
Abstract
Aging is a gradual and irreversible pathophysiological process. It presents with declines in tissue and cell functions and significant increases in the risks of various aging-related diseases, including neurodegenerative diseases, cardiovascular diseases, metabolic diseases, musculoskeletal diseases, and immune system diseases. Although the development of modern medicine has promoted human health and greatly extended life expectancy, with the aging of society, a variety of chronic diseases have gradually become the most important causes of disability and death in elderly individuals. Current research on aging focuses on elucidating how various endogenous and exogenous stresses (such as genomic instability, telomere dysfunction, epigenetic alterations, loss of proteostasis, compromise of autophagy, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, deregulated nutrient sensing) participate in the regulation of aging. Furthermore, thorough research on the pathogenesis of aging to identify interventions that promote health and longevity (such as caloric restriction, microbiota transplantation, and nutritional intervention) and clinical treatment methods for aging-related diseases (depletion of senescent cells, stem cell therapy, antioxidative and anti-inflammatory treatments, and hormone replacement therapy) could decrease the incidence and development of aging-related diseases and in turn promote healthy aging and longevity.
Similar content being viewed by others
Introduction
Aging is a ubiquitous biological process that results in a progressive and irreversible decline in physical function across all organ systems that is induced by the accumulation of damage in response to a variety of stressors. Surprisingly, in 1925, research revealed that light intensity could impact the growth rate and lifespan of Drosophila.1 This finding attracted a considerable amount of research interest in the aging area and captured the curiosity and imagination of the public (Fig. 1). In addition, caloric restriction (CR) has also been found to impact aging, age-related pathologies, and longevity in mice and rats.2 These findings show that the plasticity of the aging process is critical for longevity. Approximately 30 years ago, the first long-lived strain was isolated from C. elegans, and this discovery ushered in a new era of aging research.3 For several decades, biologists have suggested that there may be an unappreciated but important link between aging and many chronic disorders in humans, and aging increases the risks of many common diseases,4 including diabetes,5 Alzheimer’s disease (AD),6 Parkinson’s disease (PD),7 cardiovascular disease,8,9 chronic obstructive pulmonary disease (COPD),10 osteoporosis (OP),11 and even osteoarthritis (OA).12 Moreover, many older patients have multiple comorbidities with advancing age, especially after the age of 60 years. These patients require combinations of different treatments to attain therapeutic effects in the long term.13 In addition, the existing therapeutic options for many age-related diseases likely affect each other.14 Since aging is one of the leading risk factors for most chronic diseases, it is anticipated that understanding the aging process will facilitate the identification of therapeutic targets for age-related diseases and the development of pharmacological agents suitable for approved clinical use in the future.15

Timeline of research on aging and aging-related diseases. RHD rheumatic heart disease, PD Parkinson’s disease, AD Alzheimer’s disease, mtDNA mitochondrial DNA, NAD+ nicotinamide adenine dinucleotide, HP heterochronic parabiosis
Cellular senescence, with major effects on the maintenance of normal tissue homeostasis, as well as pathological conditions, is a principle causative factor in the organismal aging process and facilitates aging and aging-associated diseases. Cellular senescence is a gradual decline of the proliferation and differentiation ability as well as the physiological function of cells over time. In 1956, Denham Harman proposed the free radical theory of aging, arguing that degenerative changes during aging are mediated by the harmful effects of free radicals generated during normal cellular metabolism.16 In 1961, in the continuous culture of human diploid cells, the lifespan of fibroblasts was first found to be limited.17 Thus far, a number of molecular mechanisms that regulate aging have been discovered, such as telomere dysfunction,18 loss of proteostasis,19 mitochondrial dysfunction,20 stem cell exhaustion,21 and epigenetic alterations.22 The aging process is driven by a number of complex and important pathways, and many of those drivers are associated with chronic oxidative stress caused by elevated levels of reactive oxygen species (ROS).23
Here, we will focus on recent findings on the biological processes of aging, highlight the important roles of aging in multiple aging-related diseases in humans, and discuss efficient interventions and treatments.
Molecular mechanisms of aging
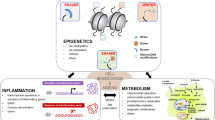
In 2013, López-Otín C et al. summarized aging in different organisms (especially mammals) as consisting of nine common features, including genomic instability, telomere attrition, epigenetic alteration, loss of proteostasis, deregulation of nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and alteration of intercellular communication.24 Over the years, the distinction between compromised autophagy and impaired proteostasis has enabled members of the aging field to propose a compromise of autophagy as the 10th hallmark of aging. This new concept was proposed by the main authors (Linda Partridge and Guido Kroemer) of a 2013 Cell paper, in addition to other aging and autophagy researchers.25 In recent years, scientists have made important achievements in research on signaling pathways and molecular mechanisms that affect the aging process. Here, we analyze and summarize these new advances at three different levels: the molecular level (genomic instability, telomere dysfunction, epigenetic alterations, loss of proteostasis, compromise of autophagy, mitochondrial dysfunction), the cellular level (cellular senescence, stem cell exhaustion, and intercellular communication), and systemic alterations (deregulated nutrient sensing). This information may broaden the understanding of multiple molecular signaling networks involved in the aging processes of organisms (Fig. 2).

The ten hallmarks of aging are subdivided into three categories: molecular hallmarks (genomic instability, telomere dysfunction, epigenetic alterations, loss of proteostasis, compromise of autophagy, and mitochondrial dysfunction), cellular hallmarks (cellular senescence, stem cell exhaustion, and altered intercellular communication), and systemic alterations (deregulated nutrient sensing). AMPK protein kinase AMP-activated catalytic subunit alpha 1, ATG5: autophagy-related 5, ATG-7 autophagy-related 7, ATP adenosine triphosphate, BECN1 Beclin 1, ER endoplasmic reticulum stress, EVs extracellular vesicles, GBA gut–brain axis, HSF-1 heat shock factor-1, HSP heat shock protein, IGF insulin-like growth factor-1, mtDNA mitochondrial DNA, mRNA messenger RNA, mTOR mechanistic target of rapamycin kinase, ncRNA noncoding RNA, OXPHOS oxidative phosphorylation, Rb retinoblastoma, ROS, reactive oxygen species, SASP senescence-associated secretory phenotype
Genomic instability
Genomic instability is an important cause of cellular senescence in many species.26 It leads to the deregulation of gene expression and consequently to impairment of cellular physiological aging, cessation of cell growth, and eventually cell death.26
Nuclear DNA damage accumulation
Endogenous DNA damage is deemed a major marker of genomic instability, especially DNA double-strand breaks.27 DNA damage activates the DNA damage response and cell cycle checkpoint pathways, such as the p53-p21 and p16INK4a-pRb pathways. Thus, the cell cycle is blocked to prevent the transmission of damaged genetic information to offspring cells.28,29,30,31 Tabula et al. performed single-cell RNA sequencing of more than 350,000 cells from male and female C57BL/6JN mice in six age groups ranging from 1 month (equivalent to human early childhood) to 30 months (equivalent to human centenarians).32 They analyzed changes in aging markers (including genomic instability) in 23 tissues and organs by Tabula Muris Senis, a tool capable of identifying aging-related changes in specific cell types.32 The researchers mapped the proportion of cells expressing each marker in all age groups and confirmed that the number of genomic mutations was positively associated with aging in all organs, with the tongue and bladder being the most affected.32 Analyses of centenarians have shown that long-lived individuals have fewer somatic and germ cell mutations than the general population, suggesting that they have more efficient DNA repair mechanisms to maintain genomic stability.33,34 Persistent foci of nuclear DNA damage in senescent cells are also known as DNA segments with chromatin alterations reinforcing senescence (DNA-SCARSs).35 DNA-SCARSs are dynamic structures that may regulate multiple aspects of senescence in cells, including growth arrest and the senescence-associated secretory phenotype (SASP).35 During cellular senescence, loss of the nuclear membrane protein Lamin B17,11,12 leads to impaired integrity of the nuclear envelope, causing chromatin fragment-containing nuclear membranes to enter the cytoplasm as cytoplasmic chromatin fragments (CCF).36 Further studies revealed that in senescent cells, CCF is recognized by cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), producing a second messenger cyclic GMP-AMP (cGAMP) that activates the stimulator of interferon genes (STING), which mediates DNA damage-induced SASP and tissue inflammatory damage in vivo.36
Mitochondrial DNA (mtDNA)
Unlike nuclear DNA, mtDNA contains only exons and lacks histone protection and an effective gene repair system. These features make mtDNA more susceptible to mutations than nuclear DNA.37 In addition, considering the role of mitochondria as cellular powerhouses, some high-energy electron leakage may occur in the respiratory chain, which constitutes a major source of oxidative stress and subsequent elevated mutation rates in mtDNA.38 Decreased mtDNA copy numbers and increased numbers of mtDNA mutations are observed in various tissues in aging organisms.39,40 Oxidative stress also leads to the release of mtDNA into the cytoplasm.40 The released mtDNA can bind to cGAS and participate in cellular senescence by activating STING. STING is closely associated with the SASP.41,42 Moreover, mtDNA is exported extracellularly and detected as circulating cell-free mtDNA in extracellular fluid and cerebrospinal fluid.43 Circulating cell-free mtDNA is a novel signal for mitochondrial communication between distal tissues and has been associated with neurological disorders and systemic inflammation.43 A recent study found that a deficiency of melatonin, an endogenous free radical scavenger that decreases with age and neurodegeneration, impairs mitochondrial homeostasis, leading to mtDNA release and activation of the cGAS/STING/IRF3 pathway, which stimulates inflammatory cytokine production.44 By supplementing with exogenous melatonin, mtDNA release, cGAS activation, and inflammatory responses were significantly ameliorated in Huntington’s disease mice.44
Nuclear architecture
Alterations in the function of the nuclear lamina may be drivers of normal aging.45 The products of the lamin A/C (LMNA) genes, mainly lamin A and C, are key components of the nuclear lamina and are essential for maintaining proper nuclear structure.45 Hundreds of LMNA gene mutations have been identified in multiple degenerative-related diseases, including neuropathies, muscular dystrophy, lipodystrophy, and Hutchinson-Gilford syndrome.45 The absence of Lamin B1 is also a well-recognized feature of cellular senescence, and Sebastian Jessberger et al. showed that the levels of lamin B1 decrease with age in humans.46 After the levels of lamin B1 are increased in aging mice, neural stem cell division is improved, and the number of new neurons increases.46
Endogenous cytoplasmic DNA
Endogenous cellular DNA is a contributing factor to aseptic inflammation, which occurs in the absence of pathogenic infection. Additionally, it is associated with the development of many chronic diseases, including cardiovascular disease and neurodegenerative disorders.47 Aseptic inflammation is an important marker of immune senescence and has been reported to be triggered by endogenous cellular DNA.47,48 A recent study identified significant increases in cytoplasmic DNA levels in the CD4+ T cells of elderly humans and mice and revealed that this increase was accompanied by elevated expression of the KU complex (heterodimeric DNA-targeting component including 70 and 83 kDa subunits, Ku70 and Ku80).48,49 Further studies revealed that KU-mediated DNA sensing promotes the activation of the AKT and mammalian target of rapamycin (mTOR) pathways. This, in turn, stimulates the proliferation and activation of CD4+ T cells, thereby enhancing aging-associated autoimmunity.48
Junk DNA
