutant p53 function, but it may be ambiguous which is more effective, whether it is mutant p53-dependent, mutant p53-independent, or both. The field of targeting mutant p53 remains a shimmering light after the dawn, and we see hope, but there is still a long road of discovery to go.
The availability of structural data on p53 mutants and their complexes with drugs, combined with data on the functional rescue of the mutants, has helped to understand the rescue mechanism and optimize drug design. For example, a special class of p53 structural mutant, Y220C, leads to a hydrophobic cavity in p53 protein structure.467 Small molecules are designed to target this cavity to stabilize the protein structure. ATO431 and PAT432 bind to an allosteric site exposed within the p53 mutant protein and help stabilize the local structure. They are likely to be effective for structural mutants or temperature-sensitive mutants, but not DNA contact mutants. Those mutations at the DNA contact surface tend to benefit from small molecules that have the ability to restore protein-DNA binding. The structures of MQ with p53 mutants R282W, R273H and R273C show that the binding of MQ with Cys277 supports the p53-DNA interface by complementing additional interactions, while MQ interaction modes exhibit large diversity by binding to some other cystines.425 In addition, structural analysis illustrates the basis of a TCR-like antibody H2-Fab specifically targeting the p53R175H peptide-HLA interaction.529 Nevertheless, structural information about p53 mutants and their complexes is still insufficient, so we need more structural insights to help target p53 mutants for rational drug design.
Challenges and perspectives
There are significant challenges in the development of drugs targeting p53. The lack of an established mechanism for protein reactivation and the lack of pockets (except in the case of Y220C) are the two main reasons. It is relatively easy to inhibit protein function by simply using compounds to occupy their active sites, but elusive as to how compound binding leads to reactivation of protein function. In addition, resistance to TP53 mutations, the off-target effects of the drug, and the possible toxic side effects of p53 accumulation in normal tissues, all make p53 difficult to drug. Currently, the structures of full-length p53 in complex with different DNA targets and full-length partner proteins remain unavailable. Certain p53 mutants cannot be expressed, and their structures are not available, which limits structure-based drug design. In recent years, there have been great advances in cryo-electron microscopy, and artificial intelligence. With these technological advances, we have reason to believe that the structural study of p53 will make more progress in the near future, which will provide a structural basis for the development of p53-targeted drugs.
For p53-targeted therapy, several other factors also need to be considered. First, TP53 mutations are heterogeneous and not all mutations are equal. Hence a one-drug-fits-all approach may not be feasible for targeting TP53 mutations.12,576,577 Therefore, different p53-targeting drugs may be required for different p53 mutants. Second, p53-targeted therapy alone may not be sufficient to treat cancer. Combination therapy, such as simultaneous blockade of the MDM2-p53 pathway and p53-BCL-2 pathway, may have a synthetic lethal mechanism. Third, there are some other new therapeutic directions that may be explored, such as targeting p53 mRNA, targeting disordered structural domains, targeting mutant protein for degradation, and genome editing using CRISPR-Cas9. CRISPR-Cas9 based gene editing technology has been applied to cancer treatment.578,579 With the advancement of CRISPR-Cas9 technology, the correction of TP53 mutations using CRISPR-Cas9 may become an effective cancer treatment option in the future.
For decades, there has been a lack of effective progress in the development of drugs targeting p53, and p53 was once considered to be an undruggable target. As technology advances, many undruggable targets are becoming druggable, such as KRAS.580,581 We have reason to believe that drugs targeting p53 will also make progress. Given the prevalence of TP53 mutations in human cancers, drugs targeting p53 may bring a breakthrough in cancer therapy.
References
Kastenhuber, E. R. & Lowe, S. W. Putting p53 in context. Cell 170, 1062–1078 (2017).
Levine, A. J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 578, 82–93 (2020).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer. 18, 89–102 (2018).
Sullivan, K. D., Galbraith, M. D., Andrysik, Z. & Espinosa, J. M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143 (2018).
Sager, R. Tumor suppressor genes: the puzzle and the promise. Science 246, 1406–1412 (1989).
Muller, P. A. J. & Vousden, K. H. p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 (2013).
Meek, D. W. Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer 9, 714–723 (2009).
Wong, K. B. et al. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 96, 8438–8442 (1999).
Kitayner, M. et al. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 22, 741–753 (2006).
Joerger, A. C. & Fersht, A. R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 97, 1–23 (2007).
Loh, S. N. Arsenic and an old place: rescuing p53 mutants in cancer. Cancer Cell 39, 140–142 (2021).
Linzer, D. I. & Levine, A. J. Characterization of a 54 K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 (1979).
Lane, D. P. & Crawford, L. V. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263 (1979).
Parada, L. F. et al. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312, 649–651 (1984).
Eliyahu, D., Michalovitz, D. & Oren, M. Overproduction of p53 antigen makes established cells highly tumorigenic. Nature 316, 158–160 (1985).
Lane, D. P. & Benchimol, S. p53: oncogene or anti-oncogene? Genes Dev. 4, 1–8 (1990).
Nigro, J. M. et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708 (1989).
Cunningham, J. et al. Expression of p53 and 17p allelic loss in colorectal carcinoma. Cancer Res 52, 1974–1980 (1992).
Hinds, P., Finlay, C. & Levine, A. J. Mutation is required to activate the p53 gene for cooperation with the ras oncogene and transformation. J. Virol. 63, 739–746 (1989).
Eliyahu, D. et al. Meth A fibrosarcoma cells express two transforming mutant p53 species. Oncogene 3, 313–321 (1988).
Donehower, L. A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Olivero, C. E. et al. p53 Activates the Long Noncoding RNA Pvt1b to Inhibit Myc and Suppress Tumorigenesis. Mol. Cell. 77, 761–774 (2020).
Yonish-Rouach, E. et al. The role of p53 as a transcription factor in the induction of apoptosis. Behring Inst. Mitt. 97, 60–71 (1996).
Wei, C.-L. et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 (2006).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014).
Lakin, N. & Jackson, S. Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655 (1999).
Gu, B. & Zhu, W. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8, 672–684 (2012).
DeHart, C., Chahal, J., Flint, S. & Perlman, D. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell Proteom. 13, 1–17 (2014).
Chen, L., Liu, S. & Tao, Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct. Target Ther. 5, 90 (2020).
Kruse, J.-P. & Gu, W. SnapShot: p53 posttranslational modifications. Cell 133, 930–30 (2008).
Aubrey, B. et al. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113 (2018).
Vaddavalli, P. & Schumacher, B. The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet 38, 598–612 (2022).
Ma, M. et al. p53 positively regulates the proliferation of hepatic progenitor cells promoted by laminin-521. Signal Transduct. Target Ther. 7, 290 (2022).
Mihara, M. et al. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 11, 577–590 (2003).
Chipuk, J. E. et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014 (2004).
Miyashita, T. & Reed, J. C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299 (1995).
Seoane, J., Le, H.-V. & Massagué, J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419, 729–734 (2002).
Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 29, 946–960 (2022).
Hafner, A., Bulyk, M., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 (2019).
Sherr, C. J. & McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2, 103–112 (2002).
Sancar, A. DNA repair in humans. Annu. Rev. Genet. 29, 69–105 (1995).
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kaçmaz, K. & Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (2004).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Liu, Y. & Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 85, 4–32 (2021).
Blagih, J., Buck, M. D. & Vousden, K. H. p53, cancer and the immune response. J. Cell Sci. 133, jcs237453 (2020).
Spike, B. T. & Wahl, G. M. p53, stem cells, and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer 2, 404–419 (2011).
White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 6, a026120 (2016).
Mrakovcic, M. & Fröhlich, L. p53-mediated molecular control of autophagy in tumor cells. Biomolecules 8, 14 (2018).
Williams, A. B. & Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 6, a026070 (2016).
Adimoolam, S. & Ford, J. M. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. USA 99, 12985–12990 (2002).
Zebian, A. et al. XPC multifaceted roles beyond DNA damage repair: p53-dependent and p53-independent functions of XPC in cell fate decisions. Mutat. Res. Rev. Mutat. Res. 789, 108400 (2022).
