
Abstract
Having a hypoxic microenvironment is a common and salient feature of most solid tumors. Hypoxia has a profound effect on the biological behavior and malignant phenotype of cancer cells, mediates the effects of cancer chemotherapy, radiotherapy, and immunotherapy through complex mechanisms, and is closely associated with poor prognosis in various cancer patients. Accumulating studies have demonstrated that through normalization of the tumor vasculature, nanoparticle carriers and biocarriers can effectively increase the oxygen concentration in the tumor microenvironment, improve drug delivery and the efficacy of radiotherapy. They also increase infiltration of innate and adaptive anti-tumor immune cells to enhance the efficacy of immunotherapy. Furthermore, drugs targeting key genes associated with hypoxia, including hypoxia tracers, hypoxia-activated prodrugs, and drugs targeting hypoxia-inducible factors and downstream targets, can be used for visualization and quantitative analysis of tumor hypoxia and antitumor activity. However, the relationship between hypoxia and cancer is an area of research that requires further exploration. Here, we investigated the potential factors in the development of hypoxia in cancer, changes in signaling pathways that occur in cancer cells to adapt to hypoxic environments, the mechanisms of hypoxia-induced cancer immune tolerance, chemotherapeutic tolerance, and enhanced radiation tolerance, as well as the insights and applications of hypoxia in cancer therapy.
Similar content being viewed by others


Introduction
Cancer occurrence is markedly associated with increasing age, as patients aged over 60 years are more than twice as likely to develop invasive cancers than younger patients. The World Health Organization (WHO) estimates that by 2050, the proportion of the world population aged over 60 years will have increased from 12 to 22%, totaling over 2 billion individuals.1 In the United States, cancer is the second leading cause of death after heart disease. In humans aged over 60 years, cancer is the leading cause of death.2 Due to the aging of the global population, cancer has become a global public health issue, with a massive economic burden and complex treatment challenges. The distribution of oxygen partial pressure in tumor tissues is of particular interest to radiologists because the radiosensitivity of tumor tissues depends on the tissue O2 tension. Tumor cells living under adequate hypoxic or hypoxic conditions are relatively resistant to radiation.3
In 1953, Gray et al. found that well-oxygenated tumor cells responded trice as well to radiotherapy than hypoxic cells.4 Tumor hypoxia was first proposed in 1955 by Thomlinson et al. in a study of tumor tissues from patients with lung cancer, and scientists have confirmed, through over 60 years of clinical and experimental evidence, that the hypoxic state is a widespread trait in a variety of solid tumors. The expression of key genes such as hypoxia-inducible factors (HIFs) and their various subunits, as well as the molecular regulatory mechanisms, were also explored under hypoxic conditions.5,6,7,8,9,10,11,12,13,14 William Kaelin, Peter Ratcliffe, and Gregg Semenza have been awarded the 2019 Nobel Prize in Physiology or Medicine for the contributions of several scientists who have discovered how human and animal cells sense and adapt to oxygen supply (Fig. 1).15 Hypoxia is present in 90% of solid tumors, which is considered a hallmark of cancer.16,17 It is difficult to determine the hypoxic state in tumors due to variations in oxygen content between tissues, as well as differences in tumor size and measurement methods, and tissue oxygenation is highly variable, also within the same organ.18 However, the available results indicate that the measurement of tumor partial pressure of oxygen (pO2) in patients (polarographic technique) has demonstrated the presence of low values (<10 mmHg) in several different tumor types, including pancreatic cancer, head and neck tumors, breast cancer, cervical cancer, and melanoma.19,20,21,22 Intra-tumor hypoxia is linked to decreased disease-free survival outcomes in several cancers including prostate, cervical cancer, and head and neck squamous cell carcinoma (HNSCC).23,24,25,26 The hypoxic environment alters the expression levels of genes that modulate metabolism and other processes. Moreover, hypoxic signaling interacts with other cellular pathways to alter cancer cell malignant behaviors and is closely associated with cancer cell proliferation, migration, invasion and angiogenesis, and affects cancer treatment outcomes.27 This article focuses on the unique hypoxic microenvironment in cancer, the possible mechanisms by which cells undergo transformation and malignancy, and the potential applications of these aspects.

A historical and chronological figure of major events in tumor hypoxia research
Factors contributing to hypoxia in cancer
The vasculature ensures the presence of tissue oxygen and energy substances. Vasculature endothelial cells (ECs) of cancer tissues or pre-neoplastic tissues are exposed to harmful substances, including various high-risk carcinogenic factors, such as drugs, carcinogens, pathogenic microorganisms, and the uniquely acidified tumor microenvironment (TME), which damage the shapes of ECs (for example, edema), resulting in dysregulated functions of cancer vasculature.28 Cancer cells are characterized by high proliferative rates and active metabolism; they also consume high amounts of energy to support their increased cell proliferation and growth rates.29 When metabolic oxygen demands exceed supply, the oxygen-deficient areas of cancer cells are exacerbated and they are change their metabolism.30 Abnormal vascular structures and patterns that are due to dysregulated angiogenesis contribute highly to hypoxia. Although the expression of erythropoietin (EPO) and angiogenic factors are increased under hypoxic conditions, which promote the proliferation of vascular ECs, the arrangement is disorganized and non-functional vessels form.31,32,33,34 Cancer rapidly grows such that the cancerous area is often deficient of vasculature and becomes hypoxic.35 In the TME, chronic hypoxia occurs as the distance of cancer cells from blood vessels increases and O2 diffusion decreases, which has been further validated in a mouse model of breast cancer.36 Hypoxia can occur in tissues more than 100–200 µM from a functional blood supply, a phenomenon that is prevalent in solid tumors.37 Additionally, using a model of rat brain gliom, Julien et al. found that the peritumor vasculature is vaso-responsive to hypoxia through alpha-smooth muscle actin, which can further exacerbate hypoxia in areas of tumor tissue with principle blood vessels.38 Some blood vessels become abnormal and malfunction, resulting in hypoxia.39 Non-cancer components and functions are greatly altered, including the activation and proliferation of stromal cells (for example, stellate cells and cancer-associated fibroblasts) and increased stromal components (for example, fibrin),40,41 leading to remodeling of cancer morphology, such as vascular compression. This can result in impaired circulation and inadequate oxygen supply, further leading to thrombosis and increased tissue hypoxia.42 Physiological effects of various factors, such as magnesium, change with the changing hypoxic environment. Under normoxic conditions, magnesium stimulates angiogenesis, whereas under hypoxic conditions, Mg inhibits angiogenesis.43 The above factors can cause hypoxia in the TME (Fig. 2).

Potential factors contributing to tumor hypoxia. Carcinogenic factors, such as drug, carcinogen, and microbiota dysbiosis, impair EC shape and function in the vascular system. TME is remodeled by tumor cells, stromal cells and stromal components (e.g., fibrin), resulting in vascular deformation due to pressure. High metabolism in cancer cells, such as increased nucleic acid synthesis and increased protein anabolism, leads to relative hypoxia. Dysregulated proliferation and alignment of vascular ECs result in the formation of non-functional blood vessels. With the increased distance between tumor cells and blood vessels, O2 diffusion decreases and leads to hypoxia
Hypoxic factors are prevalent in most solid tumors. Some tumors contain transient hypoxic cells, others contain chronic hypoxic cells, and others contain both components. This may include three different cell populations. The first type is chronically hypoxic cells. If left in situ, the cells die. These “doomed” cells survive alone after the necessary excision in the cell survival assay and does not affect the response of the tumor when left in situ, which is considered the main cause of cell necrosis in the central region of solid tumors. The second type is the chronically hypoxic cells that are viable if left in situ. These cancer cells are stimulated by hypoxia to promote proliferation, alter gene expression, and enhance cellular drug resistance and radioresistance.16,44,45,46,47,48,49 The third type is transient hypoxic cells, which are expressed closer to functional blood vessels, where the duration of hypoxia is short. Based on current evidence, tumor cells adapt to hypoxia by altering their signaling pathways. Hypoxia promotes malignant behavior of cancer cells, including proliferation, migration, infestation and epithelial-mesenchymal transition (EMT), and enhances immunotherapy, chemotherapy, and radiotherapy tolerance.
Hypoxia and carcinogenesis
Hypoxia and genomic damage
Hypoxia damages the cellular genome and drives carcinogenesis. In in vitro and in vivo hypoxic cancer models, gene mutation frequencies of hypoxic cancer cells were increased by 2- to 5-fold.50,51,52 In tissue culture and animal models, gene amplification and mutations were associated with induction of DNA strand breaks during hypoxia, including DNA double-strand break (DSB) and single-strand break (SSB).53,54,55 In addition, hypoxic conditions (<5%) could improve the efficiency of induced pluripotent stem cell (iPSC) generation from mouse and human somatic cells.56 The hypoxic signaling microenvironment maintains stem cell self-renewal by facilitating the reprogramming process.57,58,59 The extrapolation of these iPSC studies provides insight into cancer stem cell (CSC), which can also exist in hypoxic ecological niches.60 Hypoxia is known to promote and maintain CSC phenotypes.61 CSCs are thought to have the potential to form tumor that will develop into cancer, especially when they metastasize with the cancer and will give rise to novel sources of cancer.60 Both mutation due to cellular genomic damage and the maintenance of CSCs are closely related to the production of large amounts of reactive oxygen species (ROS) under hypoxic conditions.62,63 The aforementioned mutations and breaks stimulate expression of oncogenes, thereby inducing the formation of cancer cell variants that have the potential to metastasize and grow.64
Hypoxia and genetic repair
Hypoxia activates ataxia telangiectasia mutated (ATM) and ATM and RAD3-related (ATR) checkpoints. ATM and ATR belong to the phosphatidylinositol 3-kinase-like protein kinase (PIKK) family and are the main members of the DNA damage checkpoints. They are activated by different types of DNA damage and regulate cell cycle checkpoints by phosphorylating their corresponding downstream proteins (CHK1 and CHK2). The specificities and functionalities of ATM and ATR during DNA damage differ, with ATM being mainly involved in DNA DSB repair. After hypoxia-mediated DNA double-strand breaks, the MRE11-RAD50-NBS1 (MRN) complex activates ATM and is autophosphorylated at serine 367 (ser367), serine 1893 (ser1893), serine 1981 (ser1981), and serine 2996 (ser2996), thereby inducing MRE11-RAD50-NBS1 (MRN) complex-associated recruitment of various complex phosphorylation cascades, such as p53 (cancer suppressor), CHK1, and CHK2, to the DNA DSB sites. These effects can be blocked by the inhibition of CDK2 activity. During G1/S or G2/M cell cycle progression, cells have more time to repair DNA damage before entering mitosis. ATR kinase phosphorylates p53 and CHK1 under extreme hypoxia (oxygen concentration <0.02%).65 Once ATR is activated, it phosphorylates and inactivates CDC25 by phosphorylating CHK1 and CHK2, thereby resulting in failure to activate CDK2. These lesions can block G1/S or G2/M cell cycle progression by phosphorylating CDK1.65,66,67 In glioma mice models, inactivation of the ATM/CHK2/p53 pathways promoted cancer formation.68 Hypoxia cannot induce G2 arrest in CHK2-deficient cells, but these cells can undergo apoptosis under hypoxia.67,69 Persistent hypoxia enhances DNA misreplication and DNA strand breaks, resulting in mutant cell phenotypes.70 Non-homologous end joining (NHEJ) and homologous recombination (HR) regulate the repair of human DNA DSB.71,72,73,74 DNA DSB are the most severe and extensive types of damage; HR can accurately repair these damages, especially in the S/G2 cell cycle stage. NHEJ is the simplest mechanism of DNA DSB repair and can act in all cell cycle phases, except for the M phase. In hypoxic conditions, HR repairs the lesions less frequently, whereas NHEJ activities are unaffected.75,76 The mismatch repair (MMR) pathway can also be deregulated under hypoxic conditions. MMR is a DNA repair pathway that targets replication-related errors and primarily functions to correct the misintegration of nucleotides during DNA synthesis, thereby preventing permanent DNA damage in dividing cells. Hypoxia decreases the expression of MLH1 and MSH2, which induces mutations and dinucleotide repeat instability. These pathways may lead to sustained damage to intracellular RNA, resulting in cell transformation, including carcinogenesis.77,78
Hypoxia signaling pathways and cancer cells
HIFs and cancer cells
To survive under hypoxic conditions, cancer cells reprogram their metabolism, protein synthesis, and cell cycle progression through the synergy of transcription factors.79 One of the main reasons why cancer cells can survive in hypoxic environments is the activation of HIFs, which reprogram metabolism, protein synthesis, and cell cycle progression.79,80 The HIF family has two distinct subunits: α (HIF-1α, HIF-2α, and HIF-3α) and β (HIF-1β). HIF-1α is widely expressed in all body tissues, whereas HIF-2α and HIF-3α only occur in specific tissues.27 HIF-1α is an oxygen-unstable protein that becomes stable in response to hypoxia, iron chelators, and divalent cations.81,82 In cell culture under hypoxic conditions, HIF-1α mRNA levels did not change, but HIF-1α protein levels increased.83 HIF-1β is constitutively expressed in mammalian cells under normoxic conditions.84 Owing to the presence of the oxygen-dependent proline hydroxylase family (PHD), under sufficient oxygen conditions, the HIF-α protein is hydroxylated and interacts with von Hippel-Lindau tumor suppressor protein (pVHL) to promote HIF-1α ubiquitin-proteasomal degradation.85,86,87 Ectopic expression of PHD1 inhibited HIF-1α and suppressed tumor growth.88 However, under hypoxic conditions, enzymatic activity of PHD is inhibited, preventing HIF-α hydroxylation and ubiquitin-mediated proteasomal degradation, leading to abnormal accumulation of HIF-α in cells. Significantly elevated mRNA of HIFs was detected after 1 h at O2 concentrations <10%.89 In addition, high expression of the CSN subunit CSN5 stabilizes HIF-1α aerobically by inhibiting HIF-1α prolyl-564 hydroxylation.90 Also under normoxic conditions, elevated intracellular ROS induced sustained expression of HIF-1α protein.91 The accumulated HIF-1α binds to HIF-1β and enters the nucleus to bind the hypoxia response element (HRE) in the promoter region of the target gene to reduce cellular oxygen consumption.92,93 HIF-3α exerts the opposite effects in cancer cells by impairing angiogenesis, proliferation, and metabolism.94 Biologically, HIF is regulated by multiple signaling pathways, including the PI3K-mTOR signaling pathway;95,96,97,98 JAK-STAT3 signaling pathway;99 NF-κB pathway;100,101 mitogen-activated protein kinase (MAPK) pathway;102,103 Wnt/β-catenin pathway;104 Notch pathway;105 cancer suppressor gene deletion, such as p53,106 phosphatase, and tensin homolog (PTEN);107 and IDH1-R132H-FAT1-ROS-HIF-1α signaling pathway (Fig. 3).108 Mechanistically, HIF regulates cancer cell growth by regulating genes encoding enzymes that hydrolyze sugars, angiogenic signaling genes, and apoptosis/stress response genes.109

Biological changes in cancer cells adapt to hypoxia. Hypoxia promotes carcinogenesis by inducing DNA strand breaks, including DNA DSB and SSB, and by weakening DNA repair pathways, such as HR and MMR. HIF-1α is upgraded by PI3K-mTOR, JAK-STAT3, NF-κB, MAPK, Wnt/β-catenin, and Notch pathway. Deletion of tumor suppressor genes, such as p53, PTEN, and ROS production, also contributes to the upregulation of HIF-1α. The loss of pVHL function under hypoxic conditions indirectly leads to HIF-1α accumulation. HIF-1α dimerizes with HIF-1β and enters the nucleus to bind to HRE, which regulates various downstream target genes (Table 1) to promote cancer cell proliferation, migration, invasion, EMT and angiogenesis
Hypoxia and cancer cell behaviors
Among various cancers, patients whose primary cancers are hypoxic at diagnosis are more likely to have local recurrence and metastatic site recurrence, regardless of whether the initial treatment is surgery or radiation therapy. Cancer patients with conditions such as anemia or chronic obstructive pulmonary disease tend to have poorer prognostic outcomes and may have cancer-related hypoxia, at least in part, owing to increased propensity to develop metastatic disease.26,110 Young et al. reported that exposure of cancer cells to a hypoxic environment in vitro or in vivo enhanced spontaneous metastasis.111,112 Similar results were obtained in mouse models of fibrosarcoma and cervical cancer.53,113 To an extent, hypoxic conditions eliminate tumor cells that are sensitive to hypoxia, and the surviving tumor cells adapt to hypoxia through their own molecular transformation.114 Several early experiments have supported that hypoxia-induced increases in HIF-1α expression can drive the metastatic phenotype by upregulating genes involved in the metastatic cascade, such as urokinase-type plasminogen activator receptor (uPAR), matrix metalloproteinase 1 (MMP1), chemokine receptor 4 (CXCR4), osteopontin (OPN), known as secreted phosphoprotein 1 (SPP1), lysine oxidase (LOX), interleukin 8 (IL-8), and vascular endothelial growth factor (VEGF).115,116,117,118,119,120,121,122,123 Hypoxia-exposed cancer cells are selective for the loss of p53 functions or increased expressions of the p53 negative regulator (MDM2), leading to increased resistance to apoptosis and increased metastasis.124,125 Other experiments did not find correlation between hypoxia and p53.126 HIF-1α binds and activates the MAX interactor-1 (MXI1), a repressor of the c-MYC transcriptional activity, and reduces the expressions of c-MYC, a factor that encodes mitochondrial DNA replication and promotes mitochondrial biogenesis,127,128 which inhibits cancer cell mitochondrial biogenesis and cellular oxygen consumption, resulting in cancer growth and survival in a hypoxic environment.129 Unfortunately, MYC overexpression occurs in 70% of human cancers.130 HIF-2α promotes the expressions of the MYC and E2F target genes, which are involved in lipoprotein metabolism and ribosome biosynthesis.131 Conversely, MYC maintains cancer stem cell self-renewal properties by selectively binding the promoter and activating the HIF-2α stemness pathway.132 Both HIF-1α and MYC can enhance the glycolytic pathway and drive cancer proliferation and progression.79,133 Under hypoxic conditions, HIF-1α directly and positively regulates ephrin A3 (EFNA3) expressions. Ephrin type-A receptor 2 (EphA2), a key functional mediator downstream of EFNA3, promotes sterol regulatory element binding protein (SREBP1) maturation, which drives the self-renewal, proliferation, and migration of hepatocellular carcinoma (HCC) cells.35 Under the effects of HIF-1α, prostate intraepithelial neoplasia (PIN) cells highly express transglutaminase 2 (TGM2) and exhibit impaired androgen signaling, enhancing malignant progression.23 In addition, some factors, such as Orai1;134 phospholipase D2 (PLD2);135 annexin A3 (ANXA3);136 CXCR4;137 cysteine-rich protein 2 (CSRP2);138 hematopoietic pre-B cell leukemia transcription factor-interacting protein (HPIP);139 twist family bHLH transcription factor 2 (TWIST2);140 and non-coding RNAs, such as long-stranded non-coding RNA (lncRNA) PVT1,141 lncRNA-GAPLINC,142 and LncRNA-MTA2TR;143 and microRNAs, such as miR-525-5p,144 miR-301a-3p,145,146 and miR-141-3p,147 play important roles in regulating hypoxia-induced cancer cell proliferation, migration, invasion, and angiogenesis in the TME under hypoxic conditions (Table 1).
