
Abstract
The ribosome is a multi-unit complex that translates mRNA into protein. Ribosome biogenesis is the process that generates ribosomes and plays an essential role in cell proliferation, differentiation, apoptosis, development, and transformation. The mTORC1, Myc, and noncoding RNA signaling pathways are the primary mediators that work jointly with RNA polymerases and ribosome proteins to control ribosome biogenesis and protein synthesis. Activation of mTORC1 is required for normal fetal growth and development and tissue regeneration after birth. Myc is implicated in cancer development by enhancing RNA Pol II activity, leading to uncontrolled cancer cell growth. The deregulation of noncoding RNAs such as microRNAs, long noncoding RNAs, and circular RNAs is involved in developing blood, neurodegenerative diseases, and atherosclerosis. We review the similarities and differences between eukaryotic and bacterial ribosomes and the molecular mechanism of ribosome-targeting antibiotics and bacterial resistance. We also review the most recent findings of ribosome dysfunction in COVID-19 and other conditions and discuss the consequences of ribosome frameshifting, ribosome-stalling, and ribosome-collision. We summarize the role of ribosome biogenesis in the development of various diseases. Furthermore, we review the current clinical trials, prospective vaccines for COVID-19, and therapies targeting ribosome biogenesis in cancer, cardiovascular disease, aging, and neurodegenerative disease.
Similar content being viewed by others

Introduction
The primary function of the ribosome is to synthesize proteins with mRNA as a template and amino acids as raw materials.1 Studies have demonstrated that the ribosome affects the rate of protein synthesis and plays a role in cell proliferation, differentiation, apoptosis, and transformation.2,3,4 When the ribosome is abnormal, it can severely affect the cell fate, causing various ribosome-related diseases such as COVID-19 virus infection, bacterial resistance, cardiovascular diseases (CVD), blood diseases, neurodegenerative diseases, and cancer.5,6,7 This review focuses on ribosome abnormalities to explore the molecular mechanisms of common diseases. Along this line, we discussed how the ribosome biogenesis pathway could be targeted for therapeutic purposes. For example, ribosomal protein S6 kinase 1 (S6K1) and nucleolin (Ncl) are drug targets for the treatment of cardiac hypertrophy and myocardial infarction (MI).
Ribosomes and ribosome biogenesis
Eukaryotic ribosome
Ribosomes comprise four ribosomal RNAs (rRNAs) and 80 ribosomal proteins (RPs). The eukaryotic ribosome includes two subunits, the 40S small subunit, and the 60S large subunit, which combine to form the 80S ribosome with translational activity.8 The 40S subunit is responsible for recognizing and binding messenger RNA (mRNA), while the function of the 60S subunit is to form peptide bonds. Hence, the ribosome is known as the “protein synthesis factory.”
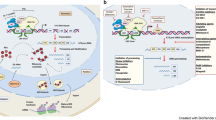
Ribosome biogenesis is the process of assembling the ribosome complex.9 This highly coordinated process is closely associated with protein synthesis, cell proliferation, differentiation, and apoptosis.10 Specifically, the process demands the organized synthesis of four rRNAs, 80 RPs, and ~70 small nucleolar RNAs.11,12 Within the nucleolus, the 47S pre-rRNA is transcribed from rDNA and cleaved to yield the 18S, 5.8S, and 28S rRNA. After synthesis in the cytoplasm, the RPs are imported to the nucleus to join the rRNAs. After modifications by snoRNAs, the rRNAs exit from the nucleolus. The 60S ribosome subunit is formed when the 5.8S, 28S rRNA is joined by the 5S rRNA in the nucleus and exported to the cytoplasm, while the 40S ribosome subunit is formed from the 18S rRNA. In the nucleolus of eukaryote cells, rRNA, RPs, small nucleolar RNAs, and other closely-related proteins participate in the generation of ribosome precursor 60S (pre-60S) and precursor 40S (pre-40S). The final ribosome assembly and maturation are completed when these molecules are exported to the cytoplasm (Fig. 1a).13

Schematic of ribosome biogenesis. a Eukaryotic ribosome biogenesis is a highly orchestrated process involving RNA Pol I, Pol II, and Pol III, which are responsible for transcribing rDNA to rRNA, producing 47S pre-rRNA in the nucleolus. The 4 rRNAs then assemble together with RPs to form a small ribosomal subunit (40S) and a large ribosomal subunit (60S). After assembly, the ribosome complex is exported from the nucleolus to the cytoplasm to form mature ribosomes for protein synthesis. b Bacterial ribosome biogenesis starts with the transcription of the precursors of 23S, 16S, and 5S rRNAs, and some tRNAs in the cytoplasm. The rRNAs then assemble with RPs to form a 30S small subunit and a 50S large subunit. After assembly, the ribosome complex forms mature ribosomes for protein synthesis. RP ribosome protein, RNA pol Ι/II/III RNA polymerase Ι/II/III, RNAP RNA polymerase, rDNA ribosomal DNA, rRNA, ribosomal RNA, tRNA transfer RNA
The maturation needs a series of chemical modifications of precursor rRNAs (pre-rRNAs). RNA polymerase I (Pol I) is required to process pre-47S transcripts into 5.8S, 18S, and 28S rRNA, and RNA polymerase III (Pol III) is responsible for the transcription of 5S rRNA.13 RPs are assembled into pre-rRNA transcripts and accelerate the formation of mature rRNA.14,15 Some RPs have other functions besides making ribosomes, including replication of DNA and cell division.16,17 Of note, ribosome dysfunction can lead to cellular dysfunction and eventually induce various diseases.
Bacteria ribosome
Bacteria do not have a nucleus but have a cell wall, cell membrane, cytoplasm, and ribosomes. The bacterial ribosome is formed by the 30S, and 50S subunits, including three types of rRNA (23S, 16S, and 5S rRNA) and 54 RPs.18 Biogenesis of the bacterial ribosome starts when the primary rRNAs are transcribed, including 23S, 16S, and 5S rRNA precursors and some transfer RNAs (tRNAs).19 This process requires the participation of RNA polymerase (RNAP), a multi-subunit enzyme that synthesizes all types of RNA and catalyzes the transcription process of rDNA.20 Precursors of rRNAs and tRNAs fold into a unique structure, then excised from their primary transcripts. The 16S rRNA then assembles with RPs to form a 30S small subunit, while the 23S and 5S rRNAs assemble to form a 50S large subunit. The bacterial ribosomes are synthesized in the cytoplasm. After assembly, the ribosome complex forms the 70S mature ribosome rather than 80S due to tertiary structure (Fig. 1b).
Ribosome biogenesis is the process of creating ribosomes in a highly regulated manner. This process involves the synthesis of rRNAs and many RPs, which are required for proliferation and cell division.
Signaling pathways involved in ribosome biogenesis
Ribosome biogenesis is regulated by several signaling pathways, among which mammalian targets of rapamycin (mTOR), myelocytomatosis oncogene (Myc), and noncoding RNA (ncRNA) are the most important regulators.
mTOR
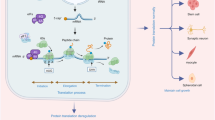
mTOR is a serine/threonine kinase that regulates ribosome biogenesis. mTOR is assembled into multiple complexes, such as mTOR complex 1/2 (mTORC1/2).21 mTORC1 senses environmental cues to promote cellular anabolism and inhibit catabolism. The mTORC1 pathway regulates pre-rRNA transcription, rRNA synthesis, and RP expression. mTORC1 positively regulates rDNA transcription by activating S6K1 and promotes the synthesis of ribosomal 40S subunits through RPS6. mTORC1 promotes the initiation and elongation of mRNA through eIF4E and eIF2α. mTORC1 also induces translation initiation by inactivating 4E-BPs. The RP assembly factor Urb1 is a downstream target of mTORC1 signaling. mTORC1 promotes the translation of mRNA by inhibiting the binding of LARP1 and PABPC1 (Fig. 2).22

mTORC1 and ribosome biogenesis. Through phosphorylation, mTORC1 activates S6K1, which enhances rDNA transcription. mTORC1phosphorylates RPS6 to promote the synthesis of ribosomal 40S subunits, actively regulates the formation of RPS10 and RPL26, and promotes the initiation and elongation of mRNA through eIF4E and eIF2α, respectively. mTORC1 also phosphorylates and inactivates 4E-BPs to induce translation initiation. The RP assembly factor Urb1 is a downstream target of mTORC1 signaling. mTORC1 inhibits the binding of LARP1 and PABPC1 to promote mRNA translation and protein synthesis. mTORC1 mammalian target of rapamycin C1, rDNA ribosomal DNA, S6K1 S6 kinase 1, 4E-BP 4E-binding protein
Ribosomal protein S6 kinase 1 (S6K1) is under the control of mTORC1 signaling.23 By activating the mTORC1/S6K1 pathway, checkpoint kinase 1 stimulates myocardial regeneration.3 Positive regulation of mTORC1 is required for normal fetal growth and development,24 and decreased expression of the RPs RPL26 and RPS10 in the placenta leads to intrauterine growth restriction in pregnant women. In addition, Urb1, an assembly factor of RPs, functions as a downstream target of mTORC1 signaling to promote the formation of the digestive systems in zebrafish.25 Therefore, the mTORC1/S6K1/RPL26/RPS10 ribosome biogenesis pathway plays an essential role in tissue development, growth, and regeneration.
mTORC1/S6K1 also regulates mRNA translation through multiple factors. The eukaryotic elongation factor 2 (eEF2) mediates ribosomal translocation.26 Its activity is inhibited by phosphorylation. S6K1 can enhance the activity of eEF2 by reducing its phosphorylation.27 The eukaryotic initiation factor 4G (eIF4G) phosphorylation stimulates protein synthesis. Vary et al.28 showed that mTORC1/S6K1 stimulates protein synthesis during skeletal muscle regeneration by increasing the phosphorylation of eIF4G. La-related protein 1 (LARP1) is an RNA-binding protein that regulates RP production. Smith et al.29 showed that inhibition of mTORC1 increases the binding of LARP1 to poly (A)-binding protein cytoplasmic 1 (PABPC1), resulting in translational inhibition of mRNA.
Moreover, 4E-binding protein 1 (4E-BP1), a key translation initiation factor downstream of mTORC1, is involved in metabolism, translation, and cell growth.30 Phosphorylation of 4E-BP1 results in the dissociation of 4E-BP1 from eIF4E, and association with eIF4G.31 eIF4G acts as a docking site to assemble other initiation factors, including eIF4A, which is an RNA helicase that interacts with eIF3 to recruit 40S ribosomes and other important initiation factors to the 5’ region of mRNA and begin protein synthesis.32
Importantly, inhibition of abnormal ribosome biogenesis using drugs that inhibit mTORC1/ribosomal protein S6 (RPS6) signaling is an effective strategy to treat cancer.33 Calvisi et al.34 showed that the activity of mTORC1 and RPS6 is higher in human hepatocellular carcinoma tissue than in the normal liver and promotes tumor progression. In addition, the mTORC1 inhibitor rapamycin significantly inhibits mTORC1 activity in cancer and RPS6 phosphorylation levels in bladder cancer. Similarly, rapamycin effectively inhibits the proliferation of lymphoma cells by inactivating the mTORC1/RPS6 signaling pathway and downregulating the expression of phosphorylated RPS6.35 PIK3CA mutations are common in some cancers. The mTORC1 inhibitor everolimus effectively inhibits cancer cell proliferation and differentiation in PIK3CA mutant colorectal cancer by reducing the phosphorylation level of RPS6.36
In summary, ribosome biogenesis regulated by mTORC1 plays a key role in tissue regeneration and cancer development.
Myc
Myc is one of the proto-oncogenes involved in abnormal ribosome biogenesis.37 An abnormal increase in nuclear size and quantity caused by Myc can be seen in most cancers. Mechanistically, Myc mainly regulates ribosome biogenesis through RNA Pol I–III-mediated rDNA transcription through UBF and SL1, rRNA processing and transcription of RPS and RPL, and the transcription of 5S rRNA and tRNA (Fig. 3).38,39

Myc regulates ribosome biogenesis. Myc directly regulates rRNA processing, ribosome assembly, translocation from the nucleus to the cytoplasm, and the early steps of mRNA translation. Myc upregulates the transcriptional levels of these factors by recruiting cofactors and remodeling chromatin structure. Myc promotes RNA Pol I-mediated rDNA transcription by binding to UBF and SL1. After transcription, the 47S pre-rRNA is processed into mature 5.8S, 18S, and 28S rRNA. The Myc–STP5/SEC complex stimulates rRNA processing and transcription of RPS and RPL for export in an RNA Pol II-dependent manner. Myc binds transcription factor IIIB (TFIIIB) and activates the transcription of 5S rRNA and tRNA mediated by RNA Pol III. Finally, Myc stimulates RP synthesis through these RNA Pol I–III pathways. Myc myelocytomatosis oncogene, SL1 selectivity factor 1, TFIIIB transcription factor IIIB, UBF upstream binding factor
Myc promotes the transcription of rDNA in an RNA Pol I-dependent manner by recruiting upstream binding factor (UBF) and selectivity factor 1 (SL1) and enhancing their expression. SL1 is a transcription factor that facilitates the transcription of rRNA by recruiting RNA Pol I.40 Myc recruits RNA Pol I to target promoters by interacting with the TBP-associated factor, a component of the SL1 complex.41 CX-5461, a small molecule inhibitor of RNA Pol I, represses the proliferation of neuroblastoma cells42 and myeloma by inhibiting Myc expression and activating p53, leading to cancer cell apoptosis.43 Therefore, targeting ribosome biogenesis provides a new opportunity to treat tumors expressing high levels of Myc.
Upon binding to the initiation factor factor-IA (TIF-IA, also named RRN3), RNA Pol I is activated and recruited to the ribosomal DNA promoter.44 In medulloblastoma cells with high Myc expression, the increased rRNA synthesis is associated with increased expression of RNA Pol I-specific transcription initiation factor RRN3/TIF-IA.45 Interestingly, repression of rDNA transcription by CX-5461 is irreversible, and the RNA Pol I-RRN3 complex remains irreversibly locked in the pre-initiation complex even after drug removal. Furthermore, c-Myc overexpression is sufficient to stimulate the biogenesis 47S pre-rRNA, total RNA, and protein synthesis in skeletal muscle without activating mTORC1.46
Myc promotes uncontrolled cancer cell growth by enhancing RNA Pol II activity to maintain rapidly-progressive transcriptional elongation.47 The super-extension complex (SEC) is required for robust and efficient transcription by RNA Pol II. Abnormal SEC activity can induce cancer. In a Myc-driven mouse cancer model, disruption of the SEC protein downregulates Myc-dependent transcriptional programs in cancer cells, reducing ribosome biogenesis and slowing cancer progression.48 This suggests that small molecules targeting SEC can be used to treat Myc-induced cancer. Furthermore, overexpression of the human RNA Pol II-associated factor 1 complex (hPAF1C) is associated with the development of various cancers. Recent studies have demonstrated that hPAF1C is positively correlated with Myc, and non-small cell lung cancer patients with low hPAF1C expression have better overall survival. It has been shown that hPAF1C can promote lung cancer cell proliferation by enhancing Myc transcription.49
RNA Pol III employs specific transcription factors to drive the target genes.50 Myc increases the expression of 43% of the genes encoding RNA Pol III-specific subunits.51 Myc enhances the function of 5S rRNA and RNA pol III in various cell types.52,53,54 An increase in Myc expression results in enhanced 5S rRNA synthesis in cardiomyocytes and hepatocytes.54,55 The effect of Myc on RNA Pol III-dependent transcription is epigenetically regulated.56 Myc is also recruited to Pol III target genes through the RNA Pol III-specific transcription factor TFIIIB/C in different cell types.57 TFIIIC recruits Myc and the related histone acetyltransferase to promote binding to TFIIIB.58 Thus, Myc regulates ribosome biogenesis by interacting with RNA Pol III at the promoters of its target genes.