Almost 50% of the human genome is made up of repetitive DNA that does not encode proteins.50 These DNA sequences are often considered to be “junk DNA” in the human genome.50 Research has shown that one of the junk DNA sequences, VNTR2-1, actually functions to enhance telomerase gene activity and that the telomerase gene is more active in people with longer VNTR2-1 sequences.51 Notably, however, a shorter sequence does not necessarily mean a shorter lifespan. Instead, it implies lower telomerase gene activity and a shorter telomere length, which may reduce the likelihood of developing cancer.51 This finding suggests that junk DNA also contributes to the genetic diversity of aging.51
Telomere dysfunction
Telomeric DNA shortens with increasing numbers of cell divisions, and when it shortens to the Hayflick limit, telomere dysfunction causes a DNA damage response. This, in turn, induces cell cycle arrest and proinflammatory factor expression, ultimately leading to organismal aging.52 Telomere length is controlled by telomerase activity, and when telomerase activity is enhanced and the integrity of chromosomes is improved, the lifespan of an organism is prolonged.52
Telomeres
A telomere is a nuclear protein structure formed by telomeric DNA and binding proteins.52 In mammals, telomeres are formed by a highly conserved hexameric tandemly repeated DNA sequence (TTAGGG).52 They are located at the end of each chromosome arm and are designed to maintain genomic stability.53 The association between telomeric sequences and the shelterin complex leads to a stalled replication fork.53 However, once telomere shortening exceeds a critical level, the proteins forming the shelterin complex can no longer associate with telomeric sequences and interact with capping chromosome ends.52 Thus, a major limiting factor for telomere function is telomere length. Numerous studies have confirmed that telomere length is inversely proportional to age.54,55,56,57,58 However, notably, telomere length is highly variable in individuals of the same or similar ages.59
Telomerase
Telomerase activity is highest in human embryonic tissues and decreases progressively with age.60 In the classic view, telomerase protects certain frequently dividing cells in normal tissues, such as embryonic cells, sperm cells, adult stem cells, and immune cells, but it is inactive in other cells.61 Research from the University of Maryland has revealed that telomerase is reactivated in normal adult cells at critical moments during the aging process to mitigate the effects of aging, allowing cells to gradually die.61 The same research has also revealed that human cells express telomerase as they approach the telomere critical length.61 In addition, human skin cells that fail to express telomerase reach the telomere critical length more quickly and show more DNA damage than those that successfully express telomerase.61 The intervening role of telomerase in aging-related diseases has been demonstrated.62,63,64 In a mouse model of Hutchinson-Gilford progeria syndrome (HGPS), overexpression of telomerase mRNA reduced markers of inflammation and DNA damage in endothelial cells of various organs and prolonged the lifespan of HGPS mice.62 Aging is one of the independent determinants of hepatic telomere decompensation, and in hepatocellular carcinoma (HCC) cell lines, silencing telomerase reverse transcriptase inhibits cell proliferation by shortening telomeres, reducing DNA damage, and inducing apoptosis.63 However, overexpression of telomerase occurs in ~90% of cancers, making scientists wary of telomerase-related antiaging therapies.65
Epigenetic alterations
Epigenetics refers to changes in phenotype or gene expression caused by mechanisms other than changes in DNA sequence.66 Epigenetic mechanisms include DNA methylation, histone modifications, chromatin remodeling, and transcriptional alteration by noncoding RNAs (ncRNAs).66
DNA methylation
DNA methylation regulates gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factors to DNA.66 Methylation at the fifth position of cytosine (5mC) in DNA is a main epigenetic modification in mammals.67 5-Hydroxymethylcytosine (5hmC) is a stable DNA base modification that results from 5-methylcytosine via the actions of the ten-eleven translocation protein family.68 Both of these modifications function as epigenetic markers.67,68 Compared to quiescent or proliferating cells, senescent cells exhibit fewer cytosine modifications.68 A recent study has reported that 5mC deamination and oxidative damage are major contributors to somatic mutagenesis, which scales with lifespan across mammals.69 It is well-established that DNA methylation changes with age in a process termed “epigenetic drift”.70 DNA methylation decreases during aging, mainly in the regions comprising heterochromatin repeats, and hypermethylation occurs in the regions of promoter CpGs.66 In 2013, Steve Horvath noted the correlation between methylation and age and thus described an epigenetic clock. This clock measures our biological age and predicts our lifespan.71 In 2018, Levine developed a second epigenetic clock, the “DNAm PhenoAge”. This clock confirmed the intrinsic links between the epigenetic clock and some aging mechanisms, such as the activation of proinflammatory and interferon pathways, transcriptional and translational mechanisms, and the DNA damage response.72 A growing number of studies have confirmed that aging-related changes can be determined using DNA methylation.73,74,75,76,77 For instance, Shireby et al. showed that epigenetic clocks can provide insight into the aging process of the human brain and predict the risk of dementia.73 In addition, the biological clocks of people with fatty liver move more quickly than normal, while those of centenarians move more slowly.74 In a study that included “Diet, Physical Activity, and Mammography” (DAMA), improved dietary habits slows the DNA mGrimAge clock and promotes healthy aging in the women tested.75 Blood-DNA methylation is sensitive to physiological changes that occur in multiple organ systems, and Belsky et al. identified a blood-DNA-methylation measure, DunedinPoAm, a method that estimates the rate of aging in subjects in the years prior to the measurement.77 They found that DunedinPoAm was associated with an increased risk of chronic disease incidence and mortality in older men.77 Moreover, adolescents raised in families with lower socioeconomic status exhibited faster DunedinPoAm, which often predicted a shorter healthy lifespan.77
Histone modification
Histone modifications include several different types: acetylation, methylation, phosphorylation, ubiquitination, glycosylation, ADP-ribosylation, deamination, and proline isomerization.78 Among them, acetylation and methylation are the two most characteristic modifications associated with senescence.78 In the presence of histone methyltransferases or histone demethylases, the methylation levels of histones are altered, and these alterations participate in transcriptional activation or transcriptional repression.79 In general, methylation at lysine 4 of histone 3 (H3K4), H3K36, and H3K79 promotes transcriptional activation, while methylation at H3K27 and H4K20 causes transcriptional repression.79 In contrast, trimethylation, dimethylation, and single methylation at H3K9 (H3K9me3, H3K9me2, and H3K9me, respectively) have different effects, and their functions depend on the methylated sites and methylation types.80 Studies have confirmed that histone methylation is altered during aging.81 For example, models of premature aging diseases, such as Hutchinson-Gilford syndrome and Werner syndrome, exhibit loss of heterochromatin and a decrease in H3K9me3 or SUV39H (an H3K9me3 histone methyltransferase).79,82 Histone acetyltransferases or histone deacetylases catalyze histone acetylation or deacetylation reactions. Histone acetyltransferases are usually transcriptionally activators, and histone deacetylases exert transcriptional repression functions.81 Histone acetyltransferases and histone deacetylases play key roles in longevity.83 For example, deletion of the histone acetyltransferase gene GCN5 shortens the replicative lifespan of yeast.81 Given the sophisticated links between aging and histone modifications, reversal of aging via intervention in histone modifications could be a potential therapeutic strategy.
Chromatin remodeling
Epigenetic studies have confirmed the widespread loss of histones and local and global remodeling of chromatin with age.84,85 For instance, the epigenomes of senescent cells show loss of chromatin rigidity, increased entropy, disorganization of the epigenome, reduced compartmentalization, convergent changes in genome-wide epigenetic signatures, and reduced polarity.86 Researchers have defined this phenomenon as convergent alteration of the epigenomic landscape during aging.86 Chromatin remodellers alter chromatin structure and nucleosome position through an ATP-dependent enzyme similar to helicase, enabling regulatory proteins to contact DNA.87 Liu et al. found that the Switch/sucrose non-fermentable (SWI2/SNF2) complex core structural domain contacts each other through two induced Brace helices, anchoring chromatin remodellers to fixed nucleosome positions and initiating substrates for remodeling reactions.87 Under senescence-induced mitochondrial stress, impaired tricarboxylic acid cycle leads to reduced acetyl coenzyme (acetyl-CoA) production, which induces nuclear accumulation of histone deacetylase and homeobox protein dve-1 and reduces histone acetylation and chromatin reorganization in C. elegans.85 Conversely, the addition of nutrients that promote acetyl-CoA production is sufficient to delay the lifespan of C. elegans after mitochondrial stress occurs.85 These findings provide new insights into chromatin remodeling. Chromatin accessibility is also a common feature of active regulatory elements, including enhancers, promoters, insulators, and chromatin-binding factors, to which transcription factors can be recruited by DNA-specific interactions.88 Aging cells present generally elevated chromatin accessibility, but the genome-wide profile differs depending on the stimulus.35 During human umbilical vein endothelial cell (HUVEC) senescence, chromatin accessibility, mainly referring to intergenic chromatin along with increased accessibility regions (IARs) or decreased accessibility regions (DARs), is redistributed.89 This process is mainly initiated by activating transcription factor 3 (ATF3) in the Jun proto-oncogene, AP-1transcription factor subunit (AP-1) transcription factor family, in which low DNA methylation enhances the binding affinity of AP-1, further increasing chromatin accessibility, facilitating chromatin rebuilding and driving the senescence program in HUVEC.89 In addition to structural abnormalities, numerical abnormalities such as aneuploidy and heteroploidy also contribute to aging.90,91 Numerical abnormalities, also known as aneuploidy, involve autosomes (chromosomes 1–22) as well as gonosomes (X or Y chromosomes).91,92 Unlike in young people, chromosomal deletions are significantly increased in cultured lymphocytes from the elderly.92,93 It has been reported that abnormal constitutional (i.e., meiotic recombination) and acquired (i.e., telomere attrition) have important effects on the generation of aneuploidy.91 On the one hand, according to the non-random loss hypothesis, the organism itself has a subset of autosomes with innate differences in acquired aneuploidy frequency, and the tendency of this chromosome to acquire aneuploidy increases with age.91 On the other hand, differences in telomere attrition rates due to oxidative stress can lead to chromosome loss, and the specific mechanism may be related to the localization of polymers in mitotic cells.94,95
Transcriptional alterations
Single-stranded RNAs (ssRNAs, including short ncRNAs (small interfering RNAs (siRNAs), microRNAs (miRNAs), circular RNAs (circRNAs), PIWI-interacting RNAs (piRNAs), endogenous siRNAs (endo-siRNAs), and long ncRNAs (lncRNAs)) mainly function as probes, antisense (AS) probes, miRNA analogs, and miRNA inhibitors and have great potential for gene therapy and molecular diagnosis.96,97 Double-stranded RNA (dsRNA) is produced by cells in the normal process of gene expression and is mainly derived from primary transcripts, which are either present in mature species or removed by RNA processing.98 Increasing evidence suggests that ssRNAs and dsRNA generated from the unstable genome are related to the aging process.99,100 For example, the levels of exosomal miRNAs secreted by hypothalamic brainstem/progenitor cells decrease during aging, while the aging rate is significantly slowed after treatment with exosomes secreted by healthy hypothalamic brainstem/progenitor cells.101 In addition, abundant circRNAs bearing miRNA response elements (MREs) may sponge miRNAs and hence lead to target mRNA repression.102 Recent profiling of genome-wide circRNAs has demonstrated that the expression of circRNAs is enhanced during aging in the brains of multiple organisms, but no other class of transcripts has been demonstrated to exhibit a correlation with senescence as strong as that of circRNAs.103 This may be because age-related elevations in global circRNA levels might originate from the high stability of circRNAs.103 Furthermore, the accumulation of dsRNA consisting of Alu sequences causes geographic atrophy, an advanced form of age-related macular degeneration caused by degeneration of the retinal pigment epithelium (RPE).99 In addition, due to changes in heterochromatin, neurodegeneration-associated proteins are tightly related to the derepression of repetitive element transcription.100 Saldi et al. reported that this derepression results in an elevation in the level of intracellular dsRNA, which activates innate immune responses and induces the neuroinflammation found in nearly all age-related neurodegenerative diseases.100