Hypoxia and cancer cell metabolism
Hypoxia and glycolysis
The majority of cancer cells increase glucose uptake and rely on glycolysis through a phenomenon known as the “Warburg effect”.30,148,149 The “Warburg effect,” activated by MYC and HIF-1 in response to growth factors and hypoxia, is aerobic glycolysis with the purpose of meeting the nutrient and energy requirements for rapid genome replication. HIF-1α is the main transcription factor that promotes Warburg-like metabolism.131,133,150 HIF-1α stimulates various enzymes such as glycolysis regulator phosphoglycerate mutase 1 (PGAM1), pyruvate kinase M (PKM), recombinant phosphoglycerate kinase 1 (PGK1), lactate dehydrogenase A (LDHA), lactate dehydrogenase C (LDHC), and lactate dehydrogenase-5 (LDH-5) to induce anaerobic metabolic shifts that lead to energy production.151,152,153,154,155,156 HIF-2α promotes MYC target gene expression and enhances constitutive expression of LDHA.131,157 In addition, HIF-1α inactivates pyruvate dehydrogenase (PDH) by activating pyruvate dehydrogenase kinase 1 (PDK1), which in turn fails to convert pyruvate to acetyl-CoA, preventing the entry of pyruvate into the tricarboxylic acid (TCA) cycle. This leads to lactate accumulation that increases intracellular adenosine triphosphate (ATP) levels and reduces hypoxic ROS production, thus rescuing these cells from hypoxia-induced apoptosis.158,159,160
Lactate and H+ generated by glycolysis cross cell membranes through monocarboxylate transport proteins (that is, MCT1/4), sodium hydrogen (Na+/H+) exchanger (NHE) isoform 1 (NEH1), and carbonic anhydrase 9 (CAR9), contributing to cellular pH homeostasis and driving cancer proliferation and progression.79,133,161,162,163 Interestingly, cancer cells relying on MCT1 can autonomously consume lactate, predominating as a carbon source for the TCA cycle.164 Pyruvate is converted from glycolysis to lactate, while lactate is used as a respiratory fuel to support the energy and synthetic functions of the TCA cycle, which is significant for the proliferation of cancer cells.164
Hypoxia and lipid metabolism
Lipid metabolism confers aggressiveness to malignant tumors by promoting membrane formation, energy storage, and production of signaling molecules, as well as by providing an important source of ATP production through fatty acid oxidation (FAO).165 Lipids are composed of triglycerides and lipoids such as phospholipids, cholesterol, and cholesteryl esters. Lipid metabolism involves lipid synthesis, storage, and degradation. Fatty acid (FA) are essential for lipid biosynthesis and are dependent on the activities of fatty acid synthase (FASN), adenosine triphosphate citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturases (SCD). Endogenous FA biogenesis constitutes an oncogenic stimulus that drives malignant tumor progression. HIF-1 significantly upregulates sterol regulatory element-binding protein (SREBP)-1, a major transcriptional regulator of the FASN gene, which in turn promotes FASN expression.166 Activation of FASN under hypoxic stress promotes de novo lipid synthesis and cell survival.167,168 ACLY is the main enzyme responsible for the production of acetyl-CoA in the cytosol in most tissues, and its product, acetyl-CoA, is used to provide a variety of biosynthetic pathways, including lipid synthesis and cholesterol synthesis. ACLY is responsible for catalyzing the conversion of citrate and CoA to acetyl-CoA and oxaloacetate.169 Studies have suggested that miR-27 and miR-195 are also upregulated in hypoxia-induced cardiomyocytes, suppressing the expression of ACLY and ACC.170,171 The mechanism of ACLY regulation in tumor cells under hypoxic conditions is still unclear. ACC (including ACC1 and ACC2) is the rate-limiting enzyme in the FA synthesis pathway.165 Under hypoxic conditions, the inhibition of ACC1 and ACLY increases the level of α-ketoglutarate by decreasing the level and activity of ETV4 to prevent hypoxia-induced apoptosis.172 ACC2 is hydroxylated by PHD3, which inhibits FAO. Under hypoxic conditions, PHD3 loss reduces ACC2 hydroxylation and promotes FAO to provide energy.173,174 SCD is a membrane protein of the endoplasmic reticulum that catalyzes the formation of monounsaturated FA (MUFA) from saturated FA, promotes tumor progression, and is associated with tumor recurrence and poor prognosis.175,176,177 The main products of SCD are palmitoleic and oleic acids, providing key substrates for the production of complex lipids such as triglycerides, phospholipids, and cholesterol esters. Intermittently hypoxic mouse hepatocytes upregulate SCD1 mRNA and protein by increasing SREBP-1 and serum monounsaturated FAs.178 HIF-1/2α promotes the progression of clear cell renal cell carcinoma by inducing SCD1 expression.179,180
Glutamine is the most abundant amino acid in blood and has been identified as necessary to promote mitochondrial metabolism in rapidly dividing cancer cells.181 HIF-1 activation leads to a significant decrease in the activity of the mitochondrial enzyme complex α-ketoglutarate dehydrogenase (αKGDH), which inhibits the oxidation of α-ketoglutarate (α-KG) as a product of the metabolic conversion of glutamine to succinate, its reductive carboxylation to isocitrate by isocitrate dehydrogenase (IDH), and then oxidation to citrate.182,183 HIF promotes glutamine-derived citrate conversion to cytoplasmic acetyl-CoA, which increases FA synthesis.184
Interestingly, the lipid catabolic metabolism also contributes to cancer metastasis. Under hypoxic conditions, the main enzymes associated with lipid catabolism in tumor cells are phospholipase A2 (PLA2), phospholipase D (PLD), and carnitine palmitoyltransferase 1 (CPT1).165 PLA2 catalyzes the hydrolysis of glycerophospholipid (GPL) to produce lysophospholipid (Lyso-PL).185 Lysophosphatidic acid stimulates PLA2 phosphorylation in a HIF-1α-dependent manner, promoting ovarian cancer cell metastasis in vivo.186 PLD hydrolyzes phosphatidylcholine (PC) to produce phosphatidic acid (PA).187 Activation of the PLD1/AKT pathway increases proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) in HCC.188 HIF-1α up-regulates the expression of PLD2 in colon cancer cells under hypoxic conditions.135 In turn, PLD regulates HIF-1α at the translational level in a vHL non-dependent manner in renal cancer cells.189,190 Prostate cancer cells with CPT1A overexpression showed enhanced proliferation, stemness, and tumor growth compared to controls under hypoxic conditions, mildly affecting the angiogenic response.191 Breast cancer cells expressing CPT1C showed increased FAO, ATP production, and resistance to glucose deprivation or hypoxia.192 However, experimental results have shown that HIF-1α and HIF-2α inhibit CPT1A expression in clear cell renal cell carcinoma, reduce FAs transport into mitochondria, and result in FAs being stored in lipid droplets. Compared with normal tissues, CPT1A expression and activity were reduced in clear cell renal cell carcinoma patient samples.193 Similar results were observed in gastric cancer tissue.194 Cancer cells optimize their requirements for rapid growth and aggressive progression by fine-tuning the lipid anabolic/catabolic switch. However, the dominant role of this fine-tuning mechanism remains unclear. Glucose and lipid metabolism in cancer cells under hypoxic conditions is showed in Fig. 4.

Glucose and lipid metabolism in cancer cells under hypoxic conditions. Glucose is taken up by cancer cells via GLUT1 and glycolysed to pyruvate via PKM, PGK1 and PGAM1. Hypoxic cancer cells promote pyruvate glycolysis to lactate by upregulating LDHA, LDHC and LDH-5, and the lactate produced is excreted outside the cell via MCT1/4. In addition, HIF-1α inactivates PDH by activating PDK1, which in turn fails to convert pyruvate to acetyl-CoA, preventing the entry of pyruvate into the TCA cycle. Cytoplasmic citrate is catalyzed by ALCY to acetyl-CoA, and acetyl-CoA catalyzed by ACC to malonyl-CoA. Acetyl-CoA and malonyl-CoA are catalyzed to FA via FASN upregulated by SREBP-1. SCD1 upregulated by SREBP-1 catalyzes the formation of MUFA from saturated FA. PHD3 loss reduces ACC2 hydroxylation and promotes FAO to provide energy. α-KG as a product of glutamine is reduced and carboxylated to isocitrate by IDH, and then oxidation to citrate. PLD hydrolyzes PC to produce PA. PLA2 catalyzes the hydrolysis of GPL to produce Lyso-PL
Hypoxia and angiogenesis
In the early 1970s, Folkman et al. popularized the concept of tumor angiogenesis, presenting that growing tumor cells must replenish their own blood supply to maintain oxygen and nutrients.195 The accumulated experimental results have shown that hypoxia favors EC proliferation and migration. Deletion of p53 in cancer cells increases HIF-1α levels and enhances transcriptional activation of HIF-1-dependent VEGF and erythropoietin (EPO) in response to hypoxia, thus promoting EC proliferation, migration, and angiogenesis.96,106,196,197,198,199 Hypoxia-induced E74-like ETS transcription factor 3 (ELF3) mediates increased secretions of insulin like growth factor (IGF1) and VEGF to promote EC proliferation, migration, and angiogenesis.200 HIF-1α mediates β-adrenergic receptor pro-angiogenesis.39,201,202,203 In addition, hypoxia stimulates the production of hyaluronic acid (a major component of the vascular basement membrane) and hyaluronidase activity, thus possibly promoting angiogenesis as a compensatory mechanism for hypoxia.204 However, tumor angiogenesis is not necessarily equivalent to tumor blood supply, as the discontinuous basement membrane of immature neovascularization allows for plasma and protein extravasation, further elevation of intra-tumor interstitial pressure, persistent vascular collapse, and poor nutrient delivery.205 In addition, some tumors exhibit an inability to maintain vascular survival, a condition that explains the well-formed, invasive peripheral and centrally necrotic hypoxic regions found in several highly angiogenic tumors.206 Therefore, in most cancers, despite the high vascular density, the neointima that forms are typically twisted and dysregulated, not functional for blood transport, and less efficient in oxygen and nutrient transport as well as drug delivery.31,32,33,34,203 Traditional anti-angiogenic strategies have attempted to reduce the vascular supply to the tumor, but their success has been limited by insufficient efficacy or the development of drug resistance. Normalization of vascular abnormalities may still be beneficial for tumor treatment with the available therapies.31
Hypoxia and immune tolerance
Cancer immunotherapy has emerged as a promising strategy for the treatment of various cancers by stimulating the immune system of the patient.207 Programmed death receptor 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA4) blocking antibodies, which are drugs approved by the Food and Drug Administration (FDA), have shown promising results in clinical trials for the treatment of various cancers. For some cancers, including breast, pancreatic, colorectal, and prostate cancers, a high proportion of non-responders has been reported, one of the main causes of which is hypoxic stress.208 Hypoxia-induced HIF-1α can promote programmed death ligand-1 (PD-L1) expression in cancer cells and suppress immune effects.209 Moreover, hypoxia plays a central role in cancer progression and resistance to therapy by promoting various changes in the biology of stromal cells in the TME, including immune cells. The main mediators of transcriptional hypoxic responses are HIF-1α and HIF-2α, which induce gene transcription, leading to hypoxic responses, and are involved in the regulation of carcinogenesis as well as stromal responses.210 HIF-1α has important functional roles in both innate and adaptive immune cells, including macrophages,211 neutrophils,212 dendritic cells (DCs),213 and lymphocytes.214 HIF-2α has been associated with macrophage NO homeostasis.215 In addition, deletion of the aryl hydrocarbon nuclear translocator (ARNT)/HIF-1β gene in CD8+ T cells impairs the expression of cytolytic effector molecules, including perforins and granzymes.216
Hypoxia and T cells
A distinctive feature of T cells is the markedly increased glucose uptake through glucose transporter 1 (GLUT1) as they respond to immune challenges and differentiate into cytotoxic T lymphocytes (CTLs).217,218 Activated lymphocytes generate energy largely through upregulation of aerobic glycolysis.217 Glycolysis requires T cells to activate and maintain the expression of glycolytic enzymes, for example, pyruvate kinase (PK) and lactate dehydrogenase (LDH), and also requires T cells to maintain high levels of glucose uptake by maintaining the expression of glucose transporters. Relatively high levels of exogenous glucose are required to maintain the CTL transcriptional program.219,220 HIF-1α enhances the glycolytic activity of but not HIF-2α.221 Elevated HIF-α expression of T cells in hypoxic environments is regulated by multiple pathways, including the PI3K/mTOR-dependent pathway,98 protein kinase C (PKC) and Ca2+/calcineurin,222 STAT3 dependence,223 NF-κB,100 MAPK pathways,103 and T cell antigen receptors (TCRs) (Fig. 5).98 Deletion of VHL impairs HIF-1α and HIF-2α degradation, which significantly elevates the expression of various members of the CTL secretory granzyme family, including perforin and tumor necrosis factor α (TNF-α), thereby sustaining CTL effector functions.224 In natural killer (NK) cells, APOBEC3G triggered site-specific editing of C-to-U RNA under hypoxia. These effects have been shown to occur independently of HIF-1α, suggesting the existence of other potential regulatory mechanisms.225 HIF-α activities in CD8+ tumor-infiltrating lymphocytes (TILs) promote their accumulation and anticancer activities,226 as well as the production of interferon-gamma (IFN-γ) by CD4+ T cells.227 HIF-1α deficiency attenuates the ability of some CD8+ T cells to cross the endothelial barrier and reduces the abundance of infiltrating CD8+ T cells in the TME. This is because VEGF-A facilitates the recruitment of immune cells.228 VEGF-A derived from T cells promotes VE-cadherin endocytosis in ECs, causing EC adhesion junction disruption and vascular homeostasis imbalances, resulting in higher expression of the adhesion molecule (VCAM-1), which mediates immune cell adhesion to the vascular endothelium. This finding suggests that T cell-derived VEGF-A affects T cell homing through the endothelial barrier. VEGF-A levels in cancers are inversely correlated with CD8+ T cell levels.221 Loss of HIF-1α decreases the ratio of CD8+ to FOXP3+ cells in TILs.229 After antigenic restimulation, the generation of effector cytokines from HIF-1α mutant T cells was suppressed.221 Ectopic HIF-2α (but not HIF-1α) mediates the extensive changes in gene expression by altering the CD8+ T cell transcription factor network, regardless of VHL inhibition, including increased perforin, granzyme B, IL-2, integrins, and CXCR4. In addition, the increase in co-inhibitors such as lymphocyte activation gene-3 (LAG-3) and CTLA-4 and the decrease in IFN-γ expression is also mediated. Overall, these molecular expression changes enhance cytotoxic differentiation and lytic functions against cancer targets.230

Hypoxia remodels CTL immune effect. HIF-1α in CTL is upgraded by TCR-PKC and Ca2+/calcineurin, PI3K/mTOR, NF-κB, NF-κB, JAK-STAT3, MAPK pathway. Deletion of pVHL impaired HIF-1α degradation. HIF-1α and HIF-1β dimerization stimulates downstream factor expression, such as perforin, IFN-γ, TNF-α, which can enhance the antitumor efficacy of CTL. However, some experiments elaborate the opposite results. Hypoxia inhibits the expression of IFN-γ, TNF-α, granzyme B, IL-2, perforin and NKG2D, while promoting PD-L1,PD-1 and CTLA-4 expression. CTL increases glucose uptake via GLUT1, which is metabolized to lactate by glycolytic enzymes PK and LDH. Lactate acidifies the TME, which inhibits CTL activation
However, the antitumor effects of CTLs under hypoxic conditions remain unclear. It has been reported that hypoxia impairs T cell function by reducing the levels of IFN-γ, IL-2, and NK group 2 member D (NKG2D).231,232 This is more in line with the actual tumor, where CD8+ and CD4+ T cells are significantly reduced in hypoxic areas of the tumor. T cell proliferations were strongly correlated with oxygen concentrations and were significantly suppressed under hypoxia (pO2 = 1%), likely due to hypoxia-associated changes in Ca2+ homeostasis in T cells.233 HIF-1α inhibits IFN-γ, TNF-α, and granzyme B expressions in CD8+, CD4+ T, and NK cells, which induces resistance to PD-1/PD-L1 blockade and suppresses T cell toxicity.234 Hyperoxia treatment suppressed the expressions of adenosine, an immunosuppressive factor,235 and reactive nitrogen production,236 and increased IFN-γ levels and perforin granules.237 Sustained hypoxic stimulation induces mitochondrial stress, resulting in the loss of mitochondrial functions, elevated ROS levels in T cells, T cell failure,238 and impeded anti-PD-1 efficacy.239 Because of the Warburg effect in cancer cells, which “ferments” glucose into lactic acid, lactic acid is transported outside the cell to prevent excess lactic acid accumulation. This stabilizes the intracellular pH of cancer cells while acidifying the extracellular environment.30,133 HIF-1α induces high expression of tumor-associated carbonic anhydrase (CA), such as CA9 and CA12, which catalyze the reversible hydration of CO2 to carbonic acid.240 In hypoxic environments, the pH of the extracellular environment of cancer cells can be as low as 5.8–6.5,208 which inhibits CTL activation, proliferation, and cytokine production, and also triggers T cell apoptosis.241