Sugasawa, K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat. Res. 685, 29–37 (2010).
el-Deiry, W. S. et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993).
Narasimha, A. M. et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 3, e02872 (2014).
Prall, O. W. et al. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 272, 10882–10894 (1997).
Harper, J. W. et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 (1993).
Stevaux, O. & Dyson, N. J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 14, 684–691 (2002).
Basak, S. et al. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol. Cell. 30, 303–314 (2008).
Doumont, G., Martoriati, A. & Marine, J.-C. PTPRV is a key mediator of p53-induced cell cycle exit. Cell Cycle 4, 1703–1705 (2005).
Fischer, M., Quaas, M., Steiner, L. & Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res 44, 164–174 (2016).
Taylor, W. R. & Stark, G. R. Regulation of the G2/M transition by p53. Oncogene 20, 1803–1815 (2001).
Hermeking, H. et al. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell. 1, 3–11 (1997).
Wang, X. W. et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc. Natl. Acad. Sci. USA 96, 3706–3711 (1999).
Ohki, R. et al. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J. Biol. Chem. 275, 22627–22630 (2000).
Eischen, C. M. Genome Stability Requires p53. Cold Spring Harb. Perspect. Med. 6, a026096 (2016).
Oda, E. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 (2000).
Nakano, K. & Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7, 683–694 (2001).
Chen, Y. et al. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res 41, 8368–8376 (2013).
Wei, H. et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 12, 2280 (2021).
Leu, J. I. J. et al. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 6, 443–450 (2004).
Bommer, G. T. et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307 (2007).
Tarasov, V. et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6, 1586–1593 (2007).
Wu, G. S., Kim, K. & el-Deiry, W. S. KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. Adv. Exp. Med. Biol. 465, 143–151 (2000).
Maecker, H. L., Koumenis, C. & Giaccia, A. J. p53 promotes selection for Fas-mediated apoptotic resistance. Cancer Res 60, 4638–4644 (2000).
Müller, M. et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med 188, 2033–2045 (1998).
Allen, M. A. et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 3, e02200 (2014).
Cordaux, R. & Batzer, M. A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703 (2009).
Tiwari, B., Jones, A. E. & Abrams, J. M. Transposons, p53 and Genome Security. Trends Genet 34, 846–855 (2018).
Harris, C. R. et al. p53 responsive elements in human retrotransposons. Oncogene 28, 3857–3865 (2009).
Haoudi, A., Semmes, O. J., Mason, J. M. & Cannon, R. E. Retrotransposition-Competent Human LINE-1 Induces Apoptosis in Cancer Cells With Intact p53. J. Biomed. Biotechnol. 2004, 185–194 (2004).
Zuckerman, V. et al. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J. Pathol. 219, 3–15 (2009).
Schmitt, C. A. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346 (2002).
Mijit, M. et al. Role of p53 in the regulation of cellular senescence. Biomolecules 10, 420 (2020).
Abuetabh, Y. et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 54, 1658–1669 (2022).
Capuozzo, M. et al. p53: from fundamental biology to clinical applications in cancer. Biology 11, 1325 (2022).
Kortlever, R. M., Higgins, P. J. & Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 877–884 (2006).
Pearson, M. et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406, 207–210 (2000).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Devic, S. Warburg effect—a consequence or the cause of carcinogenesis? J. Cancer 7, 817–822 (2016).
Matoba, S. et al. p53 regulates mitochondrial respiration. Science 312, 1650–1653 (2006).
Bensaad, K. et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 (2006).
Zhang, C. et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 108, 16259–16264 (2011).
Jiang, P. et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 13, 310–316 (2011).
Li, Q. et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat. Commun. 11, 1456 (2020).
Schwartzenberg-Bar-Yoseph, F., Armoni, M. & Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 64, 2627–2633 (2004).
Boidot, R. et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res 72, 939–948 (2012).
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012).
Zawacka-Pankau, J. et al. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: targeting Warburg effect to fight cancer. J. Biol. Chem. 286, 41600–41615 (2011).
Zhang, C. et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 4, 2935 (2013).
Basu, S. et al. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. 32, 230–243 (2018).
Wang, P.-Y. et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med. 368, 1027–1032 (2013).
Snaebjornsson, M. T., Janaki-Raman, S. & Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 31, 62–76 (2020).
Moon, S.-H. et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 176, 564–580 (2019).
Zhu, Y. et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology, (2022).
Kruiswijk, F., Labuschagne, C. F. & Vousden, K. H. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405 (2015).
Spinelli, J. B. et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946 (2017).
Zhang, T. et al. Metformin inhibits the urea cycle and reduces putrescine generation in colorectal cancer cell lines. Molecules 26, 1990 (2021).
Li, L. et al. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 567, 253–256 (2019).
Cheung, E. C. & Vousden, K. H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 22, 280–297 (2022).
Liu, B., Chen, Y. & St Clair, D. K. ROS and p53: a versatile partnership. Free Radic. Biol. Med. 44, 1529–1535 (2008).
Hussain, S. P. et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res 64, 2350–2356 (2004).
Sablina, A. A. et al. The antioxidant function of the p53 tumor suppressor. Nat. Med. 11, 1306–1313 (2005).
Humpton, T. J. & Vousden, K. H. Regulation of cellular metabolism and hypoxia by p53. Cold Spring Harb. Perspect. Med 6, a026146 (2016).
Liu, G. & Chen, X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 21, 7195–7204 (2002).
Kim, J. et al. Wild-Type p53 Promotes cancer metabolic switch by inducing PUMA-dependent suppression of oxidative phosphorylation. Cancer Cell. 35, 191–203 (2019).
Suzuki, S. et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 107, 7461–7466 (2010).
Gallo, O. et al. Down-regulation of nitric oxide synthase-2 and cyclooxygenase-2 pathways by p53 in squamous cell carcinoma. Am. J. Pathol. 163, 723–732 (2003).
Italiano, D., Lena, A. M., Melino, G. & Candi, E. Identification of NCF2/p67phox as a novel p53 target gene. Cell Cycle 11, 4589–4596 (2012).
Faraonio, R. et al. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 281, 39776–39784 (2006).
Reddy, B. A. et al. Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol. Cell. 53, 458–470 (2014).
Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Yan, H.-F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target Ther. 6, 49 (2021).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Wang, S.-J. et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 17, 366–373 (2016).
Liu, Y. & Gu, W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29, 895–910 (2022).
Li, T. et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 (2012).
Wang, Y. et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 20, e47563 (2019).
Ou, Y. et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl Acad. Sci. USA 113, E6806–E6812 (2016).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Liu, J. et al. The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 21, 8387 (2020).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Tarangelo, A. et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575 (2018).
Venkatesh, D., Stockwell, B. R. & Prives, C. p21 can be a barrier to ferroptosis independent of p53. Aging 12, 17800–17814 (2020).
Lujambio, A. et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013).
Menendez, D. et al. p53-responsive TLR8 SNP enhances human innate immune response to respiratory syncytial virus. J. Clin. Invest 129, 4875–4884 (2019).
Guo, G. et al. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res 77, 2292–2305 (2017).
Zhou, X. et al. Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting antitumor immunity. Cancer Disco. 11, 3090–3105 (2021).
Wang, B., Niu, D., Lai, L. & Ren, E. C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 4, 2359 (2013).
Xiao, Y. et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 13, 758 (2022).
Dong, Z.-Y. et al. Potential predictive value of and mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res 23, 3012–3024 (2017).
Ubertini, V. et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 34, 2493–2504 (2015).
Cooks, T. et al. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 9, 771 (2018).
Cooks, T. et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 23, 634–646 (2013).
Di Minin, G. et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 56, 617–629 (2014).
Song, H. et al. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl Acad. Sci. USA 109, 17531–17536 (2012).
Ouadah, Y. et al. Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell 179, 403–416 (2019).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Cicalese, A. et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138, 1083–1095 (2009).
Zhou, Z., Flesken-Nikitin, A. & Nikitin, A. Y. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 67, 5683–5690 (2007).
Flores, I. & Blasco, M. A. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS One 4, e4934 (2009).
Zheng, H. et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133 (2008).