Loss of proteostasis
Aging and various neurodegenerative diseases (e.g., AD and Huntington’s chorea) are mostly associated with impaired proteostasis.104 Proteostasis disruption triggers adaptive changes in the cell.104 To cope with this situation, cells have developed multiple mechanisms to reduce misfolding and remove misfolded proteins.104 One of these mechanisms is the development of chaperones, which bind to unfinished peptide chains. This prevents the peptides from folding prematurely and helps them fold into the correct shape.104 Chaperones also reduce the denaturation of proteins that occurs when cells experience heat shock; hence, these proteins are also called heat shock proteins.105,106,107
The endoplasmic reticulum (ER) initiates the unfolded protein response (UPR), which contributes to protein degradation and selective translation.108 Once the degradation and recycling systems downstream of the UPR (including the ubiquitin–proteasome system (UPS) and autophagy–lysosome system) become disordered, loss of proteostasis eventually occurs.108 Activating transcription factors (ATF3 and ATF4) that regulate UPR pathways play a key role in the aging process.89,109 In macrophages, inhibition of ATF3 leads to an elevated percentage of senescent macrophages in response to Pseudomonas aeruginosa PAO1 infection.109 ATF3 is also reported to remodel accessibility in senescence‐specific increased accessibility regions (IARs) in HUVEC senescence.89 Sun et al. reported that loss of ATF4 diminishes hematopoietic stem cell (HSC) function with an aging-like phenotype and impairs leukemogenesis by targeting HIF1α and p16Ink4a.110 During aging, proteasomal targets accumulate due to reduced ubiquitination and subsequent degradation.107 An elevation in the expression of the proteasomal target intermediate filament protein ifb-2 (IFB-2) has been shown to enhance the loss of intestinal integrity and bacterial colonization, while upregulation of epidermal growth factor receptor pathway substrate 8 (EPS8) hyperactivates AKT serine/threonine kinase 1 (RAC) in muscle cells and neurons.107 Hence, reducing the levels of age-dysregulated proteasomal targets can improve longevity. Koyuncu S et al. have reported that ubiquitin-coding gene expression is not downregulated in aged wild nematodes. This finding suggests that differences in ubiquitination levels are not due to differences in ubiquitin-protein expression but rather are due to loss of ubiquitination modifications.107 Insulin-like growth factor-1 (IGF-1) is another important anabolic growth factor that promotes protein synthesis via p70S6 kinase and p90 ribosomal S6 kinase and inhibits protein degradation mainly by suppressing proteasomal–lysosomal protein degradation.111 IGF-1 is a known driver of aging.112 In worms and flies, inhibition of the IGF-1 signaling pathways has been demonstrated to increase lifespan.113 In addition, in cultured cardiomyocytes, IGF-1 induces senescence, and a PI3K inhibitor abolishes this effect.112 However, a decrease in the expression of IGF-1 leads to declines in skeletal muscle quality and strength during aging.114 It has been suggested that IGF-1 contributes to skeletal muscle protein synthesis via the PI3K/Akt/mTOR and PI3K/Akt/GSK3β pathways, which then leads to UPS-mediated protein degradation.115 Thus, maintaining some level of IGF-1 is important for health, and extreme reductions in IGF-1 levels are detrimental for mammals.116 Small ubiquitin-related modifier (SUMO) is a newly discovered ubiquitin-like molecule.117 SUMO modification is similar to ubiquitination, but SUMO proteins do not mediate the degradation of target proteins. Instead, SUMO modification increases stability.117 Emerging evidence shows that alterations in global protein sumoylation and changes in the sumoylation pathway are extensively involved in the process of organismal aging.118,119 For instance, silencing of the sole SUMO gene (smo-1) results in a shortened lifespan, while overexpression of smo-1 extends the lifespan in C. elegans.120
In addition, ribosomes have been shown to play an important regulatory role in proteostasis.121 A recent study has suggested that as cells age, ribosomal translation “pauses” increase in frequency. This change leads to ribosome-associated quality control overload and de novo peptide aggregation, thus exacerbating protein aggregation.121
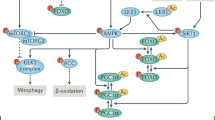
Compromise of autophagy
Among the molecular changes associated with aging, alterations in autophagy have become recognized as important features of aging in different species.25 Autophagy is a highly conserved process that degrades cellular components, including defective organelles and misfolded protein aggregates, in lysosomes.25 It is mainly initiated by mTOR inhibition or adenosine monophosphate-activated protein kinase (AMPK) activation. There is increasing evidence that autophagic activity decreases with age in different tissues in different species.25,122 Lysosomal protein hydrolysis is reduced during aging, which impairs autophagy, exacerbates cellular damage, and promotes the development of age-related diseases.123 For example, in human neuronal cells, the expression of autophagy-related genes (e.g., ATG5, ATG-7, and BECN1) decreases with age.124,125 Conversely, increased autophagy is associated with delayed aging.125 For example, increased lifespan in C. elegans is associated with increased expression of the autophagy genes ATG-1, ATG-7, and ATG-18.25 These findings suggest that the abundance of autophagy-related proteins gradually decreases with age and that translocation to lysosomes is reduced, implying that impaired autophagy is a major feature of organismal aging.25
Recently, researchers found much higher levels of transition metals (e.g., iron and copper) in senescent cells than in nonsenescent or immortalized cells (Fig. 3).126,127 Protein degradation pathways, such as autophagy and the UPS pathway, participate in the regulation of ferroptosis.128 For instance, iron accumulation is due to defective autophagic degradation of ferritin in lysosomes.126 In addition, mitochondrial ferritin (FTMT) accumulates on the outer membranes of defective mitochondria, which then promotes mitophagy, a specific form of autophagy that modulates the turnover of damaged and dysfunctional mitochondria, via specific interaction with the autophagic cargo receptor nuclear receptor coactivator 4 (NCOA4) coupled to the LC3-II double-membrane phagophore.129 In addition, enhanced levels of copper in senescent mouse embryonic fibroblasts (MEFs) and astrocytes have been reported to be accompanied by elevations in the levels of high-affinity copper transport protein 1 and reductions in the levels of copper-transporting ATPase 1 (Atp7a) (a copper exporter).130,131 Further research has shown that a lack of functional Atp7a blocks autophagic–lysosomal degradation of copper in senescent MEFs, thereby promoting aging-associated degenerative disease.131

Iron and copper accumulate in senescent cells. In senescent cells, iron accumulation is due to defective autophagic degradation of ferritin by lysosomes. In addition, in aging cells, FTMT accumulates on the outer membranes of defective mitochondria and promotes mitophagy by specifically interacting with the autophagic cargo receptor NCOA4 coupled to the LC3-II double-membrane phagophore. Furthermore, in senescent cells, reductions in the levels of Atp7a (a copper exporter) block autophagic–lysosomal degradation of copper. Atp7a copper transporter copper-transporting ATPase 1, Ctr1 copper transporter 1, FTMT mitochondrial ferritin, LC3 I cytosolic form of LC3, LC3-II LC3-phosphatidylethanolamine conjugate, mtROS mitochondrial ROS, MVB multivesicular body, NCOA4 nuclear receptor coactivator 4, TFR transferrin receptor
Mitochondrial dysfunction
With aging, mitochondria become highly susceptible to morphological changes. These changes result in reduced function due to oxygen radical damage, which eventually causes the aging of the organism.132
Reactive oxygen species (ROS)
Mitochondria are major sources of ROS.133 In each individual, there is a concentration threshold between beneficial and detrimental ROS, named the redox-stress signaling threshold (RST), below which redox stress is beneficial.134 In C. elegans, starvation (or heat stress or exercise) stimulation increases RST, and increasing RST improves Redox-stress response capacity (RRC) and health span, suggesting that increasing RST values through early stimulation can effectively delay aging.134 Moreover, in various tissues of mice, naked mole rates (NMR), and bats, the mild depolarization of mitochondria has been proven to inhibit the production of mitochondrial ROS (mROS).135 In different organs of aging mice (skeletal muscle, diaphragm, heart, spleen, and brain), the mild depolarization of mitochondria generally disappeared.135 In long-lived NMR and bats, however, the mild depolarization of mitochondria remained unchanged.135 This result shows that ROS-mediated protein damage caused by the disappearance of mild depolarization of mitochondria is one of the main causes of aging in short-lived mice, and mild depolarization of mitochondria is crucial to the mitochondrial antiaging system.135 ROS have been reported to be important for maintaining tissue-specific physiology through reversible modification of protein cysteine residues, and aging-induced dysregulation of ROS and redox signaling causes a decline in tissue physiological function.136,137 Xiao et al. performed an in-depth analysis of the cysteine oxidation networks, the Oximouse dataset, a dataset that quantifies the percentage of reversible modifications at approximately 171,000 cysteine sites in ten tissues of young and aged mice.138 They found that aging tissues did not show an overall increase in protein oxidation levels; instead, cysteine oxidation networks were radically remodeled in all aging tissues.138 Unlike the conventional notion advocated that ROS-driven protein modifications increase with age, Oximouse’s results found that different redox signaling networks are selectively altered in different tissues.138
Mitochondrial energy metabolism disorder
Aging-related mutations in mtDNA cause defects in mitochondrial oxidative phosphorylation (OXPHOS) functions.139 In mice, doxycycline-induced mutations reduce mtDNA levels, alter mitochondrial gene expression, and destabilize the complexes involved in OXPHOS in mitochondria, in turn promoting skin aging and hair loss.140 Age-dependent decreases in NAD+ levels have been identified in several pathologies.141 In preclinical models, supplementation with NAD(+) extends health span and improves several conditions, such as premature aging diseases (Cockayne syndrome, CS) and neurodegenerative diseases.142,143 Mechanistically, replenishing intracellular NAD+ promotes DNA repair and enhances mitochondrial quality via mitophagy.144 In the mitochondria, NADH is oxidized into NAD+ by the electron transport chain (ETC), and this process is coupled to ATP synthesis.145 However, mitochondrial dysfunction may lead to ATP depletion and cellular senescence by decreasing the NAD+/NADH ratio in the cytoplasm and promoting ROS production, a process known as mitochondrial dysfunction-associated senescence (MiDAS).146 Even though the NAD precursors has been viewed as an antiaging drug, the paradoxical results are reported. It is suggested that the supplement of NAD precursors alone does not improve the insulin sensitivity, mitochondrial respiration, energy metabolism, ectopic lipid accumulation, and plasma inflammatory markers of healthy overweight or obese individuals, which may be due to the insufficient supplement time and dose of NAD, resulting in the limited improvement of metabolism in overweight people.147,148,149
Mitochondrial quality control imbalance
Mitochondrial quality control is an important factor in the maintenance of mitochondrial function that mainly includes the biogenesis of mitochondria and the biodegradation of damaged mitochondria.150 Mitophagy, a selective type of autophagy, specifically degrades damaged or redundant mitochondria within the cell.151 Changes in the expression of mitophagy-related proteins can affect the degradation of damaged mitochondria and are closely related to cellular senescence.151 The mitochondrial-derived vesicle pathway is a newly identified mitochondrial quality control pathway that helps maintain stable mitochondrial function during the early stages of cellular stress and plays an important role in mitochondrial oxidative stress.152,153
The above results suggest that the dynamic balance between mitochondrial biogenesis and degradation is essential for mitochondrial quality control. A reduction in the level of biogenesis and/or degradation can cause cellular senescence; thus, mitochondrial quality control is a target of interest for antiaging actions.