Noncoding RNAs (ncRNAs)
Recent studies have shown that ncRNAs such as microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs) are involved in regulating ribosome biogenesis (Fig. 4). miR-424-5p downregulates mature rRNA levels by targeting the RNA Pol I pre-priming complex factors POLR1A and UBTF. miR-20a, miR-194, and miR-206 affect processing of 47S pre-RNA by inhibiting Ncl expression. miR-595 inhibits the synthesis of the mature 60S subunit by reducing the expression of RPL27A. lncRNA inhibits translation by reducing pre-rRNA levels through modification of rDNA chromatin. circRNA recruits the 40S ribosomal subunit to facilitate the initiation of protein synthesis.

ncRNA and ribosome biogenesis. a There is a strong link between miRNA and ribosome biogenesis. miR-424-5p is elevated and targets the RNA Pol I pre-priming complex factors POLR1A and UBTF, which in turn downregulate mature rRNA levels. miR-20a, miR-194, and miR-206 inhibit Ncl expression by binding to the 3’UTR of Ncl, thereby affecting the subsequent splicing and processing of 47S pre-RNA. miR-595 inhibits ribosome biogenesis by reducing the expression of RPL27A and the synthesis of the mature 60S subunit. b Increased expression of lncRNA inhibits rDNA transcription through modification of rDNA chromatin, resulting in reduced pre-rRNA levels, mature rRNA levels, and overall translation. c circRNA recruits the 40S ribosomal subunit and initiates mRNA translation and protein synthesis. lncRNA long noncoding RNA, Ncl nucleolin, rDNA ribosomal RNA, pol Ι RNA polymerase Ι
miRNAs are a type of short ncRNAs that regulate mRNA stability.59 Deregulation of miRNAs is involved in developing myelodysplastic syndrome (MDS) and Diamond-Blackfan anemia (DBA). The activation of miR-595, decreased ribosome protein RPL27A expression,60,61 and reduction of mature 60S subunits60 have been reported in patients with MDS, while the differential expression of some miRNAs has been found in the zebrafish model of DBA.62 Patients with chronic obstructive pulmonary disease show increased expression of miR-424-5p, which targets the RNA Pol I pre-priming complex factors POLR1A and UBTF. Upregulation of miR-424-5p is implicated in muscle wasting by inhibiting rRNA synthesis.63
lncRNAs also play important roles in ribosome biogenesis and serve as therapeutic targets or biomarkers in certain diseases. lncRNAs are >200 nt in length and highly diverse in characteristics, localization, and mode of action.64 Overexpression of lncRNA reduces pre-rRNA levels, mature rRNA, and overall translation.65 Some lncRNAs control the transcriptional activity of pre-rRNA by modifying rDNA chromatin, while small nucleolar RNA-terminal lncRNA is involved in nucleolar localization by enhancing pre-rRNA transcription.66 An lncRNA-mediated reduction of ribosome biogenesis may cause neurodegenerative diseases. Of note, the level of lncRNA is elevated in the mouse model of Alzheimer’s disease and is associated with nucleolar stress and reduced rRNA production.67,68
circRNAs are widely present in mammalian cells,69 and were previously thought to act only as miRNA sponges to refine the miRNA-mRNA axis.70,71 Emerging evidence suggests that circRNAs have protein-coding potential because they are associated with polysomes, including the initiation codon AUG and open reading frames.72,73,74 circRNA can engage the 40S ribosomal subunit and start protein translation.75 Holdt et al.76 found that circANRIL prevents atherosclerosis by inhibiting the proliferation of vascular smooth muscle cells via controlling rRNA maturation and ribosome biogenesis.
In summary, mTORC1 is involved in multiple steps in ribosome biogenesis, including the synthesis of rRNA, ribosome proteins, and the processing of precursors of rRNA. Myc overexpression increases ribosome biogenesis and is implicated in cancer cell growth. lncRNA-mediated reduction of ribosome biogenesis may cause neurodegenerative diseases. Deregulation of miRNAs contributes to the development of blood diseases. Thus, ribosome biogenesis is a potential druggable pathway for treating neurodegenerative and blood disorders.
COVID-19 and ribosome biogenesis
The coronavirus disease 2019 (COVID-19) pandemic is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Fig. 5a). SARS-CoV-2 regulates ribosome biogenesis in host cells through multiple pathways. Nsp1 is used by the SARS-CoV-2 virus to block the entry of host mRNA while inducing the cleavage of host mRNAs.

a SARS-CoV-2 regulates ribosome biogenesis in host cells through multiple pathways. Nsp1 is used by the SARS-CoV-2 virus to ensure its own replication and spread in the human host. The 5’UTR of viral Nsp1 is a key factor in directing ribosomes to viral transcripts and blocking host cell mRNA translation. Nsp1 blocks host mRNA translation and enhance the synthesis of viral proteins. Nsp1 alters the balance between viral and host cellular mRNAs through the cleavage of host mRNAs, leading to the degradation of host mRNAs by cellular nucleases. b Nsp1 acts as a gatekeeper to help SARS-CoV-2 evasion. Early in infection, Nsp1 binds to the 40S subunit with the carboxy-terminal domain of Nsp1. The viral mRNA transcript forms a translation initiation complex with the 40S-Nsp1 complex in its 5’UTR SL1. The carboxy-terminal domain of Nsp1 is removed to open the ribosome biogenesis channel for viral mRNA. The translation initiation, elongation, and termination further proceed. Upon termination, the viral mRNA is released, and the Nsp1 carboxy terminus refolds to prevent any de novo translation of cellular mRNA. c Mechanisms of bacterial resistance. Reducing the concentration of the harmful drug in the bacterium by decreasing the permeability of the bacterial outer membrane. The antimicrobial substance in the bacterial cell is removed by the efflux pumps. Mutation or modification of the target structure to reduce the affinity for antibiotics. Enzymatic degradation of antibiotics. Nsp1 nonstructural protein 1
It was shown that SARS-CoV-2 infection affects ribosome biogenesis77 and leads to immune evasion and viral replication.78
Immune evasion via cap structure of SARS-CoV-2 RNA
One of the mechanisms whereby the virus evades the immune system is through 2’-O-methylation of RNA by adding a methyl group to the 2’ hydroxyl of the ribose moiety, which is often found in the crucial regions of the ribosome.79 The methyltransferase activity of the RNA 2’-O-methyltransferase fibrillarin (FBL) is required for viral infection, which forms a strong rationale for developing FBL inhibitors as antiviral agents.80 More recent studies have demonstrated that SARS-CoV-2 evades the innate immune system by encoding its viral 2′-O-MTase, which is responsible for forming the cap structure at the 5′-end of SARS-CoV-2 RNA.81,82
Replication of SARS-CoV-2 RNA via IRES83: An internal ribosome entry site (IRES) is an element within RNA that initiates translation in a cap-independent manner. The location for IRES elements is often in the 5′UTR, but can occur elsewhere within the mRNA.84 Viral mRNAs use IRES to compete with host mRNA for ribosomes and translation factors. Small molecules that can change the structure of IRES may interfere with viral mRNA translation and block infection.83 The IRES elements in different RNA viruses differ in sequence, secondary RNA structure, and host factor requirement to recruit ribosome subunits.85,86 For example, type III IRES-mediated translation initiation requires the participation of multiple protein factors.87,88 These studies demonstrated that the IRES element mediates viral protein translation through interactions with various translation initiation factors in eukaryotic cells.
The SARS-CoV-2 genome is a positive-sense single-stranded RNA with a 5’ cap followed by an untranslated region.89,90 Due to the 5’ end cap and 3’ poly(A) tail, SARS-CoV-2 directly utilizes a cap-dependent mechanism to initiate translation after infecting host cells.91 In addition, the 5’UTR contains a region with high CG content, which can form an IRES to recruit host ribosomes to translate its RNA in a cap-independent manner.92 By taking advantage of these features, SARS-CoV-2 can replicate efficiently in host cells.93 With the assistance of IRES, SARS-CoV-2 can efficiently compete with the host cells for initiation factors and translation machinery to produce viral proteins. Therefore, drugs targeting IRES may block viral infection. Prunin is one such compound that inhibits the IRES activity of human enterovirus A71, and has shown promising results in treating hand, foot, and mouth disease.94 Islam et al.95 detected three specific mutations in the IRES that can destroy its secondary structure, thereby reducing the function of the virus. These results may guide the development of drugs targeting the IRES to slow the spread of SARS-CoV-2.
In summary, both cap-dependent and cap-independent mechanisms are utilized by the SARS-CoV-2 genome to translate viral proteins. Further study of the activity, mechanism of action, and inhibition mode of IRES elements in the 5’UTR of the SARS-CoV-2 genome can provide valuable information for developing therapeutic drugs.
Production of viral proteins via ribosome-frameshift
Viruses must use the host cell’s ribosomes to synthesize their proteins. Normally, the ribosomes move along the mRNA, reading three bases at a time and synthesizing an in-frame protein. Sometimes, ribosomes miss a base or two, generating a frameshift, resulting in a dysfunctional protein. The coronaviruses rely on frameshift to hijack the cell’s translation process and produce viral proteins.96 Using cryo-electron microscopy, Bhatt et al. discovered that the viral RNA promotes a frameshift by forming a pseudoknot structure at the entrance of the ribosomal mRNA channel. It has been shown that merafloxacin reduces the titer of new coronavirus by inhibiting the frameshift efficiency.97
Nsp1 is a potential vaccine target
The nonstructural proteins (Nsp1–Nsp16) facilitate virus replication by regulating the RNA-directed RNA polymerase and helicase.98 The Nsp1 of SARS-CoV-2 has been identified as a virulence factor because it helps to produce viral protein while inhibiting the protein synthesis of infected cells. Using high-resolution cryo-electron microscopy, Thoms et al.99 demonstrated that the Nsp1 protein could shut down the translation of host mRNA by preventing the binding of mRNA with the ribosomal 40S subunit. Understanding the Nsp1 inhibitory mechanism has been helpful for the design of anti-SARS-CoV-2 drugs. In addition, Nsp1 inhibits intracellular antiviral defense signaling pathways by blocking the retinoic acid-inducible gene I-dependent innate immune responses.99,100,101
Moreover, Nsp1 acts as a gatekeeper to restrict translation to only viral transcripts. The release of the Nsp1 is controlled by viral SL1. In coronaviruses, the viral transcripts contain three hairpins SL1, SL2, and SL3, at the 5′end.102 SARS-CoV-2 also contains these hairpins.91 More specifically, SL1 acts on the Nsp1 carboxy-terminal domain to allow viral RNAs to enter the ribosome while Nsp1 is still bound on the ribosome. SL1 plays a critical role in the completion of the viral infection. SL1 is regarded as the real "Soft Rib" of SARS-CoV-2.103 Therefore, targeting SL1 during SARS-CoV-2 infection would be a precise approach to shut down viral translation (Fig. 5b). SL1 acts as a switch to shift the translation machinery to the virus through its interaction with Nsp1.104
Other Nsps as potential vaccine targets: Upon infection, the Nsps produced by SARS-CoV-2 inhibit interferon response by blocking the ribosome channel to disrupt protein translation (Nsp1), suppressing global mRNA splicing (Nsp16), and blocking protein trafficking to the membrane (Nsp8 and Nsp9).105 Moreover, Nsp6 and Nsp13 antagonize IFN-I production by suppressing interferon regulatory factor 3.106 Thus, IFN can be used as an indicator of disease severity and potential treatment to enhance the host immune response to viral infection.107
Through RNA sequencing, ribosome profiling, and metabolic labeling of newly synthesized RNA, Finkel et al.108 showed that although viral transcripts are not preferentially translated, the virus dominates the mRNA pool in infected cells by inducing the degradation of host mRNAs. Virus inhibits the translation of innate immune genes by blocking nuclear mRNA export, preventing cellular mRNA from reaching the ribosomes.
Another study showed that herpes simplex virus type 1 (HSV-1) utilizes γ34.5 to promote its replication by inducing the cytoplasmic translocation of nucleolar protein NOP53, which in turn facilitates the recruitment of PP1αto dephosphorylate eukaryotic initiation factor eIF2α for viral translation.109
In contrast to the common belief that viral replication relies on increased ribosome abundance, Bianco et al.77 showed that human cytomegalovirus (HCMV) productive growth was enhanced by restricting ribosome biogenesis. The impairment of ribosome biogenesis leads to reduced interferon beta (IFNB1) mRNA accumulation, leading to enhanced replication of HCMV.
The clinical application of ribosome protein (RP): To investigate whether ribosome biogenesis affects the innate immune response in patients with SARS-CoV-2 infection, Yang et al.110 compared the cellular and molecular characteristics of patients with long-duration of viral shedding (LD) with those of short-duration patients. They demonstrated that LD is associated with reduced levels of RP genes, decreased numbers of natural killer cells, and CD14+ monocytes but increased regulatory T cells. These results suggest that immunosuppression and low expression of RPs are associated with the persistence of SARS-CoV-2 infection in COVID-19 patients. However, it needs to be clarified whether the decreased level of RPs in SARS-CoV-2/Nsp1/RP is a cause or a consequence of viral persistence and whether specific RPs can be used as a clinical indicator for virus persistence.
In summary, these studies uncovered how viruses promote their growth by taking over the translation machinery while suppressing the host’s innate immune response.
Bacteria and ribosome biogenesis
The number of antibiotic drugs approved by the Food and Drug Administration (FDA) of the United States has been increasing in recent years.111 However, after repeated use, bacteria become resistant to treatment. The rate of the development of drug-resistant bacteria is much faster than antibiotic development.112,113 With the in-depth study of ribosome structure, antibiotics targeting bacterial ribosomal protein synthesis have been developed.