Liu, Y. et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48 (2009).
Cheng, T. et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808 (2000).
van Os, R. et al. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells 25, 836–843 (2007).
Shetzer, Y., Molchadsky, A., & Rotter, V. Oncogenic mutant p53 gain of function nourishes the vicious cycle of tumor development and cancer stem-cell formation. Cold Spring Harb. Perspect. Med. 6, a026203 (2016).
Escoll, M. et al. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 36, 3515–3527 (2017).
Loizou, E. et al. A gain-of-function p53-Mutant oncogene promotes cell fate plasticity and myeloid leukemia through the pluripotency factor FOXH1. Cancer Disco. 9, 962–979 (2019).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Wang, S.-P. et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 11, 694–704 (2009).
Kim, N. H. et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 195, 417–433 (2011).
Dong, P. et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 32, 3286–3295 (2013).
Chang, C.-J. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 13, 317–323 (2011).
Wang, J. et al. Autophagy augments the self-renewal of lung cancer stem cells by the degradation of ubiquitinated p53. Cell Death Dis. 12, 98 (2021).
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
Abida, W. M. & Gu, W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 68, 352–357 (2008).
Crighton, D. et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006).
Mrschtik, M. & Ryan, K. M. Another DRAM involved in autophagy and cell death. Autophagy 12, 603–605 (2016).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Kenzelmann Broz, D. et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031 (2013).
Zheng, W. et al. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 59, 651–660 (2020).
Liu, Y.-J. et al. The noncanonical role of the protease cathepsin D as a cofilin phosphatase. Cell Res. 31, 801–813 (2021).
Wu, G. S., Saftig, P., Peters, C. & El-Deiry, W. S. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 16, 2177–2183 (1998).
Ikeguchi, M. et al. Correlation between cathepsin D expression and p53 protein nuclear accumulation in oesophageal squamous cell carcinoma. J. Clin. Pathol. 55, 121–126 (2002).
Yeo, S. Y. et al. Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. Elife 5, e07101 (2016).
Kim, J., Kundu, M., Viollet, B. & Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Budanov, A. V. & Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460 (2008).
Kon, N. et al. mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev. 35, 59–64 (2021).
Chang, H. W. et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 38, 3729–3742 (2019).
Fernández, Á. F. et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018).
Lee, E. F. et al. Structural insights into BCL2 pro-survival protein interactions with the key autophagy regulator BECN1 following phosphorylation by STK4/MST1. Autophagy 15, 785–795 (2019).
Feng, W., Huang, S., Wu, H. & Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 372, 223–235 (2007).
Zheng, R. et al. The status of p53 in cancer cells affects the role of autophagy in tumor radiosensitisation. J. BUON 19, 336–341 (2014).
Maiuri, M. C. et al. Autophagy regulation by p53. Curr. Opin. Cell Biol. 22, 181–185 (2010).
Livesey, K. M. et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 72, 1996–2005 (2012).
Livesey, K. M. et al. Direct molecular interactions between HMGB1 and TP53 in colorectal cancer. Autophagy 8, 846–848 (2012).
Yang, Y. et al. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 34, 688–p700 (2020).
Agupitan, A. D. et al. P53: a guardian of immunity becomes its saboteur through mutation. Int. J. Mol. Sci. 21, 3452 (2020).
Levine, A. P53 and the immune response: 40 years of exploration-a plan for the future. Int. J. Mol. Sci. 21, 541 (2020).
Min, E.-Y. et al. The effects of fucodian on senescence are controlled by the p16INK4a-pRb and p14Arf-p53 pathways in hepatocellular carcinoma and hepatic cell lines. Int. J. Oncol. 45, 47–56 (2014).
Winkler, L. et al. Functional elements of the cis-regulatory lincRNA-p21. Cell Rep. 39, 110687 (2022).
Giacomelli, A. O. et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387 (2018).
Wellenstein, M. D. et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542 (2019).
Yang, Y. et al. Targeting the miR-34a/LRPPRC/MDR1 axis collapse the chemoresistance in P53 inactive colorectal cancer. Cell Death Differ. 29, 2177–2189 (2022).
Makino, Y. et al. Constitutive activation of the tumor suppressor p53 in hepatocytes paradoxically promotes non-cell autonomous liver carcinogenesis. Cancer Res 82, 2860–2873 (2022).
Hamarsheh, S. A., Groß, O., Brummer, T. & Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 11, 5439 (2020).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
de Andrade, K. C. et al. Cancer incidence, patterns, and genotype-phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: an observational cohort study. Lancet Oncol. 22, 1787–1798 (2021).
Freed-Pastor, W. A. & Prives, C. Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 (2012).
Joerger, A. C. & Fersht, A. R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016).
Duffy, M. J., Synnott, N. C., O’Grady, S. & Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 79, 58–67 (2022).
Baugh, E. H. et al. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160 (2018).
Bullock, A. N., Henckel, J. & Fersht, A. R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 (2000).
Mantovani, F., Collavin, L. & Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212 (2019).
Yue, X. et al. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J. Mol. Biol. 429, 1595–1606 (2017).
Hainaut, P. & Pfeifer, G. P. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 6, a026179 (2016).
Petitjean, A. et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 (2007).
Petitjean, A. et al. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26, 2157–2165 (2007).
Olivier, M. et al. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 19, 607–614 (2002).
Pan, J.-W. et al. The molecular landscape of Asian breast cancers reveals clinically relevant population-specific differences. Nat. Commun. 11, 6433 (2020).
Matozaki, T. et al. p53 gene mutations in human gastric cancer: wild-type p53 but not mutant p53 suppresses growth of human gastric cancer cells. Cancer Res 52, 4335–4341 (1992).
Isaacs, W. B., Carter, B. S. & Ewing, C. M. Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res 51, 4716–4720 (1991).
Menon, A. G. et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc. Natl. Acad. Sci. USA 87, 5435–5439 (1990).
Maxwell, K. N. et al. Inherited TP53 variants and risk of prostate cancer. Eur. Urol. 81, 243–250 (2022).
Malkin, D. et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238 (1990).
McBride, K. et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 11, 260–271 (2014).
Baker, S. J. et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244, 217–221 (1989).
Kashkin, K. N., Fleĭshman, E. V., Chumakov, P. M. & Perevoshchikov, A. G. Genetic alterations in the region of the p53 gene on human chromosome 17 in colorectal cancer. Genetika 34, 1049–1055 (1998).
Cooper, M. J. et al. p53 mutations in bladder carcinoma cell lines. Oncol. Res. 6, 569–579 (1994).
Goh, A. M., Coffill, C. R. & Lane, D. P. The role of mutant p53 in human cancer. J. Pathol. 223, 116–126 (2011).
Dittmer, D. et al. Gain of function mutations in p53. Nat. Genet. 4, 42–46 (1993).
Ghosh, M. et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell. 39, 494–508 (2021).
Sethi, N. S. et al. Early TP53 alterations engage environmental exposures to promote gastric premalignancy in an integrative mouse model. Nat. Genet. 52, 219–230 (2020).
Donehower, L. A. et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28, 3010 (2019).
Hanel, W. & Moll, U. M. Links between mutant p53 and genomic instability. J. Cell. Biochem. 113, 433–439 (2012).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Livingstone, L. R. et al. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 70, 923–935 (1992).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010).
Tomasini, R., Mak, T. W. & Melino, G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol. 18, 244–252 (2008).
Baslan, T. et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 608, 795–802 (2022).
Chan-Seng-Yue, M. et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 52, 231–240 (2020).
Mueller, S. et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 554, 62–68 (2018).
Maddipati, R. et al. Levels regulate metastatic heterogeneity in pancreatic adenocarcinoma. Cancer Disco. 12, 542–561 (2022).
Boettcher, S. et al. A dominant-negative effect drives selection of missense mutations in myeloid malignancies. Science 365, 599–604 (2019).
Zhu, J. et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 (2015).
Xiong, S. et al. Differential gain-of-function activity of three p53 hotspot mutants in vivo. Cancer Res 82, 1926–1936 (2022).
Song, H., Hollstein, M. & Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 9, 573–580 (2007).