Hypoxia induces cancer cells to express chemokines such as CC chemokine ligand 28 (CCL28) to recruit regulatory T cells (Tregs).229 HIF-1α promotes Treg polarization and significantly contributes to colorectal cancer development and progression. This mechanism had no significant effect on the inhibition of Foxp3 expressions. Inhibiting Treg HIF-1α expression suppressed cancer growth.242 However, the HIF-1α-dependent transcriptional program contributes to helper T cell (Th) 17 development by mediating glycolytic activities. Moreover, HIF-1α can activate RORvt and form a tertiary complex with retinoic acid-related orphan receptor γt (ROR-γt), as well as p300, and recruit it to the IL17 promoter. In addition, HIF-1α attenuates Treg development and differentiation by binding to Foxp3 and targeting it for proteasomal degradation.223,243,244 Tregs express immunosuppressive factors,245 immunosuppressive adenosine,246 and cytokines such as IL-10,247 IL-17,248 and IL-35247 to inhibit effector T cell toxicity; therefore, they are closely associated with poor cancer prognosis.249 In addition, Tregs inhibit the expressions of nutrient transporters on CD8+ T cells, limiting nutrient uptake by T cells during activation, thereby suppressing T cell proliferation.250 Respiratory hyperoxia therapy may reduce the immunosuppressive effects of Tregs in the TME.251 Amphiregulin (Areg) is an epidermal growth factor receptor (EGFR) ligand, and Areg-EGFR promotes HIF-1α levels, leading to cancer progression.252 The lack of HIF-1α decreases the ratio of IFN-γ+ to FOXP3+ cells in CD4+ TILs, as HIF-1α deficiency reduces FOXP3 proteasomal degradation and cannot efficiently bind the IFN-γ promoter to drive Th1 responses.227
Hypoxia and B cells
The differentiation process of mammalian B cells includes pro-B, pre-B, immature B, and mature B cells. HIF-1α activity, which is regulated by miR-582, is high in human and murine bone marrow pro-B and pre-B cells, promoting cell proliferation, differentiation, apoptosis, and gene rearrangement.253 However, reduced immature B cell stages and sustained high activities of HIF-1α reduced the abundance of surface B cell receptors (BCR), CD19, and B cell activator receptors and increased the expression of the pro-apoptotic factor (BIM), preventing normal B cell development.254 Although hypoxia impairs B cell proliferation, it simultaneously alters B cell metabolism and promotes antigen-mediated differentiation.255 HIF transcription factors suppress the activities of prolyl hydroxydioxygenase, which stabilizes HIF by hydroxylating HIF-1α and HIF-2α in conjunction with pVHL to disrupt HIF. Therefore, reducing antigen-specific B cells in germinal centers (GCs; produce long-lived plasma cells and memory B cells) disrupts high-affinity IgG production and shifts to IgG2c, early memory B cells, and recall antibody responses. In contrast, sustained hypoxia or HIF induction induced by VHL deficiency inhibits mTOR complex 1 (mTORC1) activity in B lymphoblastoid cells, which impairs B cell clonal expansion, activation-induced cytosine deaminase (AID) expression, and the ability to produce IgG2c and high-affinity antibodies.256 HIF-1α is closely correlated with B-cell lymphoma (BCL-6). Upregulated BCL-6 promotes the immortalization of mouse embryonic fibroblasts and primary B cells by elevating cyclin D1 levels.257 HIF-1α binds CXC chemokine receptor type 4 (CXCR4) to promote B-cell viability.258 The immune effects of B cells on cancers have not been conclusively established. Large numbers of CD20+ B cell follicles and Foxp3+ low-infiltrating cells are associated with cancer survival and better recurrence-free survival outcomes in cancer patients, including gastric and pancreatic cancers.259,260 Activation of HIF in B cells modulates immune responses by inducing VEGF to increase lymphatic and endothelial vessel formation and enhance DC maturation and antigen presentation.261 Cancer-promoting effects of B cells have been reported.262 Hypoxia activates HIF-1α and induces autocrine transforming growth factor-beta (TGF-β) signaling, promoting myofibroblast activation, CXCL13 induction, B lymphocyte recruitment,263 and factor MYC secretion, which favors B cell proliferation and survival,264 and drives cancer recurrence.265 HIF-1α-dependent glycolysis facilitates CD1dhiCD5+ B cell expansion and promotes IL-10 expressions.266 B cell-derived IL-35 is associated with attenuated macrophage as well as inflammatory T cell viabilities and inhibited functions of B cells as antigen presenting cells (APCs).267 IL-10 and IL-35 promoted cancer cell metastasis,268 and inhibited anticancer immune responses in mice.269,270,271 In addition, one of the mechanisms of origin of extracellular immunosuppressive adenosine relies on CD73+ and CD19+ extracellular vesicles (EVs) from B, which is regulated by HIF-1α.272,273 Regulatory B cells (Bregs) are a functional B cell subpopulation with the key function of secreting IL-10, thereby preventing the production of cytokines.274 HIF-1α is a tumor suppressor in some cancers.275 For instance, pancreatic-specific HIF-1α deficiency significantly accelerated Kras (G12D)-driven pancreatic adenoma formation with a significant increase in intrapancreatic B lymphocytes.276 Depletion of B cells increases exocrine tissue regeneration owing to a significant decrease in overall immune infiltrations and fibrosis.277 Pancreatitis-induced tissue hypoxia and HIF-1α accumulation-induced B-cell depletion enhances pancreatic regeneration. B cell depletion in mice with pancreatitis significantly enhanced tissue regeneration and chemotherapeutic drug resistance.278
Hypoxia and macrophages
Hypoxia, HIF-1α, and tumor secretion of multiple chemokines facilitate cancer-associated macrophage (TAM) recruitment to the TME.279,280,281 TAMs constitute a plastic and heterogeneous cell population and can account for up to 50% of certain solid tumors.282 TAMs promote cancer progression by creating an immunosuppressive microenvironment,283 and enhancing the progression and metastasis of various cancers.284,285,286 Hypoxia-induced HIF-1α promotes the adaptation of TAMs to the hypoxic environment and is associated with cancer prognosis.287 Lipopolysaccharides (LPS) promote HIF-1α expression by stimulating the expression of Toll-like receptors in macrophages.288 Hypoxia (1.5% oxygen) leads to inadequate T-cell responses by increasing macrophage pro-inflammatory responses, including TGF-β, increased platelet-derived growth factor (PDGF), phagocytosis maintenance, and reduced antigen presentation. Hypoxia enhances fibrosis by promoting pro-fibrotic cytokine responses and isolating fibroblasts in the vicinity of granulomas.289 The hypoxic environment leads to elevated HIF-1α levels in macrophages and promotes the expression of inducible nitric oxide synthase (iNOS), which rapidly blocks T cell proliferation through NO and subsequent peroxynitrite formation.290,291,292 IL-15Rα+ TAMs downregulate CX3CL1 expression in cancer cells through the non-transcriptional activity of HIF-1α, reducing CD8+ T cells and/or increasing CD4+ T cells to reduce the ratio of CD8+ T cells to CD4+ T cells and thus weaken anti-cancer immunity.293 In addition, macrophages express HIF-2α under hypoxic conditions and inactivate drug-induced chemoresistance to 5-FU through HIF-2α-mediated specific overexpression of dihydropyrimidine dehydrogenase (DPD).294,295 Furthermore, acidic TME affect TAMs. Lactate, converted from pyruvate in cancer cells, plays a key signaling role by inducing HIF-1α-mediated expression of vascular endothelial growth factor in other processes.296,297,298,299 In addition, M2-like TAM promotes cancer progression by remodeling the extracellular microenvironment.282
Hypoxia and NK cells
NK cells are bone marrow-derived and account for 10–18% of peripheral blood mononuclear lymphocytes.300 NK cells can kill various cancer targets, including cancer cells with low expressions MHC-I.301 Some cancer cells secrete chemokines to recruit NK cells. For instance, HCC cells are often in hypoxic microenvironments, an environment that helps CD103+ DCs to take up and clear cancer DNA, which is enhanced by blocking cell surface protein (CD47). By secreting IL-12 and CXCL9, activated CD103+ DCs induce NK cell recruitment; upregulate the expression of granzyme B, NKG2D, IFN-γ and TNF-α; and downregulate the expression of NKG2A, which in turn enhances antitumor effects. However, under hypoxic conditions, most HCC cells highly express the CD47 protein, which is associated with poor prognostic outcomes.302,303 Expression of NKG2D receptors, intracellular perforin, and granzyme B in NK cells is severely impaired under hypoxic conditions, which suppresses NK cell immunotoxicity.304 Hypoxia promotes cancer cell metalloproteinase ADAM10 expression through HIF-1α, leading to shedding of NK cell activation ligand (MICA) from cancer cell surfaces, thereby attenuating NK cell-mediated lysis.291 In an inflammatory model, elevated NK cell HIF-1α levels stimulated the release of IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF), enhancing antimicrobial defenses while promoting M1 macrophage polarization, leading to slow angiogenesis and wound healing.305 NK cell counts and cytotoxic effects were similarly suppressed in a chronic hypoxia mouse model, accompanied by inhibited IFN-γ secretion by NK cells. NK cells also secrete large amounts of MMP-9, which affects blood vessel remodeling.306 Although the absence of NK cell HIF-1α inhibits tumor growth, it is associated with various challenges. For example, deletion of HIF-1α in NK cells suppressed the expression of the angiostatic soluble form of VEGF receptor 1 (sVEGFR1) in cancer, increasing VEGF bioavailability in the TME and leading to non-productive angiogenesis. The immature vascular phenotype promotes cancer cell intravasation and distant pulmonary metastases from melanoma in vivo.202 Moreover, cancer cells influence the cytotoxic effects of NK cells. Upregulated HIF in the TME increases PD-L1 expression, decreases autophagic MHC-I expression, and inhibits NK cell recognition.103,221 HIF-2α promotes IL-10 release by activating the STAT3 signaling pathway in HCC cells, thereby inhibiting the killing activities of NK cells, which in turn promotes HCC recurrence and metastasis.307 HIF-2α stimulates ITPR1 expression in hypoxic cancer cells, activates autophagy in NK cells, inhibits granzyme B activity, and impairs the killing sensitivity of NK cells.308 Activated autophagy in hypoxic cancer cells selectively degrades the pro-apoptotic NK-derived serine protease GZMB/granzyme B, leading to the suppression of NK-mediated target cell apoptosis.309
Hypoxia and neutrophils
Neutrophils are the major players in the innate immune system. Neutrophil infiltration is uncommon in normal tissues.310 Neutrophils are glycolytic cells that derive little ATP from oxidative phosphorylation and require gluconeogenesis to generate intracellular glycogen stores to kill bacteria.311 In a hypoxic environment, neutrophils reduce glycogen recycling, leading to impaired functions,311 and consume extracellular proteins to promote their own central carbon metabolism to maintain their functions.312 Infiltrating neutrophils in the TME, known as tumor-associated neutrophils (TANs), promote cancer growth and is strongly associated with poor prognosis.313 For instance, neutrophil-derived ROS damages hepatocyte DNA to drive HCC development.314 Neutrophils can eliminate bacteria from the body, forming an immune barrier that allows cancers to start growing back;315,316 therefore, they are closely correlated with prognostic outcomes of cancer patients.310,317 Elimination of neutrophil infiltration leads to cancer regression and prolonged survival outcomes.318 Sequencing of pancreatic single-cell transcriptome revealed that BHLHE40 is a key regulator of neutrophil polarization toward the TAN-1 phenotype, which has pro-cancer and immunosuppressive functions. However, the associated mechanism should be investigated.319 Neutrophils have an extensive mitochondrial network that uses the glycolytic product (glycerol-3-phosphate) to maintain polarized mitochondria and produce ROS to regulate HIF-1α stability.320 Neutrophil extracellular traps (NETs) promote cancer cell colonization by enhancing migration, invasion and cancer cell stemness, leading to poor cancer prognosis. Interestingly, NET formation is strongly associated with elevated HIF-1α levels in neutrophils. Downregulation of neutrophil HIF-1α can effectively inhibit NET-mediated circulating tumor cell (CTC) metastasis and prolong the median survival of mice with breast cancer lung metastasis.321 However, some experiments have shown the anticancer effects of NK cells. Hypoxia favors neutrophil viability and function.322 Hypoxia recruits polymorphonuclear neutrophils (PMNs), which are the main effector cells against endometrial adenocarcinoma growth, and induces cancer cell detachment from the basement membrane.323 Upon relief of tumor hypoxia, recruitment of PMNs to the TME is significantly reduced; however, the recruited cells can efficiently kill cancer cells by releasing NADPH oxidase-associated MMP-9 and ROS.324
Hypoxia and MDSCs
Myeloid-derived suppressor cells (MDSCs) can suppress immune cell activity. Hypoxia and HIF-1α drive MDSCs recruitment to the TME.280,281 This is because cancer cells in hypoxic areas express various cytokines, such as chemokine (C-C motif) ligand 26, G-CSF, and IL-6, to recruit MDSCs.297,325,326 Functionally, MDSCs enhance the evasion of cancer cells from immune surveillance and promote cancer drug resistance.327 Inhibiting the infiltration of MDSCs improves anti-tumor effects.328 In an HCC model, Chiu et al. found that hypoxia-induced HIF-1α promotes the expression of ENTPD2/CD39L1, an indicator of poor HCC prognosis, which is thought to convert extracellular ATP to 5’-AMP, preventing the differentiation of MDSCs, but maintaining their survival.329 In addition, under hypoxic conditions, MDSCs express multiple immunosuppressive factors. For instance, PD-L1 is secreted by hypoxia-induced HIF-1α, but not HIF-2α in MDSCs,330 arginase and NO are promoted by HIF-1α-induced miR-210 expression,331,332 surface ectonucleotidases CD39 and CD73,333 TGF-β1, and exosomes, such as S100A9, RAR-related orphan receptor alpha (RORA), and PTEN,281,334,335 promote cancer cell stemness and growth and inhibit CTL function. Hypoxia through HIF-1α dramatically alters the function of MDSCs in the TME and redirects their differentiation toward TAMs, although the macrophage subtype has not been clearly established.331 These effects may be attributed to the ability of MDSCs to express SIRT1,336 a factor that regulates the glycolytic activities of MDSCs and affects the functional differentiation of MDSCs,337 and some miR-29a-containing exosomes.338
Hypoxia and ILCs
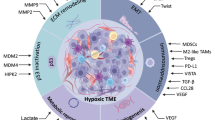
Innate lymphocytes (ILCs) consist of NK cells ILC1, ILC2, and ILC3, and are involved in the immune response to virus, bacteria, parasites, and transformed cells.339,340 Their roles in cancer immunity and immunotherapy have not been established.341,342 Specific changes in cancer cytokines alter ILC composition in cancers by inducing plasticity and altering ILC functions.343,344 IL-15 promoted ILC1 granzyme A expressions and cytotoxicity, induced the apoptosis of murine leukemia stem cells (LSC), maintained anticancer immunity, and was positively correlated with survival outcomes.345,346,347 IL33-activated ILC2 selectively expresses chemokine ligand 5 (CCL5), which recruits CD103+ DCs into the TME and activates CD8+ T cells to lyse cancer cells.342 IL-33 results in massive amplifications of ILC2 and produces CXCR2 ligands from these cells, which enhances cancer cell-specific apoptosis through CXCR2.348 Administration of hypoxic ILC2 resulted in a higher cancer volume, suggesting that hypoxia-exposed ILC2 enhanced the progression of pancreatic cancer cells. These outcomes were attributed to the conversion of ILC2 to ILCregs under hypoxic conditions, which inhibited T cell infiltration and IFN-γ expression by secreting IL-10.349 IL-25 promotes intra-tumoral ILC2 infiltration and enhancing the ability of cancer-infiltrating MDSCs to suppress antitumor immunity and reduce survival outcomes in colorectal cancer (CRC) patients.350 ILC3 have antitumor effects. Their intrinsic disruption in CRC drives dysfunctional adaptive immunity, cancer progression, and immunotherapeutic resistance.351 The production of chemokines (CCL20) and pro-inflammatory cytokines (IL-1β) at the tumor site leads to ILC3 recruitment and activation. ILC3 secrete the chemokine CXCL10, which recruits CD4+ and CD8+ T cells and promotes antitumor immune responses.352 IL-22, secreted by ILC3 cells, is required for initiation of DNA damage responses (DDR) after DNA damage. Stem cells that lose IL-22 signaling and are exposed to carcinogens escape DDR-controlled apoptosis, develop more mutations, and are more likely to cause colon cancer.353 The interaction between immune cells and TME is showed in Fig. 6.