Mechanism of ribosome-targeting antibiotics
Structural basis of the bacterial ribosome
The bacterial ribosome comprises a 30S small subunit and a 50S large subunit, including 3 rRNAs and 54 ribosomal proteins.18,114 Antibiotics targeting the 50S subunit act on the peptidyl transferase center (PTC), the GTP hydrolase association center, the nascent peptide chain channel, or interfere with protein synthesis by incorporation into the extended peptide chain.115 For the 30S subunit, antibiotics prevent protein synthesis by inhibiting the formation of the ribosomal initiation complex, interfering with tRNA binding, and interfering with tRNA site transfer during translation.19,116
Antibiotics targeting tRNA
Tetracycline is a broad-spectrum antibiotic that blocks bacterial protein synthesis.117 It binds to the 30S ribosome subunit and prevents aminoacyl-tRNA binding to the ribosome A-site.112,118,119 Although tetracycline is widely used in the clinic, drug resistance due to mutations within the ribosomal binding sites limits its clinical efficacy.120 Due to the emergence of drug-resistant bacteria, the second (doxycycline) and third generation (glycylcyclines) of tetracyclines were produced. The glycylcycline antibiotic tigecycline has a similar mechanism of action as tetracycline but with higher affinity to the 30S ribosome, thus, blocking bacteria protein synthesis more efficiently.121,122,123
Aminoglycoside antibiotics target the aminoacyl group of the 16S ribosomal RNA on the 30S ribosome subunit, preventing the correct positioning of aa-tRNA at the A and P positions, resulting in a misreading of the genetic code and abnormal protein synthesis.119 Macrolide antibiotics target bacterial translation by binding to the P site in the 50S ribosome subunit. Mupirocin blocks bacterial protein synthesis by inhibiting isoleucine tRNA synthetase.124 Under the action of these antibiotics, the bacterial translation process is impaired, resulting in the death of the bacteria.125,126
Targeting peptidyl transferase center
The peptidyl transferase is an aminoacyl transferase that catalyzes the reaction to form peptide bonds between two adjacent amino acids. This is a process of adding an amino acid to the growing polypeptide chain during protein synthesis. The peptidyl transferase center (PTC) is situated in the large subunit of the ribosome. Chloramphenicol and macrolides are two examples of antibiotics targeting the PTC. Chloramphenicol prevents peptide bond formation by binding to the 23S rRNA of the 50S ribosome subunit, whereas macrolides do so by binding to the P site of the 50S subunit. The position where chloramphenicol binds to PTC overlaps with a part of the acyl group at the A-site of tRNA (Cam1).127,128 The binding site (Cam2) of chloramphenicol to the 50S subunit of the halophilic archaea is located deeper in the exit channel, overlapping the erythromycin binding site.129 The binding site of chloramphenicol to the archaeal 50S subunit is not in Cam1, which is in line with the finding that higher concentrations of chloramphenicol are needed to inhibit archaeal growth than bacterial growth. Chloramphenicol and erythromycin directly inhibit the biosynthesis of the 50S subunit.130 Erythromycin does not inhibit PTC but interferes with aminoacyl translocation, preventing the move of tRNA from the A-site to the P site of the ribosome.
Molecular mechanisms of bacterial resistance
At present, it is generally believed that there are four main categories of molecular mechanisms by which bacteria develop drug resistance:131,132 (1) Reduction of intracellular drug concentration; (2) Modification of drug target due to mutation or modification of target genes; (3) Overexpression and protection of target genes; (4) Inactivation of the drug due to hydrolyzation (Fig. 5c). Thus, drug resistance is achieved by reducing the permeability of the bacterial outer membrane, decreasing the concentration of the antimicrobial substance by efflux pump, reducing the affinity for antibiotics through mutations, and degrading the antibiotics.
Reducing intracellular drug concentration
Gram-negative bacteria contain an outer membrane that protects the bacteria from external toxic agents. Therefore, Gram-positive bacteria are more susceptible to antibiotics than Gram-negative bacteria.133 Another way of keeping drug concentration low is to increase its efflux by acquiring genes encoding an efflux pump through chromosome mutation. The upregulation of the efflux pump causes drug resistance to most ribosome-targeting antibiotics.
Mutation or modification of target gene
Mutations in target genes can lead to reduced drug affinity.115,116 The ribosome-targeted antibiotics interact with rRNA, so mutations in rRNA nucleotides can change the conformation of the drug-binding site, resulting in tolerance. Most bacteria have more than one copies of rRNA operons, which means that only the same mutations that occur in all of the operons will the bacteria become resistant to the drug, which is a rare situation. It mainly occurs in Mycoplasma pneumoniae and Mycobacterium tuberculosis because these pathogens only have one or two rRNA operons. rRNA methyltransferases—mediated methylation of rRNA is one of the most common modifications. The methyltransferase KgmA and KamA–mediated methylation confer aminoglycoside tolerance.134 Cfr methylates nucleotide A2503 of 23S rRNA,135 a modification that promotes bacterial resistance to chloramphenicol, SA antibiotics, lincosamides,136 and some intracyclic macrocyclic Ester antibiotic resistance.137 Methylation of A2058 of 23S rRNA is responsible for bacterial resistance to macrolides, lincosamide, and streptogramins B.
Overexpression and protection of target genes
Overexpression of target genes leads to resistance to certain antibiotics because these target genes can sequester drugs. Antibiotic resistance caused by target gene overexpression has not been demonstrated in vivo, but they have been confirmed by in vitro experiments. Overexpression of 16S rRNA h34 resulted in bacterial resistance to spectinomycin.138 Likewise, overexpression of EF-Tu restored protein translation in the presence of EF-Tu inhibitors.139
Hydrolyzing or modifying enzymes that inactivate antibiotics
Certain enzymes encoded by bacteria are capable of modifying or degrading antibiotics.140,141 These enzymes include hydroxylases (beta-lactamases, esterases, epoxide hydroxylase) (phenicols), transferases (acetyltransferases, phosphotransferases, nucleotidyltransferases, glycosyltransferases, ADP-ribosyltransferases, S-transferases) (aminoglycosides and macrolides),142,143 and redox enzymes (monoxygenases, lyases) (tetracycline144and tigecycline145). Esterases EreA and EreB hydrolyze macrolide ester bonds, leading to resistance to macrolide antibiotics.141 For aminoglycosides, drug inactivation due to modification of their chemical structure by bacterial enzymes is the main mechanism of drug resistance. The amide and hydroxyl groups of aminoglycosides can be modified by acetyltransferase, nucleotidyl transferase, and phosphotransferase.
Ribosome biogenesis is an attractive target for developing new antibiotics. Antibiotic resistance mechanisms include degradation and efflux of the drug and mutation of target genes. Future work should focus on developing antibiotics targeting new sites on the ribosome.
Ribosomes and cardiovascular diseases (CVDs)
In 2020, the World Health Organization proclaimed that the mortality caused by CVDs is significantly higher than that caused by tumors and other diseases.146 Many studies have pointed out that ribosome dysfunction triggers various CVDs.3,4 Here, we discuss the specific regulatory mechanisms of rDNA, rRNA, RPs, and Ncl in CVDs (Table 1).
Ribosome and cardiac hypertrophy
Cardiac hypertrophy is an adaptive response that may occur after pressure or volume overload, inflammatory cardiomyopathy, and long-term stimulation, ultimately leading to heart failure.147,148 However, the molecular mechanisms underlying cardiac hypertrophy are currently unclear. Recent studies have suggested that ribosome biogenesis plays an important role in cardiac hypertrophy by promoting or inhibiting the pathological process.149,150
The rDNA transcription rate is positively correlated with the degree of phosphorylation of RNA Pol I and the upstream binding factor (UBF). Studies have shown that the hypophosphorylated form of UBF blocks rDNA transcription by disrupting the UBF/SL1 complex.151 Others reported that the proliferation rate of cultured neonatal cardiomyocytes is correlated with the phosphorylation of UBF.152 Endothelin-1-induced cardiac hypertrophy is associated with UBF hyperphosphorylation.153 Moreover, enhanced rDNA transcript levels are associated with increased UBF protein levels under α1 adrenergic receptor- or stress-load-induced cardiac hypertrophy. Further studies have confirmed that high UBF expression and activity stimulate ribosome biogenesis during cardiac hypertrophy.154,155
rRNA not only acts as the central scaffold for ribosomal subunits but also serves as the center for catalytic activity in ribosome biogenesis.156,157 The abnormal pre-rRNA processing causes defects in ribosome structure and function, disrupts cardiac protein balance, and induces cardiac hypertrophy. Rackham et al.158 reported that when the endoribonuclease component of the RNase P complex, MRPP3, is knocked out in the heart, the mice develop severe cardiomyopathy and the lifespan is shortened. The loss of MRPP3 causes defects in rRNA processing, leading to the production of immature rRNAs and a reduction of RPs. Their findings revealed that rRNA processing acts as a "bridge" to link transcription and translation by regulating ribosome assembly. In addition, RNA Pol I is primarily responsible for the processing of pre-47S transcripts into 5.8S, 18S, and 28S rRNA, and then generating mature rRNAs.15 Studies have demonstrated that TAF1A gene mutation of RNA Pol I cause gene-specific nucleolar defects and impair rRNA synthesis in cardiomyocytes, eventually leading to decreased ventricular systolic function, dilated cardiomyopathy (DCM), and heart failure.9,159,160
S6K1-dependent activation of RP synthesis is crucial for cardiomyocyte proliferation and growth. S6K1 promotes rDNA transcription in cardiomyocytes, accelerating the pathological evolution of cardiac hypertrophy.161 This suggests that S6K1 is a key factor in regulating ribosome biogenesis and cardiomyocyte growth. The above studies suggest that aberrant activation of S6K1 increases protein synthesis and growth in hypertrophic cardiomyocytes. S6K1 is an important drug target for the treatment of cardiac hypertrophy (Fig. 6a). Thus, abnormal upregulation of UBF, RNA Pol I, and S6K1 induces cardiac hypertrophy.

a (i) Ribosome dysfunction and cardiac hypertrophy. Increased UBF activity and abnormal activation of RNA Pol I and S6K1 induce cardiac hypertrophy. (ii) Ribosome dysfunction and MI. Downregulation of Ncl, rRNA, RPL9, and RPL26 leads to the failure of pre-rRNA processing, ultimately causing ribosome dysfunction and MI. b Ribosomes and atherosclerosis. Left: The over-activation of ribosome biogenesis results in abnormal proliferation of VSMCs and atherosclerosis. The expression of BOP1 and PES1 is increased in atherosclerotic patients, and both promote rRNA maturation by promoting pre-rRNA splicing and ribosome biogenesis. Right: RPL13 inhibits macrophage-induced inflammation and atherosclerosis. A high-fat diet promotes atherosclerosis in ApoE−/− mice, accompanied by higher levels of inflammatory cytokines. RPL13 inhibits the inflammatory response by inhibiting the translation of inflammatory genes such as CCL22, CXCL13a, and CCR3. BOP1 blocking of proliferation 1, PES1 pescadillo homolog 1
Ribosomes and myocardial infarction (MI)
MI is caused by occlusion of the coronary artery. Although intracoronary vascular surgery can prolong the life of patients,162 postoperative ischemia-reperfusion injuries can cause further damage to the heart. Moreover, insufficient cardiac regeneration eventually leads to ventricular remodeling and heart failure.163,164 Many studies have confirmed that ribosome dysfunction plays an important role in MI.165,166,167 Downregulation of Ncl, rRNA, RPL9, and RPL26 is involved in MI-induced ribosome dysfunction (Fig. 6a).
Recent bioinformatics analysis showed that the expression levels of ribosomal proteins L9 (RPL9) and L26 (RPL26) are downregulated in patients with MI compared with controls.168 Therefore, RPL9 and RPL26 are potential targets for diagnosing or treating MI.
Ncl has many functions in cell survival and growth pathways. Some studies have found that knockout of Ncl in mice aggravates cardiac function after MI and reduces the survival rate. Mechanistically, Ncl improves cardiac function during MI by enhancing M2 macrophage polarization.169 In mouse models of MI, autophagy-related gene deletion or drug inhibition aggravates cardiac dysfunction and myocardial remodeling.170 Deng et al.166 found that the Ncl/autophagy signaling pathway is upregulated in the infarcted myocardium. Moreover, Nicorandil treatment affects the TGF-β/Smad signaling pathway by up-regulating the Ncl/autophagy axis, reducing cardiac remodeling after MI, and improving cardiac function. These studies suggest that Ncl is a potential target for the treatment of MI.
Ribosomes and atherosclerosis
Atherosclerosis is caused by lipid deposition in the intima of the arterial wall and increasing its thickening. This is the common pathological basis of CVDs, such as MI and coronary heart disease.171
Abnormal proliferation of vascular smooth muscle cells (VSMCs) accelerates the process of atherosclerosis by promoting arterial intimal hyperplasia. Therefore, inhibiting the abnormal proliferation of VSMCs is the current method to inhibit neointimal hyperplasia.172,173,174 Ribosome biogenesis regulates cell proliferation and differentiation. It is known that ribosome biogenesis includes three key steps: rRNA synthesis, ribosomal protein (RP) synthesis, and their assembly into the mature ribosome.175,176 The maturation of rRNA during ribosome assembly is closely associated with the PES-BOP1-WDR12 (PeBoW) complex, which binds to pre-rRNA and promotes 47S pre-rRNA into mature 28S and 5.8S rRNA. The core regulator of the PeBoW complex may be involved in the production of 28S and 5.8S rRNA.177 Jia et al.177,178 found that BOP1 is increased in the coronary arteries of patients with atherosclerosis and is accompanied by the neointimal hyperplasia caused by VSMC proliferation (Fig. 6b). Therefore, increased expression of BOP1 and PES1 promotes ribosome biogenesis leading to abnormal proliferation of VSMCs and atherosclerosis. Holdt et al.76 confirmed that circARNL occupies the pre-rRNA site of another member of the PeBoW complex, PES1, and inhibits the cleavage of pre-rRNA by exonuclease, thereby inhibiting the proliferation of VSMCs by inhibiting ribosome biogenesis and delaying the atherosclerotic process. Importantly, it has been reported that curcumin effectively inhibits the proliferation of VSMCs and slows the development of atherosclerosis. The mechanism is that curcumin promotes DNMN2 expression, which in turn elevates the levels of 18S rRNA methylation.179 The above reports suggest that suppressing ribosome biogenesis may be a new approach for inhibiting the proliferation of VSMCs.
Atherosclerosis is also a chronic inflammatory disease, as an inflammatory response accompanies its onset.180 Macrophages are one of the cells responsible for atherosclerosis; they form foam cells by phagocytosing lipid droplets and accelerating the progression of atherosclerosis.181,182 Basu et al.183 reported that macrophage-specific deletion of RPL13 increases the susceptibility of ApoE knockout mice to high-fat diet-induced atherosclerosis. The protective effect of RPL13 on atherosclerosis may be related to its translational silencing activity: the deletion of RPL13 promotes the translation of mRNAs for CCL22, CXCL13, and CCR3 in macrophages, causing increased expression of inflammatory cytokines. In summary, RPL13 inhibits macrophage-induced inflammation and atherosclerosis by inhibiting the translation of inflammatory genes such as CCL22, CXCL13a, and CCR3.
Since atherosclerosis is a chronic process, the time from its onset to its detection may take a long time. The use of ribosome biogenesis as a predictive factor for atherosclerosis has emerged. Wang et al.184 analyzed the plasma samples of patients with familial hypercholesterolemia and found that RPL9, RPL35, RPS7, and RPL23 showed a downward trend. The expressed genes were mainly involved in the ribosomal and oxidative phosphorylation pathways. Jiménez et al.185 found that m6A in the 18S rRNA component is significantly reduced in both early and late atherosclerosis samples by mass spectrometry, suggesting that 18S rRNA has abnormal methylation modification in early and late atherosclerosis. Therefore, various components of the ribosome may be used for the early detection of atherosclerosis.