Restle, A. et al. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucleic Acids Res 36, 5362–5375 (2008).
Dong, P. et al. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol. Cancer 8, 103 (2009).
Powell, E., Piwnica-Worms, D. & Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Disco. 4, 405–414 (2014).
Brosh, R. & Rotter, V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 (2009).
Oren, M. & Kotler, E. p53 mutations promote proteasomal activity. Nat. Cell Biol. 18, 833–835 (2016).
van Oijen, M. & Slootweg, P. Gain-of-function mutations in the tumor suppressor gene p53. Clin. Cancer Res. 6, 2138–2145 (2000).
Halevy, O., Michalovitz, D. & Oren, M. Different tumor-derived p53 mutants exhibit distinct biological activities. Science 250, 113–116 (1990).
Redman-Rivera, L. N. et al. Acquisition of aneuploidy drives mutant p53-associated gain-of-function phenotypes. Nat. Commun. 12, 5184 (2021).
Jiang, Z. et al. Immunogenomics analysis reveals that TP53 mutations inhibit tumor immunity in gastric cancer. Transl. Oncol. 11, 1171–1187 (2018).
Lyu, H. et al. Correlate the mutation and the mutation with immune signatures in head and neck squamous cell cancer. Comput Struct. Biotechnol. J. 17, 1020–1030 (2019).
Eastham, J. A. et al. Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1, 1111–1118 (1995).
Lang, G. A. et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872 (2004).
Olive, K. P. et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860 (2004).
Kim, M. P. et al. Oncogenic recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Disco. 11, 2094–2111 (2021).
Zheng, S. et al. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene 26, 6896–6904 (2007).
Robey, R. W. et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–464 (2018).
Chan, K.-T. & Lung, M. L. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother. Pharmacol. 53, 519–526 (2004).
Alam, S. K. et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 23, 707–722 (2016).
Wong, R. P. C. et al. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 6, 1054–1061 (2007).
Masciarelli, S. et al. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 33, 1601–1608 (2014).
Joerger, A. C. & Fersht, A. R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 77, 557–582 (2008).
Chillemi, G. et al. Structural evolution and dynamics of the p53 proteins. Cold Spring Harb. Perspect. Med. 7, a028308 (2017).
Joerger, A. C. & Fersht, A. R. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2, a000919 (2010).
Bell, S. et al. p53 contains large unstructured regions in its native state. J. Mol. Biol. 322, 917–927 (2002).
Krois, A. S., Dyson, H. J. & Wright, P. E. Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 115, E11302–E11310 (2018).
Walker, K. K. & Levine, A. J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl Acad. Sci. USA 93, 15335–15340 (1996).
Kussie, P. H. et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 (1996).
Popowicz, G. M. et al. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 6, 2386–2392 (2007).
Lee, C. W., Martinez-Yamout, M. A., Dyson, H. J. & Wright, P. E. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 49, 9964–9971 (2010).
Krois, A. S. et al. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl Acad. Sci. USA 113, E1853–E1862 (2016).
Feng, H. et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 17, 202–210 (2009).
Miller Jenkins, L. M. et al. Characterization of the p300 Taz2-p53 TAD2 complex and comparison with the p300 Taz2-p53 TAD1 complex. Biochemistry 54, 2001–2010 (2015).
Bochkareva, E. et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 102, 15412–15417 (2005).
Rowell, J. P. et al. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 20, 2014–2024 (2012).
Di Lello, P. et al. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 22, 731–740 (2006).
Ecsédi, P. et al. Structure determination of the transactivation domain of p53 in complex with S100A4 using Annexin A2 as a crystallization chaperone. Structure 28, 943–953 (2020).
Okuda, M. & Nishimura, Y. Extended string binding mode of the phosphorylated transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 136, 14143–14152 (2014).
Lee, C. W. et al. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl Acad. Sci. USA 107, 19290–19295 (2010).
Raj, N. & Attardi, L. D. The transactivation domains of the p53 protein. Cold Spring Harb. Perspect. Med 7, a026047 (2017).
Dumont, P. et al. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33, 357–365 (2003).
Ruaro, E. M. et al. A proline-rich motif in p53 is required for transactivation-independent growth arrest as induced by Gas1. Proc. Natl. Acad. Sci. USA 94, 4675–4680 (1997).
Jiang, M. et al. p53 binds the nuclear matrix in normal cells: binding involves the proline-rich domain of p53 and increases following genotoxic stress. Oncogene 20, 5449–5458 (2001).
Golubovskaya, V. M. et al. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem. J. 411, 151–160 (2008).
Yang, L., Li, Y., Bhattacharya, A. & Zhang, Y. PEPD is a pivotal regulator of p53 tumor suppressor. Nat. Commun. 8, 2052 (2017).
Bergamaschi, D. et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 38, 1133–1141 (2006).
Zilfou, J. T. et al. The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol. Cell. Biol. 21, 3974–3985 (2001).
Cho, Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 (1994).
Iwafuchi-Doi, M. & Zaret, K. S. Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692 (2014).
Clarke, C. L. & Graham, J. D. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PLoS One 7, e35859 (2012).
Nishimura, M., Takizawa, Y., Nozawa, K. & Kurumizaka, H. Structural basis for p53 binding to its nucleosomal target DNA sequence. PNAS Nexus 1, pgac177 (2022).
el-Deiry, W. S. et al. Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 (1992).
Funk, W. D. et al. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 12, 2866–2871 (1992).
Friedman, P. N., Chen, X., Bargonetti, J. & Prives, C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. USA 90, 3319–3323 (1993).
Riley, T., Sontag, E., Chen, P. & Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 (2008).
Brázda, V. & Coufal, J. Recognition of local DNA structures by p53 protein. Int. J. Mol. Sci. 18, 375 (2017).
Brázda, V. & Fojta, M. The rich world of p53 DNA binding targets: the role of DNA structure. Int. J. Mol. Sci. 20, 5605 (2019).
Klein, C. et al. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. J. Biol. Chem. 276, 49020–49027 (2001).
Rippin, T. M., Freund, S. M. V., Veprintsev, D. B. & Fersht, A. R. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 319, 351–358 (2002).
Ho, W. C., Fitzgerald, M. X. & Marmorstein, R. Structure of the p53 core domain dimer bound to DNA. J. Biol. Chem. 281, 20494–20502 (2006).
Chen, Y., Dey, R. & Chen, L. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure 18, 246–256 (2010).
Kitayner, M. et al. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 17, 423–429 (2010).
Golovenko, D. et al. New insights into the role of DNA shape on its recognition by p53 proteins. Structure 26, 1237–1250 (2018).
Petty, T. J. et al. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 30, 2167–2176 (2011).
Vainer, R. et al. Structural basis for p53 Lys120-acetylation-dependent DNA-binding mode. J. Mol. Biol. 428, 3013–3025 (2016).
Iwabuchi, K. et al. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA 91, 6098–6102 (1994).
Baldock, R. A. et al. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with γH2AX. Cell Rep. 13, 2081–2089 (2015).
Gorina, S. & Pavletich, N. P. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 274, 1001–1005 (1996).
Derbyshire, D. J. et al. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 21, 3863–3872 (2002).
Samuels-Lev, Y. et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell. 8, 781–794 (2001).
Chen, S. et al. iASPP mediates p53 selectivity through a modular mechanism fine-tuning DNA recognition. Proc. Natl. Acad. Sci. USA 116, 17470–17479 (2019).
Follis, A. V. et al. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat. Struct. Mol. Biol. 21, 535–p543 (2014).
Lilyestrom, W. et al. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 20, 2373–2382 (2006).
Martinez-Zapien, D. et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545 (2016).
Scheffner, M. et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136 (1990).
Jeffrey, P. D., Gorina, S. & Pavletich, N. P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267, 1498–1502 (1995).
Lee, W. et al. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Biol. 1, 877–890 (1994).
Clore, G. M. et al. Interhelical angles in the solution structure of the oligomerization domain of p53: correction. Science 267, 1515–1516 (1995).
Kamada, R. et al. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 106, 598–612 (2016).