Changes in cytokine secretion by innate immune cells under hypoxic conditions. M1 microphage secretes iNOS, and M2 macrophages secrete IL-10, EGF, VEGF, MMP, DPD, PDGF and TGF-β to suppress immune responses. Lactate induces M2-like polarization. HIF-1α inhibits NK cell expression of perforin, NKG2D, GM-CSF, granzyme B and IFN-γ. Hypoxia induces neutrophils to produce ROS and NETs, which promote tumorigenesis and metastasis. HIF-1α induces MDSCs to secrete cytokines NO, arginase, PD-L1, RORA, PTEN, CD39, CD73 and S100A9, which inhibit immune response and promote the tumor cell stemness and growth. ILC1 secretes granzyme A and maintains antitumor efficacy. ILC2 enhances immune response through selective expression of CCL5, CXCR2. Hypoxia induces IL-10 expression in ILC2 and suppresses immunity. ILC3 secretes CXCL10 and IL-22, improving antitumor immune response
Hypoxia and chemotherapeutic resistance
In vivo experiments have shown that hypoxia increases the tolerance of cancer cells to drug toxicity.354,355,356 Hypoxia induces high expression of drug resistance genes in cancer cells, increases chemotherapeutic drug efflux, and reduces intracellular drug concentrations. Once they are combined with anticancer drugs, P-glycoprotein (PGP), known as multidrug resistance protein 1 (MDR1), and breast cancer resistance protein (BCRP) are ATP-binding cassette (ABC) transporters and energy-dependent drug excretion pumps that can pump drugs out of the cell by providing energy through ATP.357 Therefore, the intracellular concentration of drugs continues to decrease, which weakens their cytotoxic effects until they are dissipated, and drug resistance occurs. ROS generated under hypoxic conditions induce PGP and BCRP expression through HIF-1α and increase cancer cell drug resistance.358 HIF-1α induces multidrug resistance-associated protein 1 (MRP1) expressions and enhances drug resistance in colon cancer cells.359 Moreover, HIF-1α enhances the inactivation of chemotherapeutic drugs and reduces their cytotoxic effects. Cytidine deaminase (CDD), an important metabolic enzyme involved in drug resistance development in cancer cells, is derived from cancer cells or bacteria within cancer cells. Elevated CDD levels promote the metabolism of gemcitabine to its inactive form.360 Moreover, solute carrier (SLC) transport proteins are primarily involved in the uptake of small molecules into cells, and their absence affects the uptake of chemotherapeutic agents into cancer cells, leading to drug resistance.361 The role of hypoxia in relation to CDD and SLC has not been fully established. Recently, alternative resistance pathways have been identified in hypoxic cancer cells. For instance, cancer and stromal cells in the TME, such as cancer-associated fibroblasts, secrete various cytokines, including IL-6, which induces HIF-1α expressions to regulate downstream chemotherapeutic resistance genes, such as olfactomedin 4 (OLFM4), pyruvate kinase muscle 1 (PKM1) that enhances mitochondrial oxidative phosphorylation (OXPHOS), and expressions of non-coding genes, for example, miR-27a that increases PGP expression, to promote chemoresistance acquisition in cancer cells.362,363,364,365,366,367 HIF-1α and cancer-associated fibroblast-secreted TGF-β signaling synergistically promote GLI family zinc finger 2 (GLI2) expressions through SMAD3, inducing CRC cell stemness and chemoresistance.368 Upregulated HIF-2α promotes sorafenib resistance in hypoxic HCC cells by activating the TGF-α/EGFR and COX-2 pathways.369,370 Under normoxic conditions, autophagy activation did not counteract cisplatin-induced stress, leading to cell death, whereas under hypoxic conditions, autophagy induction was enhanced, resolving cisplatin-induced stress and inhibiting the cisplatin-induced BCL2 interacting protein 3 (BNIP3) death pathway, allowing cell survival.371 Downregulation of BNIP3 may contribute to the resistance of pancreatic cancer to hypoxia-induced cell death.372 However, in non-small lung cancer samples, high expression of BNIP3 was significantly associated with HIF-1α and poorer overall survival was associated.373 Due to fibrosis and formation of non-functional vessels in the TME, drug diffusion and delivery to cells away from functional vessels is decreased374, and this may trigger drug resistance in cancer cells. In addition, an altered pH gradient in the TME can attenuate drug action in cells (for example, alkylating agents and antimetabolites) to enhance drug resistance.374 The mechanisms of hypoxia-enhanced tumor chemotherapy resistance is showed in Fig. 7.

Hypoxia enhances tumor chemotherapy resistance. IL-6 stimulates HIF-1α expression, which increases PGP expression through upregulation of miR-27a. Chemotherapeutic agents are transported by HIF-1α-induced intracellular MRP1 and excreted from cells via PGP and BCRP. HIF-1α induces OLFM4 to enhance cancer cell chemoresistance. HIF-1α enhances chemotherapeutic resistance in cancer cells by promoting PKM1, enhancing mitochondrial OXPHOS. Under hypoxic conditions, cancer cells induce enhanced autophagy and inhibit the chemotherapeutic drug-induced BNIP3 death pathway to resist drug toxicity. HIF-1α synergizes with TGF-β to promote GLI2 expression through SMAD3, inducing cancer cell stemness and chemoresistance. HIF-2α upregulation promotes the ability of hypoxic cancer cells to resist drug toxicity by activating the TGF-α/EGFR pathway and COX-2. In addition, CDD and SLC play important roles in the drug resistance of cancer cells, the effect of hypoxic conditions on CDD and SLC is unclear
Hypoxia and radiation resistance
Radiotherapy directly destroys macromolecule fixation and leads to DNA damage through the generation of free radicals, such as hydroxyl (OH•) and hydrogen (H•) radicals, by ionizing radiation (IR), a process known as “oxygen fixation theory”. The damage caused by radiotherapy is permanent and irreversible. Hypoxia reduces the effectiveness of radiotherapy and free radicals become unstable, leading to limited extent of their damage, and most of the damage can be easily repaired.375,376 Classical in vitro and in vivo experiments have shown that at O2 (pO2) partial pressures below 10 mmHg, tumor cells can acquire radiobiologic hypoxia and thus become relatively resistant to radiotherapy. For instance, at 1 mm Hg, cancer cells are three times more resistant to radiation than ordinary oxygen cells.16 Several preclinical and clinical trials have demonstrated that the number of lethal DNA DSB formed under hypoxic conditions is also reduced by 2 to 3-fold and hypoxia can alter the expression and function of DNA DSB-associated genes. Under hypoxic conditions, additional DNA repair pathways can be activated.16,376,377,378,379 HIF-1α stimulates DNA-dependent protein kinase (DNA-PK) expression and repairs DNA DSB caused by ionizing radiation.380 Radiation upregulates HIF-1 expression and enhances its activity in cancer cells.381 Elevated HIF-1α levels confer radioresistance via various pro-cancer mechanisms. HIF-1α promotes ATP metabolism, and p53 activation and stimulates EC survival, thereby mediating the ultimate cancer radiation responses.382,383 The serine peptidase inhibitor Kazal type 1 (SPINK1) is secreted in a HIF-dependent paracrine manner to reduce radiation-induced DNA damage and enhance radiation resistance in adjacent cancer cells through EGFR and nuclear factor erythroid 2-related factor 2 (NRF2).384 Radiation-induced EC death leads to secondary tumor cell killing.385 The vascular system also affects radiation therapy. Radiation-induced secretion of VEGF, FGF, and PDGF by cancers protects the cancer vasculature from radiation-mediated cytotoxicity and enhances the radioresistance of ECs.386,387,388 Thus, VEGF, FGF, and PDGF inhibitors can significantly improve the efficiency of radiotherapy.388,389 The radiosensitivity of hypoxic cancer cells was showed to be significantly increased after reoxygenation.390 Elevated H2O2 levels in cancer tissues exacerbate hypoxia-induced resistance to radiotherapy. Reducing oncological cellular H2O2 or using it to generate oxygen through chemical reactions may improve radiotherapeutic outcomes.391 However, these mechanisms require further evaluation (Fig. 8).

Hypoxia enhances the ability of the tumor to resist radiotherapy. Radiotherapy induces cancer cell death by directly damaging DNA through the production of free radicals such as OH• and H•. SPINK1, secreted by hypoxic cancer cells, upregulates EGFR and NRF2 antioxidant response in adjacent cancer cells to reduce radiation-induced DNA damage, thereby inducing cancer radioresistance. VEGF, FGF and PDGF secreted by hypoxic cancer cells enhanced the radiation resistance of ECs. HIF-1α stimulates DNA-PK expression and repairs DNA DSB
Hypoxia-mediated therapy
Targeted therapy
Approximately 80–90% of deaths among cancer patients are directly or indirectly attributed to drug resistance. Progress in the development of new drugs is also hampered by drug resistance, which has become a considerable challenge in cancer treatment.392 Hypoxia drives cancer development and progression. Therefore, it is essential to increase the oxygen concentration in TME. Oxygen delivery in the TME is increased by enhancing cancer vascular formation to elevate oxygen levels, however, the vessels formed in cancers are usually abnormal and non-functional.39 Intravenous oxygen delivery to the cancer through blood is not feasible for reasons such as the potential risk of systemic ROS exposure and the low solubility of oxygen in blood.393 Increasing oxygen delivery by increasing blood flow to the tumor through pharmacological vasodilatation methods has been proposed, but in practice it is difficult to improve or reduce blood flow to the tumor tissue.394 To overcome this challenge, nano- and bio-based technologies have been used to carry oxygen generators to generate enough oxygen in the target cancer such as pancreatic ductal adenocarcinoma, colorectal cancer, fibrosarcoma, melanoma, etc., which aids in drug delivery and significantly improves the effectiveness of chemotherapy, radiotherapy as well as immunotherapy.281,395,396,397,398 Moreover, the reoxygenation process generates large amounts of ROS, which results in DNA damage, contributing to cancer cell death.65,399 However, the damage this brings to the surrounding normal cells is a matter of concern. Second, inhibition of hypoxia-induced HIF and its downstream target genes is an effective strategy.400 For instance, 32–134D, a low-molecular-weight compound that inhibits HIF-1/2-mediated gene expression in HCC cells, combined with anti-PD-1 increased HCC eradication rates in mice (from 25 to 67%).401 HIF-1α inhibitors, such as topotecan, bortezomib (PS-341) as well as HIF-2α inhibitors, such as PT2399, PT2385 and PT2977, inhibit HIF-1/2α activities and their downstream target gene expressions.402,403 These inhibitors reduce VEGF levels in circulating tumor cells (such as neuroblastoma, multiple myeloma, HCC and renal cell carcinoma) to inhibit tumor angiogenesis,404,405,406,407,408 and enhances the effects of chemotherapeutic drugs, such as oxaliplatin, to inhibit colorectal cancer cell proliferation.409 Hypoxia in the TME provides a special environment for the effects of some drugs, called hypoxia-activated prodrugs (HAPs).395 For instance, evofosfamide (TH-302) and tarloxotinib-effector inhibited cancer growth by suppressing signaling and cell proliferation in patient-derived cancer cells in vitro and in mouse xenograft models.410,411 However, there is controversy in the clinical treatment of advanced soft tissue sarcoma.412,413 Mechanistically, DNA dioxygenase ten-eleven translocation (TET)2 is recruited by the transcription factor (HNF4α) to activate FBP1 expressions, thereby antagonizing the functions of HIF-1/2α) in metabolic reprogramming to suppress clear cell renal cell carcinoma growth.414 Clinical trials regarding hypoxia-targeted therapy are summarized in Table 2.
These clinical trials were designed using hypoxia tracers, hypoxia-activated prodrugs, and drugs targeting HIF and downstream targets. Hypoxia tracers can visualize and quantify tumor hypoxia, assisting clinicians in the non-invasive detection and assessment of tumor hypoxia levels, and with the aid of PET imaging can be used to monitor the dynamic response of patients to treatment, potentially allowing for individualized patient treatment.415,416 Hypoxia-activated prodrugs can selectively target hypoxic tumor cells, have hypoxia-selective cytotoxicity, and exhibit broad antitumor activity.412,417,418 HIF and downstream gene-targeted drugs effectively inhibited tumor even, significantly improved patient survival, and phenocopied better tolerability.419,420,421 However, these drugs have certain toxic side effects, and common adverse events include headache, fatigue, nausea, vomiting, diarrhea and dehydration, and serious adverse events include Gastrointestinal bleeding, blood clots/deep vein thrombosis, lymphocytopenia, febrile neutropenia, anemia and triggering secondary infections. Some clinical trials do not have sufficient data to analyze the safety and efficacy of drugs.
Hypoxia and immunotherapy
Hypoxia inhibits anti-cancer immune sensitivity.422 Reduction of the hypoxic environment in the TME increases the efficacy of immune checkpoint blockade (ICB) against cancers in vivo. The rational use of the pulsatile system of the TME can ameliorate hypoxia. Increasing the oxygen partial pressure through intravenous oxygenation can reverse hypoxia in the TME.423 However, some of the vessels in cancer are abnormal and non-functional. Therefore, vascular remodeling in the TME is necessary to ameliorate hypoxia. Elevated levels of Delta-like 1 (DLL1) in breast and lung cancer induce long-term cancer vascular normalization to alleviate cancer hypoxia, promote the accumulation of IFN-γ-expressing CD8+ T cells, and enhance macrophage polarization toward the M1-phenotype.424 Hyperoxia upregulates MHC-I expression in cancer cells, enhances T cell-mediated cytotoxicity,398 promotes the anti-cancer activities of NK cells,235 reduces the immunosuppressive effects of Tregs in the TME, and inhibits the production of MDSC-derived exosomes to reduce cancer cell stemness.235,281 Pro-oxidants can also be used to increase oxygen levels in the TME. Wu et al.396 developed nanoparticle-stabilized oxygen microcapsules that could be precisely targeted to pancreatic ductal adenocarcinoma (PDAC), improving hypoxia and significantly increasing the efficacy of anti-PD-1 antibodies against PDAC. Treatment with oxygen microcapsules combined with anti-PD-1 antibodies alleviates TAM infiltration, polarizes macrophages from the pro-cancer M2 phenotype to the anti-cancer M1 phenotype, and increases the proportion of TME Th1 cells and CTLs to enhance anti-cancer immune effects.396 The oxygenating agent (TH-302) eliminates hypoxia in cancers and promotes T-cell infiltration.425 The pro-oxidant adaptor (p66SHC) reduces ATP production in B cells by limiting glycolysis and compromising mitochondrial integrity, while activating AMPK to promote B cell mitochondrial autophagy and limiting plasma cell differentiation.426 Suppression of HIF pathway-mediated PD-L1 is an important measure for enhancing cancer immune responses. Inhibition of HIF-1α synthesis suppresses PD-L1 expression and induces lysosomal degradation of PD-L1, enhancing the ability of CTLs to kill cancer cells.427 The deletion of HIF-1α in NK cells significantly inhibits cancer growth and improves patient survival outcomes by activating the NF-κB pathway and stimulating the expression of effector molecules, such as IL-18.428 Single inhibition of HIF-1α in immune cells restores immune cytotoxicity; however, it also induces PD-L1 expression in cancer cells, impairing anticancer immunity.234 Therefore, multichannel combination therapy is vital for cancer treatment. Hypoxia leads to the increased expression of immune checkpoints and promotes fibrosis, thereby suppressing the efficacy of cancer immunotherapy. Increasing the oxygen concentration affects cancer immune responses by altering the extracellular matrix. For instance, hyperbaric oxygen (HBO) promotes PD-1 antibody delivery and T cell infiltration into the cancer parenchyma by depleting major components of the extracellular matrix, such as collagen and fibronectin.429 Genetic engineering approaches can be used to construct high-affinity NK (haNK) cells, which can express high-affinity CD16 receptors and IL-2, improve tolerance against acute hypoxia, and maintain the functions of NK cells to kill cancer cells.430
Hypoxia and radiotherapy
Radiotherapy is a non-invasive oncologic treatment approach for drug-resistant cancers.431 It is based on the principle of targeting cancer tissues with X-rays or gamma rays, which directly or indirectly damage biomolecules to ionize the surrounding water and generate ROS, inducing DNA damage and apoptosis.432,433 Oxygen in the TME can anchor broken ends of DNA and form stable organic peroxide groups that inhibit further DNA repair.434 However, local hypoxia in the TME markedly reduces the therapeutic efficacy of radiation therapy. In addition, radiotherapy requires high doses of X-rays to treat cancers, which can cause serious side effects.435 By utilizing the sustainable production of O2 from endogenous H2O2 decomposition, cancer hypoxia can be improved and the efficacy of radiotherapy is enhanced.436 Chai et al. found that under continuous external irradiation with a 660 nm laser and X-ray beams, cyanobacteria continuously photosynthesized and released oxygen, and the large amount of ROS produced by the two-dimensional (2D) bismuth radiosensitizer greatly enhanced the efficacy of radiation therapy and inhibited cancer growth in vivo.437 Improving hypoxia increases the sensitivity of cancer cells to radiotherapy,437 which may be related to HIF-1α inhibition in cancer cells by changing the optical redox status.438 The mechanisms involved in hypoxia-induced tolerance to radiotherapy in cancer cells should be investigated further.