Nucleolin (Ncl) and CVD
Ribosome biogenesis begins in the nucleolus, where different rRNA subunits are transcribed as a polycistronic transcript by RNA polymerase I. More than 300 non-ribosomal factors, including nucleolin, regulates pre-rRNA transcription and ribosome assembly.186 Due to the complex nature of the ribosome biogenesis process, misassembly of the ribosomes may occur. To prevent the build-up of incorrectly folded ribosomes, a quality control mechanism is in place to detect and degrade the non-functional ribosomes, and this process is regulated by non-ribosomal proteins.187 Nucleolin is also present on the cell surface and is overexpressed in cancer cells.188
Ncl regulates cellular functions by participating in multiple stages of ribosome biogenesis, such as rDNA transcription, increased RNA Pol I transcriptional activity, and ribosome assembly.189,190 In addition, as a shuttle protein, the correct subcellular localization of Ncl is critical for its biological function, and the abnormal localization of Ncl is involved in various pathological processes.190 Recent studies suggest that abnormal Ncl expression is involved in the development of CVD, including MI191, heart failure (HF)192, and atherosclerosis.193
The protein level of Ncl is significantly decreased during MI, while the overexpression of Ncl reduces the infarct area and cell death in rat hearts.194 Cardiomyocyte-specific Ncl transgenic mice are resistant to doxorubicin-induced cardiotoxicity, indicating the cardioprotective effect of Ncl in this process.195 Of note, the heart of Ncl-deficient fish exhibits a severe cardiomyocyte defect, cardiac development disorder, and abnormal ventricular remodeling and dysfunction; the mechanism involves the reduction of rRNA transcription and heterochromatin caused by reduced Ncl.196 Knockdown of Ncl expression in neonatal rat ventricular myocytes leads to an increased expression of heterochromatin marker H3K9Me3 and a decrease in the transcript levels of pre-rRNA and 18S rRNA.197
Current studies confirm that Ncl can protect against MI through myocardial ischemic preconditioning (IP).191 IP is a short period of myocardial ischemia/reperfusion (I/R), significantly reducing the damage caused by subsequent long-term I/R, and is also a powerful endogenous heart protection mechanism.198,199 Jiang et al.200 found that Ncl is an important endogenous cardioprotective factor in myocardial IP. Ncl binds to the 3’UTR of HSPA1A mRNA, a member of the heat-shock protein (HSP) family, to upregulate the expression of HSPA1A by stabilizing its mRNA. As a phosphorylated protein, Ncl participates in rRNA synthesis and cell proliferation.190 Tong et al.167 demonstrated that threonine phosphorylation at positions 76 and 84 is essential for Ncl to reduce caspase-3 activity and inhibit cardiomyocyte apoptosis. This study also found that Ncl undergoes phosphorylation modification after translocation from the nucleus to the cytoplasm.
Macrophage phenotype-switching between the pro-inflammatory M1 and the anti-inflammatory M2 phenotype exhibit plasticity during the post-MI inflammatory response and tissue repair.201 Using a mouse MI model, Tang et al.169 found that the expression of Ncl mRNA and protein decreased gradually from day 3 after MI, then increased gradually, reaching a peak on day 7. The macrophages in the myocardium switch from the M1 phenotype 2 days after MI to the M2 phenotype 5 days after MI, suggesting a potential correlation between Ncl expression and macrophage phenotype-switching. Mechanistically, Ncl promotes the polarization of M2 macrophages by binding to the 5’ UTR or translation region of Notch3 and STAT6 mRNA to promote their stabilization.
The main feature of atherosclerosis is the formation of fat-rich plaques in medium and large vessels.202 The progression of the disease is linked to the recruitment of monocytes that differentiate into macrophages, which subsequently absorb lipids to become foam cells that produce inflammatory cytokines. Thus, rapid clearance of foam cells can inhibit the inflammatory response and ultimately delay plaque development.203,204 Ncl plays an important role in regulating the progression of atherosclerosis.203 Li et al.205 reported that macrophage transformation into foam cells is associated with decreased expression of Ncl protein. Ncl enhances the stability of ABCA1, which promotes cholesterol efflux and inhibits lipid accumulation and the formation of foam cells. It has also been shown that under normal conditions, the interaction between Ncl with Dnm3os prevents its enrichment to histone H3K9ac on the promoters of pro-inflammatory genes such as IL6.193 However, Sun et al.206 showed that oxidized low-density lipoprotein (oxLDL), one of the inducers of atherosclerosis, upregulates Ncl mRNA and protein expression in vascular smooth muscle cells (VSMCs) in a dose-dependent manner. The abnormal proliferation of VSMCs is the pathological basis for the development of atherosclerosis. Ncl promotes VSMC proliferation and cell cycle changes under oxLDL treatment. Under normal circumstances, Aurora B is mainly located in the nucleus and is dynamically expressed in the G2-M phase. Aurora B is associated with Ncl proteins, and it is speculated that the combination of the two promotes cell proliferation.
In summary, ribosome biogenesis plays an essential role in developing CVDs. The specific functions of ribosome biogenesis regulators, such as Ncl, in different cardiovascular cells and diseases deserve further study.
Ribosomes, aging, and neurodegenerative diseases
Aging is a complex multifactorial, and irreversible biological process characterized by the degeneration of cellular, tissue, and organ functions over time. Among the various molecular mechanisms of aging, deterioration of the rate of protein synthesis and the function of ribosomes plays a central role.207,208,209
The evidence of the association between deregulated ribosome biogenesis and aging
Zhang et al.210 compared gene expression differences by RT-PCR differential display using epithelia dissected from age-related cataracts and normal lenses. The results revealed that human age-related cataract is correlated with decreased expression of ribosomal proteins L21, L15, L13a, and L7a.
A gradual decrease in muscle mass is a common feature in older individuals. Kirby et al.211 showed that aged muscle failed to upregulate pre-47S ribosomal RNA (rRNA) expression, suggesting a reduction of ribosome biogenesis in aged mice.
It was shown that both the translation of RP mRNA and overall mRNA translation efficiency decline with age, suggesting some defect in the process of ribosome biogenesis during aging.212 A recent study revealed that the ribosome of aging cells moves more slowly and periodically, causing the "stalling" of ribosome translation to increase, resulting in a collision and the accumulation of new peptides, thus aggravating ribosome dysfunction and aging.213 This study shed new light on the mechanism of the ribosome-stalling/collision/ribosome-dysfunction axis in age-dependent damage (Fig. 7a). The stalling of ribosomes during translation can cause protein truncation. Ribosomal Quality Control (RQC) is a mechanism to clear the stalling of ribosomes. However, the function of RQC decreased with age, leading to ribosome-stalling/collision and protein aggregation.

a Ribosome-stalling/collision/ribosome dysfunction and aging. The primary clearance pathway for ribosome collisions is the degradation of nascent peptides through ribosomal quality control (RQC). RQC decreases with age, and ribosome-stalling/collision triggers ribosome dysfunction. As a result, ribosome dysfunction leads to increased nascent polypeptides and protein aggregation. b Ribosomes and the signaling pathway of cellular senescence. Cellular DNA damage can be induced by radiation or chemotherapy, which activates p53-dependent stress responses and cause cellular senescence. SIRT1, a member of the longevity protein family, directly affects the activity of key proteins in the senescence pathway through deacetylating transcription factors. Moreover, SIRT1 recruits methylation enzymes to affect ribosome biogenesis by regulating chromatin remodeling, and abnormal ribosome biogenesis can directly affect p53 and telomerase activity to accelerate cellular senescence. Genomic instability of rDNA causes cellular senescence, and SIRT7, a member of the longevity protein family, affects rDNA stability by interacting with chromatin remodeling factors and RNA polymerase. Ncl and SIRT7 interact with proteins, and Ncl binding to telomeric reverse transcriptase affects ribosome biogenesis. Ac acetylation, CDK cyclin-dependent kinases, Me methylation, Ncl nucleolin, p phosphorylation, rDNA ribosomal DNA, RNA pol Ι RNA polymerase Ι
The potential mechanisms contributing to deregulated ribosome biogenesis and aging
rDNA genome instability due to the accumulation of DNA damage has been implicated as a causal factor in aging.214 It was shown that hematopoietic stem cells build up chromosome breaks in their rDNA genes during aging.215 Moreover, increased rDNA instability has been found in premature aging diseases, such as Bloom and Werner syndromes.6
SIRT7 can stabilize SNF2H protein at the rDNA promoter. When SIRT7 is knocked out, the copy number of rDNA is reduced by 50%.216,217,218 SIRT7 also promotes RNA polymerase binding to the rDNA promoter region and coding region by catalyzing the deacetylation of RNA pol I.219 Moreover, SIRT7 maintains rDNA repeat stability and nucleolar integrity through the recruitment of DNA methyltransferase 1 and SIRT1.219,220 The nucleolus protein u3-55k is also a deacetylation substrate for SIRT7, and SIRT7/u3-55k deacetylation is required for 18s rRNA maturation and ribosome biogenesis (Fig. 7b).221 Radiation or chemotherapy-induced cellular senescence is mediated by p53-dependent stress responses. SIRT1 recruits methylation enzymes to affect ribosome biogenesis by regulating chromatin remodeling.
Ribosome and neurodegenerative diseases
Studies have shown that most nervous system diseases are caused by abnormal protein synthesis, and ribosome dysfunction disrupts the homeostasis of neurons and glial cells.222,223
Alzheimer′s disease (AD) is a neurodegenerative disease, which is chronic and progressive, and the probability of developing dementia is as high as 60–70%.224 It is widely assumed that ribosome dysfunction in the cerebral cortex is one of the important pathological mechanisms of AD. Studies have found that protein levels of 5.8S and 5S rRNA are significantly reduced in early AD patients, suggesting that AD affects rRNA processing and maturation.225 Similarly, total rRNA levels are decreased in the brains of AD patients and are accompanied by an increase in 5S rRNA oxidation.226 This study indicated that the increased oxidation of 5S rRNA is associated with neurodegeneration in AD. Hernandez-Ortega et al.227 found that the protein levels of Ncl, and the mRNA of the upstream binding transcription factor RNA Pol I gene are decreased in late AD. In addition, alterations in the translation initiation factors eIF2α, eIF3h, eIF5, and elongation factor eEF2 have been found in AD patients.
Parkinson’s disease (PD) is an age-related neurodegenerative disorder characterized by degeneration, necrosis, and reduction of dopamine and noradrenergic neurons.228,229 Decreased rRNA synthesis and changes in nucleolar volume are physiological characteristics of the elderly, while aging is the major risk factor for PD. Reduced rRNA synthesis and nucleolar destruction have been reported in the dopaminergic neurons of PD mice, indicating that ribosome dysfunction may cause neuronal degeneration in PD patients.230 Further studies have demonstrated that rRNA synthesis and nucleolar volume are reduced in the dopaminergic neurons of PD patients, while nucleolar integrity is compromised as the disease progresses.231 The above results demonstrate that ribosome biogenesis is impaired in the neurodegenerative process of PD, and may be one of the factors that trigger neuronal cell death.
Accumulating evidence suggests that dysregulated ribosome biogenesis is involved in aging and neurodegenerative diseases. Future studies should focus on developing novel therapies to restore the function of ribosomes.
Ribosomes and cancer
Multiple oncogenic signaling pathways contribute to enhanced ribosome biogenesis and protein synthesis in cancer cells. Thus, ribosome biogenesis has emerged as a potential therapeutic target. For further information on the role of ribosome biogenesis in cancer, we refer readers to other reviews.232,233,234,235
Abnormally active ribosome biogenesis is critical for cancer cells and mediates fundamental mechanisms of cell proliferation and migration (Fig. 8).236,237 In cancer cells, Myc upregulates rDNA transcription by activating RNA pol I-mediated 47S pre-rRNA synthesis. Ncl promotes cancer growth by increasing the RNA Pol I activity. Blood diseases are associated with RPL5/11, RPS7/24, and RPS19 dysfunction, leading to the activation of p53. Abnormal rDNA transcription affects the processing of 47S pre-rRNA into mature 5.8S, 18S, and 28S rRNA, disrupting rRNA synthesis and nucleolar integrity, and ultimately inducing neuronal cell death. Many studies have demonstrated that RPs regulate the expression of oncogenes and cancer suppressor genes and therefore participate in the development and metastasis of cancers.238 The oncogene Myc acts as an active hub for the progress of cancer239 and increases the rate of ribosome biogenesis by directly activating rRNA synthesis in cancer cells.39,240,241 Meanwhile, the direct binding of Myc to rDNA activates the synthesis of 47S pre-rRNA. Of note, Myc-induced upregulation of RPL14 and RPL28 accelerates lymphoma progression. This process leads to impaired translation of the cell-cycle regulator CDK11 and genomic instability, ultimately leading to lymphatic damage.242,243 Therefore, one primary mechanism by which Myc induces cancer is the alterations of ribosome biogenesis.244

Abnormally active ribosome biogenesis is critical for cancer cell growth. The oncogene Myc upregulates rDNA transcription, activates RNA pol I-mediated 47S pre-rRNA synthesis, and induces the expression of RPL14 and RPL28 to stimulate ribosome biogenesis. In addition, Ncl enables cancer cells to differentiate and grow indefinitely by increasing the RNA Pol I activity. Blood diseases are associated with abnormal expression of RPs. RPL5/11 and RPS7/24 dysfunction and mutations in RPS19 activate p53, leading to a decline or even disappearance of erythroid cells, increasing the risk of malignant transformation. Ribosome dysfunction disrupts neuronal and glial homeostasis. Abnormal rDNA transcription affects the subsequent processing of 47S pre-rRNA into mature 5.8S, 18S, and 28S rRNA, disrupting rRNA synthesis and nucleolar integrity, and ultimately inducing neuronal cell death. Myc myelocytomatosis oncogene, Ncl nucleolin, rDNA ribosomal DNA, rRNA ribosomal RNA, RNA pol Ι RNA polymerase Ι
Ncl promotes rRNA transcription and pre-rRNA processing. Researchers found that Ncl combines with noncoding RNA cytoskeleton regulator RNA and then jointly participates in the development of colorectal cancer.245 Ncl also binds to telomerase reverse transcriptase (TERT) in the nucleoplasm and promotes its nucleolar localization, preventing cancer cells from senescence and maintaining cancer proliferation.246 Furthermore, Lee et al.247 showed that Ncl combines with astrocyte-elevated gene-1 in breast cancer to promote the growth and metastasis of breast cancer cells. Increased ribosome biogenesis in cancer cells is closely associated with continuously activated RNA Pol I/III. It promotes cell proliferation by enhancing the transcriptional activity of RNA Pol I/III and ribosome biogenesis, enabling cancer cells to differentiate and grow indefinitely.190,248
Ribosome biogenesis is an extremely energy-demanding process as it synthesizes the most abundant RNA and various proteins required for cell growth and proliferation. When ribosome biogenesis is impaired, cells must be able to sense the level of intracellular stress and slow down the cell cycle by altering ribosome biogenesis to avoid partial growth and improvised division. This task is accomplished by the tumor suppressor protein p53. Volarevic et al.249 were the first ones to describe cell cycle arrest in mouse liver cells resulting from impaired ribosome biogenesis caused by the deletion of ribosomal protein RPS6 (eS6). Since then, several studies have shown that cell cycle arrest resulting from the activation of p53 can be triggered by the interruption of ribosome biogenesis. This process has been referred to as Impaired Ribosome Biogenesis Checkpoint (IRBC).250
Ribosomopathies are a group of diseases caused by abnormalities of the ribosomal components. Such diseases include Diamon-Blackfan-Anemia, 5-q syndrome, SDS, X-linked DC, CHH, etc. Patients with ribosomopathies are at increased risk of cancer, bone marrow failure, and developmental abnormalities.251 Eμ-Myc lymphoma cells can induce short hairpin RNA expression as ribosomal protein L7a (RPL7a) or RPL11, a key component of IRBC. Only RPL7a reduction induces p53-mediated apoptosis due to the degradation of MCL-1.252 The evidence that IRBC responds to aberrant ribosome biogenesis to prevent oncogenesis was from the knock-in mice expressing mutant mouse double minute 2 (MDM2) C305F, which cannot complex with RPL5/RPL11/5S rRNA material combination. Crossing these mice with Eμ-Myc transgenic mice showed that c-Myc was upregulated and significantly accelerated the development of B-cell lymphomas.253,254 The ability of mutant MDM2 C305F to promote tumorigenesis in a c-Myc-driven cancer model is caused by the inability of the RPL5/RPL11/5S rRNA complex to upregulate p53 levels. These offer the possibility for therapeutic interventions based on IRBC-mediated p53 activation to inhibit carcinogenesis. A recent study demonstrated that IRBC could prevent cancer by reducing DNA damage and genomic instability.255 Impaired ribosome biogenesis caused by abnormal processing of pre-rRNA leads to nucleotide imbalances that can lead to replication stress, DNA damage, and genomic instability, which are hallmarks of cancer.256 Activation of IRBC prevents cancer by inducing p21-dependent G1 phase arrest, thereby preventing cells from entering the S phase in the presence of DNA damage. Many oncogenes induce nucleotide imbalance and impair ribosome biogenesis, DNA replication, and cell proliferation. Therefore, the role of IRBC in maintaining genomic stability will be crucial.255,257
Glutathione peroxidase 4 (GPX4) has emerged as a promising therapeutic target for cancer therapy, but some cancer cells are resistant to ferroptosis triggered by GPX4 inhibition. A recent study by Zhipeng et al.258 revealed the rewiring of selenoprotein hierarchies in cancer cells and identified ribosomal stall and collision during GPX4 translation as a ferroptosis vulnerability in cancer. The development of blood vessels is spatiotemporally regulated, but how this is coordinated across lineages is unclear. Katarzyna and colleagues found that the RNA helicase Ddx21, an important regulator of rRNA synthesis and ribosome biogenesis, controls Vegfc-driven developmental lymphangiogenesis by balancing endothelial ribosome biosynthesis and p53 function.259 This mechanism may be targeted in diseases of hyperlymphangiogenesis, such as cancer metastasis or lymphatic malformations. Fibrillin (FBL), a ribosomal biogenic factor, plays an important role in the early steps of ribosome biogenesis and is associated with poor prognosis in breast cancer when overexpressed. It was shown that a low level of FBL is a new independent marker of poor outcomes in breast cancer.260
Ribosome biogenesis is an essential player in cancer growth and metastasis. The development of ribosome-targeted therapy is emerging. Future works should focus on the influence of microenvironment and ribosome heterogeneity on the development of drug resistance.