Cellular senescence
Cellular senescence can be divided into two categories: replicative senescence and stress-induced premature senescence.154 Replicative senescence refers to the premature senescence that occurs after a limited number of divisions and a gradual shortening of telomeres at the ends of chromosomes. This results in cell proliferation stagnation and loss of differentiation ability.154 Stress-induced premature senescence refers to the premature senescence that occurs in response to pathological stimuli, such as DNA damage and oxidative stress.154 These two types of senescence share many regulatory molecules, and they both cause cell cycle arrest mainly through the p53/p21 and p16Ink4a/retinoblastoma protein signaling pathways.155,156 The number of senescent cells increases with age, obesity, and diabetes, and clearing senescent cells can alleviate many aging-related diseases and prolong the lifespan in mice.155,157 Senescent cells induce the formation of a complex, multicomponent SASP by secreting a range of cytokines, inflammatory factors, and adhesion factors.158 In the local microenvironment, the SASP alters the biological behavior of adjacent cells through autocrine and paracrine signaling.158 For instance, a systemic environment that shapes aging-related diseases has been reported to exist, as the infusion of young cerebrospinal fluid (CSF) improves the memory function of aged brains.159 This improvement is mainly mediated by serum response factor (SRF)-mediated oligodendrocyte progenitor cell (OPC) proliferation after exposure to young CSF.
Elderly individuals often present with chronic low-grade inflammation, which is collectively referred to as immune aging.160 In the process of aging, the numbers of monocytes/macrophages, dendritic cells, nd natural killer (NK) cells increase, possibly because of an increase in the number of aging cells, and results in increases in the numbers of macrophages and NK cells, thereby eliminating aging cells.108 Immune cells may further induce the production of more proinflammatory cytokines and aggravate the progression of aging-related diseases.108 In contrast, T-cell senescence may be one of the main features of immune senescence. Premature T-cell failure may accelerate aging in multiple organs and systems, with thymic degeneration, mitochondrial dysfunction, genetic and epigenetic alterations, and imbalance in protein homeostasis being the four main hallmarks of T-cell senescence.161 Desdín-Micó G et al. reported that in mice with mitochondrial transcription factor A (TFAM) deletion, T cells with mitochondrial dysfunction induce a variety of aging-related phenotypes, such as metabolic disorders, cognitive impairment, and cardiovascular diseases, which ultimately lead to the premature death of mice.162
Stem cell exhaustion
Stem cells, with their potential for self-renewal and multidirectional differentiation, are core components of regenerative medicine. They have been used in the treatment of a variety of diseases, including hematopoietic, central nervous system, and immune system disorders.163 Autophagy is necessary to maintain the stemness and differentiation capacity of stem cells, but autophagy is impaired during stem cell aging.164 With aging, autophagy of bone marrow mesenchymal stem cells (MSCs) and osteoblasts decreases. Activation of autophagy can alleviate the aging of bone marrow MSCs and restore osteogenic differentiation and proliferation in senescent bone marrow MSCs.165
Theodore T. Ho et al. found that hematopoietic stem cells (HSCs) that are unable to undergo cellular autophagy have a buildup of mitochondria and are in a constant state of metabolic activation, which accelerates the differentiation of myeloid cells through abnormal DNA modifications, ultimately affecting the ability of HSCs to self-enhance.166 Furthermore, recent studies have shown that the expansion, depletion or maintenance of the stem cell pool are regulated through symmetric and asymmetric division events.167,168,169 Failure to properly control cell division patterns can lead to premature depletion of the stem cell pool or to abnormal growth and differentiation disorders that accelerate cellular senescence.168 Cell polarity proteins are potential key regulators of asymmetric cell division, and a reduction in or loss of asymmetric cell division may be associated with diseases common to the aging process.169 In mice, senescence damages CD8+ T asymmetric cell division and affects long-term T-cell survival and function, but this phenotype can be reversed by inhibition of mTOR.170 In addition, cell size is also reported to be a determinant of stem cell potential during aging. Murine and human HSCs enlarge during the aging process, which may result in reduced proliferation and altered metabolism and may ultimately reduce stem cell function.171
Altered intercellular communication
Intercellular communication is typically characterized by the release of soluble factors and affects the function of neighboring cells.172 In the tissue microenvironment, the SASP has a range of negative effects on neighboring cells, the surrounding extracellular matrix and other structural components, including chronic inflammation and passive senescence of healthy cells.173 Extracellular vesicles (EVs) are lipid membrane vesicles that can be released by all cells and are well-established mediators of intercellular communication.174 Specific mesenchymal cells transfer specific processed tRNAs directly to granulocyte-/monocyte-lineage hematopoietic progenitors via EVs to promote protein translation, cell proliferation and eventual differentiation in granulocyte–macrophage progenitors.175 This unique form of stress-regulated communication may alter the physiological state of the organism in response to challenges, including infection.175 In addition, EVs exchange protein and lipid signals between endothelial cells and adipocytes, transmitting information regarding changes in the nutritional status of blood in adipose tissue.176
Senescent cells also communicate with each other and with neighboring cells in a cell-to-cell or proximal-secretory manner.177 Juxtacrine signaling is a form of intercellular communication that relies on the binding of receptors to ligands.177 For example, a juxtacrine NOTCH-JAG1 pathway induces senescence in oncogene-induced senescent cells.178 Cell‒cell fusion is a form of intercellular communication that induces senescence not only in primary cells but also in other cells.179 Bone marrow cell-derived TNF-α promotes muscle aging by affecting the fusion of muscle cells with aging muscle fibers.180 In addition to cell fusion, cytoplasmic bridges enable the intercellular exchange of biological materials, including RNA, proteins, and even organelles such as mitochondria and lysosomes.181 It was recently observed that mitochondria in senescent cells can be transferred to neighboring cells via a large number of membrane-bound intercellular bridges or tunneling nanotubes, a process that is largely dependent on signals from the mTOR pathway.182 Ma et al. suggested that CR improves the aging-disturbed immune ecosystem by reversing abnormal cell‒cell communication patterns, such as excessive proinflammatory ligand‒receptor interplay.183 Recently, bulk RNA sequencing of 17 organs and plasma proteomics at 10 ages across the lifespan of Mus musculus demonstrated how gene expression shifts in distinct tissues are tightly related to the corresponding protein levels in plasma, promoting the aging of the systemic circulation, indicating the existence of a similar yet asynchronous inter- and intra-organ progression of aging.184
Deregulated nutrient sensing
The somatotrophic axis is a neuroendocrine axis consisting of relevant hormones and receptors on the hypothalamus–pituitary–target organs that plays an important role in nutrient sensing and cellular energy perception.185 Nutrient perception refers to the ability of cells to recognize and respond to energy substrates, such as glucose, fatty acids, and ketones.185 Three key nutrient-sensing pathways are the insulin/IGF-1 signaling pathway, the mTOR pathway, and the AMPK pathway.24 In mammals, insulin/IGF-1 signaling is an important coordinator of nutrient availability with energy homeostasis and metabolic processes, which is activated by insulin-like peptide (ILP) ligands in response to nutrient availability.116 Insulin/IGF-1 signaling can initiate signal transduction via the PI3K/Akt pathway, which then phosphorylates many targets, including tuberous sclerosis complex (TSC) 1/TSC2, thereby regulating the activity of mTOR complex (mTORC) 1.116 Nutrients are key mTORC1 activators since they alone are enough to activate TORC1 in unicellular organisms, subsequently promoting anabolic processes including protein, lipid, and nucleotide synthesis and inhibiting catabolic processes such as autophagy.186 Mammalian AMPK is initiated by a falling cellular energy status and is activated by elevated AMP/ATP and ADP/ATP ratios.187 AMPK and mTOR signaling are interlinked and sense opposing nutrient states, regulate opposite metabolic processes, and regulate cell growth.188 Generally, AMPK turns off mTORC1 signaling when the energy status of cells is compromised.188 Dysregulation of insulin/IGF-1, mTOR and AMPK signaling is tightly related to human aging and age-related diseases due to nutrient insufficiency.189,190
The human microbiota contains multiple symbiotic microorganisms and participates in nutrient sensing.25 Thus far, bidirectional communication between the gut microbiota and the brain has been extensively found to occur through immune, circulatory and neural pathways in the so-called gut–brain axis (GBA).25 Disturbance along the GBA contributes to aging-related diseases.191 For instance, alterations in the gut microbiota composition enhance gut barrier permeability and systemic inflammation due to immune disorder, which then weakens the blood‒brain barrier (BBB) and induces neuroinflammation and ultimately neurodegeneration.191 Studies have found that the centenarian population has a unique intestinal flora composition from those of other populations, with certain intestinal flora and metabolites that produce unique secondary bile acids through new biosynthetic pathways.192,193 The gut microbiota plays a key role in immunity and metabolism due to its close association with other organs and tissues in the body.194 Aging-related alterations in the gut microbiota promote the development of systemic inflammation, which can have profound effects on disease, either directly or indirectly.194,195 For instance, microglia are the brain’s resident immune cells and regulate the survival of neurons and neuronal progenitor cells by secreting growth factors.196 However, the highly reactive and imbalanced state of microglia during aging causes cognitive dysfunction, including changes in brain plasticity and neurodegeneration.196 In germ-free (GF) mice, the microglial function can be improved by restoring key gut microbiota metabolites, such as short-chain fatty acids (SCFAs).197
Summary
Aging is the result of a combination of physical, environmental, and social factors, so elaborating the molecular mechanisms that trigger aging is a daunting task. Human lifespan is closely related to the reduction of tissue and organ repair and regenerative potential. Specifically, at the molecular, cellular and systematic levels, genetic, epigenetic, and environmental regulatory factors cause a reduction in the physiological reserve of the organism in response to stress through complex molecular mechanisms that work together to promote aging. Molecular mechanisms (e.g., telomere shortening, accumulation of DNA damage, metabolic alterations, and excessive ROS production) link various factors closely to the rate of aging. Overall, these mechanisms stunt cell proliferation, alter metabolism and gene expression patterns and induce high levels of ROS production, maintaining the cellular senescent phenotype. Although the number of early senescent cells is not large, they can limit the regenerative capacity of tissue stem cells and induce the accumulation of cellular damage thereby promoting age-related diseases. Current developments in high-throughput genomics, proteomics, and metabolomics allow the characterization and quantification of thousands of epigenetic markers, transcripts, proteins and metabolites, and can reveal the overall changes that occur with age in complex organisms at the molecular level. Therefore, the integration of these molecular markers and related molecular mechanisms into a comprehensive assessment of biological age to counteract age-related functional decline and morbidity is increasingly becoming a hot issue of interest for scientists.