Nano application therapy
Nanoparticles are characterized by small sizes, large surface areas, high bioavailability, and good biocompatibility, and have great potential for safely and efficiently transporting external oxygen to the hypoxic TME.439 The nanocomposite “oxygen bomb” PSPP-Au980-D is used to precisely locate the hypoxic microenvironment of pancreatic cancer, and through different nm laser irradiation, it can improve hypoxia, generate singlet oxygen, and enhance the efficacy of photodynamic therapy (PDT).440 The use of an acidic microenvironment to generate oxygen is a feasible method. For instance, an acidic environment triggers a reaction between MnO2 and H2O2, releasing large amounts of oxygen to alleviate intra-tumoral hypoxia. Fragmented human-induced pluripotent stem cells (iPSCs) release cancer-sharing antigens, which trigger strong innate and adaptive immune responses against cancers, promote dendritic cell maturation, and activate effector T and NK cells. Meanwhile, they also decreased the amount of Treg cells. iPS-MnO2@Ce6 significantly inhibited cancer growth, metastasis and reduced mortality in cancer-bearing mice models.441 Nanoparticles can also host various drugs, such as oxygenating agents, chemotherapeutic agents and immune-boosting drugs, improving the anti-cancer effects.442 Manganese dioxide based nanocarriers can enhance the anticancer effects of piggyback paclitaxel by alleviating the degree of hypoxia in the in situ glioma microenvironment.442 Porous Au@Pt core-shell nanostructures inhibit the expressions of HIF-1α and MDR1 gene by oxidizing the TME, thereby reducing the extracellular secretion of adriamycin (DOX) and enhancing the efficacy of chemo-photothermal therapy.443 When hybrid nanospheres containing Fe3+, aggregation-induced emission (AIE) photosensitizer, and Bcl-2 inhibitor of sabutoclax were consumed by cancer cells, they increased intratumoral oxygen concentration through Fe3+-mediated Fenton reactions, and intracellular PDT resistance of the AIE photosensitizer was mitigated by sabutoclax.444 Remodeling the tumor immune microenvironment by nanoparticle technology can achieve efficient cancer therapy. Mesoporous silica nanoparticles (MSNs) in combination with pro-oxidants and mitochondrial respiration inhibitors can alleviate the hypoxic environment and inhibit MDSCs infiltration.445 TK-M@Man-HMPB/HCQ alleviated hypoxic microenvironment-induced TAM polarization, promoted CTL infiltration, and significantly inhibited cancer growth.446 Novel poly (vinylpyrrolidone) (PVP)-modified BiFeO3/Bi2 WO6 (BFO/BWO) with a p-n type heterojunction reshapes the immunosuppressive TME. It triggers H2O2 catabolism to generate O2 to alleviate cancer hypoxia, enhance PDT and radiotherapy sensitivity, promote TAM polarization from the M2 to the M1 phenotype, and inhibit HIF-1α expression. This photo-activated nanoconjugated radiotherapy can activate and recruit T cells and stimulate TAMs toward the M1 phenotype, significantly reversing the immunosuppressive TME to immunoreactive TME and further enhancing the immune memory response.447 Advances in nanotechnology to improve the hypoxic TME, or to combine chemotherapy, radiotherapy, and immunotherapy, have achieved satisfactory outcomes, compared to traditional treatments, and may become a treatment measure with great potential in the future.
Biotherapy
Accumulating evidence suggests that microbiota play an important role in cancer by damaging cellular DNA, inducing transformation, activating and altering stromal cell components in the TME, and influencing cellular metabolism.448,449 The microbiota characteristics under hypoxic conditions are currently not fully understood. Recent research has demonstrated that a combination of live bacteria and treatment modalities such as surgery, chemotherapy, and radiotherapy can produce good clinical outcomes. Certain bacteria have the potential to gravitate to the hypoxic core of cancer cells and can spread and proliferate. Spores of the anaerobic bacterium Clostridium novyi-NT were used to treat the transplanted tumors in mice. In several mouse models including colorectal cancer, biliary cancer, melanoma and squamous cell carcinoma, this bacterium was found to significantly improve the efficacy of radiotherapy.450 The use of the attenuated pathogenic anaerobe Salmonella VNP20009 to target the cancer hypoxic region, equipped with photosensitizers and bromodomain and extra terminal domain (BET) protein inhibitors with mitochondrial targeting properties improved the heat elimination abilities of photothermal therapy (PTT) and significantly inhibited the expressions of PD-L1 in cancer cells, thereby enhancing and maintaining a durable immune response.451 Bioluminescent bacteria designed by transforming attenuated Salmonella typhimurium strains effectively increased PDT and promoted inflammation by converting macrophages M2 to M1, activated NK cells, CD4+ Th cells and CD8+ T cells in the TME, reduced intratumoral immunosuppressive Tregs, and upregulated the expressions of various effector cytokines.452 Salmonella preferentially localizes sites and proliferates in hypoxic cancers, due to this advantage, Salmonella is also used as a cancer targeting vector to deliver different therapeutic agents and to achieve synergistic anticancer effects. However, colonized Salmonella recruits a large number of neutrophils, thus favoring cancer growth.315,316 The elimination of Salmonella-recruited neutrophils facilitates complete cancer eradication and reduces side effects.453 Delivery of peroxidase is an effective strategy for reducing resistance to radiotherapy. The peroxidase membrane vesicles (EMs) of Escherichia coli have higher peroxidase activity than free peroxidase, which decomposes H2O2 into oxygen to alleviate cancer hypoxia. Combined with their immunostimulatory properties, EMs can effectively enhance the effects of radiation therapy and induce anticancer immune memory.391 Maximizing the use of live bacteria under hypoxic conditions has the potential to be a therapeutic modality for the treatment of cancer.
Conclusions and perspectives
Over the last 60 years, extensive progress has been made in the understanding of hypoxia-mediated signaling pathways. Hypoxia-based research has provided new insights into how hypoxia works and has laid the foundation for the development of targeted therapies that can improve patient prognosis. Carcinogenic factors and mature cancer characteristics promote hypoxia in the TME, and cancer cells adapt to hypoxia by changing their own metabolism through key genes such as HIFs. With advances in analytical tools and genomic technologies, such as pan-genomic analysis, are important for in-depth studies of HIFs.454,455 Hypoxia-induced pathways can alter the malignant behavior of cancer cells. Under hypoxic conditions, most downstream targets are HIF-dependent. Whether there are other key genes independent of HIFs requires further investigation. For instance, hypoxia-induced RNA editing by endogenous RNA editing enzyme can be mimicked by inhibiting mitochondrial respiration and occurs independently of HIF-1α to facilitate adaptation to hypoxic stress.225 Moreover, hypoxia reshapes the stromal cell properties of the TME, especially with the rise of immunotherapy, and both innate and adaptive immune cells in hypoxic environments have received extensive attention. However, despite significant breakthroughs, some results remain controversial. The reasons for this may be related to tissue specificity in addition to the experimental tools used and design bias, and mature modeling is also critical. The composition of the TME is diverse, and in addition to its own components, other foreign components may be present, such as microbiota, an emerging field that plays an important role in various cancers. Microbiota in the TME is one of the factors that form and maintain chronic hypoxia, activates HIF-1α in a non-hypoxic manner, increases HIF-1α mRNA levels, stabilizes HIF-1α protein, and induces the expression of HIF-1α regulatory genes.288,456,457 Changes in the characteristics of the microbiota in hypoxic environments and their effects on tumors are interesting and deserve further exploration. Therapeutic measures based on these characteristics, including targeted therapy, immunotherapy, chemotherapy, radiotherapy, radiotherapy, and biotherapy, offer good prospects. However, the complexity of the tumor relationship makes it inefficient to treat cancer from a single perspective. Multidisciplinary combination therapy, such as biology, chemistry, materials, machinery, electronics, artificial intelligence, and other multidisciplinary approaches for cancer (especially refractory cancer), is an interesting trend. Extensive study is still required before the development and implementation of efficacious and robust hypoxia-related precision treatment.
References
Fane, M. & Weeraratna, A. T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Tannock, I. F. Population kinetics of carcinoma cells, capillary endothelial cells, and fibroblasts in a transplanted mouse mammary tumor. Cancer Res. 30, 2470–2476 (1970).
Gray, L. H. et al. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radio. 26, 638–648 (1953).
Semenza, G. L. & Wang, G. L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 12, 5447–5454 (1992).
Wang, G. L. & Semenza, G. L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood 82, 3610–3615 (1993).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92, 5510–5514 (1995).
Iliopoulos, O. et al. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl Acad. Sci. USA 93, 10595–10599 (1996).
Semenza, G. L. et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 51, 553–555 (1997).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 (1999).
Gu, Y. Z. et al. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 7, 205–213 (1998).
Semenza, G. L. et al. Assignment of the hypoxia-inducible factor 1alpha gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics 34, 437–439 (1996).
Tian, H., McKnight, S. L. & Russell, D. W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11, 72–82 (1997).
Johnson, B. et al. The Ah receptor nuclear translocator gene (ARNT) is located on q21 of human chromosome 1 and on mouse chromosome 3 near Cf-3. Genomics 17, 592–598 (1993).
Ledford, H. & Callaway, E. Biologists who decoded how cells sense oxygen win medicine Nobel. Nature 574, 161–162 (2019).
Bristow, R. G. & Hill, R. P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192 (2008).
Ye, Y. et al. Characterization of Hypoxia-associated Molecular Features to Aid Hypoxia-Targeted Therapy. Nat. Metab. 1, 431–444 (2019).
Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Invest 132, e159839 (2022).
Lartigau, E. Radiation sensitizing agents for hypoxic cells: past, present and future. Cancer Radiother. 2, 775–780 (1998).
Koong, A. C. et al. Pancreatic tumors show high levels of hypoxia. Int J. Radiat. Oncol. Biol. Phys. 48, 919–922 (2000).
Moulder, J. E. & Rockwell, S. Hypoxic fractions of solid tumors: experimental techniques, methods of analysis, and a survey of existing data. Int J. Radiat. Oncol. Biol. Phys. 10, 695–712 (1984).
Lartigau, E. et al. Oxygenation of head and neck tumors. Cancer 71, 2319–2325 (1993).
Abu El Maaty, M. A. et al. Hypoxia-mediated stabilization of HIF1A in prostatic intraepithelial neoplasia promotes cell plasticity and malignant progression. Sci. Adv. 8, eabo2295 (2022).
Beasley, N. J. P. et al. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res. 62, 2493–2497 (2002).
Höckel, M. et al. Intratumoral pO2 predicts survival in advanced cancer of the uterine cervix. Radiother. Oncol. 26, 45–50 (1993).
Hockel, M. et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 56, 4509–4515 (1996).
Luo, Z. et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct. Target Ther. 7, 218 (2022).
Auerbach, W. & Auerbach, R. Angiogenesis inhibition: a review. Pharm. Ther. 63, 265–311 (1994).
Zhao, Y. et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol. Cancer 16, 79 (2017).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Jain, R. K. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat. Med. 7, 987–989 (2001).
Jain, R. K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62 (2005).
Baluk, P., Hashizume, H. & McDonald, D. M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet Dev. 15, 102–111 (2005).
Nagy, J. A. et al. Heterogeneity of the tumor vasculature. Semin Thromb. Hemost. 36, 321–331 (2010).
Husain, A. et al. Ephrin-A3/EphA2 axis regulates cellular metabolic plasticity to enhance cancer stemness in hypoxic hepatocellular carcinoma. J. Hepatol. 77, 383–396 (2022).
Dewhirst, M. W. et al. Quantification of longitudinal tissue pO2 gradients in window chamber tumours: impact on tumour hypoxia. Br. J. Cancer 79, 1717–1722 (1999).
Dachs, G. U., Dougherty, G. J., Stratford, I. J. & Chaplin, D. J. Targeting gene therapy to cancer: a review. Oncol. Res. 9, 313–325 (1997).
Julien, C. et al. Assessment of vascular reactivity in rat brain glioma by measuring regional blood volume during graded hypoxic hypoxia. Br. J. Cancer 91, 374–380 (2004).
Vaupel, P., Kallinowski, F. & Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465 (1989).
Vonlaufen, A. et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 (2008).
Gilkes, D. M., Semenza, G. L. & Wirtz, D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer 14, 430–439 (2014).
Vaupel, P. Hypoxia in neoplastic tissue. Microvasc. Res. 13, 399–408 (1977).
Globig, P. et al. Mg-based materials diminish tumor spreading and cancer metastases. Bioact. Mater. 19, 594–610 (2023).
Herndon, B. L., Lally, J. J. & Hacker, M. Atmospheric pressure effects on tumor growth: hypobaric anoxia and growth of a murine transplantable tumor. J. Natl Cancer Inst. 70, 739–745 (1983).
Peters, L. J. & Fletcher, G. H. Causes of failure of radiotherapy in head and neck cancer. Radiother. Oncol. 1, 53–63 (1983).
Teicher, B. A., Lazo, J. S. & Sartorelli, A. C. Classification of antineoplastic agents by their selective toxicities toward oxygenated and hypoxic tumor cells. Cancer Res. 41, 73–81 (1981).
Tannock, I. F., Guttman, P. & Rauth, A. M. Failure of 2-deoxy-D-glucose and 5-thio-D-glucose to kill hypoxic cells of two murine tumors. Cancer Res. 43, 980–983 (1983).
Song, C. W., Clement, J. J. & Levitt, S. H. Preferential cytotoxicity of 5-thio-D-glucose against hypoxic tumor cells. J. Natl Cancer Inst. 57, 603–605 (1976).
Ludwig, C. Drug resistance of hypoxic tumour cells in vitro. Cancer Treat. Rev. 11(Suppl A), 173–178 (1984).
Papp-Szabó, E., Josephy, P. D. & Coomber, B. L. Microenvironmental influences on mutagenesis in mammary epithelial cells. Int. J. Cancer 116, 679–685 (2005).
Reynolds, T. Y., Rockwell, S. & Glazer, P. M. Genetic instability induced by the tumor microenvironment. Cancer Res. 56, 5754–5757 (1996).
Li, C. Y. et al. Persistent genetic instability in cancer cells induced by non-DNA-damaging stress exposures. Cancer Res. 61, 428–432 (2001).
Cairns, R. A. & Hill, R. P. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 64, 2054–2061 (2004).
Luk, C. K., Veinot-Drebot, L., Tjan, E. & Tannock, I. F. Effect of transient hypoxia on sensitivity to doxorubicin in human and murine cell lines. J. Natl Cancer Inst. 82, 684–692 (1990).
Stoler, D. L. et al. Anoxia-inducible endonuclease activity as a potential basis of the genomic instability of cancer cells. Cancer Res. 52, 4372–4378 (1992).
Yoshida, Y. et al. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 5, 237–241 (2009).
Mathieu, J. et al. Hypoxia induces re-entry of committed cells into pluripotency. Stem Cells 31, 1737–1748 (2013).
Mohyeldin, A., Garzón-Muvdi, T. & Quiñones-Hinojosa, A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7, 150–161 (2010).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Tuy, K., Rickenbacker, L. & Hjelmeland, A. B. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. 44, 101953 (2021).
Li, Z. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009).
Hernansanz-Agustín, P. et al. Na controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586, 287–291 (2020).
Semenza, G. L. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 36, 252–259 (2017).
Niu, Y. et al. Loss-of-function genetic screening identifies aldolase a as an essential driver for liver cancer cell growth under hypoxia. Hepatology 74, 1461–1479 (2021).
Hammond, E. M., Dorie, M. J. & Giaccia, A. J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem. 278, 12207–12213 (2003).
Hammer, S. et al. Hypoxic suppression of the cell cycle gene CDC25A in tumor cells. Cell Cycle 6, 1919–1926 (2007).