Ribosome and blood diseases
Due to the rapid renewal of bone marrow hematopoietic cells, ribosome biogenesis is involved in this process.261,262 Therefore, the blood system is more vulnerable to ribosome dysfunction. It has been shown that some blood diseases are associated with ribosome dysfunction.263,264,265
Diamond-Blackfan anemia (DBA) is a bone marrow failure syndrome in which the number of erythroid precursor cells in the marrow of patients is markedly decreased or even disappears, which increases the risk of malignant transformation.266,267 Studies have shown that a mutation of ribosomal protein S19 (RPS19) involved in the assembly of the 40S small ribosome subunit is closely associated with the pathogenesis of DBA. RPS19 is critical for hematopoietic differentiation in zebrafish, and its deletion leads to abnormal apoptosis during erythropoiesis.268 The overexpression of RPS19 cDNA in bone marrow cells of DBA mice reduces apoptosis, increases the formation of erythroid colonies, and attenuates bone marrow failure.269 The mutation of RPS19 activates p53 and causes cell senescence, and the decreased red blood cells caused by RPS19 mutation can be reversed by p53 deletion.270 Furthermore, it was shown that anemia is significantly alleviated in p53-deficient mice.271 In addition, RPS7, RPS24, RPL11, and RPL5 have also been confirmed to be closely associated with DBA.272,273
Shwachman-Diamond syndrome (SDS), the inherited ribosomopathy, is characterized by bone marrow failure and a high risk of myeloid malignancies at a young age. In 2003, Boocock et al.274 reported that the mutation of the Shwachman Bodian Diamond Syndrome (SBDS) gene is the genetic basis for developing SDS. The protein is a cofactor for elongation factor-like GTPase1 (EFL1), which removes the eukaryotic translation initiation factor 6 (eIF6) from the large ribosomal subunit, allowing the 60S subunit to form an active ribosome with the 40S ribosomal subunit.275,276 In particular, mutations in other genes associated with SBDS, DNAJC21, EFL1, and SRP54, all participate in the removal of eIF6 from the 60S subunit together with SBDS.275,277 Failure to release eIF6 can cause SDS.278 In addition, SBDS coprecipitated with pre-60S subunit in a sucrose gradient and bound to 28S rRNA as a component of mature 60S ribosomal subunit.279 Therefore, mutations in the SBDS gene lead to impaired ribosome assembly. SDS cells were also found to have aberrant expression of many genes involved in rRNA and mRNA processing and reduced expression of several ribosomal protein genes, including RPS9, RPS20, RPL6, RPL15, RPL22, RPL23, and RPL29, which are involved in cell growth and survival.280
X-linked dyskeratosis congenita (X-linked DC) is associated with mutations in the DKC1 gene encoding dyskerin.281 Dyskerin is an enzyme that catalyzes the pseudouridylation of specific uridine residues in newly synthesized rRNAs.264 Pseudouridines regulate ribosome and rRNA biogenesis when associated with small nucleolar RNAs (snoRNA). Cell lines from patients with X-linked DC exhibit altered regulation of snoRNAs and defects in rRNA processing.276,282 Furthermore, NPM1 mutations found in DC patients lead to alterations in rRNA 2’-O-methylation.283
Cartilage Hair Hypoplasia (CHH) is an autosomal recessive disorder caused by mutations of the RNA component of the mitochondrial RNA processing endoribonuclease (RMRP) gene.284 RMRP is a long noncoding RNA (lncRNA) that participates in the formation of RNase-MRP complexes and tRNA maturation. RMRP in mitochondria encodes the RNA component of RNase-MRP and is classified as snoRNAs, which are involved in several steps of ribosomal RNA synthesis.285 CHH fibroblasts exhibit reduced ribosome biogenesis.276,286
These findings imply that ribosome dysfunction is involved in the pathological process of blood diseases. Therefore, an in-depth exploration of the complex interactions between ribosome biogenesis and related signaling pathways will provide a new treatment for the diseases.
Ribosome-targeted therapeutic strategies
Clinical trials have shown promise for ribosome-targeted therapeutic strategies, while the underlying mechanisms differ (Table 2).
Several new rDNA transcription inhibitors are in early clinical studies, such as CX-3543, BMH-21, and CX-5461.287,288,289, CX-3543 binds to the G4 sequence and disrupting the interaction of the rDNA G4 structure with Ncl, thereby inhibiting RNA Pol I function and inducing apoptosis in cancer cells. BMH-21 inhibits Pol I function by interacting with the rDNA backbone in GC-rich DNA sequences and also promotes the degradation of the Pol I catalytic subunit RPA194.290 CX-5461 decreases the binding affinity of the RNA Pol I complex to rDNA promoters and activates P53-dependent anticancer signaling. Xu et al.291 have shown that CX-5461 is a G-quadruplex stabilizer that kills cancer cells. DNA repair requires the BRCA and NHEJ pathways, and failure to repair leads to cell death. These data reinforce strategies for rDNA G4-targeted therapy, especially for NHEJ-deficient cancers. CX-5461 is currently in Phase I clinical trial in patients with BRCA1/2-deficient cancer (Canadian Trial, NCT02719977).
Hilton et al. reported the results of phase I trial of CX-5461 in patients with solid tumors enriched for DNA-repair deficiencies.292 The purpose of the trial was to determine the safety and best doses. They recruited 40 patients with solid malignant tumors who had previously failed treatment, including breast cancer (19 cases), ovarian cancer (seven cases), and pancreatic cancer (three cases). The recommended phase II dose of 475 mg/m2 days 1, 8, and 15 every 4 weeks were generally well tolerated. The efficacy of CX-5461 is long-lasting, and the pharmacokinetic evaluation of the trial shows that the drug can be administered once a week. In addition, the side effects and adverse reactions of CX-5461 are relatively few, and the most common adverse events are skin phototoxicity and nausea. In addition, there are a number of phase I clinical trials to evaluate the safety and tolerance of CX-5461 in the treatment of solid tumors (NCT04890613), and the efficacy of CX-5461 combined with tazopril in the treatment of castrated drug-resistant metastatic prostate cancer (NCT05425862). Although the antitumor regimen targeted by G4 DNA has been studied for many years, there are no drugs on the market, and only a handful of drugs have successfully entered clinical trials. Therefore, the performance of CX-5461 is particularly noteworthy.
In the future, we need to find an appropriate dose of ribosome biogenesis-based therapy that is efficient without causing severe side effects. Meanwhile, a more in-depth analysis of the mechanisms underlying drug resistance is needed to identify new therapeutic targets.
Conclusions
The ribosome is an evolutionarily-conserved protein synthesis machine, and ribosome biogenesis ensures ribosome homeostasis.293 Ribosome biogenesis involves rDNA transcription, pre-rRNA cleavage, modification to form mature rRNA, RP synthesis and translocation into the nucleus, and rRNA assembly into large and small subunits; this is a complex and highly energy-intensive process.176 Therefore, different steps of ribosome biogenesis can be targeted to achieve the treatment of specific diseases. We reviewed the ribosome biogenesis and mechanisms in COVID-19, aging, the cardiovascular system, neurodegenerative disease, blood diseases, and cancer. This article aims to provide a new idea for scientists to understand how “ribosomal diseases” occur and provide a theoretical basis for developing a drug to treat such diseases.
Viruses utilize the translational machinery in host cells to achieve efficient replication. Recent researches suggest targeting ribosome biogenesis and functions is a potential new approach for controlling viral infection.294 CX-5461, a small-molecule drug that inhibits ribosome biogenesis, has exhibited good application prospects for malignant proliferative cancer accompanied by inhibited RNA pol I function.175 The SARS-CoV-2 5′UTR contains a SL1 hairpin that interact with Nsp1 to allow viral translation to proceed.103 Many scholars have found that the amino-terminal portion of Nsp1 is required for escape from Nsp1 inhibition.105,295 In order to shut down host translation, the virus maintains Nsp1 on the 40S subunit ribosome. After the viral translation is completed, the carboxy-terminal domain of Nsp1 folds back into the mRNA channel to prevent the translation of host mRNA.
We need to find a strategy to inhibit cancer precisely without affecting the function of normal cells by loading therapeutic drugs into cancer-specific targets. In addition, the implementation of precision medicine is a developing trend for the hierarchical management of different patients with different diseases, or even different patients with the same illness, based on ribosome biogenesis. It has been shown that genetic differences in ribosome occupancy can affect protein translation.296,297 A thorough understanding of the consequences of genetic differences among individuals is an important step toward personalized medicine. For the treatment of persons with a viral infection, the aim is to better understand the heterogeneity among individuals and determine who is most likely to benefit from a specific treatment.298 To achieve this goal, it is critical to develop biosensors to detect biomarkers such as viral molecules, ACE2 receptor, vitamin D levels, IgG, IgM antibodies, and C-reactive protein to help medical professionals to diagnose infection, determine the risk of infection, monitor immune response, and assess disease severity.299
Technological advances opened a new door to many aspects of ribosome biogenesis. Several new technologies, such as polysome profiling, ribosome-sequence, cryo-electron microscopy, and translating ribosome affinity purification, can accurately assess ribosome morphology, structure, translocation, ribosome-“frameshift”, ribosome-stalling, and ribosome-collision.300 For example, the abnormally expressed proteins in the plasma of patients with specific diseases can be found through ribosome-sequence technology, and the abundance of ribosomal proteins can be comprehensively monitored through Ribosome-Halo technology.301 These technologies not only explore the new mechanism of ribosome biogenesis and greatly expand the understanding of “ribosome diseases”, but also provide new targets for the precise diagnosis and treatment of such diseases.
Future direction
SARS-CoV-2 hijacks the host ribosome machinery to produce its proteins. This is achieved through the interaction between the viral Nsps and the host ribosome subunits. Nsp1 binds to the 40S subunit to block the entrance for host mRNA while allowing viral mRNA to pass through and get translated. The Nsps also inhibit host mRNA nuclear export and promote its degradation. Thus, drugs or vaccines targeting viral Nsps may be a useful strategy to tackle SARS-CoV-2 infection.302 The big question is how many booster shots we need to prevent infection. To answer this question, it is necessary to identify biomarkers that can predict the success of a vaccine. The titer of neutralizing antibodies may not be a reliable marker since both humoral and cellular immune responses are involved in virus killing.303,304
Ribosome inhibitors have been among the most successful antimicrobial drugs in the past. However, the pathogens have evolved to become resistant to most antibiotics. To overcome this problem, researchers have been trying to screen new compounds from natural products of cultivatable microorganisms. However, this approach often leads to the rediscovery of an old compound. The new generation of antibiotics will rely on the discovery of new synthetic scaffolds that are designed based on high-resolution ribosome structures.115,137 Ribosome protection proteins such as the ATP-binding cassette subfamily F(ATP-F) are responsible for the resistance to a wide array of antibiotics.305 Further research on the mechanisms underlying ATP-F-mediated ribosome protection will improve our understanding of antibiotic resistance and develop more efficient therapy.
References
Djumagulov, M. et al. Accuracy mechanism of eukaryotic ribosome translocation. Nature 600, 543–546 (2021).
Lv, K. et al. HectD1 controls hematopoietic stem cell regeneration by coordinating ribosome assembly and protein synthesis. Cell Stem Cell 28, 1275–1290 (2021).
Fan, Y. et al. Phosphoproteomic analysis of neonatal regenerative myocardium revealed important roles of checkpoint kinase 1 via activating mammalian target of rapamycin C1/ribosomal protein S6 kinase b-1 pathway. Circulation 141, 1554–1569 (2020).
Domitrovic, T., Moreira, M. H., Carneiro, R. L., Ribeiro-Alves, M. & Palhano, F. L. Natural variation of the cardiac transcriptome in humans. RNA Biol. 18, 1374–1381 (2021).
Prakash, V. et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 10, 2110 (2019).
Turi, Z., Lacey, M., Mistrik, M. & Moudry, P. Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging 11, 2512–2540 (2019).
Goncalves, A. M., Pereira-Santos, A. R., Esteves, A. R., Cardoso, S. M. & Empadinhas, N. The mitochondrial ribosome: a world of opportunities for mitochondrial dysfunction toward Parkinson’s disease. Antioxid. Redox Signal 34, 694–711 (2021).
Khoshnevis, S., Dreggors-Walker, R. E., Marchand, V., Motorin, Y. & Ghalei, H. Ribosomal RNA 2’-O-methylations regulate translation by impacting ribosome dynamics. Proc Natl Acad Sci USA. 119, e2117334119 (2022).
Melnikov, S., Manakongtreecheep, K. & Soll, D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol. Biol. Evol. 35, 1588–1598 (2018).
Kang, J. et al. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct. Target Ther. 6, 323 (2021).
Norris, K., Hopes, T. & Aspden, J. L. Ribosome heterogeneity and specialization in development. Wiley Interdiscip. Rev. RNA 12, e1644 (2021).