Pathogenic and regulatory mechanisms of aging-related diseases
Aging is the most important risk factor for aging-related diseases. Therefore, the increasing age of the world population is accompanied by increases in the occurrence of various aging-related diseases. These diseases include neurodegenerative diseases, cardiovascular diseases, metabolic diseases, etc., all of which cause patients to lose normal life abilities, cause disabilities or even cause death. These diseases place a great burden on the social economy and the public health system. Next, we will focus on the regulatory mechanisms of aging and the pathogeneses of aging-related diseases.
Alzheimer’s disease (AD)
AD is a progressive neurological disorder that causes problems with memory, thinking and behavior in elderly individuals. AD commonly occurs in individuals 60 years of age and older.198 AD is caused by progressive loss of neurons in the cerebral cortex and hippocampus, abnormal deposition of amyloid β-protein (Aβ) and the formation of senile plaques. Additionally, the hyperphosphorylation of tau proteins leads to the formation of senile plaques, which lead to impaired memory and reduced cognitive function.198
DNA mutations and defects in DNA repair mechanisms are important causes of AD. When DNA damage exceeds the repair capacity, mistranslation by DNA polymerase can lead to the development of neurodegenerative diseases.199,200 Due to aging and a reduced DNA repair capacity, DNA damage increases and accumulates in neurons. This leads to enhanced cellular oxidative stress and increased inflammatory responses. These processes trigger aging-related neurodegeneration and promote neuron senescence and AD.201,202
Epigenetic modifications, such as DNA methylation, PARylation, ubiquitination, and acetylation, also play important regulatory roles in AD progression.203 The brain is composed primarily of neurons and oligodendrocytes. Neurons cannot proliferate and are sensitive to epigenetic modifications caused by aging.203 Aging has been shown to alter DNA methylation processes. This leads to DNA damage, which may be responsible for neurodegeneration. Phosphorylation or hyperphosphorylation of histone H3 and deacetylation of histone H4 can be detected in the hippocampi of early AD patients. These findings suggest that epigenetic factors play an important role in the occurrence of AD.204
An increase in misfolded proteins and aggregation of tau proteins are also involved in the development of AD. Aβ oligomerization may block synaptic plasticity and signal transduction.205 Aβ can also surround mitochondria and impair their function, leading to the release of ROS, overactivation of microglia and generation of proinflammatory factors.205 Another important change is that Aβ self-aggregates and accumulates on neuronal membranes, generates ROS, undergoes membrane lipid peroxidation, and generates 4-hydroxy-2-nonenal.206 This, in turn, impairs the functions of membrane ion kinetic ATPase and glucose and glutamate transporters and disrupts neuronal Ca2+ homeostasis. These effects result in neuronal hyperexcitability, susceptibility to excitotoxicity and metabolic exhaustion, ultimately resulting in Aβ neurotoxicity.206,207 However, some other studies found that this hypothesis may be controversial.208 Recent research has found that cellular damage with AD characteristics appears in neurons before amyloid fragment accumulation and amyloid plaque formation. Further research has revealed that these nerve cells have autophagy disorders and cannot effectively decompose Aβ, leading to the corresponding phenotype.209
Tau proteins may also aggregate and form neurofibrillary tangles.210 Neurofibrillary tangles gradually accumulate in the brain and are closely related to the prevalence of AD and the degree of disease.211 Furthermore, dominant tau mutations lead to increased tau aggregation, neuroinflammation and neurodegeneration. These pathological tau conformations can recruit native tau proteins, induce the formation of more abnormally folded tau proteins, and further promote pathological fibrillar aggregation.206
Decreased mitochondrial quality and activity are associated with normal aging, neuronal mitochondrial dysfunction and energy deficits during AD development and promote Aβ and tau pathology. New findings suggest that the autophagy/lysosomal pathway that removes damaged mitochondria (mitophagy) is also compromised in AD, leading to the accumulation of dysfunctional mitochondria. Research in animal and cellular models of AD and in patients with sporadic late-onset AD suggests that impaired mitophagy triggers Aβ and tau accumulation through increased oxidative damage and cellular energy deficit, leading to synaptic dysfunction and cognitive deficits. These changes in turn impair mitophagy. Neurons require high levels of ATP to perform their physiological functions, so mitochondrial dysfunction also contributes to the development of AD.212 Mitochondrial dysfunction promotes tau phosphorylation through the activation of AMPK and excessive mitochondrial fission, which in turn impairs ATP production.213 In addition, the PTEN-induced putative protein kinase 1 (PINK1)-Parkin pathway is important in mitophagy and in neuronal mitochondrial dynamics and function.214 Dysregulated PINK1 and Parkin functions may decrease mitochondrial function, increase Aβ aggregation in AD brain cells, and decrease mitophagy function.215 Thus, interventions that improve mitochondrial quality and function may prevent neurodegenerative processes in AD.
In addition, AD mainly manifests as a large group of SASPs caused by abnormal secretion of growth factors, cytokines, ROS and metalloproteinases. Astrocytes are the largest population of glial cells in the brain and are involved in various physiological functions of the central nervous system. Senescent astrocytes exhibit decreased normal physiological function and increased secretion of SASP factors that contribute to Aβ accumulation, tau hyperphosphorylation, neurofibrillary tangle deposition, and neurological deficits in AD. The disruption of astrocyte functions may lead to a chronic inflammatory response and central nervous system pathologies, including impaired synaptic plasticity, BBB dysfunction, glutamate excitotoxicity, and a decrease in the number and proliferation of neural stem cells, leading to the development of neurodegenerative diseases such as AD.216,217
Parkinson’s disease (PD)
PD is a chronic and progressive neurodegenerative disease with movement disorder in elderly individuals. Due to striatal dopamine deficiency, PD presents with dyskinesias, including impaired range and speed of movement, limb stiffness, or resting tremors.218 Dopaminergic neurons in PD often exhibit Lewy bodies, in which there is massive accumulation of α-synuclein (α-syn) in the cytoplasm.218
In PD patients, α-syn aggregation is widely regarded as a major causative factor. α-Syn oligomers form large, insoluble, neurotoxic fibrils called Lewy bodies. α-Syn oligomers can spread from cell-to-cell throughout the brain, thereby aggravating the progression of PD.219 Some studies have suggested that another potential mechanism for the pathological spread of α-syn is the binding of misfolded prefibrils to lymphocyte activation gene 3 (LAG3), which initiates the endocytosis, delivery, and cytotoxicity of α-syn prefibrils.220
Many studies have found that older PD patients have more severe impairment of dopamine function and higher levels of α-syn and tau proteins in the cerebrospinal fluid than younger people. To date, ~20 genetic mutations have been associated with PD, including missense mutations in SNCA (α-syn), PARK7, and LRRK2 and missense mutations or loss-of-function mutations in PINK1, PRKN, PLOG, and GBA.221,222 Therefore, recent pharmacological developments have focused on restoring striatal dopamine levels through gene- and cell-based approaches, and α-syn aggregation and cellular transport have been identified as the therapeutic targets with the greatest potential.223
Neuroinflammation has been an important target of drug intervention in neurodegenerative diseases. Increased numbers of senescent cells in PD patients are associated with increased SA-β-gal and p16 activity and sporadic α-syn deposition, leading to increased production of the proinflammatory cytokine interleukin-6 (IL-6). In addition to glial cells, fibrillar α-syn increases IL-1β secretion by interacting with TLR2, which is associated with NLRP3 inflammasome activation.224
Heart failure (HF)
Currently, over 64 million patients worldwide have HF,225 and HF is a growing area of interest. Studies have suggested that cardiac aging is a critical risk factor for impaired cardiac function and the progression of HF.226 Senescent cardiomyocytes play pivotal roles in conduction abnormalities, increased pacing frequency, mitochondrial dysfunction, increased oxidative stress, and metabolic dysfunction.227 Interestingly, senescent cardiomyocytes have been demonstrated to maintain cell-intrinsic senescence and induce neighboring healthy cell senescence via paracrine signaling. Thus, senescent cardiomyocytes can promote inflammation and dysfunction.228 They can also promote the activation of cardiac fibroblasts in a paracrine manner and induce cardiac fibrosis. The SASP of senescent cardiomyocytes promotes HF progression. Therapies targeting the SASP could therefore also be used to treat HF-related pathologies.
Research has indicated that oxidative stress plays a major role in the pathophysiology of cardiomyocyte senescence, hypertrophic remodeling and HF.229 Consistent with this notion, increased ROS levels lead to irreversible cardiomyocyte damage, senescence and death by contributing to DNA and protein oxidative damage, lipid peroxidation, mitochondrial dysfunction, and cytochrome c release. These processes are strongly associated with severe cardiac dysfunction and HF progression.230 Oxidative stress can also disrupt mitochondrial integrity and eventually trigger a vicious cycle of mitochondrial impairment and oxidative damage.231 In addition, several reports have suggested that oxidative stress in cardiomyocytes induces the premature senescence of cardiac stromal cells, increases the recruitment of CCR2+ monocytes, and eventually contributes to an excessive inflammatory response and cardiac dysfunction.232 Moreover, several studies have shown that aging-related increases in ROS levels contribute to mitochondrial dysfunction, metabolic imbalance and irreparable cardiomyocyte damage via accumulation of mtDNA damage and mutations, which could lead to HF.