Gibson, S. L., Bindra, R. S. & Glazer, P. M. Hypoxia-induced phosphorylation of Chk2 in an ataxia telangiectasia mutated-dependent manner. Cancer Res. 65, 10734–10741 (2005).
Squatrito, M. et al. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 18, 619–629 (2010).
Freiberg, R. A. et al. DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response. Mol. Cell Biol. 26, 1598–1609 (2006).
Koritzinsky, M. et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25, 1114–1125 (2006).
Weterings, E. & van Gent, D. C. The mechanism of non-homologous end-joining: a synopsis of synapsis. DNA Repair (Amst.) 3, 1425–1435 (2004).
van Gent, D. C., Hoeijmakers, J. H. & Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet 2, 196–206 (2001).
Helleday, T., Lo, J., van Gent, D. C. & Engelward, B. P. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst.) 6, 923–935 (2007).
Bindra, R. S., Crosby, M. E. & Glazer, P. M. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 26, 249–260 (2007).
Chan, N. et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 68, 605–614 (2008).
Bindra, R. S. & Glazer, P. M. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 26, 2048–2057 (2007).
Chan, N. & Bristow, R. G. “Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repair. Clin. Cancer Res. 16, 4553–4560 (2010).
Kondo, A. et al. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 61, 7603–7607 (2001).
Gordan, J. D., Thompson, C. B. & Simon, M. C. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12, 108–113 (2007).
Akakura, N. et al. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 61, 6548–6554 (2001).
Giaccia, A., Siim, B. G. & Johnson, R. S. HIF-1 as a target for drug development. Nat. Rev. Drug Disco. 2, 803–811 (2003).
Chong, T. W. et al. A mycobacterial iron chelator, desferri-exochelin, induces hypoxia-inducible factors 1 and 2, NIP3, and vascular endothelial growth factor in cancer cell lines. Cancer Res. 62, 6924–6927 (2002).
Wenger, R. H. et al. Hypoxia-inducible factor-1 alpha is regulated at the post-mRNA level. Kidney Int. 51, 560–563 (1997).
Brahimi-Horn, C., Berra, E. & Pouysségur, J. Hypoxia: the tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol. 11, S32–S36 (2001).
Jaakkola, P. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 (2001).
Latif, F. et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260, 1317–1320 (1993).
Zimmer, M., Doucette, D., Siddiqui, N. & Iliopoulos, O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL-/- tumors. Mol. Cancer Res. 2, 89–95 (2004).
Erez, N. et al. Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res. 63, 8777–8783 (2003).
Evinger, M. J. et al. Hypoxia activates multiple transcriptional pathways in mouse pheochromocytoma cells. Ann. N. Y Acad. Sci. 971, 61–65 (2002).
Bemis, L. et al. Distinct aerobic and hypoxic mechanisms of HIF-alpha regulation by CSN5. Genes Dev. 18, 739–744 (2004).
Park, J.-H. et al. Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin. Cancer Res. 9, 433–440 (2003).
Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 (2004).
Wu, D. et al. Structural integration in hypoxia-inducible factors. Nature 524, 303–308 (2015).
Ando, H. et al. A hypoxia-inducible factor (HIF)-3α splicing variant, HIF-3α4 impairs angiogenesis in hypervascular malignant meningiomas with epigenetically silenced HIF-3α4. Biochem Biophys. Res. Commun. 433, 139–144 (2013).
Düvel, K. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010).
Zhong, H. et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60, 1541–1545 (2000).
Laughner, E. et al. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 21, 3995–4004 (2001).
Nakamura, H. et al. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J. Immunol. 174, 7592–7599 (2005).
LaGory, E. L. & Giaccia, A. J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 18, 356–365 (2016).
Bollinger, T. et al. Transcription regulates HIF-1α expression in CD4(+) T cells. Immunol. Cell Biol. 94, 109–113 (2016).
van Uden, P. et al. Evolutionary conserved regulation of HIF-1β by NF-κB. PLoS Genet. 7, e1001285 (2011).
Wan, J. & Wu, W. Hyperthermia induced HIF-1a expression of lung cancer through AKT and ERK signaling pathways. J. Exp. Clin. Cancer Res. 35, 119 (2016).
Wang, Z. et al. Panaxadiol inhibits programmed cell death-ligand 1 expression and tumour proliferation via hypoxia-inducible factor (HIF)-1α and STAT3 in human colon cancer cells. Pharm. Res. 155, 104727 (2020).
Lau, M. T., Klausen, C. & Leung, P. C. K. E-cadherin inhibits tumor cell growth by suppressing PI3K/Akt signaling via β-catenin-Egr1-mediated PTEN expression. Oncogene 30, 2753–2766 (2011).
Li, Y. et al. Expression of Notch-Hif-1α signaling pathway in liver regeneration of rats. J. Int. Med. Res. 48, 300060520943790 (2020).
Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 14, 34–44 (2000).
Zundel, W. et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14, 391–396 (2000).
Li, L.-C., Zhang, M., Feng, Y.-K. & Wang, X.-J. IDH1-R132H suppresses glioblastoma malignancy through FAT1-ROS-HIF-1α signaling. Neurol. India 68, 1050–1058 (2020).
Lal, A. et al. Transcriptional response to hypoxia in human tumors. J. Natl Cancer Inst. 93, 1337–1343 (2001).
Brizel, D. M. et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 56, 941–943 (1996).
Young, S. D., Marshall, R. S. & Hill, R. P. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc. Natl Acad. Sci. USA 85, 9533–9537 (1988).
Young, S. D. & Hill, R. P. Effects of reoxygenation on cells from hypoxic regions of solid tumors: anticancer drug sensitivity and metastatic potential. J. Natl Cancer Inst. 82, 371–380 (1990).
De Jaeger, K., Kavanagh, M. C. & Hill, R. P. Relationship of hypoxia to metastatic ability in rodent tumours. Br. J. Cancer 84, 1280–1285 (2001).
Höckel, M., Schlenger, K., Höckel, S. & Vaupel, P. Hypoxic cervical cancers with low apoptotic index are highly aggressive. Cancer Res. 59, 4525–4528 (1999).
Shyu, K.-G. et al. Hypoxia-inducible factor 1alpha regulates lung adenocarcinoma cell invasion. Exp. Cell Res. 313, 1181–1191 (2007).
Dong, L. et al. Arylsulfonamide 64B inhibits Hypoxia/HIF-induced expression of c-Met and CXCR4 and reduces primary tumor growth and metastasis of uveal melanoma. Clin. Cancer Res. 25, 2206–2218 (2019).
Zhu, Y. et al. C-C chemokine receptor type 1 mediates osteopontin-promoted metastasis in hepatocellular carcinoma. Cancer Sci. 109, 710–723 (2018).
Colla, S. et al. The new tumor-suppressor gene inhibitor of growth family member 4 (ING4) regulates the production of proangiogenic molecules by myeloma cells and suppresses hypoxia-inducible factor-1 alpha (HIF-1alpha) activity: involvement in myeloma-induced angiogenesis. Blood 110, 4464–4475 (2007).
Pez, F. et al. The HIF-1-inducible lysyl oxidase activates HIF-1 via the Akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 71, 1647–1657 (2011).
Wei, H. et al. Long non-coding RNA PAARH promotes hepatocellular carcinoma progression and angiogenesis via upregulating HOTTIP and activating HIF-1α/VEGF signaling. Cell Death Dis. 13, 102 (2022).
Rofstad, E. K. Microenvironment-induced cancer metastasis. Int J. Radiat. Biol. 76, 589–605 (2000).
Chan, D. A. & Giaccia, A. J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 26, 333–339 (2007).
Shi, Q. et al. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 5, 3711–3721 (1999).
Graeber, T. G. et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379, 88–91 (1996).
Zhang, L. & Hill, R. P. Hypoxia enhances metastatic efficiency by up-regulating Mdm2 in KHT cells and increasing resistance to apoptosis. Cancer Res. 64, 4180–4189 (2004).
Nordsmark, M. et al. Hypoxia in human soft tissue sarcomas: adverse impact on survival and no association with p53 mutations. Br. J. Cancer 84, 1070–1075 (2001).
Löfstedt, T. et al. HIF-1alpha induces MXI1 by alternate promoter usage in human neuroblastoma cells. Exp. Cell Res. 315, 1924–1936 (2009).
Li, F. et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 25, 6225–6234 (2005).
Zhang, H. et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11, 407–420 (2007).
Nilsson, J. A. & Cleveland, J. L. Myc pathways provoking cell suicide and cancer. Oncogene 22, 9007–9021 (2003).
Hoefflin, R. et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 11, 4111 (2020).
Das, B. et al. Regulates the stemness pathway via and to maintain self-renewal in cancer stem cells versus non-stem cancer cells. Cancer Res. 79, 4015–4025 (2019).
Pouysségur, J. et al. ‘Warburg effect’ controls tumor growth, bacterial, viral infections and immunity - Genetic deconstruction and therapeutic perspectives. Semin Cancer Biol. 86, 334–346 (2022).
Liu, X. et al. Hypoxia-induced upregulation of Orai1 drives colon cancer invasiveness and angiogenesis. Eur. J. Pharm. 832, 1–10 (2018).
Liu, M. et al. Hypoxia-inducible factor 1-alpha up-regulates the expression of phospholipase D2 in colon cancer cells under hypoxic conditions. Med. Oncol. 32, 394 (2015).
Du, K. et al. ANXA3 is upregulated by hypoxia-inducible factor 1-alpha and promotes colon cancer growth. Transl. Cancer Res. 9, 7440–7449 (2020).
Ding, X. et al. CTHRC1 promotes gastric cancer metastasis via HIF-1α/CXCR4 signaling pathway. Biomed. Pharmacother. 123, 109742 (2020).
Hoffmann, C. et al. Hypoxia promotes breast cancer cell invasion through HIF-1α-mediated up-regulation of the invadopodial actin bundling protein CSRP2. Sci. Rep. 8, 10191 (2018).
Khumukcham, S. S. et al. A reciprocal feedback loop between HIF-1α and HPIP controls phenotypic plasticity in breast cancer cells. Cancer Lett. 526, 12–28 (2022).
Yang, J. et al. HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J. Exp. Clin. Cancer Res. 35, 26 (2016).
Wang, C., Han, C., Zhang, Y. & Liu, F. LncRNA PVT1 regulate expression of HIF1α via functioning as ceRNA for miR‑199a‑5p in non‑small cell lung cancer under hypoxia. Mol. Med. Rep. 17, 1105–1110 (2018).
Liu, L. et al. Hypoxia promotes gastric cancer malignancy partly through the HIF-1α dependent transcriptional activation of the long non-coding RNA GAPLINC. Front Physiol. 7, 420 (2016).
Zeng, Z. et al. LncRNA-MTA2TR functions as a promoter in pancreatic cancer via driving deacetylation-dependent accumulation of HIF-1α. Theranostics 9, 5298–5314 (2019).
Wu, X. et al. Fibroblast growth factor 11 (FGF11) promotes non-small cell lung cancer (NSCLC) progression by regulating hypoxia signaling pathway. J. Transl. Med. 19, 353 (2021).
Xia, X. et al. Hypoxic gastric cancer-derived exosomes promote progression and metastasis via MiR-301a-3p/PHD3/HIF-1α positive feedback loop. Oncogene 39, 6231–6244 (2020).
Zhang, K.-D. et al. MiR-301a transcriptionally activated by HIF-2α promotes hypoxia-induced epithelial-mesenchymal transition by targeting TP63 in pancreatic cancer. World J. Gastroenterol. 26, 2349–2373 (2020).
Sun, S. et al. Hypoxia-responsive miR-141-3p is involved in the progression of breast cancer via mediating the HMGB1/HIF-1α signaling pathway. J. Gene Med. 22, e3230 (2020).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Shrieve, D. C., Deen, D. F. & Harris, J. W. Effects of extreme hypoxia on the growth and viability of EMT6/SF mouse tumor cells in vitro. Cancer Res. 43, 3521–3527 (1983).
Gordan, J. D. et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14, 435–446 (2008).
Firth, J. D., Ebert, B. L., Pugh, C. W. & Ratcliffe, P. J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3’ enhancer. Proc. Natl Acad. Sci. USA 91, 6496–6500 (1994).
Semenza, G. L., Roth, P. H., Fang, H. M. & Wang, G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757–23763 (1994).
Discher, D. J. et al. Hypoxia regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J. Biol. Chem. 273, 26087–26093 (1998).
Liu, X. et al. Chromatin remodeling induced by ARID1A loss in lung cancer promotes glycolysis and confers JQ1 vulnerability. Cancer Res. 82, 791–804 (2022).
Koslowski, M. et al. Multiple splice variants of lactate dehydrogenase C selectively expressed in human cancer. Cancer Res. 62, 6750–6755 (2002).
Koukourakis, M. I. et al. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br. J. Cancer 89, 877–885 (2003).
Osthus, R. C. et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800 (2000).
Kim, J.-w, Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Dupuy, F. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015).
Papandreou, I. et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 (2006).
Gordan, J. D. et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 11, 335–347 (2007).
Lee, P., Chandel, N. S. & Simon, M. C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020).
Gupta, V. K. et al. Hypoxia-driven oncometabolite L-2HG maintains stemness-differentiation balance and facilitates immune evasion in pancreatic cancer. Cancer Res. 81, 4001–4013 (2021).
Faubert, B. et al. Lactate metabolism in human lung tumors. Cell 171, 358–371.e9 (2017).
Luo, X. et al. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 16, 76 (2017).
Furuta, E. et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 68, 1003–1011 (2008).
Gao, X. et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960 (2016).
Ni, T. et al. Oroxylin A suppresses the development and growth of colorectal cancer through reprogram of HIF1α-modulated fatty acid metabolism. Cell Death Dis. 8, e2865 (2017).
Bauer, D. E. et al. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 (2005).
Lin, J. et al. Hypoxia-induced miR-27 and miR-195 regulate ATP consumption, viability, and metabolism of rat cardiomyocytes by targeting PPARγ and FASN expression. Aging (Albany NY) 13, 10158–10174 (2021).
Haraguchi, M. et al. Snail modulates cell metabolism in MDCK cells. Biochem. Biophys. Res. Commun. 432, 618–625 (2013).
Keenan, M. M. et al. ACLY and ACC1 Regulate Hypoxia-Induced Apoptosis by Modulating ETV4 via α-ketoglutarate. PLoS Genet. 11, e1005599 (2015).
Yoon, H. et al. PHD3 loss promotes exercise capacity and fat oxidation in skeletal muscle. Cell Metab. 32, 215–228.e7 (2020).
German, N. J. et al. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol. Cell 63, 1006–1020 (2016).
Ackerman, D. et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 24, 2596–2605.e5 (2018).
Lai, K. K. Y. et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 152, 1477–1491 (2017).
Luis, G. et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 43, 102006 (2021).
Li, J. et al. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ. Res. 97, 698–706 (2005).
Melana, J. P. et al. The hypoxic microenvironment induces stearoyl-CoA desaturase-1 overexpression and lipidomic profile changes in clear cell renal cell carcinoma. Cancers 13, 2962 (2021).
Zhang, Y. et al. Positive feedback loop and synergistic effects between hypoxia-inducible factor-2α and stearoyl-CoA desaturase-1 promote tumorigenesis in clear cell renal cell carcinoma. Cancer Sci. 104, 416–422 (2013).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
Sun, R. C. & Denko, N. C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 19, 285–292 (2014).
Holleran, A. L., Briscoe, D. A., Fiskum, G. & Kelleher, J. K. Glutamine metabolism in AS-30D hepatoma cells. Evidence for its conversion into lipids via reductive carboxylation. Mol. Cell Biochem. 152, 95–101 (1995).
Gameiro, P. A. et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 17, 372–385 (2013).
Dennis, E. A. et al. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185 (2011).
Kim, K.-S. et al. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 66, 7983–7990 (2006).
Selvy, P. E., Lavieri, R. R., Lindsley, C. W. & Brown, H. A. Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119 (2011).
Yao, B. et al. Hypoxia-induced cofilin 1 promotes hepatocellular carcinoma progression by regulating the PLD1/AKT pathway. Clin. Transl. Med. 11, e366 (2021).
Toschi, A. et al. HIF alpha expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene 27, 2746–2753 (2008).
Han, S. et al. Phospholipase D activates HIF-1-VEGF pathway via phosphatidic acid. Exp. Mol. Med. 46, e126 (2014).
Rios-Colon, L. et al. Carnitine palmitoyltransferase 1 regulates prostate cancer growth under hypoxia. Cancers 13, 6302 (2021).
Zaugg, K. et al. Carnitine palmitoyltransferase 1 C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 25, 1041–1051 (2011).
Du, W. et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 8, 1769 (2017).
Ezzeddini, R. et al. Downregulation of fatty acid oxidation by involvement of HIF-1α and PPARγ in human gastric adenocarcinoma and related clinical significance. J. Physiol. Biochem. 77, 249–260 (2021).
Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971).
Richard, D. E., Berra, E. & Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J. Biol. Chem. 275, 26765–26771 (2000).
Xu, T. et al. HIF-1alpha/VEGF pathway mediates 1,3,6,8-tetrabromo-9 H-carbazole-induced angiogenesis: a potential vascular toxicity of an emerging contaminant. J. Hazard Mater. 432, 128718 (2022).