Doudna, J. A. & Rath, V. L. Structure and function of the eukaryotic ribosome: the next frontier. Cell 109, 153–156 (2002).
Bassler, J. & Hurt, E. Eukaryotic ribosome assembly. Annu. Rev. Biochem. 88, 281–306 (2019).
Liu, S. et al. RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 184, 1314–1329 (2021).
Heiss, F. B., Daiss, J. L., Becker, P. & Engel, C. Conserved strategies of RNA polymerase I hibernation and activation. Nat. Commun. 12, 758 (2021).
Pecoraro, A., Pagano, M., Russo, G. & Russo, A. Ribosome biogenesis and cancer: overview on ribosomal proteins. Int. J. Mol. Sci. 22, 5496 (2021).
Petelski, A. A. & Slavov, N. Analyzing ribosome remodeling in health and disease. Proteomics. 20, 2000039 (2020).
Rundlet, E. J. et al. Structural basis of early translocation events on the ribosome. Nature 595, 741–745 (2021).
Wood, W. N., Mohler, K., Rinehart, J. & Ibba, M. Deacylated tRNA accumulation is a trigger for bacterial antibiotic persistence independent of the stringent response. mBio. 12, e0113221 (2021).
Pilotto, S. et al. Structural basis of RNA polymerase inhibition by viral and host factors. Nat. Commun. 12, 5523 (2021).
Deleyto-Seldas, N. & Efeyan, A. The mTOR-autophagy axis and the control of metabolism. Front. Cell Dev. Biol. 9, 655731 (2021).
Iadevaia, V., Liu, R. & Proud, C. G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin Cell Dev. Biol. 36, 113–120 (2014).
Ben-Sahra, I., Howell, J. J., Asara, J. M. & Manning, B. D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013).
Rosario, F. J., Powell, T. L., Gupta, M. B., Cox, L. & Jansson, T. mTORC1 transcriptional regulation of ribosome subunits, protein synthesis, and molecular transport in primary human trophoblast cells. Front. Cell Dev. Biol. 8, 583801 (2020).
He, J. et al. Ribosome biogenesis protein Urb1 acts downstream of mTOR complex 1 to modulate digestive organ development in zebrafish. J. Genet. Genomics 44, 567–576 (2017).
Vasamsetti, B. M. K., Liu, Z., Park, Y. S. & Cho, N. J. Muscarinic acetylcholine receptors regulate the dephosphorylation of eukaryotic translation elongation factor 2 in SNU-407 colon cancer cells. Biochem. Biophys. Res. Commun. 516, 424–429 (2019).
Fujita, S. et al. Nutrient signalling in the regulation of human muscle protein synthesis. J. Physiol. 582, 813–823 (2007).
Vary, T. C., Anthony, J. C., Jefferson, L. S., Kimball, S. R. & Lynch, C. J. Rapamycin blunts nutrient stimulation of eIF4G, but not PKCepsilon phosphorylation, in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 293, E188–E196 (2007).
Smith, E. M. et al. The mTOR regulated RNA-binding protein LARP1 requires PABPC1 for guided mRNA interaction. Nucleic Acids Res. 49, 458–478 (2021).
Gallagher, E. E. et al. Consideration of binding kinetics in the design of stapled peptide mimics of the disordered proteins eukaryotic translation initiation factor 4E-binding protein 1 and eukaryotic translation initiation factor 4G. J. Med. Chem. 62, 4967–4978 (2019).
Murooka, T. T., Rahbar, R., Platanias, L. C. & Fish, E. N. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood 111, 4892–4901 (2008).
Zhou, H. & Huang, S. The complexes of mammalian target of rapamycin. Curr. Protein Pept. Sci. 11, 409–424 (2010).
Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757 (2018).
Calvisi, D. F. et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071–1083 (2011).
Zhou, L. H., Zhu, X. P., Xiao, H. F., Xin, P. L. & Li, C. T. [Effects of mTOR inhibitor rapamycin on Burkitt’s lymphoma cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 25, 1397–1405 (2017).
Fricke, S. L. et al. MTORC1/2 inhibition as a therapeutic strategy for PIK3CA mutant cancers. Mol. Cancer Ther. 18, 346–355 (2019).
Popay, T. M. et al. MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. Elife. 10, e60191 (2021).
Morcelle, C. et al. Oncogenic MYC induces the impaired ribosome biogenesis checkpoint and stabilizes p53 independent of increased ribosome content. Cancer Res. 79, 4348–4359 (2019).
Destefanis, F., Manara, V. & Bellosta, P. Myc as a regulator of ribosome biogenesis and cell competition: a link to cancer. Int. J. Mol. Sci. 21, 4037 (2020).
van Riggelen, J., Yetil, A. & Felsher, D. W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 (2010).
Grandori, C. et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 7, 311–318 (2005).
Hald, O. H. et al. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 38, 2800–2813 (2019).
Lee, H. C. et al. RNA polymerase I inhibition with CX-5461 as a novel therapeutic strategy to target MYC in multiple myeloma. Br. J. Haematol. 177, 80–94 (2017).
Li, F. et al. Rrn3 gene knockout affects ethanol-induced locomotion in adult heterozygous zebrafish. Psychopharmacol. 239, 621–630 (2022).
Lewinska, A., Klukowska-Rotzler, J., Deregowska, A., Adamczyk-Grochala, J. & Wnuk, M. c-Myc activation promotes cofilin-mediated F-actin cytoskeleton remodeling and telomere homeostasis as a response to oxidant-based DNA damage in medulloblastoma cells. Redox Biol. 24, 101163 (2019).
Mori, T. et al. c-Myc overexpression increases ribosome biogenesis and protein synthesis independent of mTORC1 activation in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 321, E551–E559 (2021).
Baluapuri, A. et al. MYC recruits SPT5 to RNA polymerase II to promote processive transcription elongation. Mol. Cell 74, 674–687 (2019).
Liang, K. W. et al. Targeting processive transcription elongation via SEC disruption for MYC-induced cancer therapy. Cell. 175, 766-779.e17 (2018).
Zhi, X. et al. Human RNA polymerase II associated factor 1 complex promotes tumorigenesis by activating c-MYC transcription in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 465, 685–690 (2015).
Verosloff, M. S. et al. RNA sequence and structure determinants of Pol III transcriptional termination in human cells. J. Mol. Biol. 433, 166978 (2021).
Campbell, K. J. & White, R. J. MYC regulation of cell growth through control of transcription by RNA polymerases I and III. Cold Spring Harb. Perspect. Med. 4, a018408 (2014).
Felton-Edkins, Z. A. et al. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle 2, 181–184 (2003).
Raha, D. et al. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl Acad. Sci. USA 107, 3639–3644 (2010).
Li, Z. & Hann, S. R. Nucleophosmin is essential for c-Myc nucleolar localization and c-Myc-mediated rDNA transcription. Oncogene 32, 1988–1994 (2013).
Shukla, S. K. & Kumar, V. Hepatitis B virus X protein and c-Myc cooperate in the upregulation of ribosome biogenesis and in cellular transformation. FEBS J. 279, 3859–3871 (2012).
Park, J. L. et al. Epigenetic regulation of RNA polymerase III transcription in early breast tumorigenesis. Oncogene 36, 6793–6804 (2017).
Gomez-Roman, N., Grandori, C., Eisenman, R. N. & White, R. J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 421, 290–294 (2003).
Kenneth, N. S. et al. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc. Natl Acad. Sci. USA 104, 14917–14922 (2007).
McCool, M. A., Bryant, C. J. & Baserga, S. J. MicroRNAs and long non-coding RNAs as novel regulators of ribosome biogenesis. Biochem. Soc. Trans. 48, 595–612 (2020).
Alkhatabi, H. A. et al. RPL27A is a target of miR-595 and may contribute to the myelodysplastic phenotype through ribosomal dysgenesis. Oncotarget 7, 47875–47890 (2016).
Starczynowski, D. T. et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 16, 49–58 (2010).
Wan, Y. et al. Transcriptome analysis reveals a ribosome constituents disorder involved in the RPL5 downregulated zebrafish model of Diamond-Blackfan anemia. BMC Med. Genomics 9, 13 (2016).
Connolly, M. et al. miR-424-5p reduces ribosomal RNA and protein synthesis in muscle wasting. J. Cachexia Sarcopenia Muscle 9, 400–416 (2018).
Quinodoz, S. A. et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790 (2021).
Li, D. et al. Activity dependent LoNA regulates translation by coordinating rRNA transcription and methylation. Nat. Commun. 9, 1726 (2018).
Xing, Y. H. et al. SLERT regulates DDX21 rings associated with Pol I transcription. Cell 169, 664–678 (2017).
Pietrzak, M., Rempala, G., Nelson, P. T., Zheng, J. J. & Hetman, M. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS ONE. 6, e22585 (2011).
Sia, P. I. et al. Role of the nucleolus in neurodegenerative diseases with particular reference to the retina: a review. Clin. Exp. Ophthalmol. 44, 188–195 (2016).
Tang, X. et al. Review on circular RNAs and new insights into their roles in cancer. Comput. Struct. Biotechnol. J. 19, 910–928 (2021).
Kleaveland, B., Shi, C. Y., Stefano, J. & Bartel, D. P. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174, 350–362 (2018).
Lei, B., Tian, Z., Fan, W. & Ni, B. Circular RNA: a novel biomarker and therapeutic target for human cancers. Int. J. Med Sci. 16, 292–301 (2019).
Abe, N. et al. Rolling circle translation of circular RNA in living human cells. Sci. Rep. 5, 16435 (2015).
Chen, X. et al. circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 6, 34985 (2016).
Wilusz, J. E. Circular RNAs: unexpected outputs of many protein-coding genes. RNA Biol. 14, 1007–1017 (2017).
Chen, C. Y. & Sarnow, P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268, 415–417 (1995).
Holdt, L. M. et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun. 7, 12429 (2016).
Bianco, C. & Mohr, I. Ribosome biogenesis restricts innate immune responses to virus infection and DNA. Elife. 8, e49551 (2019).
Prendecki, M. et al. Humoral and T-cell responses to SARS-CoV-2 vaccination in patients receiving immunosuppression. Ann. Rheum. Dis. 80, 1322–1329 (2021).
Dimitrova, D. G., Teysset, L. & Carre, C. RNA 2’-O-methylation (Nm) modification in human diseases. Genes. 10, 117 (2019).
Li, P. et al. RNA 2’-O-methyltransferase fibrillarin facilitates virus entry into macrophages through inhibiting type I interferon response. Front. Immunol. 13, 793582 (2022).
Krafcikova, P., Silhan, J., Nencka, R. & Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 11, 3717 (2020).
Viswanathan, T. et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 11, 3718 (2020).
Sorokin, I. I. et al. Non-canonical translation initiation mechanisms employed by eukaryotic viral mRNAs. Biochemistry 86, 1060–1094 (2021).
Kirby, M. P. et al. The hinge region of the Israeli acute paralysis virus internal ribosome entry site directs ribosomal positioning, translational activity, and virus infection. J. Virol. 96, e0133021 (2022).
Martinez-Salas, E., Francisco-Velilla, R., Fernandez-Chamorro, J. & Embarek, A. M. Insights into structural and mechanistic features of viral IRES elements. Front. Microbiol. 8, 2629 (2017).
Arhab, Y., Bulakhov, A. G., Pestova, T. V. & Hellen, C. U. T. Dissemination of internal ribosomal entry sites (IRES) between viruses by horizontal gene transfer. Viruses. 12, 612 (2020).
Gosavi, D. et al. Insights into the secondary and tertiary structure of the bovine viral diarrhea virus internal ribosome entry site. RNA Biol. 19, 496–506 (2022).
Sims, S., Michaelsen, K., Burkhard, S. & Fraefel, C. In vitro comparison of the internal ribosomal entry site activity from rodent hepacivirus and pegivirus and construction of pseudoparticles. Adv. Virol. 2021, 5569844 (2021).
Miao, Z., Tidu, A., Eriani, G. & Martin, F. Secondary structure of the SARS-CoV-2 5’-UTR. RNA Biol. 18, 447–456 (2021).
Verma, R. et al. RNA-Protein Interaction Analysis of SARS-CoV-2 5’ and 3’ Untranslated Regions Reveals a Role of Lysosome-Associated Membrane Protein-2a during Viral Infection. mSystems. 6, e0064321 (2021).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921 (2020).
Wang, Y. et al. Human SARS-CoV-2 has evolved to reduce CG dinucleotide in its open reading frames. Sci. Rep. 10, 12331 (2020).
Wang, Y. et al. Author correction: human SARS-CoV-2 has evolved to reduce CG dinucleotide in its open reading frames. Sci. Rep. 11, 12141 (2021).
Gunaseelan, S. et al. Prunin suppresses viral IRES activity and is a potential candidate for treating enterovirus A71 infection. Sci. Transl. Med. 11, eaar5759 (2019).
Islam, A. & Khan, M. A. SARS-CoV-2 mutations altering regulatory properties: deciphering host’s and virus’s perspectives. Gene Rep. 24, 101236 (2021).
Bhatt, P. R. et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 372, 1306–1313 (2021).
Sun, Y., Abriola, L., Surovtseva, Y. V., Lindenbach, B. D. & Guo, J. U. Restriction of SARS-CoV-2 replication by targeting programmed -1 ribosomal frameshifting in vitro. Proc Natl Acad Sci U S A. 118, e2023051118 (2020).
da Silva, S. J. R., Alves da Silva, C. T., Mendes, R. P. G. & Pena, L. Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J. Med Virol. 92, 1427–1429 (2020).
Thoms, M. et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369, 1249–1255 (2020).
Schubert, K. et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 27, 959–966 (2020).
Zhang, K. et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci Adv. 7, eabe7386 (2021).
Sola, I., Almazan, F., Zuniga, S. & Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288 (2015).
Tidu, A. et al. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA 27, 253–264 (2020).
Vora, S. M. et al. Targeting stem-loop 1 of the SARS-CoV-2 5’ UTR to suppress viral translation and Nsp1 evasion. Proc Natl Acad Sci USA 119, e2117198119 (2022).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339 (2020).
Xia, H. et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 33, 108234 (2020).
Zhou, Q. et al. Interferon-alpha2b treatment for COVID-19. Front. Immunol. 11, 1061 (2020).
Finkel, Y. et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature 594, 240–245 (2021).
Meng, W., Han, S. C., Li, C. C., Dong, H. J. & Wang, X. J. Multifunctional viral protein gamma34.5 manipulates nucleolar protein NOP53 for optimal viral replication of HSV-1. Cell Death Dis. 9, 103 (2018).
Yang, B. et al. Clinical and molecular characteristics of COVID-19 patients with persistent SARS-CoV-2 infection. Nat. Commun. 12, 3501 (2021).
Andrei, S., Droc, G. & Stefan, G. FDA approved antibacterial drugs: 2018-2019. Discoveries. 7, e102 (2019).
Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Huemer, M., Mairpady Shambat, S., Brugger, S. D. & Zinkernagel, A. S. Antibiotic resistance and persistence-Implications for human health and treatment perspectives. EMBO Rep. 21, e51034 (2020).
Huter, P., Muller, C., Arenz, S., Beckert, B. & Wilson, D. N. Structural basis for ribosome rescue in bacteria. Trends Biochem. Sci. 42, 669–680 (2017).
Lin, J., Zhou, D., Steitz, T. A., Polikanov, Y. S. & Gagnon, M. G. Ribosome-targeting antibiotics: modes of action, mechanisms of resistance, and implications for drug design. Annu. Rev. Biochem. 87, 451–478 (2018).