The heart is a well-recognized organ with extremely active energy metabolism. Due to the inefficiency of the heart in storing ATP, cardiomyocytes must continuously generate ATP. Recent studies have suggested that cardiomyocyte senescence could have major adverse effects on multiple aspects of energy metabolism, reduce the heart rate and result in HF. However, miR-195 could regulate the metabolism of the failing myocardium by altering the Sirtuin 3 (Sirt3) expression and the mitochondrial protein acetylation.233 Interestingly, activation of p53 signaling inhibits glucose transport and glycolysis via GLUT1 and GLUT4, which is the major glucose transporters in cells. Additionally, it causes the senescent phenotype in cardiomyocytes during aging.234
Epigenetic alterations have also been increasingly recognized as major contributors to the initiation and progression of cardiomyocyte senescence and HF. For example, recent studies have shown that overexpression of the histone demethylase KDM4D in cardiomyocytes through upregulation of genes involved in proliferation and the cell cycle can delay cell cycle exit and profoundly promote cardiomyocyte proliferation.235 Furthermore, interestingly, second-generation sequencing analysis has revealed that changes in the accumulation of m6A RNA methylation exceed changes in both mouse and human gene expression during the pathophysiological processes present in HF.236 Fibroblast growth factor 20 is a member of the fibroblast growth factor family and plays key roles in regulating cell autophagy, inflammation, senescence and apoptosis. For instance, fibroblast growth factor 20 reduces pathological cardiac hypertrophy by activating the signaling pathway of the deacetylase SIRT1, inducing deacetylation of FOXO1 and reducing oxidative stress.237
In addition to senescent cardiomyocytes, senescent nonmyocytes in the heart, such as endothelial cells, can also be observed in HF.238 Myocardial hypertrophy and interstitial fibrosis have been found to occur in aging-accelerated mice at 24 weeks of age. These conditions could lead to systolic and diastolic dysfunction and thereby drive HF. Moreover, the senescence-associated hallmarks of endothelial cells, including p53 acetylation and senescence-associated β-galactosidase (SA-β-gal) activity, are significantly upregulated.239 Accumulating evidence suggests that increased expression of p53 and p16, which are markers of aging, can induce cellular senescence and atrial fibrillation. In addition, a study has shown that in atrial appendages of patients undergoing cardiac procedures, increased levels of endothelial nitric oxide synthase can promote many cardiovascular phenotypes, including atrial fibrillation, during endothelial cell dysfunction.240
Similarly, multiple types of chemotherapeutic agents, including anthracyclines, can result in the senescence of many types of cells in the heart in the clinical management of cancer. This effect is achieved via the induction of severe DNA damage and cardiac mitochondrial dysfunction in cells, which ultimately leads to HF. For example, SIRT1 mRNA and protein levels are decreased, and the activation of AMPK is inhibited. This, in turn, enhances inflammatory stimulation in doxorubicin-induced senescent vascular smooth muscle cells (VSMCs).241 The accumulation of senescent cardiomyocytes promotes cardiac aging and the development of HF; however, autophagy-regulating protease 4a performs essential biological functions that facilitate mitochondrial function and subsequently inhibit doxorubicin-induced cardiomyocyte senescence.242
Atherosclerosis
Vascular aging refers to aging-induced structural and functional changes that occur in the vasculature. Dysfunction of the vasculature contributes to aging-related diseases such as atherosclerosis, giant cell arteritis and AD, and is one of the leading causes of morbidity and mortality in elderly individuals.
Endothelial cells tightly regulate vasodilation by secreting vasoactive substances and growth factors. Senescent endothelial cells can be observed in atherosclerosis. The increased production of endothelin-1 and decreased production of nitric oxide in senescent endothelial cells lead to vascular inflammation and impaired vasodilation, compromise vascular endothelial integrity, and lead to vascular aging and atherosclerosis. Therefore, the accumulation of senescent endothelial cells can lead to vascular dysfunction, and vice versa.243
Oxidative stress is one of the main mechanisms driving atherosclerosis. Endothelial senescence can be triggered by oxidative stress or vascular inflammation. Nuclear factor E2-related factor 2 (Nrf2) is a key transcription factor that regulates hundreds of antioxidant genes and cytoprotective genes. Studies have shown that Nrf2 function is defective in atherosclerosis, hypertension and HF, and these conditions increase oxidative stress and accelerate aging.244 In addition, small extracellular vesicles derived from MSCs attenuate oxidative stress-induced endothelial cell senescence and stimulate angiogenesis through miR-146a/Src.245
Epigenetic changes, such as miRNA binding or histone acetylation, also contribute to endothelial cell senescence. An increasing amount of evidence suggests that miRNAs play important roles in the pathogenesis of vascular aging and atherosclerosis, and identification of aging-related miRNAs may provide opportunities for the treatment of cardiovascular disease. For example, in ApoE−/− mice, miR-217 causes endothelial cell dysfunction and exacerbates atherosclerosis by downregulating endothelial nitric oxide synthase.246 In endothelial cells, miR-217 can also stimulate the senescent phenotype by downregulating SIRT1 expression.247 In the circulating blood of patients with atherosclerosis and hypertension, the numbers of some exosomes containing miRNAs that can induce endothelial dysfunction are significantly increased,248 which can increase the risk of atherosclerosis.249 Exosomes can accelerate the aging process by carrying miRNAs that promote cellular senescence to different tissues and organs, including those of the cardiovascular system. These aging-related exosomes may become biomarkers for some aging-related diseases and provide new targets for the treatment of these diseases in the future. In addition, the levels of SIRT1 and SIRT6, protein deacetylases that play key roles in regulating DNA damage repair, maintaining telomere length and metabolic homeostasis,250 are decreased in atherosclerosis.251 SIRT6 deficiency promotes endothelial cell senescence, leading to impaired vasodilation, vascular dysfunction and atherosclerosis.
Metabolic factors such as hyperuricemia and dysregulation of the renin–angiotensin system can also promote endothelial cell senescence. Prohibitin-1 is highly expressed in endothelial cells. This protein is mainly located in the inner mitochondrial membrane and plays important roles in mitochondrial biogenesis and the maintenance of mitochondrial function.252 Knockdown of prohibitin-1 leads to increased mitochondrial ROS production, which in turn leads to cellular senescence, cell migration, and impaired angiogenesis. This suggests that prohibitin-1 is involved in cardiovascular disease.253 Klotho is also a key molecule associated with aging. Klotho expression is reduced in mouse models of premature aging, which results in the development of atherosclerosis and greatly shortens lifespan. However, overexpression of Klotho prolongs lifespan.254
Similar to endothelial cell senescence, VSMC senescence also contributes to atherosclerosis.255 VSMCs coordinate with endothelial cells to control blood pressure, vascular tone, and blood flow.256 Thus, senescent VSMCs play an important role in the development of atherosclerosis. Studies have shown that both p16 and p21 expression and SA-β-gal activity are increased in plaque VSMCs.257 Telomere shortening, DNA damage, oxidative stress and epigenetic changes can induce VSMC senescence. For example, sustained DNA damage signaling promotes the secretion of pro-osteogenic cytokines, leading to VSMC senescence, mineralization, and subsequent vascular calcification.258 Plaque VSMCs also exhibit reduced SIRT6 expression, which can lead to hyperacetylation of H3K9 and H3K27. This, in turn, leads to telomeric DNA damage and VSMC senescence.259 Senescent VSMCs exhibit increased expression of inflammatory cytokines, such as C-C motif chemokine ligand 2 (CCL2), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein-1α/β and CCL3/4. These cytokines promote the recruitment of monocytes, macrophages and lymphocytes, thereby accelerating the risks of plaque growth and rupture.260 In addition, senescent VSMCs secrete IL-1, IL-6, and IL-8, and exhibit upregulated TLR4-mediated signaling, and downregulated expression of anti-inflammatory factors.261 The upregulation of IL-1α expression results in the activation of the SASP of neighboring cells and increased secretion of IL-6, suggesting that senescent VSMCs can induce local inflammation through paracrine effects.262 Accumulation of prelamin A in VSMCs leads to the clinical manifestations of Hutchinson-Gilford progeria syndrome. Patients with this syndrome suffer from severe atherosclerosis, which accelerates aging and causes premature death.263
Immune system functions decline with increasing age, and a decline in immune function is a major risk factor for some cardiovascular and neurodegenerative diseases. Blood vessels are special sites for the immune system. Blood vessels consist of endothelial cells, VSMCs, macrophages, dendritic cells, fibroblasts and pericytes. Thus, the senescence of multiple cells can affect vascular homeostasis. Increased proportions of Th17 and regulatory T cells are observed in atherosclerotic plaques.264 The imbalance between Th17 and regulatory T cells enhances autoimmunity and increases autoantibody production, which may cause further tissue damage, ultimately leading to immunosenescence and atherosclerosis.
Circulating endothelial progenitor cells (EPCs) are generated in the bone marrow and are important for maintaining endothelial integrity.265 Many studies have shown the presence of senescent EPCs in various cardiovascular diseases, such as atherosclerosis, hypertension, ischemic heart disease and HF, and these senescent stem cells are dysfunctional in repairing endothelial damage.266 EPCs also show dysfunction in diabetes due to abnormal glucose metabolism, which may be one of the reasons why diabetic patients are prone to complicated cardiovascular disease.267
Type 2 diabetes mellitus (T2DM)
T2DM is a global health problem, especially for older adults. This disease is characterized by defective insulin secretion, hyperglycemia and hyperlipidemia. The incidence of T2DM is growing rapidly for people over the age of 65.268 In 2019, 111 million T2DM cases were reported for people in this age group.269 Aging and obesity are the predominant risk factors for T2DM, and an increased number of senescent β-cells is associated with the pathogenesis of T2DM.157
Pancreatic β-cells secrete insulin and maintain the balance of blood glucose and lipids. The senescence of β-cells leads to β-cell dysfunction, which impairs insulin secretion and the homeostasis of glucose and lipid metabolism.270 Telomere attrition is a hallmark of aging and is also a surrogate marker of senescent β-cells in T2DM.271 In pancreatic β-cells from T2DM patients, the telomere length is shortened, which may impair the proliferation and insulin secretion of β-cells.272 In mice with short telomeres, insulin secretion and glucose intolerance are impaired. The expression of p16(INK4a) and the number of senescent β-cells are increased in pancreatic islets.273 The loss of β- cell mass leads to fasting hyperglycemia and impairs mitochondrial membrane integrity.273 In a cancer-resistant mouse model, telomerase reverse transcriptase overexpression has been found to improve glucose tolerance.274
Elevated levels of plasma free fatty acids (FFAs) and glucose result in inflammatory factor and ROS accumulation, ER stress and mitochondrial dysfunction, which impair the proliferation of β-cells and adipose cells.270,275,276 ROS lead to the formation of advanced glycation end products and disturb proteostasis.277 Inflammation also leads to the dysfunction of β-cells and adipose cells. Circulating FFAs and glucose activate the TLR4-MyD 8 pathway and stimulate the production of proinflammatory factors and chemokines in β-cells, such as IL-1β, IL-6, IL-8, CCL2, and CXCL1.278,279 IL-1β attenuates insulin secretion and stimulates the activation of resident immune cells.278 The chemokines CCL2 and CXCL1 promote the recruitment of monocytes within the islets of T2DM patients and the differentiation of these cells into macrophages. In the islets of T2DM patients, macrophage infiltration is increased. These macrophages are prone to polarize toward the proinflammatory (M1) type.280,281
Insulin resistance is a major factor for the pathogenesis of T2DM, which accelerates β-cell senescence. In one T2DM animal model, senescent cells accumulated, proliferation was diminished, and the levels of senescence markers were increased in β-cells and adipose cells. These findings suggest that β-cell and adipose cell senescence might be associated with insufficient insulin secretion and the pathogenesis of T2DM.282,283,284 Senescent β-cells produce the SASP factors CCL4 and IL-6 and affect resident cells. Conditioned medium from β-gal-positive cells increases the expression of p16 in healthy β-cells.283 The β-cell SASP involves many proinflammatory factors that can lead to the senescence and dysfunction of neighboring cells through paracrine actions.285 Multiple SASP factors are transcriptionally upregulated in models of β-cell senescence, aging, insulin resistance, and T2DM.