Wu, C. et al. Injectable conductive and angiogenic hydrogels for chronic diabetic wound treatment. J. Control Release 344, 249–260 (2022).
Hlatky, L., Tsionou, C., Hahnfeldt, P. & Coleman, C. N. Mammary fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cancer Res. 54, 6083–6086 (1994).
Seo, S. H. et al. Hypoxia-induced ELF3 promotes tumor angiogenesis through IGF1/IGF1R. EMBO Rep. 23, e52977 (2022).
Amato, R. et al. HIF-1-dependent induction of β3 adrenoceptor: evidence from the mouse retina. Cells 11, 1271 (2022).
Krzywinska, E. et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 8, 1597 (2017).
Carmeliet, P. & Jain, R. K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Disco. 10, 417–427 (2011).
Gao, F. et al. Hypoxia-induced alterations in hyaluronan and hyaluronidase. Adv. Exp. Med. Biol. 566, 249–256 (2005).
Duffy, J. P., Eibl, G., Reber, H. A. & Hines, O. J. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol. Cancer 2, 12 (2003).
Giatromanolaki, A. et al. ‘Invading edge vs. inner’ (edvin) patterns of vascularization: an interplay between angiogenic and vascular survival factors defines the clinical behaviour of non-small cell lung cancer. J. Pathol. 192, 140–149 (2000).
Wu, Y., Biswas, D. & Swanton, C. Impact of cancer evolution on immune surveillance and checkpoint inhibitor response. Semin Cancer Biol. 84, 89–102 (2022).
Chouaib, S., Noman, M. Z., Kosmatopoulos, K. & Curran, M. A. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 36, 439–445 (2017).
Ding, X.-C. et al. The relationship between expression of PD-L1 and HIF-1α in glioma cells under hypoxia. J. Hematol. Oncol. 14, 92 (2021).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Cramer, T. et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 (2003).
Walmsley, S. R. et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 201, 105–115 (2005).
Jantsch, J. et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 180, 4697–4705 (2008).
McNamee, E. N., Korns Johnson, D., Homann, D. & Clambey, E. T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 55, 58–70 (2013).
Takeda, N. et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 24, 491–501 (2010).
Finlay, D. K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012).
Fox, C. J., Hammerman, P. S. & Thompson, C. B. Fuel feeds function: energy metabolism and the T-cell response. Nat. Rev. Immunol. 5, 844–852 (2005).
Maciver, N. J. et al. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 84, 949–957 (2008).
Cham, C. M. & Gajewski, T. F. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677 (2005).
Cham, C. M., Driessens, G., O’Keefe, J. P. & Gajewski, T. F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 38, 2438–2450 (2008).
Palazon, A. et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32, 669–683.e5 (2017).
Lukashev, D. et al. Differential regulation of two alternatively spliced isoforms of hypoxia-inducible factor-1 alpha in activated T lymphocytes. J. Biol. Chem. 276, 48754–48763 (2001).
Dang, E. V. et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011).
Doedens, A. L. et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 14, 1173–1182 (2013).
Sharma, S. et al. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 20, 37 (2019).
Liikanen, I. et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8 + T cells. J. Clin. Invest. 131, e143729 (2021).
Lee, J. H., Elly, C., Park, Y. & Liu, Y.-C. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity 42, 1062–1074 (2015).
Melder, R. J. et al. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat. Med. 2, 992–997 (1996).
Facciabene, A. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230 (2011).
Veliça, P. et al. Modified hypoxia-inducible factor expression in CD8 T cells increases antitumor efficacy. Cancer Immunol. Res. 9, 401–414 (2021).
Lukashev, D. et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J. Immunol. 177, 4962–4965 (2006).
Park, J. H. et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat. Immunol. 22, 336–346 (2021).
Robbins, J. R. et al. Hypoxia modulates early events in T cell receptor-mediated activation in human T lymphocytes via Kv1.3 channels. J. Physiol. 564, 131–143 (2005).
Ma, S. et al. Hypoxia induces HIF1α-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat. Commun. 13, 4118 (2022).
Hatfield, S. M. et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 7, 277ra230 (2015).
Nagaraj, S. et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 13, 828–835 (2007).
Ohta, A. et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl Acad. Sci. USA 103, 13132–13137 (2006).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021).
Zandberg, D. P. et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J. Immunother. Cancer 9, e002088 (2021).
Watson, P. H. et al. Carbonic anhydrase XII is a marker of good prognosis in invasive breast carcinoma. Br. J. Cancer 88, 1065–1070 (2003).
Fischer, K. et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007).
Zhang, Y. et al. YYFZBJS inhibits colorectal tumorigenesis by enhancing Tregs-induced immunosuppression through HIF-1α mediated hypoxia in vivo and in vitro. Phytomedicine 98, 153917 (2022).
Konieczny, P. et al. Interleukin-17 governs hypoxic adaptation of injured epithelium. Science 377, eabg9302 (2022).
Shi, L. Z. et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Deaglio, S. et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 (2007).
Jarnicki, A. G., Lysaght, J., Todryk, S. & Mills, K. H. G. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8 + regulatory T cells. J. Immunol. 177, 896–904 (2006).
Hsu, T.-S. et al. HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 11, 5005 (2020).
Sato, E. et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl Acad. Sci. USA 102, 18538–18543 (2005).
Byrnes, J. R. et al. Hypoxia is a dominant remodeler of the effector T cell surface proteome relative to activation and regulatory T cell suppression. Mol. Cell Proteom. 21, 100217 (2022).
Sitkovsky, M. V. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 30, 102–108 (2009).
Roy, S. et al. EGFR-HIF1α signaling positively regulates the differentiation of IL-9 producing T helper cells. Nat. Commun. 12, 3182 (2021).
Li, X. et al. miR-582 negatively regulates pre-B cell proliferation and survival through targeting Hif1α and Rictor. Cell Death Dis. 13, 107 (2022).
Burrows, N. et al. Dynamic regulation of hypoxia-inducible factor-1α activity is essential for normal B cell development. Nat. Immunol. 21, 1408–1420 (2020).
Jellusova, J. et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 18, 303–312 (2017).
Cho, S. H. et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 537, 234–238 (2016).
Bos, R. et al. Protein expression of B-cell lymphoma gene 6 (BCL-6) in invasive breast cancer is associated with cyclin D1 and hypoxia-inducible factor-1alpha (HIF-1alpha). Oncogene 22, 8948–8951 (2003).
Jang, J.-W., Thuy, P. X., Lee, J.-W. & Moon, E.-Y. CXCR4 promotes B cell viability by the cooperation of nuclear factor (erythroid-derived 2)-like 2 and hypoxia-inducible factor-1α under hypoxic conditions. Cell Death Dis. 12, 330 (2021).
Hennequin, A. et al. Tumor infiltration by Tbet+ effector T cells and CD20 + B cells is associated with survival in gastric cancer patients. Oncoimmunology 5, e1054598 (2016).
Muthalagu, N. et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Disco. 10, 872–887 (2020).
Shrestha, B. et al. B cell-derived vascular endothelial growth factor A promotes lymphangiogenesis and high endothelial venule expansion in lymph nodes. J. Immunol. 184, 4819–4826 (2010).
Gunderson, A. J. et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Disco. 6, 270–285 (2016).
Ammirante, M. et al. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl Acad. Sci. USA 111, 14776–14781 (2014).
Le, A. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012).
Sander, S. et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 22, 167–179 (2012).
Meng, X. et al. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 9, 251 (2018).
Shen, P. et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370 (2014).
Wang, J. et al. Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 75, 306–315 (2015).
Zhang, Y. et al. Hypoxia enhances IL-10-producing B cell generation through upregulating high-mobility group B1 on tumor cell-released autophagosomes. Immunol. Lett. 216, 36–42 (2019).
Turnis, M. E. et al. Interleukin-35 Limits Anti-Tumor. Immunity Immunity 44, 316–329 (2016).
Sawant, D. V. et al. Adaptive plasticity of IL-10 and IL-35 T cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 20, 724–735 (2019).
Conter, L. J., Song, E., Shlomchik, M. J. & Tomayko, M. M. CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS One 9, e92009 (2014).
Zhang, F. et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8 T Cell Responses. Immunity 50, 738–750.e7 (2019).
Mauri, C. & Bosma, A. Immune regulatory function of B cells. Annu Rev. Immunol. 30, 221–241 (2012).
Kaelin, W. G. Common pitfalls in preclinical cancer target validation. Nat. Rev. Cancer 17, 425–440 (2017).
Lee, K. E. et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Disco. 6, 256–269 (2016).
Lee, K. E. et al. Hif1α deletion limits tissue regeneration via aberrant B cell accumulation in experimental pancreatitis. Cell Rep. 23, 3457–3464 (2018).
Kim, R. et al. Early tumor-immune microenvironmental remodeling and response to first-line fluoropyrimidine and platinum chemotherapy in advanced gastric cancer. Cancer Disco. 12, 984–1001 (2022).
Dong, F. et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and IL8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res. 81, 5876–5888 (2021).
Chaturvedi, P., Gilkes, D. M., Takano, N. & Semenza, G. L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl Acad. Sci. USA 111, E2120–E2129 (2014).
Wang, Y. et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv. Sci. (Weinh.) 6, 1901278 (2019).
Vitale, I. et al. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50 (2019).
Mantovani, A. et al. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Chen, X.-J. et al. A novel lymphatic pattern promotes metastasis of cervical cancer in a hypoxic tumour-associated macrophage-dependent manner. Angiogenesis 24, 549–565 (2021).
Piao, H. et al. A positive feedback loop between gastric cancer cells and tumor-associated macrophage induces malignancy progression. J. Exp. Clin. Cancer Res. 41, 174 (2022).
Ruffell, B. & Coussens, L. M. Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015).
Cowman, S. J. et al. Macrophage HIF-1α is an independent prognostic indicator in kidney cancer. Clin. Cancer Res. 26, 4970–4982 (2020).
Blouin, C. C., Pagé, E. L., Soucy, G. M. & Richard, D. E. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood 103, 1124–1130 (2004).
Jeny, F. et al. Hypoxia promotes a mixed inflammatory-fibrotic macrophages phenotype in active sarcoidosis. Front Immunol. 12, 719009 (2021).
Doedens, A. L. et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 70, 7465–7475 (2010).
Graham, C. et al. Mechanisms of hypoxia-induced immune escape in cancer and their regulation by nitric oxide. Redox Biol. 5, 417 (2015).
Melillo, G. et al. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 182, 1683–1693 (1995).
Zhang, W. et al. Crosstalk between IL-15Rα tumor-associated macrophages and breast cancer cells reduces CD8 T cell recruitment. Cancer Commun. (Lond.) 42, 536–557 (2022).
Malier, M. et al. Hypoxia drives dihydropyrimidine dehydrogenase expression in macrophages and confers chemoresistance in colorectal cancer. Cancer Res. 81, 5963–5976 (2021).
Onita, T. et al. Hypoxia-induced, perinecrotic expression of endothelial Per-ARNT-Sim domain protein-1/hypoxia-inducible factor-2alpha correlates with tumor progression, vascularization, and focal macrophage infiltration in bladder cancer. Clin. Cancer Res. 8, 471–480 (2002).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Chiu, D. K.-C. et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 64, 797–813 (2016).
Xu, Y. et al. PINK1 deficiency in gastric cancer compromises mitophagy, promotes the Warburg effect, and facilitates M2 polarization of macrophages. Cancer Lett. 529, 19–36 (2022).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Oberg, H. H. et al. Tribody [(HER2)xCD16] is more effective than trastuzumab in enhancing γδ T cell and natural killer cell cytotoxicity against HER2-expressing cancer cells. Front Immunol. 9, 814 (2018).
Rodig, S. J. et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 10, eaar3342 (2018).
Wang, S. et al. Blocking CD47 promotes antitumour immunity through CD103 dendritic cell-NK cell axis in murine hepatocellular carcinoma model. J. Hepatol. 77, 467–478 (2022).
André, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743.e13 (2018).
Sarkar, S. et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 8, e64835 (2013).
Sobecki, M. et al. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nat. Commun. 12, 4700 (2021).
Ormiston, M. L. et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 126, 1099–1109 (2012).
Cui, C. et al. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 38, 229 (2019).
Messai, Y. et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 74, 6820–6832 (2014).
Viry, E. et al. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 10, 173–175 (2014).
Reid, M. D. et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol. 24, 1612–1619 (2011).
Sadiku, P. et al. Neutrophils fuel effective immune responses through gluconeogenesis and glycogenesis. Cell Metab. 33, 1062–1064 (2021).
Watts, E. R. et al. Hypoxia drives murine neutrophil protein scavenging to maintain central carbon metabolism. J. Clin. Invest 131, e134073 (2021).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015).
Wilson, C. L. et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 6, 6818 (2015).
Tyrkalska, S. D. et al. Neutrophils mediate Salmonella Typhimurium clearance through the GBP4 inflammasome-dependent production of prostaglandins. Nat. Commun. 7, 12077 (2016).
Westphal, K. et al. Containment of tumor-colonizing bacteria by host neutrophils. Cancer Res. 68, 2952–2960 (2008).
Tian, S. et al. Tumour-associated neutrophils secrete AGR2 to promote colorectal cancer metastasis via its receptor CD98hc-xCT. Gut 71, 2489–2501 (2022).
Steele, C. W. et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845 (2016).
Wang, L. et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut 0, 1–14 (2022)..
Willson, J. A. et al. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood 139, 281–286 (2022).
Zhang, Y. et al. Neutrophil cyto-pharmaceuticals suppressing tumor metastasis via inhibiting hypoxia-inducible factor-1α in circulating breast cancer cells. Adv. Health. Mater. 11, e2101761 (2022).
Monceaux, V. et al. Anoxia and glucose supplementation preserve neutrophil viability and function. Blood 128, 993–1002 (2016).
Blaisdell, A. et al. Neutrophils oppose uterine epithelial carcinogenesis via debridement of hypoxic tumor cells. Cancer Cell 28, 785–799 (2015).
Mahiddine, K. et al. Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J. Clin. Invest. 130, 389–403 (2020).
Chafe, S. C. et al. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. 75, 996–1008 (2015).
Hong, J. et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J. Natl Cancer Inst. 107, djv209 (2015).
Horikawa, N. et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br. J. Cancer 122, 778–788 (2020).
Gunda, V. et al. Anti-PD-1/PD-L1 therapy augments lenvatinib’s efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int J. Cancer 144, 2266–2278 (2019).
Chiu, D. K.-C. et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 8, 517 (2017).
Noman, M. Z. et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 211, 781–790 (2014).
Corzo, C. A. et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med 207, 2439–2453 (2010).
Noman, M. Z. et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. 75, 3771–3787 (2015).
Li, J. et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6, e1320011 (2017).
Wang, Y. et al. Hypoxia and TGF-β1 lead to endostatin resistance by cooperatively increasing cancer stem cells in A549 transplantation tumors. Cell Biosci. 5, 72 (2015).
Guo, X. et al. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten pathways. Oncogene 37, 4239–4259 (2018).
Liu, G. et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 74, 727–737 (2014).
Yu, Q., Dong, L., Li, Y. & Liu, G. SIRT1 and HIF1α signaling in metabolism and immune responses. Cancer Lett. 418, 20–26 (2018).
Guo, X. et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J. Cancer 144, 3111–3126 (2019).
Jacquelot, N., Seillet, C., Vivier, E. & Belz, G. T. Innate lymphoid cells and cancer. Nat. Immunol. 23, 371–379 (2022).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Salimi, M. et al. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer 18, 341 (2018).
Moral, J. A. et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579, 130–135 (2020).
Ren, G. et al. Transcription factors TCF-1 and GATA3 are key factors for the epigenetic priming of early innate lymphoid progenitors toward distinct cell fates. Immunity 55, 1402–1413 (2022).
Heinrich, B. et al. The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut 71, 1161–1175 (2022).
Li, Z. et al. ILC1s control leukemia stem cell fate and limit development of AML. Nat. Immunol. 23, 718–730 (2022).
Kansler, E. R. et al. Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat. Immunol. 23, 904–915 (2022).
Song, J. et al. Requirement of RORα for maintenance and antitumor immunity of liver-resident natural killer cells/ILC1s. Hepatology 75, 1181–1193 (2022).
Kim, J. et al. Intratumorally establishing type 2 innate lymphoid cells blocks tumor growth. J. Immunol. 196, 2410–2423 (2016).
Ye, L. et al. Hypoxia-reprogrammed regulatory group 2 innate lymphoid cells promote immunosuppression in pancreatic cancer. EBioMedicine 79, 104016 (2022).
Jou, E. et al. An innate IL-25-ILC2-MDSC axis creates a cancer-permissive microenvironment for mutation-driven intestinal tumorigenesis. Sci. Immunol. 7, eabn0175 (2022).
Goc, J. et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 184, 5015–5030 (2021).
Bruchard, M. et al. Recruitment and activation of type 3 innate lymphoid cells promote antitumor immune responses. Nat. Immunol. 23, 262–274 (2022).
Gronke, K. et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566, 249–253 (2019).
Wilson, R. E., Keng, P. C. & Sutherland, R. M. Drug resistance in Chinese hamster ovary cells during recovery from severe hypoxia. J. Natl Cancer Inst. 81, 1235–1240 (1989).