Wilson, D. N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12, 35–48 (2014).
Fischbach, M. A. & Walsh, C. T. Antibiotics for emerging pathogens. Science 325, 1089–1093 (2009).
Brodersen, D. E. et al. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103, 1143–1154 (2000).
Wohlgemuth, I. et al. Translation error clusters induced by aminoglycoside antibiotics. Nat. Commun. 12, 1830 (2021).
Mutuku, C., Gazdag, Z. & Melegh, S. Occurrence of antibiotics and bacterial resistance genes in wastewater: resistance mechanisms and antimicrobial resistance control approaches. World J. Microbiol. Biotechnol. 38, 152 (2022).
Olson, M. W. et al. Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob. Agents Chemother. 50, 2156–2166 (2006).
Grossman, T. H. et al. Target- and resistance-based mechanistic studies with TP-434, a novel fluorocycline antibiotic. Antimicrob. Agents Chemother. 56, 2559–2564 (2012).
Jenner, L. et al. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl Acad. Sci. USA 110, 3812–3816 (2013).
Yanagisawa, T., Lee, J. T., Wu, H. C. & Kawakami, M. Relationship of protein structure of isoleucyl-tRNA synthetase with pseudomonic acid resistance of Escherichia coli. A proposed mode of action of pseudomonic acid as an inhibitor of isoleucyl-tRNA synthetase. J. Biol. Chem. 269, 24304–24309 (1994).
Yarlagadda, V. et al. Resistance-guided discovery of elfamycin antibiotic producers with antigonococcal activity. ACS Infect. Dis. 6, 3163–3173 (2020).
Leiva, L. E. et al. Modulation of Escherichia coli translation by the specific inactivation of tRNA(Gly) under oxidative stress. Front. Genet. 11, 856 (2020).
Schlunzen, F. et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 413, 814–821 (2001).
Bulkley, D., Innis, C. A., Blaha, G. & Steitz, T. A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl Acad. Sci. USA 107, 17158–17163 (2010).
Hansen, J. L., Moore, P. B. & Steitz, T. A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 330, 1061–1075 (2003).
Champney, W. S. The other target for ribosomal antibiotics: inhibition of bacterial ribosomal subunit formation. Infect. Disord. Drug Targets 6, 377–390 (2006).
Yelin, I. & Kishony, R. Antibiotic resistance. Cell 172, 1136–1136 (2018).
Christaki, E., Marcou, M. & Tofarides, A. Antimicrobial resistance in bacteria: mechanisms, evolution, and persistence. J. Mol. Evol. 88, 26–40 (2020).
Fernandez, L. & Hancock, R. E. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 25, 661–681 (2012).
Smith, A. M., Jain, M., Mulroney, L., Garalde, D. R. & Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE. 14, e0216709 (2019).
Tsai, K. et al. Directed evolution of the rRNA methylating enzyme Cfr reveals molecular basis of antibiotic resistance. Elife. 11, e70017 (2022).
Long, K. S., Poehlsgaard, J., Kehrenberg, C., Schwarz, S. & Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 50, 2500–2505 (2006).
Mitcheltree, M. J. et al. A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 599, 507–512 (2021).
Gc, K., To, D., Jayalath, K. & Abeysirigunawardena, S. Discovery of a novel small molecular peptide that disrupts helix 34 of bacterial ribosomal RNA. RSC Adv. 9, 40268–40276 (2019).
Jimbo, H., Izuhara, T., Hihara, Y., Hisabori, T. & Nishiyama, Y. Light-inducible expression of translation factor EF-Tu during acclimation to strong light enhances the repair of photosystem II. Proc. Natl Acad. Sci. USA 116, 21268–21273 (2019).
Lopatkin, A. J. et al. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science. 371, eaba0862 (2021).
Schaenzer, A. J. & Wright, G. D. Antibiotic resistance by enzymatic modification of antibiotic targets. Trends Mol. Med. 26, 768–782 (2020).
Rajesh, T. et al. Phosphorylation of chloramphenicol by a recombinant protein Yhr2 from Streptomyces avermitilis MA4680. Bioorg. Med. Chem. Lett. 23, 3614–3619 (2013).
Wright, G. D. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57, 1451–1470 (2005).
Rudra, P., Hurst-Hess, K., Lappierre, P. & Ghosh, P. High levels of intrinsic tetracycline resistance in Mycobacterium abscessus are conferred by a tetracycline-modifying monooxygenase. Antimicrob. Agents Chemother. 62, e00119–e00118 (2018).
Volkers, G., Palm, G. J., Weiss, M. S., Wright, G. D. & Hinrichs, W. Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett. 585, 1061–1066 (2011).
De la Corte-Rodriguez, H., Rodriguez-Merchan, E. C., Alvarez-Roman, M. T. & Jimenez-Yuste, V. Applying World Health Organization 2020 guidelines on physical activity and sedentary behavior to people with hemophilia. Expert Rev. Hematol. 14, 429–436 (2021).
Packer, M. Differential pathophysiological mechanisms in heart failure with a reduced or preserved ejection fraction in diabetes. JACC Heart Fail. 9, 535–549 (2021).
Gao, R. et al. Long noncoding RNA cardiac physiological hypertrophy-associated regulator induces cardiac physiological hypertrophy and promotes functional recovery after myocardial ischemia-reperfusion injury. Circulation 144, 303–317 (2021).
Liao, H. et al. GPR39 promotes cardiac hypertrophy by regulating the AMPK-mTOR pathway and protein synthesis. Cell Biol. Int. 45, 1211–1219 (2021).
Gelinas, R. et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 9, 374 (2018).
Russell, J. & Zomerdijk, J. C. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 30, 87–96 (2005).
Zeitz, M. J. & Smyth, J. W. Translating translation to mechanisms of cardiac hypertrophy. J. Cardiovasc. Dev. Dis. 7, 9 (2020).
Luyken, J., Hannan, R. D., Cheung, J. Y. & Rothblum, L. I. Regulation of rDNA transcription during endothelin-1-induced hypertrophy of neonatal cardiomyocytes. Hyperphosphorylation of upstream binding factor, an rDNA transcription factor. Circ. Res. 78, 354–361 (1996).
Hannan, R. D., Luyken, J. & Rothblum, L. I. Regulation of ribosomal DNA transcription during contraction-induced hypertrophy of neonatal cardiomyocytes. J. Biol. Chem. 271, 3213–3220 (1996).
Hannan, R. D., Luyken, J. & Rothblum, L. I. Regulation of rDNA transcription factors during cardiomyocyte hypertrophy induced by adrenergic agents. J. Biol. Chem. 270, 8290–8297 (1995).
Liu, Y. et al. Dependence of genome size and copy number of rRNA gene on cell volume in dinoflagellates. Harmful Algae 109, 102108 (2021).
Nieto, B. et al. Efficient fractionation and analysis of ribosome assembly intermediates in human cells. RNA Biol. 18, 182–197 (2021).
Rackham, O. et al. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 16, 1874–1890 (2016).
Pugh, T. J. et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16, 601–608 (2014).
Zhang, Z., Liu, R., Townsend, P. A. & Proud, C. G. p90(RSK)s mediate the activation of ribosomal RNA synthesis by the hypertrophic agonist phenylephrine in adult cardiomyocytes. J. Mol. Cell Cardiol. 59, 139–147 (2013).
Kang, J. et al. Amino acid-dependent signaling via S6K1 and MYC is essential for regulation of rDNA transcription. Oncotarget 7, 48887–48904 (2016).
Dagenais, G. R. et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet 395, 785–794 (2020).
Riquelme, J. A. et al. Therapeutic targeting of autophagy in myocardial infarction and heart failure. Expert Rev. Cardiovasc. Ther. 14, 1007–1019 (2016).
Lonborg, J. T. Targeting reperfusion injury in the era of primary percutaneous coronary intervention: hope or hype? Heart 101, 1612–1618 (2015).
Tran, Q. K. Reciprocality between estrogen biology and calcium signaling in the cardiovascular system. Front. Endocrinol. 11, 568203 (2020).
Deng, H. F. et al. Nicorandil alleviates cardiac remodeling and dysfunction post -infarction by up-regulating the nucleolin/autophagy axis. Cell Signal 92, 110272 (2022).
Tong, Z. et al. Phosphorylation of nucleolin is indispensable to upregulate miR-21 and inhibit apoptosis in cardiomyocytes. J. Cell Physiol. 234, 4044–4053 (2019).
Xue, J. et al. The identification and validation of hub genes associated with acute myocardial infarction using weighted gene co-expression network analysis. J. Cardiovasc. Dev. Dis. 9, 30 (2022).
Tang, Y. et al. Nucleolin improves heart function during recovery from myocardial infarction by modulating macrophage polarization. J. Cardiovasc. Pharm. Ther. 26, 386–395 (2021).
Kanamori, H. et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 300, H2261–H2271 (2011).
Zhang, X., Ren, Z., Xu, W. & Jiang, Z. Necroptosis in atherosclerosis. Clin. Chim. Acta 534, 22–28 (2022).
Aherrahrou, R. et al. Genetic regulation of atherosclerosis-relevant phenotypes in human vascular smooth muscle cells. Circ. Res. 127, 1552–1565 (2020).
Basatemur, G. L., Jorgensen, H. F., Clarke, M. C. H., Bennett, M. R. & Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 16, 727–744 (2019).
Breznak, S. M., Kotb, N. M. & Rangan, P. Dynamic regulation of ribosome levels and translation during development. Semin. Cell Dev. Biol. 136, 27–37 (2022).
Zisi, A., Bartek, J. & Lindstrom, M. S. Targeting ribosome biogenesis in cancer: lessons learned and way forward. Cancers. 14, 2126 (2022).
Klinge, S. & Woolford, J. L. Jr. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 20, 116–131 (2019).
Jia, F. et al. BOP1 knockdown attenuates neointimal hyperplasia by activating p53 and inhibiting nascent protein synthesis. Oxid. Med. Cell Longev. 2021, 5986260 (2021).
Strezoska, Z., Pestov, D. G. & Lau, L. F. Bop1 is a mouse WD40 repeat nucleolar protein involved in 28S and 5. 8S RRNA processing and 60S ribosome biogenesis. Mol. Cell Biol. 20, 5516–5528 (2000).
Lewinska, A. et al. Curcumin induces oxidation-dependent cell cycle arrest mediated by SIRT7 inhibition of rDNA transcription in human aortic smooth muscle cells. Toxicol. Lett. 233, 227–238 (2015).
Fonseca, F. A. & Izar, M. C. Role of inflammation in cardiac remodeling after acute myocardial infarction. Front. Physiol. 13, 927163 (2022).
Xiang, P., Blanchard, V. & Francis, G. A. Smooth muscle cell-macrophage interactions leading to foam cell formation in atherosclerosis: location, location, location. Front. Physiol. 13, 921597 (2022).
Yang, K., Xiao, Q., Niu, M., Pan, X. & Zhu, X. Exosomes in atherosclerosis: convergence on macrophages. Int. J. Biol. Sci. 18, 3266–3281 (2022).
Basu, A. et al. Ribosomal protein L13a deficiency in macrophages promotes atherosclerosis by limiting translation control-dependent retardation of inflammation. Arterioscler. Thromb. Vasc. Biol. 34, 533–542 (2014).
Wang, H. X. & Zhao, Y. X. Prediction of genetic risk factors of atherosclerosis using various bioinformatic tools. Genet. Mol. Res. 15 (2016).
Quiles-Jimenez, A. et al. N6-methyladenosine in RNA of atherosclerotic plaques: An epitranscriptomic signature of human carotid atherosclerosis. Biochem. Biophys. Res. Commun. 533, 631–637 (2020).
Altvater, M., Schutz, S., Chang, Y. & Panse, V. G. Dissecting ribosome assembly and transport in budding yeast. Methods Cell Biol. 122, 437–461 (2014).
Sergeeva, O. et al. Modification of adenosine196 by Mettl3 methyltransferase in the 5’-external transcribed spacer of 47S pre-rRNA affects rRNA maturation. Cells. 9, 1061 (2020).
Yazdian-Robati, R. et al. Therapeutic applications of AS1411 aptamer, an update review. Int. J. Biol. Macromol. 155, 1420–1431 (2020).
Tagliaferro, M. et al. Nucleolar localization of the ErbB3 receptor as a new target in glioblastoma. BMC Mol. Cell Biol. 23, 13 (2022).
Jia, W., Yao, Z., Zhao, J., Guan, Q. & Gao, L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 186, 1–10 (2017).
Chen, C. et al. LncRNA H19 is involved in myocardial ischemic preconditioning via increasing the stability of nucleolin protein. J. Cell Physiol. 235, 5985–5994 (2020).
Rosello-Lleti, E. et al. Influence of heart failure on nucleolar organization and protein expression in human hearts. Biochem. Biophys. Res. Commun. 418, 222–228 (2012).
Das, S. et al. Diabetes mellitus-induced long noncoding RNA Dnm3os regulates macrophage functions and inflammation via nuclear mechanisms. Arterioscler. Thromb. Vasc. Biol. 38, 1806–1820 (2018).
Jiang, B. et al. Nucleolin protects the heart from ischaemia-reperfusion injury by up-regulating heat shock protein 32. Cardiovasc. Res. 99, 92–101 (2013).
Sun, H. et al. Nucleolin protects against doxorubicin-induced cardiotoxicity via upregulating microRNA-21. J. Cell Physiol. 233, 9516–9525 (2018).
Monte, E. et al. Systems proteomics of cardiac chromatin identifies nucleolin as a regulator of growth and cellular plasticity in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 305, H1624–H1638 (2013).
Bakkenist, C. J. & Kastan, M. B. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair 36, 8–12 (2015).
Murry, C. E., Jennings, R. B. & Reimer, K. A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136 (1986).
Shi, W. & Vinten-Johansen, J. Endogenous cardioprotection by ischaemic postconditioning and remote conditioning. Cardiovasc. Res. 94, 206–216 (2012).
Jiang, B. et al. Nucleolin involved in myocardial ischaemic preconditioning via post-transcriptional control of HSPA1A expression. Cardiovasc. Res. 102, 56–67 (2014).
Ma, Y., Mouton, A. J. & Lindsey, M. L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28 (2018).
Libby, P. et al. Atherosclerosis. Nat. Rev. Dis. Prim. 5, 56 (2019).
Chen, W. et al. Extracellular vesicle YRNA in atherosclerosis. Clin. Chim. Acta 517, 15–22 (2021).
Wolf, D. & Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 124, 315–327 (2019).
Li, Y. et al. Nucleolin protects macrophages from oxLDL-induced foam cell formation through up-regulating ABCA1 expression. Biochem. Biophys. Res. Commun. 486, 364–371 (2017).
Sun, H. et al. Nucleolin regulates the proliferation of vascular smooth muscle cells in atherosclerotic via Aurora B. J. Cell Mol. Med. 25, 751–762 (2021).
Bjedov, I. & Rallis, C. The target of rapamycin signalling pathway in ageing and lifespan regulation. Genes. 11, 1043 (2020).
McKendry, J., Stokes, T., McLeod, J. C. & Phillips, S. M. Resistance exercise, aging, disuse, and muscle protein metabolism. Compr. Physiol. 11, 2249–2278 (2021).
van Gastel, J. et al. Aging-related modifications to G protein-coupled receptor signaling diversity. Pharm. Ther. 223, 107793 (2021).