Higashimoto, Y. et al. Unfolding, aggregation, and amyloid formation by the tetramerization domain from mutant p53 associated with lung cancer. Biochemistry 45, 1608–1619 (2006).
Stommel, J. M. et al. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18, 1660–1672 (1999).
Chène, P. The role of tetramerization in p53 function. Oncogene 20, 2611–2617 (2001).
Laptenko, O., Tong, D. R., Manfredi, J. & Prives, C. The tail that wags the dog: how the disordered c-terminal domain controls the transcriptional activities of the p53 tumor-suppressor protein. Trends Biochem. Sci. 41, 1022–1034 (2016).
Tong, Q. et al. Structural plasticity of methyllysine recognition by the tandem tudor domain of 53BP1. Structure 23, 312–321 (2015).
Liou, S.-H. et al. Structure of the p53/RNA polymerase II assembly. Commun. Biol. 4, 397 (2021).
Veprintsev, D. B. et al. Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. USA 103, 2115–2119 (2006).
Tidow, H. et al. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 104, 12324–12329 (2007).
Arlt, C., Ihling, C. H. & Sinz, A. Structure of full-length p53 tumor suppressor probed by chemical cross-linking and mass spectrometry. Proteomics 15, 2746–2755 (2015).
Thornton, J. M., Laskowski, R. A. & Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 27, 1666–1669 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Callaway, E. What’s next for AlphaFold and the AI protein-folding revolution. Nature 604, 234–238 (2022).
Buel, G. R. & Walters, K. J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 29, 1–2 (2022).
Mullard, A. What does AlphaFold mean for drug discovery? Nat. Rev. Drug. Discov. 20, 725–727 (2021).
Humphreys, I. R. et al. Computed structures of core eukaryotic protein complexes. Science 374, eabm4805 (2021).
Binder, J. L. et al. AlphaFold illuminates half of the dark human proteins. Curr. Opin. Struct. Biol. 74, 102372 (2022).
Manfredi, J. J. Mdm2 and MdmX: partners in p53 destruction. Cancer Res 81, 1633–1634 (2021).
Freedman, D. A., Wu, L. & Levine, A. J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 55, 96–107 (1999).
Deng, L. et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target Ther. 5, 11 (2020).
Linke, K. et al. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 15, 841–848 (2008).
Iyappan, S. et al. Turning the RING domain protein MdmX into an active ubiquitin-protein ligase. J. Biol. Chem. 285, 33065–33072 (2010).
Wade, M., Wang, Y. V. & Wahl, G. M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20, 299–309 (2010).
Bhattacharya, S., Chakraborty, D., Basu, M. & Ghosh, M. K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct. Target Ther. 3, 17 (2018).
Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013).
Sun, D. et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 57, 1454–1472 (2014).
Tovar, C. et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res 73, 2587–2597 (2013).
Ding, Q. et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 56, 5979–5983 (2013).
Khurana, A. & Shafer, D. A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther. 12, 2903–2910 (2019).
Fan, X. et al. MDM2 inhibitor RG7388 potently inhibits tumors by activating p53 pathway in nasopharyngeal carcinoma. Cancer Biol. Ther. 20, 1328–1336 (2019).
Cui, Y., Zhou, J. & Rong, F. Combination of metformin and RG7388 enhances inhibition of growth and induction of apoptosis of ovarian cancer cells through the PI3K/AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 533, 665–671 (2020).
Berberich, A. et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res 25, 253–265 (2019).
Skalniak, L. et al. Prolonged idasanutlin (RG7388) treatment leads to the generation of p53-mutated cells. Cancers (Basel) 10, 396 (2018).
Rew, Y. & Sun, D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J. Med. Chem. 57, 6332–6341 (2014).
Her, N.-G. et al. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 9, 792 (2018).
Sahin, I. et al. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Disco. 6, 57 (2020).
Erba, H. P. et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 3, 1939–1949 (2019).
Wang, S. et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res 74, 5855–5865 (2014).
de Jonge, M. et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 76, 144–151 (2017).
de Weger, V. A. et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 120, 286–293 (2019).
Jung, J. et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat. Commun. 7, 12609 (2016).
Holzer, P. et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 58, 6348–6358 (2015).
Weisberg, E. et al. Inhibition of wild-type p53-expressing AML by the novel small molecule HDM2 inhibitor CGM097. Mol. Cancer Ther. 14, 2249–2259 (2015).
Maser, T. et al. The MDM2 inhibitor CGM097 combined with the BET inhibitor OTX015 induces cell death and inhibits tumor growth in models of neuroblastoma. Cancer Med 9, 8144–8158 (2020).
Portman, N. et al. MDM2 inhibition in combination with endocrine therapy and CDK4/6 inhibition for the treatment of ER-positive breast cancer. Breast Cancer Res 22, 87 (2020).
Carvajal, L. A. et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 10, eaao3003 (2018).
Saleh, M. N. et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-Type. Clin Cancer Res 27, 5236–5247 (2021).
Kannan, S., Partridge, A. W., Lane, D. P. & Verma, C. S. The dual interactions of p53 with MDM2 and p300: implications for the design of MDM2 inhibitors. Int. J. Mol. Sci. 20, 5996 (2019).
Stein, E. M. et al. Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin. Cancer Res 28, 870–881 (2022).
Stachyra-Valat, T. et al. Abstract 1239: NVP-HDM201: Biochemical and biophysical profile of a novel highly potent and selective PPI inhibitor of p53-Mdm2. Cancer Res 76, 1239–1239 (2016).
Ravandi, F. et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res 48, 92–100 (2016).
Wagner, A. J. et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J. Clin. Oncol. 35, 1304–1311 (2017).
Kang, M. H. et al. Initial testing (stage 1) of MK-8242-A novel MDM2 inhibitor-by the pediatric preclinical testing program. Pediatr. Blood Cancer 63, 1744–1752 (2016).
Cornillie, J. et al. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin. Transl. Oncol. 22, 546–554 (2020).
Zhou, J. et al. The ubiquitin ligase MDM2 sustains STAT5 stability to control T cell-mediated antitumor immunity. Nat. Immunol. 22, 460–470 (2021).
Aguilar, A. et al. Discovery of 4-((3’R,4’S,5’R)-6″-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2″-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3″-indoline]-5’-carboxamido)bicyclo[2.2.2]octane-1-carboxylicAcid (AA-115/APG-115): a potent and orally active murine double minute 2 (MDM2) inhibitor in clinical development. J. Med. Chem. 60, 2819–2839 (2017).
Yi, H. et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 37, 97 (2018).
Takahashi, S. et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 112, 2361–2370 (2021).
Han, X., Wei, W. & Sun, Y. PROTAC degraders with ligands recruiting MDM2 E3 ubiquitin ligase: an updated perspective. Acta Mater. Med 1, 244–259 (2022).
Wang, B. et al. Development of selective small molecule MDM2 degraders based on nutlin. Eur. J. Med. Chem. 176, 476–491 (2019).
Wang, B. et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: Lessons and discoveries. Eur. J. Med. Chem. 219, 113425 (2021).
Li, Y. et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 62, 448–466 (2019).
Terzian, T. et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 22, 1337–1344 (2008).
de Andrade, K. C. et al. The TP53 database: transition from the international agency for research on cancer to the US national cancer institute. Cell Death Differ. 29, 1071–1073 (2022).
Yu, X., Narayanan, S., Vazquez, A. & Carpizo, D. R. Small molecule compounds targeting the p53 pathway: are we finally making progress? Apoptosis 19, 1055–1068 (2014).
Miller, J. J., Gaiddon, C. & Storr, T. A balancing act: using small molecules for therapeutic intervention of the p53 pathway in cancer. Chem. Soc. Rev. 49, 6995–7014 (2020).
Lopes, E. A., Gomes, S., Saraiva, L. & Santos, M. M. M. Small molecules targeting mutant P53: a promising approach for cancer treatment. Curr. Med. Chem. 26, 7323–7336 (2019).
Silva, J. L. et al. Recent synthetic approaches towards small molecule reactivators of p53. Biomolecules 10, 635 (2020).