Skarsgard, L. D., Vinczan, A., Skwarchuk, M. W. & Chaplin, D. J. The effect of low pH and hypoxia on the cytotoxic effects of SR4233 and mitomycin C in vitro. Int J. Radiat. Oncol. Biol. Phys. 29, 363–367 (1994).
Yokoi, K. & Fidler, I. J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 10, 2299–2306 (2004).
Sakata, K. et al. Hypoxia-induced drug resistance: comparison to P-glycoprotein-associated drug resistance. Br. J. Cancer 64, 809–814 (1991).
Pinzón-Daza, M. L. et al. Oxidative stress promotes doxorubicin-induced Pgp and BCRP expression in colon cancer cells under hypoxic conditions. J. Cell Biochem. 118, 1868–1878 (2017).
Lv, Y. et al. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. Onco Targets Ther. 8, 1941–1948 (2015).
Geller, L. T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 (2017).
El-Gebali, S., Bentz, S., Hediger, M. A. & Anderle, P. Solute carriers (SLCs) in cancer. Mol. Asp. Med. 34, 719–734 (2013).
Gao, X.-Z. et al. Blocking OLFM4/HIF-1α axis alleviates hypoxia-induced invasion, epithelial-mesenchymal transition, and chemotherapy resistance in non-small-cell lung cancer. J. Cell Physiol. 234, 15035–15043 (2019).
Wang, K. et al. Interleukin-6 contributes to chemoresistance in MDA-MB-231 cells via targeting HIF-1α. J. Biochem. Mol. Toxicol. 32, e22039 (2018).
Okazaki, M. et al. The effect of HIF-1α and PKM1 expression on acquisition of chemoresistance. Cancer Manag Res. 10, 1865–1874 (2018).
Taniguchi, K. et al. PKM1 is involved in resistance to anti-cancer drugs. Biochem. Biophys. Res. Commun. 473, 174–180 (2016).
Jain, S. et al. Metabolic targeting of cancer by a ubiquinone uncompetitive inhibitor of mitochondrial complex I. Cell Chem. Biol. 29, 436–450.e15 (2022).
Zhao, Q. et al. HIF-1α induces multidrug resistance in gastric cancer cells by inducing MiR-27a. PLoS ONE 10, e0132746 (2015).
Tang, Y.-A. et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl Acad. Sci. USA 115, E5990–E5999 (2018).
Zhao, D. et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal 26, 1030–1039 (2014).
Zhao, C.-X., Luo, C.-L. & Wu, X.-H. Hypoxia promotes 786-O cells invasiveness and resistance to sorafenib via HIF-2α/COX-2. Med. Oncol. 32, 419 (2015).
Wu, H.-M. et al. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 5, 12291 (2015).
Okami, J., Simeone, D. M. & Logsdon, C. D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 64, 5338–5346 (2004).
Giatromanolaki, A. et al. BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin. Cancer Res. 10, 5566–5571 (2004).
Shannon, A. M., Bouchier-Hayes, D. J., Condron, C. M. & Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 29, 297–307 (2003).
Telarovic, I., Wenger, R. H. & Pruschy, M. Interfering with tumor hypoxia for radiotherapy optimization. J. Exp. Clin. Cancer Res. 40, 197 (2021).
Mukherjee, B. et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 69, 4252–4259 (2009).
Olive, P. L. & Banáth, J. P. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J. Radiat. Oncol. Biol. Phys. 58, 331–335 (2004).
Brown, J. M., Evans, J. & Kovacs, M. S. The prediction of human tumor radiosensitivity in situ: an approach using chromosome aberrations detected by fluorescence in situ hybridization. Int J. Radiat. Oncol. Biol. Phys. 24, 279–286 (1992).
Zhang, H., Koch, C. J., Wallen, C. A. & Wheeler, K. T. Radiation-induced DNA damage in tumors and normal tissues. III. Oxygen dependence of the formation of strand breaks and DNA-protein crosslinks. Radiat. Res. 142, 163–168 (1995).
Um, J. H. et al. Association of DNA-dependent protein kinase with hypoxia inducible factor-1 and its implication in resistance to anticancer drugs in hypoxic tumor cells. Exp. Mol. Med. 36, 233–242 (2004).
Moeller, B. J., Cao, Y., Li, C. Y. & Dewhirst, M. W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5, 429–441 (2004).
Unruh, A. et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 22, 3213–3220 (2003).
Moeller, B. J. et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 8, 99–110 (2005).
Suwa, T. et al. SPINK1 as a plasma marker for tumor hypoxia and a therapeutic target for radiosensitization. JCI Insight 6, e148135 (2021).
Semenza, G. L. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell 5, 405–406 (2004).
Nilsson, M. B. et al. Altered regulation of HIF-1α in naive- and drug-resistant EGFR-mutant NSCLC: implications for a vascular endothelial growth factor-dependent phenotype. J. Thorac. Oncol. 16, 439–451 (2021).
Gorski, D. H. et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 59, 3374–3378 (1999).
Ning, S., Laird, D., Cherrington, J. M. & Knox, S. J. The antiangiogenic agents SU5416 and SU6668 increase the antitumor effects of fractionated irradiation. Radiat. Res. 157, 45–51 (2002).
Lund, E. L., Bastholm, L. & Kristjansen, P. E. Therapeutic synergy of TNP-470 and ionizing radiation: effects on tumor growth, vessel morphology, and angiogenesis in human glioblastoma multiforme xenografts. Clin. Cancer Res. 6, 971–978 (2000).
Koritzinsky, M., Wouters, B. G., Amellem, O. & Pettersen, E. O. Cell cycle progression and radiation survival following prolonged hypoxia and re-oxygenation. Int J. Radiat. Biol. 77, 319–328 (2001).
Zai, W. et al. Membrane vesicles as a catalase carrier for long-term tumor hypoxia relief to enhance radiotherapy. ACS Nano 15, 15381–15394 (2021).
Yuan, R. et al. Natural products to prevent drug resistance in cancer chemotherapy: a review. Ann. N. Y Acad. Sci. 1401, 19–27 (2017).
Secomb, T. W. et al. Analysis of the effects of oxygen supply and demand on hypoxic fraction in tumors. Acta Oncol. 34, 313–316 (1995).
Jirtle, R. L. Chemical modification of tumour blood flow. Int J. Hyperth. 4, 355–371 (1988).
Wilson, W. R. & Hay, M. P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 (2011).
Wu, J. et al. Oxygen microcapsules improve immune checkpoint blockade by ameliorating hypoxia condition in pancreatic ductal adenocarcinoma. Bioact. Mater. 20, 259–270 (2023).
Ma, J., Chen, C.-S., Blute, T. & Waxman, D. J. Antiangiogenesis enhances intratumoral drug retention. Cancer Res. 71, 2675–2685 (2011).
Hatfield, S. M. et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med (Berl.) 92, 1283–1292 (2014).
Kim, B.-M. et al. Reoxygenation following hypoxia activates DNA-damage checkpoint signaling pathways that suppress cell-cycle progression in cultured human lymphocytes. FEBS Lett. 581, 3005–3012 (2007).
Gao, P. et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12, 230–238 (2007).
Salman, S. et al. HIF inhibitor 32–134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J. Clin. Invest. 132, e156774 (2022).
Beppu, K. et al. Topotecan blocks hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res. 65, 4775–4781 (2005).
Shin, D. H. et al. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 111, 3131–3136 (2008).
Cho, H. et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 539, 107–111 (2016).
Chen, W. et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117 (2016).
Courtney, K. D. et al. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin. Cancer Res. 26, 793–803 (2020).
Chen, M. et al. HIF-2α-targeted interventional chemoembolization multifunctional microspheres for effective elimination of hepatocellular carcinoma. Biomaterials 284, 121512 (2022).
Hammers, H. J. et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 Study. J. Clin. Oncol. 35, 3851–3858 (2017).
Wei, T.-T. et al. Metabolic targeting of HIF-1α potentiates the therapeutic efficacy of oxaliplatin in colorectal cancer. Oncogene 39, 414–427 (2020).
Guo, H. et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat. Commun. 10, 278 (2019).
Estrada-Bernal, A. et al. Tarloxotinib is a hypoxia-activated Pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin. Cancer Res. 27, 1463–1475 (2021).
Chawla, S. P. et al. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J. Clin. Oncol. 32, 3299–3306 (2014).
Tap, W. D. et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 18, 1089–1103 (2017).
Zhang, X. et al. TET2 Suppresses VHL deficiency-driven clear cell renal cell carcinoma by inhibiting HIF signaling. Cancer Res. 82, 2097–2109 (2022).
Zegers, C. M. L. et al. In vivo quantification of hypoxic and metabolic status of NSCLC tumors using [18 F]HX4 and [18 F]FDG-PET/CT imaging. Clin. Cancer Res. 20, 6389–6397 (2014).
Zegers, C. M. L. et al. Evaluation of tumour hypoxia during radiotherapy using [F]HX4 PET imaging and blood biomarkers in patients with head and neck cancer. Eur. J. Nucl. Med Mol. Imaging 43, 2139–2146 (2016).
Yang, S. et al. Selectively Potentiating Hypoxia Levels by Combretastatin A4 Nanomedicine: Toward Highly Enhanced Hypoxia-Activated Prodrug Tirapazamine Therapy for Metastatic Tumors. Adv. Mater. 31, e1805955 (2019).
Brenner, A. et al. Hypoxia-activated evofosfamide for treatment of recurrent bevacizumab-refractory glioblastoma: a phase I surgical study. Neuro Oncol. 20, 1231–1239 (2018).
Rini, B. I. et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. 21, 95–104 (2020).
Agulnik, M. et al. A phase II study of tivozanib in patients with metastatic and nonresectable soft-tissue sarcomas. Ann. Oncol.: Off. J. Eur. Soc. Med. Oncol. 28, 121–127 (2017).
Courtney, K. D. et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J. Clin. Oncol. 36, 867–874 (2018).
Jayaprakash, P., Vignali, P. D. A., Delgoffe, G. M. & Curran, M. A. Hypoxia reduction sensitizes refractory cancers to immunotherapy. Annu Rev. Med. 73, 251–265 (2022).
Khan, M. S. et al. Oxygen-carrying micro/nanobubbles: composition, synthesis techniques and potential prospects in photo-triggered theranostics. Molecules 23, 2210 (2018).
Zhang, N. et al. DLL1 orchestrates CD8 T cells to induce long-term vascular normalization and tumor regression. Proc. Natl Acad. Sci. USA 118, e2020057118 (2021).
Jayaprakash, P. et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Invest 128, 5137–5149 (2018).
Onnis, A. et al. The pro-oxidant adaptor p66SHC promotes B cell mitophagy by disrupting mitochondrial integrity and recruiting LC3-II. Autophagy 14, 2117–2138 (2018).
Zhao, J. et al. Corrigendum to “Arenobufagin, isolated from toad venom, inhibited epithelial-to-mesenchymal transition and suppressed migration and invasion of lung cancer cells via targeting IKKβ/NFκB signal cascade”. J. Ethnopharmacol. 265, 113313 (2021).
Ni, J. et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α unleashes NK cell activity. Immunity 52, 1075–1087.e8 (2020).
Liu, X. et al. Hyperbaric oxygen boosts PD-1 antibody delivery and T cell infiltration for augmented immune responses against solid tumors. Adv. Sci. (Weinh.) 8, e2100233 (2021).
Solocinski, K. et al. Overcoming hypoxia-induced functional suppression of NK cells. J. Immunother. Cancer 8, e000246 (2020).
Schaue, D. & McBride, W. H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 12, 527–540 (2015).
Baumann, M. et al. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 16, 234–249 (2016).
Chen, X., Song, J., Chen, X. & Yang, H. X-ray-activated nanosystems for theranostic applications. Chem. Soc. Rev. 48, 3073–3101 (2019).
Semenza, G. L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547 (2011).
De Ruysscher, D. et al. Radiotherapy toxicity. Nat. Rev. Dis. Prim. 5, 13 (2019).
Hua, Y. et al. Composition-dependent enzyme mimicking activity and radiosensitizing effect of bimetallic clusters to modulate tumor hypoxia for enhanced cancer therapy. Adv. Mater. 34, e2203734 (2022).
Chai, R. et al. Oxygen-evolving photosynthetic cyanobacteria for 2D bismuthene radiosensitizer-enhanced cancer radiotherapy. Bioact. Mater. 17, 276–288 (2022).
Schwartz, D. L. et al. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 9, 2057–2067 (2010).
Zhang, C. & Pu, K. Molecular and nanoengineering approaches towards activatable cancer immunotherapy. Chem. Soc. Rev. 49, 4234–4253 (2020).
Zhang, S. et al. An NIR-II photothermally triggered “Oxygen Bomb” for hypoxic tumor programmed cascade therapy. Adv. Mater. 34, e2201978 (2022).
Liu, Y. et al. Human iPS cells loaded with MnO-based nanoprobes for photodynamic and simultaneous enhanced immunotherapy against cancer. Nanomicro Lett. 12, 127 (2020).
Wang, Y. et al. CXCR4-guided liposomes regulating hypoxic and immunosuppressive microenvironment for sorafenib-resistant tumor treatment. Bioact. Mater. 17, 147–161 (2022).
Sun, J. et al. A porous bimetallic Au@Pt core-shell oxygen generator to enhance hypoxia-dampened tumor chemotherapy synergized with NIR-II photothermal therapy. ACS Nano 16, 10711–10728 (2022).
Shi, L. et al. Hybrid nanospheres to overcome hypoxia and intrinsic oxidative resistance for enhanced photodynamic therapy. ACS Nano 14, 2183–2190 (2020).
Zuo, H. et al. Circumventing myeloid-derived suppressor cell-mediated immunosuppression using an oxygen-generated and -economized nanoplatform. ACS Appl. Mater. Interfaces 12, 55723–55736 (2020).
Hou, L. et al. Hybrid-membrane-decorated prussian blue for effective cancer immunotherapy via tumor-associated macrophages polarization and hypoxia relief. Adv. Mater. 34, e2200389 (2022).
Yang, Z. et al. Reshaping the tumor immune microenvironment based on a light-activated nanoplatform for efficient cancer therapy. Adv. Mater. 34, e2108908 (2022).
Chen, Z. et al. The microbiota and aging microenvironment in pancreatic cancer: cell origin and fate. Biochim. Biophys. Acta Rev. Cancer 1877, 188826 (2022).
Chen, Z. et al. Association of the microbiota and pancreatic cancer: opportunities and limitations. Front Immunol. 13, 844401 (2022).
Bettegowda, C. et al. Overcoming the hypoxic barrier to radiation therapy with anaerobic bacteria. Proc. Natl Acad. Sci. USA 100, 15083–15088 (2003).
Chen, W. et al. Dual drugs decorated bacteria irradiate deep hypoxic tumor and arouse strong immune responses. Biomaterials 286, 121582 (2022).
Yang, Z. et al. Engineering bioluminescent bacteria to boost photodynamic therapy and systemic anti-tumor immunity for synergistic cancer treatment. Biomaterials 281, 121332 (2022).
Mi, Z. et al. “Trojan Horse” Salmonella enabling tumor homing of silver nanoparticles via neutrophil infiltration for synergistic tumor therapy and enhanced biosafety. Nano Lett. 21, 414–423 (2021).
Grampp, S. et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 7, 13183 (2016).
Grampp, S. et al. Multiple renal cancer susceptibility polymorphisms modulate the HIF pathway. PLoS Genet. 13, e1006872 (2017).
Hartmann, H. et al. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134, 756–767 (2008).
Holden, V. I. et al. Bacterial siderophores that evade or overwhelm lipocalin 2 induce hypoxia inducible factor 1α and proinflammatory cytokine secretion in cultured respiratory epithelial cells. Infect. Immun. 82, 3826–3836 (2014).
Acknowledgements
Not applicable.
Funding
This article was supported by National Natural Science Foundation of China (82260555), Natural Science Foundation of Gansu Province (22JR11RA033), The First Hospital of Lanzhou University Intra-Hospital Fund Youth Fund (ldyyyn2020-93), Lanzhou Science and Technology Development Guiding Plan Project (2022-ZD-99).
Author information
Authors and Affiliations
Contributions
C.Z., H.F.F., and Z.W.C. conceived the review. C.Z., H.F.F., D.Y., S.H.Q., and Z.W.C. undertook the initial research. C.Z. and H.F.F. were involved in writing, Z.W.C. reviewed the manuscript, and all authors contributed to the final version. H.F.F. contributed equally to this work and should be considered co-first author. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Han, F., Du, Y. et al. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Sig Transduct Target Ther 8, 70 (2023). https://doi.org/10.1038/s41392-023-01332-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01332-8
This article is cited by
-
tRNA-derived small RNAs in human cancers: roles, mechanisms, and clinical application
Molecular Cancer (2024)
-
ANGPTL4 regulates ovarian cancer progression by activating the ERK1/2 pathway
Cancer Cell International (2024)
-
Lactylation stabilizes DCBLD1 activating the pentose phosphate pathway to promote cervical cancer progression
Journal of Experimental & Clinical Cancer Research (2024)
-
Cancer-associated fibroblast-secreted FGF7 as an ovarian cancer progression promoter
Journal of Translational Medicine (2024)
-
Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies
Signal Transduction and Targeted Therapy (2024)