Zhang, W. et al. Decreased expression of ribosomal proteins in human age-related cataract. Invest Ophthalmol. Vis. Sci. 43, 198–204 (2002).
Kirby, T. J. et al. Blunted hypertrophic response in aged skeletal muscle is associated with decreased ribosome biogenesis. J. Appl. Physiol. 119, 321–327 (2015).
Hu, Z. et al. Ssd1 and Gcn2 suppress global translation efficiency in replicatively aged yeast while their activation extends lifespan. Elife. 7, e35551 (2018).
Stein, K. C., Morales-Polanco, F., van der Lienden, J., Rainbolt, T. K. & Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 601, 637–642 (2022).
Vijg, J. & Suh, Y. Genome instability and aging. Annu Rev. Physiol. 75, 645–668 (2013).
Flach, J. et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014).
Paredes, S. et al. The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability. J. Biol. Chem. 293, 11242–11250 (2018).
Smirnov, E., Chmúrčiaková, N. & D, C. Human rDNA and cancer. Cells 10, 3452 (2021).
Lee, J. W. & Ebb, O. Genomic instability and cellular senescence: lessons from the budding yeast. Front. Cell Dev. Biol. 8, 619126 (2021).
Kumari, P., Tarighi, S., Braun, T. & Ianni, A. SIRT7 acts as a guardian of cellular integrity by controlling nucleolar and extra-nucleolar functions. Genes. 12, 1361 (2021).
Ianni, A., Hoelper, S., Krueger, M., Braun, T. & E, B. Sirt7 stabilizes rDNA heterochromatin through recruitment of DNMT1 and Sirt1. Biochem. Biophys. Res. Commun. 492, 434–440 (2017).
Chen, S. et al. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat. Commun. 7, 10734 (2016).
Drokhlyansky, E. et al. The human and mouse enteric nervous system at single-cell resolution. Cell 182, 1606–1622 (2020).
Scheurer, L. et al. Expression of immunoglobulin constant domain genes in neurons of the mouse central nervous system. Life Sci. Alliance. 4, e202101154 (2021).
Qazi, T. J., Quan, Z., Mir, A. & Qing, H. Epigenetics in Alzheimer’s disease: perspective of DNA methylation. Mol. Neurobiol. 55, 1026–1044 (2018).
Ding, Q., Markesbery, W. R., Chen, Q., Li, F. & Keller, J. N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 25, 9171–9175 (2005).
Ding, Q. et al. Increased 5S rRNA oxidation in Alzheimer’s disease. J. Alzheimers Dis. 29, 201–209 (2012).
Hernandez-Ortega, K., Garcia-Esparcia, P., Gil, L., Lucas, J. J. & Ferrer, I. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 26, 593–605 (2016).
Tolosa, E., Garrido, A., Scholz, S. W. & Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 20, 385–397 (2021).
Wang, Q. et al. The role of gut dysbiosis in Parkinson’s disease: mechanistic insights and therapeutic options. Brain 144, 2571–2593 (2021).
Healy-Stoffel, M., Ahmad, S. O., Stanford, J. A. & Levant, B. Altered nucleolar morphology in substantia nigra dopamine neurons following 6-hydroxydopamine lesion in rats. Neurosci. Lett. 546, 26–30 (2013).
Parlato, R. & Liss, B. How Parkinson’s disease meets nucleolar stress. Biochim. Biophys. Acta 1842, 791–797 (2014).
Figueiredo, V. C. & McCarthy, J. J. Targeting cancer via ribosome biogenesis: the cachexia perspective. Cell Mol. Life Sci. 78, 5775–5787 (2021).
Pellegrino, S., Terrosu, S., Yusupova, G. & Yusupov, M. Inhibition of the eukaryotic 80S ribosome as a potential anticancer therapy: a structural perspective. Cancers. 13, 4392 (2021).
Elhamamsy, A. R., Metge, B. J., Alsheikh, H. A., Shevde, L. A. & Samant, R. S. Ribosome biogenesis: a central player in cancer metastasis and therapeutic resistance. Cancer Res. 82, 2344–2353 (2022).
El Khoury, W. & Nasr, Z. Deregulation of ribosomal proteins in human cancers. Biosci. Rep. 41, BSR20211577 (2021).
van den Akker, G. G. H., Caron, M. M. J., Peffers, M. J. & Welting, T. J. M. Ribosome dysfunction in osteoarthritis. Curr. Opin. Rheumatol. 34, 61–67 (2022).
Challa, S. et al. Ribosome ADP-ribosylation inhibits translation and maintains proteostasis in cancers. Cell 184, 4531–4546 (2021).
Jung, J. H. et al. Colocalization of MID1IP1 and c-Myc is Critically Involved in Liver Cancer Growth via Regulation of Ribosomal Protein L5 and L11 and CNOT2. Cells. 9, 985 (2020).
Dang, C. V. MYC on the path to cancer. Cell 149, 22–35 (2012).
Gaviraghi, M., Vivori, C. & Tonon, G. How cancer exploits ribosomal RNA biogenesis: a journey beyond the boundaries of rRNA transcription. Cells. 8, 1098 (2019).
Ji, H. et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 6, e26057 (2011).
Barna, M. et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975 (2008).
Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 69, 8839–8843 (2009).
Nait Slimane, S. et al. Ribosome biogenesis alterations in colorectal cancer. Cells. 9, 2361 (2020).
Wang, X. et al. The long non-coding RNA CYTOR drives colorectal cancer progression by interacting with NCL and Sam68. Mol. Cancer 17, 110 (2018).
Khurts, S. et al. Nucleolin interacts with telomerase. J. Biol. Chem. 279, 51508–51515 (2004).
Lee, S. J. et al. Identification of nucleolin as a novel AEG-1-interacting protein in breast cancer via interactome profiling. Cancers. 13, 2842 (2021).
Raska, I., Shaw, P. J. & Cmarko, D. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell Biol. 18, 325–334 (2006).
Volarevic, S. et al. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288, 2045–2047 (2000).
Gentilella, A. et al. Autogenous control of 5’TOP mRNA stability by 40S ribosomes. Mol. Cell 67, 55–70 (2017).
Nakhoul, H. et al. Ribosomopathies: mechanisms of disease. Clin. Med. Insights Blood Disord. 7, 7–16 (2014).
Domostegui, A. et al. Impaired ribosome biogenesis checkpoint activation induces p53-dependent MCL-1 degradation and MYC-driven lymphoma death. Blood 137, 3351–3364 (2021).
Macias, E. et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18, 231–243 (2010).
Liu, S., Tackmann, N. R., Yang, J. & Zhang, Y. Disruption of the RP-MDM2-p53 pathway accelerates APC loss-induced colorectal tumorigenesis. Oncogene 36, 1374–1383 (2017).
Pelletier, J. et al. Nucleotide depletion reveals the impaired ribosome biogenesis checkpoint as a barrier against DNA damage. EMBO J. 39, e103838 (2020).
Danilova, N. & Gazda, H. T. Ribosomopathies: how a common root can cause a tree of pathologies. Dis. Model Mech. 8, 1013–1026 (2015).
Bursac, S., Prodan, Y., Pullen, N., Bartek, J. & Volarevic, S. Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer 7, 57–76 (2021).
Li, Z. et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat. Chem. Biol. 18, 751–761 (2022).
Koltowska, K. et al. The RNA helicase Ddx21 controls Vegfc-driven developmental lymphangiogenesis by balancing endothelial cell ribosome biogenesis and p53 function. Nat. Cell Biol. 23, 1136–1147 (2021).
Nguyen Van Long, F. et al. Low level of Fibrillarin, a ribosome biogenesis factor, is a new independent marker of poor outcome in breast cancer. BMC Cancer 22, 526 (2022).
Signer, R. A., Magee, J. A., Salic, A. & Morrison, S. J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 (2014).
Choijilsuren, H. B., Park, Y. & Jung, M. Mechanisms of somatic transformation in inherited bone marrow failure syndromes. Hematol. Am. Soc. Hematol. Educ. Program 2021, 390–398 (2021).
Khajuria, R. K. et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173, 90–103 (2018).
Garus, A. & Autexier, C. Dyskerin: an essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA 27, 1441–1458 (2021).
Chang, M., Johnston, S., Seilie, A. M., Hergott, D. & Murphy, S. C. Application of dried blood spot sample pooling strategies for Plasmodium 18S rRNA biomarker testing to facilitate identification of infected persons in large-scale epidemiological studies. Malar. J. 20, 391 (2021).
Da Costa, L., Leblanc, T. & Mohandas, N. Diamond-Blackfan anemia. Blood 136, 1262–1273 (2020).
Dorn, K. M. et al. Diamond-Blackfan anemia: a case report and review of the literature. Neonatology 118, 500–504 (2021).
Uechi, T. et al. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum. Mol. Genet. 17, 3204–3211 (2008).
Jaako, P. et al. Gene therapy cures the anemia and lethal bone marrow failure in a mouse model of RPS19-deficient Diamond-Blackfan anemia. Haematologica 99, 1792–1798 (2014).
Danilova, N., Sakamoto, K. M. & Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112, 5228–5237 (2008).
McGowan, K. A. et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 40, 963–970 (2008).
Da Costa, L., Narla, A. & Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Res. 7, F1000 (2018).
Zhu, Y. et al. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35, 316–326 (2009).
Boocock, G. R. et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 33, 97–101 (2003).
Stepensky, P. et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in aShwachman-Diamond like syndrome. J. Med. Genet. 54, 558–566 (2017).
Venturi, G. & Montanaro, L. How altered ribosome production can cause or contribute to human disease: the spectrum of ribosomopathies. Cells. 9, 2300 (2020).
Carapito, R. et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J. Clin. Invest 127, 4090–4103 (2017).
Tan, S. et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 134, 277–290 (2019).
Ganapathi, K. A. et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood 110, 1458–1465 (2007).
Rujkijyanont, P., Adams, S. L., Beyene, J. & Dror, Y. Bone marrow cells from patients with Shwachman-Diamond syndrome abnormally express genes involved in ribosome biogenesis and RNA processing. Br. J. Haematol. 145, 806–815 (2009).
Nguyen, T. A., Lehr, A. W. & Roche, K. W. Neuroligins and neurodevelopmental disorders: X-linked genetics. Front. Synaptic Neurosci. 12, 33 (2020).
Ruggero, D. et al. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299, 259–262 (2003).
Nachmani, D. et al. Germline NPM1 mutations lead to altered rRNA 2’-O-methylation and cause dyskeratosis congenita. Nat. Genet. 51, 1518–1529 (2019).
Welting, T. J., van Venrooij, W. J. & Pruijn, G. J. Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res. 32, 2138–2146 (2004).
Narla, A. & Ebert, B. L. Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205 (2010).
Steinbusch, M. M. F. et al. Expression of RMRP RNA is regulated in chondrocyte hypertrophy and determines chondrogenic differentiation. Sci. Rep. 7, 6440 (2017).
Bywater, M. J. et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 22, 51–65 (2012).
Drygin, D. et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 69, 7653–7661 (2009).
Drygin, D. et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430 (2011).
Wei, T. et al. Small-molecule targeting of RNA polymerase I activates a conserved transcription Elongation checkpoint. Cell Rep. 23, 404–414 (2018).
Xu, H. et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 8, 14432 (2017).
Hilton, J. et al. Results of the phase I CCTG IND.231 trial of CX-5461 in patients with advanced solid tumors enriched for DNA-repair deficiencies. Nat. Commun. 13, 3607 (2022).
An, H. & Harper, J. W. Ribosome abundance control via the ubiquitin-proteasome system and autophagy. J. Mol. Biol. 432, 170–184 (2020).
Dong, H. J., Zhang, R., Kuang, Y. & Wang, X. J. Selective regulation in ribosome biogenesis and protein production for efficient viral translation. Arch. Microbiol. 203, 1021–1032 (2021).
Shi, M. et al. SARS-CoV-2 Nsp1 suppresses host but not viral translation through a bipartite mechanism. Preprint at bioRxiv (2020).
Zhang, H. et al. Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in human aortic dissection. BMC Genomics 22, 724 (2021).
Cenik, C. et al. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 25, 1610–1621 (2015).
Beitler, J. R. et al. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir. Med. 10, 107–120 (2022).
Vasquez, V. & Orozco, J. Detection of COVID-19-related biomarkers by electrochemical biosensors and potential for diagnosis, prognosis, and prediction of the course of the disease in the context of personalized medicine. Anal. Bioanal. Chem. 16, 1–29 (2022).
VanInsberghe, M., van den Berg, J., Andersson-Rolf, A., Clevers, H. & van Oudenaarden, A. Single-cell Ribo-seq reveals cell cycle-dependent translational pausing. Nature 597, 561–565 (2021).
An, H., Ordureau, A., Korner, M., Paulo, J. A. & Harper, J. W. Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature 583, 303–309 (2020).
Wei, J. & Hui, A. Review of ribosome interactions with SARS-CoV-2 and COVID-19 mRNA vaccine. Life . 12, 57 (2022).
Callaway, E. Scientists identify long-sought marker for COVID vaccine success. Nature. 2 (2021).
Khoury, D. S. et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 27, 1205–1211 (2021).
Ero, R., Yan, X. F. & Gao, Y. G. Ribosome protection proteins-"new" players in the global arms race with antibiotic-resistant pathogens. Int. J. Mol. Sci. 22, 5356 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, No. 81870194 to Y.Li, No. 91849122 to Y.Li, NSFC, Nos. 81873528, 81670358 to Y.-H.S., No. U1601227 to X.-Y.Y.), Jiangsu Province Peak of Talent in Six Industries (BU24600117 to Y.Li.), the project for the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Translational Research Grant of NCRCH (2020WSB07). The Introduction Project of Clinical Medicine Expert Team for Suzhou (No. SZYJTD201704). The Fundamental Research Funds for the Central Universities (2019PT350005 to X.P.), National Natural Science Foundation of China (81970444 to X.P.), Beijing Municipal Science and Technology Project (Z201100005420030 to X.P.), National high-level talents special support plan (2020-RSW02 to X.P.), CAMS Innovation Fund for Medical Sciences (2021-I2M-1-065 to X.P.). We apologize in advance to colleagues whose work was not directly cited in this Review because of space limitations.
Author information
Authors and Affiliations
Contributions
L.J., Y.L., X.-Y.Y., and X.P. collected references and wrote parts of the paper. L.J., Y.Z., and J.T. collected references and drew the figures and tables. Y.-H.S. and Y.L. conceived, wrote, and revised the paper. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiao, L., Liu, Y., Yu, XY. et al. Ribosome biogenesis in disease: new players and therapeutic targets. Sig Transduct Target Ther 8, 15 (2023). https://doi.org/10.1038/s41392-022-01285-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01285-4
This article is cited by
-
In-depth metaproteomics analysis of tongue coating for gastric cancer: a multicenter diagnostic research study
Microbiome (2024)
-
Protein neddylation and its role in health and diseases
Signal Transduction and Targeted Therapy (2024)
-
Enhancing the productivity and proliferation of CHO-K1 cells by oncoprotein YAP (Yes-associated protein)
Applied Microbiology and Biotechnology (2024)
-
Weighted gene co-expression network analysis revealed T cell differentiation associated with the age-related phenotypes in COVID-19 patients
BMC Medical Genomics (2023)
-
Transcriptome driven discovery of novel candidate genes for human neurological disorders in the telomer-to-telomer genome assembly era
Human Genomics (2023)