Aside from β-cells, adipose tissue is the most important energy reservoir and endocrine organ, and it regulates the homeostasis of lipid and glucose metabolism. Many factors can lead to the senescence of adipose cells, such as telomere attrition, DNA damage, mitochondrial dysfunction, ROS, ER stress and inflammation. Adipose tissue contains a large number of immune cells, which are affected by different physiological environments, lifestyle factors, caloric intake and aging. Adipose cell senescence triggers inflammation and insulin resistance in other metabolic organs, which lead to T2DM.286 Moreover, changes in the composition of the gut microbiota are linked to the onset of T2DM through decreased glucose tolerance and insulin resistance.287
Nonalcoholic fatty liver disease (NAFLD)
NAFLD has been linked to aging-related chronic liver disease, and the major characteristic of NAFLD is hepatocellular fat accumulation.288 NAFLD is classified into two categories according to liver pathology: nonalcoholic fatty liver (NAFL), which is also referred to as simple hepatic steatosis, and nonalcoholic steatohepatitis (NASH). NASH is associated with inflammation and fibrosis and can further progress to advanced cirrhosis and even hepatocellular carcinoma.289 Hepatocellular fat deposition results from an imbalance caused by enhanced de novo lipogenesis (DNL) and lipid absorption and decreased fatty acid oxidation and lipid secretion. NAFL progresses to NASH in approximately 10–25% of patients. Although the pathogenesis of NASH is not fully understood, lipotoxicity, oxidative stress, apoptosis, and inflammation have been suggested to promote the progression from NAFL to NASH.288,289
Genetic and epigenetic changes affect the pathogenesis of NAFLD. Genomic studies have reported that multiple single-nucleotide polymorphism (SNPs) are independently associated with the development and progression of NAFLD. In a cohort of individuals from the UK and Finland, the rs762623 variant within CDKN1A was found to be significantly associated with the progression of NAFLD.290 Additionally, multiple genome-wide association studies (GWASs) have verified that the rs738409 variant in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene is associated with NAFLD progression.291 Epigenetic DNA methylation has also been verified to be associated with NAFLD progression. Loomba et al. found that a set of 152 differentially methylated CpG islands in the peripheral blood-DNA of NASH patients correlate with the severity of hepatic fibrosis and that this DNA methylation signature is associated with the age-related acceleration of NASH in patients.74 However, demethylation of histone H3K9 on the promoter of PPARγ2 may induce hepatic steatosis through upregulation of hepatic PPARγ2 expression.292
Impairment of mitochondrial function is a major factor that contributes to NAFLD.293,294 Hepatocytes are rich in mitochondria, which are responsible for energy metabolism. Mitochondrial dysfunction has been linked to a reduction in fatty acid β-oxidation (FAO) that is due to decreased carnitine palmitoyl transferase-1 (CPT-1) activity and decreased fatty acid clearance, resulting in the pathogenesis of NAFL.295 Moreover, impaired hepatic FAO is related to the progression of NAFLD in patients with obesity. During NAFLD, the impairment of FAO inhibits the activity of PPARα, resulting in hepatic lipid accumulation and inflammation.296 Liver mitochondria from aged mice produce more ROS and have a greater extent of dysfunction than those from young mice. The increased ROS levels may cause further FAO damage, which aggregates hepatic steatosis.289 Mitochondrial ROS production is linked to a reduced mitochondrial metabolic rate and decreased ETC activity.297 Impaired ETC activity may decrease ATP production and mitochondrial NAD+ levels, leading to p53-dependent cellular senescence.298 NAD+ plays a crucial role in the development of age-related NAFLD. Experiments in aged mice have verified that supplementation with the NAD+ precursor nicotinamide riboside (NR) alleviate hepatic lipid accumulation, improve liver function, and ameliorate mitochondrial dysfunction and fibrosis. These experiments have also demonstrated that these effects are SIRT2- and irisin-dependent.299,300,301
Chronic ER stress and UPR signaling play a major role in the aging process and are involved in NAFLD.302 The ER is the major organelle for lipid synthesis in hepatocytes, and a high concentration of intracellular lipids can activate ER stress by disrupting Ca2+ homeostasis. The accumulation of free cholesterol or phosphatidylcholine may affect sarcoplasmic reticulum/ER Ca2+-ATPase (SERCA) activity by changing the free cholesterol (FC)/phospholipid ratio of the ER membrane. This leads to decreased intra-ER Ca2+ concentrations and induces ER stress.302 Lipids can also directly activate ER stress via UPR sensors, especially IRE1ɑ and PERK. The transmembrane domains of IRE1ɑ and PERK may recognize the change in the unsaturated/saturated acyl chain ratio of the ER membrane.303 Upon ER stress, activated IRE1ɑ and PERK can stimulate proinflammatory and proapoptotic pathways. These pathways induce inflammatory reactions and hepatocyte apoptosis and promote NASH progression.304 Activated hepatic IRE1ɑ can increase the release of ceramide-rich EVs via XBP1-induced transcription of serine palmitoyl transferase, leading to the recruitment of monocyte-derived macrophages to the liver and to NASH in mice.305 In addition, cysteine-rich with EGF-like domains 2 (Creld2), which is a target of the UPR sensor ATF6, promotes tolerance to ER stress by augmenting protein folding. One study has verified that Creld2 deficiency results in dysregulation of the UPR and causes NASH during ER stress conditions in male mice.306
Autophagic function declines with increasing age. Lipophagy is a process of macroautophagy in which lipid droplets are selected for autophagic degradation.307 Lipophagy impairment has also been verified to occur in a cohort of NAFLD patients via histomorphological and molecular analyses.308 Chaperone-mediated autophagy (CMA) is a form of autophagy that regulates lipid metabolism by targeting the lipid droplet-associated protein perilipin 2/3 for degradation and then promotes lipophagy.309 Inhibition of key components in autophagy leads to lipid deposition in both mouse livers and hepatocytes. Both autophagic and CMA activity are impaired with aging, leading to lipid accumulation in various organs, including the liver.310 Furthermore, the accumulation of damaged mitochondria is a hallmark of NASH.311 Mitophagy, a form of autophagy that selectively targets abnormal mitochondria for lysosomal degradation, is important in hepatic homeostasis. With aging, impairment of hepatic autophagy in the fatty liver may fail to remove damaged mitochondria, therefore leading to activation of the mitochondrial death pathway and causing NASH via oxidative stress and apoptosis.295
In general, the liver has a remarkable capacity for regeneration and restoration. However, age-mediated changes impair the hepatic regenerative capacity. Senescent hepatocytes exhibit telomere attrition, which correlates with the progression of NAFLD.298,312 To date, the precise relations between cellular senescence and NAFLD have not been established, and cellular senescence may be both the cause and consequence of NAFLD. Senescence markers (p16, p21, and p53) have been identified in hepatocytes of NAFLD, and hepatocyte senescence correlates with the progression of NAFLD.312,313 Deletion of p16Ink4a-expressing senescent hepatocytes in INK-ATTAC mice significantly reduces hepatic lipid accumulation.314,315 Cyclin-dependent kinase-4 (CDK4) may be inhibited by p16 to maintain the senescent phenotype. However, the levels of CDK4 in NAFLD patients are increased in an age-dependent manner.316 In addition, senescent hepatocytes exhibit specific metabolic dysregulation and mitochondrial dysfunction, which have been linked to NAFL pathogenesis.312
Seventy percent of the blood that the liver receives is supplied from the intestine via the portal vein; thus, bacteria-derived molecules influence hepatic metabolism and the pathogenesis and progression of NAFLD through enterohepatic circulation.317,318 There is an association between gram-negative bacteria and the progression of NAFLD with fibrosis. Advanced NAFLD patients exhibit significant dysbiosis with increased abundances of Bacteroides, Escherichia, and Ruminococcus and a decreased abundance of Prevotella bacteria.318 Studies have shown that impairment of intestinal permeability leads to increased liver inflammation under high-fat diet administration. When microbial products reach the liver via the portal vein, toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs) in both parenchymal and nonparenchymal cells can be activated and trigger the innate immune system.319
Under insulin-resistant conditions, the amounts of nonesterified fatty acids (NEFAs) in the circulation that are derived from white adipose tissue lipolysis increase, resulting in fat overload in the liver.291 Moreover, hepatic fat overload causes mitochondrial dysfunction, worsening hepatic insulin resistance. Interestingly, under conditions of NAFLD-related hepatic insulin resistance, hepatic DNL remains activated in the absence of hepatic gluconeogenesis inhibition.320 Recently, after comparing obese NAFLD patients with obese-only patients, Horst discovered that the hepatic DNL increase is independent of hepatic insulin resistance and that the underlying molecular mechanism involves the activation of carbohydrate response element-binding protein (ChREBP). Activated ChREBP stimulates the glycolytic pathway, leading to an increase in the metabolic precursors for DNL.321,322 Additionally, insulin resistance may promote oxidative stress and inflammation, resulting in the progression of NAFLD. However, NAFLD further worsens hepatic and systemic insulin resistance.323
Human studies have revealed that there is a positive association between the number of macrophages and NAFLD severity. Hepatic macrophages include both resident macrophages (Kupffer cells (KCs)) and infiltrating monocyte-derived macrophages.324,325 Deletion of KCs prevents the progression of NAFLD, and inhibition of monocytes in the liver via blockade of C-C motif chemokine receptor 2 (CCR2) improves NASH.326 During NAFLD, activated KCs present M1-like proinflammatory activity and secrete cytokines. These cytokines induce inflammation and promote monocyte infiltration into the liver.327 Recent studies have found that monocyte-derived KCs appear in the liver during NASH. Further studies have shown that the self-renewal of resident KCs is impaired in NASH mice and that monocyte-derived KCs are generated to maintain KC numbers.328,329 Although the transcriptomic landscape between the two types of KCs is different, the lipotoxicity gene signature remains constant. This suggests that the cellular stress signature probably drives both the death of resident KCs and the generation of monocyte-derived KCs.328,329,330 In addition, monocyte-derived KCs localize to fibrotic areas in the liver and localize close to desmin+ hepatic stellate cells (HSCs) during NASH in mice, suggesting that they may participate in hepatic fibrosis.331
Therefore, aging-related hepatic fat metabolic imbalance results in NAFL. Hepatic accumulation of lipids induces mitochondrial dysfunction and ER stress and further causes oxidative stress, hepatocytic senescence, inflammation, and fibrosis. These factors lead to the progression of NAFLD. Insulin resistance and dysbiosis of the gut microbiota accelerate the development and progression of NAFLD. Impaired hepatic lipid metabolism and inflammation further worsen insulin resistance and dysbiosis of the gut microbiota and promote the progression of atherosclerosis (Fig. 4).