Binayke, A. et al. Awakening the guardian of genome: reactivation of mutant p53. Cancer Chemother. Pharmacol. 83, 1–15 (2019).
Bykov, V. J. N. et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8, 282–288 (2002).
Wang, T., Lee, K., Rehman, A. & Daoud, S. S. PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem. Biophys. Res. Commun. 352, 203–212 (2007).
Wiman, K. G. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene 29, 4245–4252 (2010).
Bykov, V. J. N. et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 24, 3484–3491 (2005).
Zandi, R. et al. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 17, 2830–2841 (2011).
Saha, M. N. et al. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 12, 2331–2341 (2013).
Müller, M. et al. Combining APR-246 and HDAC-inhibitors: a novel targeted treatment option for neuroblastoma. Cancers (Basel) 13, 4476 (2021).
Liu, D. S. H. et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 64, 1506–1516 (2015).
Mohell, N. et al. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 6, e1794 (2015).
Kobayashi, T. et al. APR-246 induces apoptosis and enhances chemo-sensitivity via activation of ROS and TAp73-Noxa signal in oesophageal squamous cell cancer with TP53 missense mutation. Br. J. Cancer 125, 1523–1532 (2021).
Ceder, S. et al. Mutant p53-reactivating compound APR-246 synergizes with asparaginase in inducing growth suppression in acute lymphoblastic leukemia cells. Cell Death Dis. 12, 709 (2021).
Nahi, H. et al. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br. J. Haematol. 127, 285–291 (2004).
Nahi, H. et al. Mutated and non-mutated TP53 as targets in the treatment of leukaemia. Br. J. Haematol. 141, 445–453 (2008).
Chipuk, J. E., Maurer, U., Green, D. R. & Schuler, M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4, 371–381 (2003).
Lambert, J. M. R. et al. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 29, 1329–1338 (2010).
Ali, D. et al. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 86, 206–215 (2011).
Li, X.-L. et al. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 6, 36689–36699 (2015).
Patyka, M. et al. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 7, 60245–60269 (2016).
Menichini, P. et al. Antitumor effects of PRIMA-1 and PRIMA-1 (APR246) in hematological malignancies: still a mutant P53-dependent affair? Cells. 10, (2021).
Tessoulin, B. et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 124, 1626–1636 (2014).
Mlakar, V. et al. PRIMA-1-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J. Exp. Clin. Cancer Res. 38, 69 (2019).
Peng, X. et al. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 4, e881 (2013).
Bykov, V. J. N. et al. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 6, 21 (2016).
Haffo, L. et al. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 8, 12671 (2018).
Birsen, R. et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 107, 403–416 (2022).
Liu, D. S. et al. Inhibiting the system x/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 8, 14844 (2017).
Fujihara, K. M. et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci. Adv. 8, eabm9427 (2022).
Teoh, P. J. et al. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 7, 61806–61819 (2016).
Ali, D. et al. Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br. J. Haematol. 174, 117–126 (2016).
Sallman, D. A. et al. Eprenetapopt (APR-246) and Azacitidine in -Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 39, 1584–1594 (2021).
Cluzeau, T. et al. Eprenetapopt plus azacitidine in -mutated myelodysplastic syndromes and acute myeloid leukemia: a phase II study by the groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 39, 1575–1583 (2021).
Lehmann, S. et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 30, 3633–3639 (2012).
Gourley, C. et al. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J. Clin. Oncol. 34, 5571–5571 (2016).
Mishra, A. et al. Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell transplantation for -mutant acute myeloid leukemia and myelodysplastic syndromes. J. Clin. Oncol., JCO2200181 (2022).
Maslah, N. et al. Synergistic effects of PRIMA-1 (APR-246) and 5-azacitidine in -mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 105, 1539–1551 (2020).
Welch, J. S. et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med. 375, 2023–2036 (2016).
Lambert, J. M. R. et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 (2009).
Sun, X. Z. et al. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid. Redox Signal 5, 655–665 (2003).
Wassman, C. D. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 4, 1407 (2013).
Degtjarik, O. et al. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ). Nat. Commun. 12, 7057 (2021).
Soignet, S. L. et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 339, 1341–1348 (1998).
Zhang, X.-W. et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 328, 240–243 (2010).
Liu, Q., Hilsenbeck, S. & Gazitt, Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 101, 4078–4087 (2003).
Zheng, T. et al. Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in Hepatocellular Carcinoma. Mol. Cancer 13, 133 (2014).
Li, Y. et al. Arsenic trioxide induces apoptosis and G2/M phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and thereby regulate p53 activation. Cancer Lett. 284, 208–215 (2009).
Chen, S. et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 39 (2021).
Tang, Y. et al. Repurposing antiparasitic antimonials to noncovalently rescue temperature-sensitive p53 mutations. Cell Rep. 39, 110622 (2022).
Liarte, D. B. & Murta, S. M. F. Selection and phenotype characterization of potassium antimony tartrate-resistant populations of four New World Leishmania species. Parasitol. Res. 107, 205–212 (2010).
Dumetz, F. et al. Molecular preadaptation to antimony resistance in leishmania donovani on the indian subcontinent. mSphere. 3 (2018).
Wang, B. et al. The antiparasitic drug, potassium antimony tartrate, inhibits tumor angiogenesis and tumor growth in nonsmall-cell lung cancer. J. Pharmacol. Exp. Ther. 352, 129–138 (2015).
Zhang, Y.-K. et al. Establishment and characterization of arsenic trioxide resistant KB/ATO cells. Acta Pharm. Sin. B 7, 564–570 (2017).
Lecureur, V., Lagadic-Gossmann, D. & Fardel, O. Potassium antimonyl tartrate induces reactive oxygen species-related apoptosis in human myeloid leukemic HL60 cells. Int. J. Oncol. 20, 1071–1076 (2002).
Lecureur, V. et al. Potassium antimonyl tartrate induces caspase- and reactive oxygen species-dependent apoptosis in lymphoid tumoral cells. Br. J. Haematol. 119, 608–615 (2002).
Butler, J. S. & Loh, S. N. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 42, 2396–2403 (2003).
Bullock, A. N. et al. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl Acad. Sci. USA 94, 14338–14342 (1997).
Yu, X., Vazquez, A., Levine, A. J. & Carpizo, D. R. Allele-specific p53 mutant reactivation. Cancer Cell 21, 614–625 (2012).
Yu, X. et al. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 5, 8879–8892 (2014).
Yu, X. et al. Thiosemicarbazones functioning as zinc metallochaperones to reactivate mutant p53. Mol. Pharmacol. 91, 567–p575 (2017).
Blanden, A. R. et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 87, 825–831 (2015).
Na, B. et al. BRCA1Therapeutic targeting of and mutant breast cancer through mutant p53 reactivation. NPJ breast cancer 5, 14 (2019).
Yu, X. et al. Zinc metallochaperones reactivate mutant p53 using an ON/OFF switch mechanism: a new paradigm in cancer therapeutics. Clin. Cancer Res. 24, 4505–4517 (2018).
Zaman, S. et al. Combinatorial therapy of zinc metallochaperones with mutant p53 reactivation and diminished copper binding. Mol. Cancer Ther. 18, 1355–1365 (2019).
Salim, K. Y., Maleki Vareki, S., Danter, W. R. & Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 7, 41363–41379 (2016).
Lindemann, A. et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 25, 5650–5662 (2019).
Bormio Nunes, J. et al. Cancer cell resistance against the clinically investigated thiosemicarbazone COTI-2 is based on formation of intracellular copper complex glutathione adducts and ABCC1-mediated efflux. J. Med. Chem. 63, 13719–13732 (2020).
Synnott, N. C., O’Connell, D., Crown, J. & Duffy, M. J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 179, 47–56 (2020).
Guo, Y., Zhu, X. & Sun, X. COTI-2 induces cell apoptosis in pediatric acute lymphoblastic leukemia via upregulation of miR-203. Bioengineered 11, 201–208 (2020).
Maleki Vareki, S., Salim, K., Danter, W. & Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS One 13, e0191766 (2018).
Foster, B. A., Coffey, H. A., Morin, M. J. & Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 (1999).
Rippin, T. M. et al. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129 (2002).
Wischhusen, J. et al. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 22, 8233–8245 (2003).
Wang, W., Takimoto, R., Rastinejad, F. & El-Deiry, W. S. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol. Cell. Biol. 23, 2171–2181 (2003).
Zache, N. et al. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2, 70–80 (2008).
Bykov, V. J. N. et al. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 280, 30384–30391 (2005).
Bou-Hanna, C. et al. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 359, 211–217 (2015).
Saha, M. et al. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 110, 2224–2231 (2014).
Durairaj, G. et al. Discovery of compounds that reactivate p53 mutants in vitro and in vivo. Cell Chem Biol. 29 (2022).
Kaar, J. L. et al. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 19, 2267–2278 (2010).
Bauer, M. R. et al. Targeting cavity-creating p53 cancer mutations with small-molecule stabilizers: the Y220X paradigm. ACS Chem. Biol. 15, 657–668 (2020).
Basse, N. et al. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56 (2010).
Bykov, V. J. N. & Wiman, K. G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 588, 2622–2627 (2014).
Boeckler, F. M. et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 105, 10360–10365 (2008).
Liu, X. et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044 (2013).
Wilcken, R. et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 134, 6810–6818 (2012).
Joerger, A. C. et al. Exploiting transient protein states for the design of small-molecule stabilizers of mutant p53. Structure 23, 2246–2255 (2015).
Bauer, M. R. et al. Harnessing fluorine-sulfur contacts and multipolar interactions for the design of p53 mutant Y220C rescue drugs. ACS Chem. Biol. 11, 2265–2274 (2016).
Baud, M. G. J. et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 152, 101–114 (2018).
Bauer, M. R. et al. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 11, 2491–2504 (2019).
Raghavan, V., Agrahari, M. & Gowda, D. Virtual screening of p53 mutants reveals Y220S as an additional rescue drug target for PhiKan083 with higher binding characteristics. Comput. Biol. Chem. 80, 398–408 (2019).
Miller, J. et al. Multifunctional compounds for activation of the p53-Y220C mutant in cancer. Chemistry 24, 17734–17742 (2018).
Dong, T. et al. Hybrid molecular dynamics for elucidating cooperativity between halogen bond and water molecules during the interaction of p53-Y220C and the PhiKan5196 complex. Front. Chem. 8, 344 (2020).
Synnott, N. C. et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 414 (2018).
Bauer, M. R., Joerger, A. C. & Fersht, A. R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 113, E5271–E5280 (2016).
Dumble, M. et al. Abstract LB006: PC14586: The first orally bioavailable small molecule reactivator of Y220C mutant p53 in clinical development. Cancer Res 81, LB006–LB006 (2021).
Dumbrava, E. E. et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 40, 3003–3003 (2022).
Wang, G. & Fersht, A. R. First-order rate-determining aggregation mechanism of p53 and its implications. Proc. Natl. Acad. Sci. USA 109, 13590–13595 (2012).
Stenger, J. E. et al. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J. 13, 6011–6020 (1994).
Wang, G. & Fersht, A. R. Propagation of aggregated p53: Cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 112, 2443–2448 (2015).
Levy, C. B. et al. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 43, 60–64 (2011).
Xu, J. et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295 (2011).
Eisenberg, D. & Jucker, M. The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012).
Nelson, R. et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature 435, 773–778 (2005).
Ghosh, S. et al. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry 53, 5995–6010 (2014).
Lei, J. et al. Self-aggregation and coaggregation of the p53 core fragment with its aggregation gatekeeper variant. Phys. Chem. Chem. Phys. 18, 8098–8107 (2016).
Soragni, A. et al. A designed inhibitor of p53 Aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell. 29 (2016).
Lei, J. et al. Molecular dynamics study on the inhibition mechanisms of ReACp53 peptide for p53-R175H mutant aggregation. Phys. Chem. Chem. Phys. 23, 23032–23041 (2021).
Zhang, Y. et al. Therapeutic potential of ReACp53 targeting mutant p53 protein in CRPC. Prostate Cancer Prostatic Dis. 23, 160–171 (2020).
Neal, A. et al. Combining ReACp53 with carboplatin to target high-grade serous ovarian cancers. Cancers (Basel). 13 (2021).
Zhang, Y. et al. Proteomic identification of ERP29 as a key chemoresistant factor activated by the aggregating p53 mutant Arg282Trp. Oncogene 36, 5473–5483 (2017).
Wang, G. & Fersht, A. R. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA 114, E2634–E2643 (2017).
Palanikumar, L. et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 12, 3962 (2021).
Kumar, S. & Hamilton, A. D. α-helix mimetics as modulators of Aβ self-assembly. J. Am. Chem. Soc. 139, 5744–5755 (2017).
Kumar, S. et al. Islet amyloid-induced cell death and bilayer integrity loss share a molecular origin targetable with oligopyridylamide-based α-helical mimetics. Chem. Biol. 22, 369–378 (2015).
Miller, J. J. et al. Bifunctional ligand design for modulating mutant p53 aggregation in cancer. Chem. Sci. 10, 10802–10814 (2019).
Li, Y. & Prives, C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 26, 2220–2225 (2007).
Wiech, M. et al. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One 7, e51426 (2012).
Muller, P. et al. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 27, 3371–3383 (2008).
Tutuska, K. et al. Statin as anti-cancer therapy in autochthonous T-lymphomas expressing stabilized gain-of-function mutant p53 proteins. Cell Death Dis. 11, 274 (2020).
Alexandrova, E. M., Xu, S. & Moll, U. M. Ganetespib synergizes with cyclophosphamide to improve survival of mice with autochthonous tumors in a mutant p53-dependent manner. Cell Death Dis. 8, e2683 (2017).
Cooper, B. et al. Peptides as a platform for targeted therapeutics for cancer: peptide-drug conjugates (PDCs). Chem. Soc. Rev. 50, 1480–1494 (2021).
Saw, P., Xu, X., Kim, S. & Jon, S. Biomedical Applications of a Novel Class of High-Affinity Peptides. Acc. Chem. Res. 54, 3576–3592 (2021).
Friedler, A. et al. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 99, 937–942 (2002).
Friedler, A. et al. Structural distortion of p53 by the mutation R249S and its rescue by a designed peptide: implications for “mutant conformation”. J. Mol. Biol. 336, 187–196 (2004).
Friedler, A., Veprintsev, D., Hansson, L. & Fersht, A. Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J. Biol. Chem. 278, 24108–24112 (2003).
Issaeva, N. et al. Rescue of mutants of the tumor suppressor p53 in cancer cells by a designed peptide. Proc. Natl Acad. Sci. USA 100, 13303–13307 (2003).
Tal, P. et al. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 7, 11817–11837 (2016).
Yamada, T. et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol. Cancer Ther. 8, 2947–2958 (2009).
Taylor, B. N. et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 69, 537–546 (2009).
Yamada, T. et al. Internalization of bacterial redox protein azurin in mammalian cells: entry domain and specificity. Cell. Microbiol. 7, 1418–1431 (2005).
Bizzarri, A. R. et al. Interaction of an anticancer peptide fragment of azurin with p53 and its isolated domains studied by atomic force spectroscopy. Int J. Nanomed. 6, 3011–3019 (2011).
Signorelli, S. et al. Binding of Amphipathic Cell Penetrating Peptide p28 to Wild Type and Mutated p53 as studied by Raman, Atomic Force and Surface Plasmon Resonance spectroscopies. Biochim Biophys. Acta Gen. Subj. 1861, 910–921 (2017).
Yamada, T. et al. p28, a first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 108, 2495–2504 (2013).
Yamada, T., Das Gupta, T. K. & Beattie, C. W. p28, an anionic cell-penetrating peptide, increases the activity of wild type and mutated p53 without altering its conformation. Mol. Pharm. 10, 3375–3383 (2013).
Yamada, T., Das Gupta, T. & Beattie, C. p28-mediated activation of p53 in G2-M phase of the cell cycle enhances the efficacy of DNA damaging and antimitotic chemotherapy. Cancer Res. 76, 2354–2365 (2016).
Warso, M. A. et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 108, 1061–1070 (2013).
Lulla, R. R. et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro Oncol. 18, 1319–1325 (2016).
Jia, L. et al. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother. Pharmacol. 68, 513–524 (2011).
Hamers-Casterman, C. et al. Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 (1993).
Hu, X. et al. A novel nanobody-heavy chain antibody against Angiopoietin-like protein 3 reduces plasma lipids and relieves nonalcoholic fatty liver disease. J. Nanobiotechnology 20, 237 (2022).
Zeng, Z. et al. Activatable cancer sono-immunotherapy using semiconducting polymer nanobodies. Adv Mater. e2203246, (2022).
Hong, J. et al. Dromedary camel nanobodies broadly neutralize SARS-CoV-2 variants. Proc. Natl. Acad. Sci. USA 119, e2201433119 (2022).
Bethuyne, J. et al. A nanobody modulates the p53 transcriptional program without perturbing its functional architecture. Nucleic Acids Res. 42, 12928–12938 (2014).
Danilova, L. et al. The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: a sensitive platform for monitoring antitumor immunity. Cancer Immunol. Res 6, 888–899 (2018).
Hsiue, E. H.-C. et al. Targeting a neoantigen derived from a common mutation. Science. 371 (2021).
Malekzadeh, P. et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest 129, 1109–1114 (2019).
Nikolova, P. V. et al. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 19, 370–378 (2000).
Suad, O. et al. Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations. J. Mol. Biol. 385, 249–265 (2009).
Otsuka, K. et al. The screening of the second-site suppressor mutations of the common p53 mutants. Int. J. Cancer 121, 559–566 (2007).
Joerger, A. C., Allen, M. D. & Fersht, A. R. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 279, 1291–1296 (2004).
Odell, A. F. et al. A novel p53 mutant found in iatrogenic urothelial cancers is dysfunctional and can be rescued by a second-site global suppressor mutation. J. Biol. Chem. 288, 16704–16714 (2013).
Eldar, A. et al. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res 41, 8748–8759 (2013).
Brachmann, R. K. et al. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 17, 1847–1859 (1998).
Joerger, A. C. & Fersht, A. R. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242 (2007).
Nikolova, P. V., Henckel, J., Lane, D. P. & Fersht, A. R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. USA 95, 14675–14680 (1998).
Demir, Ö. et al. Ensemble-based computational approach discriminates functional activity of p53 cancer and rescue mutants. PLoS Comput. Biol. 7, e1002238 (2011).
Merabet, A. et al. Mutants of the tumour suppressor p53 L1 loop as second-site suppressors for restoring DNA binding to oncogenic p53 mutations: structural and biochemical insights. Biochem. J. 427, 225–236 (2010).
Chattopadhyay, G. et al. Mechanistic insights into global suppressors of protein folding defects. PLoS Genet 18, e1010334 (2022).
Kamaraj, B. & Bogaerts, A. Structure and function of p53-DNA complexes with inactivation and rescue mutations: a molecular dynamics simulation study. PLoS One 10, e0134638 (2015).
Ramani, R. G. & Jacob, S. G. Prediction of cancer rescue p53 mutants in silico using Naïve Bayes learning methodology. Protein Pept. Lett. 20, 1280–1291 (2013).
Wallentine, B. D. et al. Structures of oncogenic, suppressor and rescued p53 core-domain variants: mechanisms of mutant p53 rescue. Acta Crystallogr 69, 2146–2156 (2013).
Joerger, A. C. et al. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 280, 16030–16037 (2005).
Viadiu, H., Fronza, G. & Inga, A. Structural studies on mechanisms to activate mutant p53. Subcell. Biochem. 85, 119–132 (2014).
Selivanova, G. et al. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 3, 632–638 (1997).
Selivanova, G. et al. Reactivation of mutant p53 through interaction of a C-terminal peptide with the core domain. Mol. Cell. Biol. 19, 3395–3402 (1999).
Kaldmäe, M. et al. A “spindle and thread” mechanism unblocks p53 translation by modulating N-terminal disorder. Structure 30, (2022).
Wang, T.-H., Li, W.-T., Yu, S.-H. & Au, L.-C. The use of 10-23 DNAzyme to selectively destroy the allele of mRNA with a unique purine-pyrimidine dinucleotide. Oligonucleotides 18, 295–299 (2008).
Iyer, S. V. et al. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 7, 5401–5415 (2016).
Hoffman-Luca, C. G. et al. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin. Cancer Res 21, 2558–2568 (2015).
Decaudin, D. et al. Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur. J. Cancer. 126 (2020).
Han, X. et al. Nonsense-mediated mRNA decay: a ‘nonsense’ pathway makes sense in stem cell biology. Nucleic Acids Res. 46, 1038–1051 (2018).
Pan, Y. et al. RNA dysregulation: an expanding source of cancer immunotherapy targets. Trends Pharmacol. Sci. 42, 268–282 (2021).
Hassin, O. & Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug. Discov. (2022).
Floquet, C., Deforges, J., Rousset, J.-P. & Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res 39, 3350–3362 (2011).
Bidou, L. et al. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 14, 378–388 (2017).
Gudikote, J. P. et al. Inhibition of nonsense-mediated decay rescues p53β/γ isoform expression and activates the p53 pathway in MDM2-overexpressing and select p53-mutant cancers. J. Biol. Chem. 297, 101163 (2021).
Lindeboom, R. G. H., Vermeulen, M., Lehner, B. & Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651 (2019).
Khajavi, M., Inoue, K. & Lupski, J. R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 14, 1074–1081 (2006).
Chen, J. et al. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 23, 278–290 (2009).
Bourdon, J.-C. et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 (2005).
Senturk, S. et al. p53Ψ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc. Natl Acad. Sci. USA 111, E3287–E3296 (2014).
Khoury, M. P. & Bourdon, J.-C. p53 isoforms: an intracellular microprocessor? Genes Cancer 2, 453–465 (2011).
Bourdon, J. C. p53 isoforms change p53 paradigm. Mol. Cell Oncol. 1, e969136 (2014).
Marcel, V. et al. Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 584, 4463–4468 (2010).
Mondal, A. M. et al. Δ133p53α, a natural p53 isoform, contributes to conditional reprogramming and long-term proliferation of primary epithelial cells. Cell Death Dis. 9, 750 (2018).
Burslem, G. M. & Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020).
Li, Z. et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 575, 203–209 (2019).
Takahashi, D. et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell. 76 (2019).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Chapeau, E. A. et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf mouse model. Proc. Natl. Acad. Sci. USA 114, 3151–3156 (2017).
Wang, S. & Chen, F.-E. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem. 236, 114334 (2022).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Vlatković, N., Crawford, K., Rubbi, C. P. & Boyd, M. T. Tissue-specific therapeutic targeting of p53 in cancer: one size does not fit all. Curr. Pharm. Des. 17, 618–630 (2011).
Gallo, D. et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 604, 749–756 (2022).
Rosenblum, D. et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci Adv. 6 (2020).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Awad, M. M. et al. Acquired resistance to KRAS inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 81570537, 81974074, 82172654, 82273496 and 31900880), Hunan Provincial Science and Technology Department (2018RS3026 and 2021RC4012) and Central South University Innovation-Driven Research Programme (2023CXQD077).
Author information
Authors and Affiliations
Contributions
All authors were involved in the design of the review, and in writing the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Guo, M., Wei, H. et al. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Sig Transduct Target Ther 8, 92 (2023). https://doi.org/10.1038/s41392-023-01347-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01347-1
This article is cited by
-
p53/E2F7 axis promotes temozolomide chemoresistance in glioblastoma multiforme
BMC Cancer (2024)
-
LASS2 suppresses metastasis in multiple cancers by regulating the ferroptosis signalling pathway through interaction with TFRC
Cancer Cell International (2024)
-
Structure of the p53 degradation complex from HPV16
Nature Communications (2024)
-
Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases
Signal Transduction and Targeted Therapy (2024)
-
Identifying SLC2A6 as the novel protective factor in breast cancer by TP53-related genes affecting M1 macrophage infiltration
Apoptosis (2024)