Crosstalk between aging and NAFLD in aging-related metabolic disease. Aging is related to impaired insulin sensitivity. Under insulin-resistant conditions, the amount of NEFAs in the circulation that are derived from white adipose tissue lipolysis increases, resulting in fat overload in the liver. Aging-related impairment of autophagy and mitochondrial dysfunction reduce hepatic lipid droplet breakdown and fatty acid β-oxidation, respectively. Moreover, under conditions of aging-related obesity, hepatic DNL increases due to ChREBP pathway activation. These disorders of lipid metabolism result in the pathogenesis of NAFL. Following hepatic lipid metabolic impairment and lipid accumulation, lipotoxicity and ER stress are induced, and mitochondrial function further worsens, leading to oxidative stress, hepatocyte apoptosis, hepatocyte senescence and inflammation and thus promoting the progression of NASH. Senescent hepatocytes secrete proinflammatory cytokines (IL-6, IL-8, TNF-α, and IL-1β) that stimulate resident KCs in the liver. Activated KCs present M1-like proinflammatory activity and secrete cytokines to induce monocyte infiltration into the liver and differentiation into macrophages. Furthermore, impaired resident KCs can induce monocyte differentiation into monocyte-derived KCs to maintain the KC pool in the liver. Both resident KCs and monocyte-derived KCs interact with HSCs and activate HSCs to produce collagen. In addition, dysbiosis of the gut microbiota impairs intestinal permeability; thus, bacteria-derived molecules enter the liver via the portal vein and in turn influence hepatic metabolism and the progression of NAFLD. However, hepatic metabolic impairment and inflammation further worsen insulin resistance and dysbiosis of the gut microbiota. Moreover, hepatic lipid metabolic disorder results in hypercholesterolemia and hypertriglyceridemia, leading to accelerated progression of atherosclerosis. (Fig. 4 includes modified templates from Servier Medical Art (http://www.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.)
Osteoarthritis (OA)
OA is a chronic inflammation-related disease characterized by joint pain, cartilage loss, and joint inflammation.332 Cartilage on the joint surface provides a smooth platefor the painless movement of the joint. Cartilage loss is an important pathological feature of OA.333 More than 80% of elderly individuals over 65 years of age suffer from OA, which causes disabilities in elderly individuals and places a heavy burden on patients and society.334
Chondrocyte senescence is one of the major risk factors leading to OA. Chondrocytes maintain the stability of the joint synovium by synthesizing or degrading extracellular matrix components, such as type 2 collagen and proteoglycan polymers. With the development of OA, chondrocytes begin to degrade collagen and proteoglycans by secreting matrix metalloproteinase (MMP) 13 and ADAMTS-5, which ultimately leads to cartilage calcification.335,336,337,338 Senescent chondrocytes have been found in the cartilage tissue of joint specimens obtained during total joint replacement surgery.339 In OA patients, the number of senescent chondrocytes in articular cartilage tissue increases with age.340,341,342
Telomere attrition is associated with the pathogenesis of OA.343 The number of ultrashort telomeres is increased at the lesion site and is significantly correlated with the severity of OA at the lesion.344 Oxidative stress disturbs the balance between catabolism and anabolism in articular cartilage, which leads to matrix loss in joints and to OA pathogenesis.345 With aging, the accumulation of oxidative stress in chondrocytes reduces cell viability and sensitivity to growth factors.346 ROS can activate the MAPK and PI3K/Akt signaling pathways and upregulate p53 and p21 protein levels in chondrocytes. This leads to apoptosis and an inflammatory response.347
Mitochondrial dysfunction in chondrocytes is also related to the pathogenesis of OA. In the chondrocytes of OA patients, the numbers of mitochondria are decreased, and the integrity of the mitochondrial membrane is impaired.348 Damaged mitochondria promote ROS production and MMP13 expression, leading to loss of the cartilage matrix in OA patients.349,350
Inflammation promotes metabolic reprogramming of chondrocytes in which glycolysis is upregulated and OXPHOS is downregulated. This leads to cartilage degeneration and chondrocyte senescence.351,352 The expression of glycolysis-related genes, such as Glut-1, hexokinase II and PKM2, is increased in the chondrocytes of OA patients.353,354,355 The acidic microenvironment (pH = 6.6) in joints induced by accelerated glycolysis inhibits the synthetic activity of chondrocytes and leads to cartilage degeneration. Metabolic reprogramming also leads to mitochondrial dysfunction.351 AMPK and mTOR regulate energy metabolism and inflammation. Thus, they can maintain homeostasis and participate in the pathogenesis of OA.348,356 Inhibiting AMPK promotes the expression of the inflammatory factors TNF α and IL-1 β and the degradation of the cartilage matrix in chondrocytes.357 Cartilage-specific knockout of sirt1 accelerates the progression of OA in mice.358 Senescent chondrocytes induce senescence in other chondrocytes in cartilage by releasing SASP factors, such as IL-6, IL-1β, and MCP1. The SASP recruits macrophages to infiltrate the synovium and activate the inflammatory response, which leads to synovitis.359
Osteoporosis (OP)
OP is an aging-related bone disease that is characterized by bone mass reduction and bone microstructure damage.360 Normal bone remodeling requires a balance between bone formation driven by osteoblasts and bone resorption driven by osteoclasts. With aging, the anabolic pathway (bone formation) is downregulated, and the absorption pathway (bone resorption) is upregulated. Aging-related bone loss is caused by a reduction in the number of osteoblasts.
Genomic instability is a hallmark of aging, which also leads to aging-related bone mass loss.24 DNA damage can further lead to the senescence of osteocytes and osteoprogenitors. An abnormal DNA repair system accelerates cell senescence. In humans, abnormal DNA repair systems lead to progeria, which is characterized by abnormal bones and low bone mass.361 Telomere reverse transcriptase–knockout (Terc−/−) mice are characterized by accelerated aging bone, reduced numbers of osteoblasts, increased numbers of osteoclasts and an inflammatory bone microenvironment.362,363,364,365 In OP, excessive ROS induce the apoptosis of osteoblasts and osteocytes and inhibit mineralization and osteogenesis. This causes an imbalance between bone remodeling and bone loss.366,367,368 Accumulation of senescent cells in bones is one cause of OP. The expression level of p16 is elevated in B cells, T cells, myoid cells, osteoprogenitors, osteoblasts and osteoclasts of 24-month-old mice.369 Senescent bone cells can impair resident cells by secreting SASP components. SASP factors in the supernatant of senescent cell culture can promote the survival of osteoclast progenitor cells and inhibit osteoblast differentiation.370
MSCs located in the bone marrow and spongy bone are responsible for maintaining the balance of bone resorption and formation.371 Bone marrow MSCs are located in the bone marrow and can differentiate into osteoblasts, adipocytes or chondrocytes. Depletion of bone marrow MSCs is one of the main causes of OP in the elderly population.372 Stenderup et al. found that MSCs derived from aging individuals exhibit a decreased maximal lifespan and an increased proportion number of SA beta-gal+ cells.373 Bone marrow MSC senescence can be caused by telomere shortening, genotoxic stresses/DNA damage, strong mitogenic signals, oxidative stress, and distortions in chromatin organization.374,375,376,377,378,379 In senescent bone marrow MSCs, the ability to differentiate into adipocytes is increased, and the ability to differentiate into osteoblasts is decreased.370,380
Chronic obstructive pulmonary disease (COPD)
COPD is a lung disease characterized by the presence of chronic bronchitis or emphysema that leads to the development of airflow limitations. The incidence rate of COPD is high in elderly individuals, especially those above the age of 65 years.381,382
Age is one of the main risk factors for COPD. The structural and physiological characteristics between aged lungs and COPD lungs overlap to a considerable extent; such characteristics include increases in the size of alveoli and end-expiratory lung volume without destruction to the alveolar wall.382,383 Additionally, the clinical features of premature lung aging can predict the possibility of being diagnosed with COPD in later years.383 Senescence-accelerated mice (SAM) and Klotho gene-knockout mice show phenotypes of COPD,384,385 indicating that aging accelerates the process of COPD.
The senescence of functional cells and the exhaustion of progenitor cell groups in aged lungs lead to a decline in lung function that is closely related to the progression of COPD.383 Decreases in the number and frequency of ciliary body beats in ciliated cells in the trachea, bronchus and bronchioles decrease mucus clearance in the lungs.386 A reduction in the number of type I alveolar cells results in the obstruction of gas exchange in the alveoli.387 Decreases in the ability of goblet cells, tuft cells and club cells to remove pathogens and cell debris increase the risk of infection.388 The reduction in the repair and renewal of these functional cells after the injury is closely related to the exhaustion of lung progenitor cells. Basal cells are multipotent progenitors located in the conducting airway that can differentiate into club cells and further differentiate into ciliated cells or secretory cells (goblet cells, etc.). Research has shown that the numbers of basal and club cells decrease with age. Alveolar type 2 cells form the main progenitor cell group of the lung parenchyma and can differentiate into alveolar type 1 cells. Although the number of alveolar type 2 cells remains unchanged, the self-renewal and differentiation capacity of these cells decrease.383 In addition, the changes in immune cell function and phenotype caused by aging promote the susceptibility of elderly individuals to COPD. During aging, the concentration of alveolar macrophages (AMs) in the respiratory tract declines, and phagocytosis and the scavenging capacity are impaired. This triggers nonspecific inflammatory reactions that recruit neutrophils and dendritic cells to inflammatory sites. Insufficient clearance leads to an aggravated inflammatory response. Increases in the levels of proinflammatory cytokines, such as IL-6, are associated with increased COPD obstruction and increased risks of COPD-related complications.
Autophagy alterations contribute to COPD.389 Decreased autophagy in the lung tissue of COPD patients is related to the severity of emphysema. In the COPD mouse model, the activity of TFEB, the main transcriptional regulator of autophagy and lysosomal biogenesis, is inhibited.390 Transgenic mice with reduced proteasome activity (β5T) show an emphysema phenotype.391 Exposure to cigarette smoke may activate autophagy, resulting in ciliary dysfunction and the death of airway epithelial cells.392 Autophagy differs in different cell types and cellular environments, making it difficult to target autophagy therapeutically.392
Many studies have shown that mitochondrial dysfunction accelerates COPD. In the airways, the lungs and blood of COPD patients show mitochondrial morphological alterations, mitochondrial dysfunction, and increased ROS levels.393 The mitochondria-specific antioxidant mitoQ can reduce airway smooth muscle cell inflammation and the airway response.394 Mitochondrial autophagy is also deficient in COPD patients. The levels of PARK2 protein are decreased in COPD lungs and are positively correlated with lung function. PINK1- and PARK2-deficient mice show mitochondrial dysfunction and a COPD phenotype.395,396
Epigenetic alterations also play a role in the process of COPD. Abnormal DNA methylation has been detected in small airways397 and lymphocytes398 in COPD patients. In COPD patients, the expression levels of the deacetylases SIRT1 and HDAC2399 are decreased in macrophages and/or lung tissue. These reduced levels fail to control downstream transcription factors, such as FoxO3 and Nrf2, resulting in an abnormal immune response.400,401 Knockout of the SIRT1 gene in mouse airway epithelial cells induces a COPD phenotype.402 A variety of ncRNAs are involved in the pathogenesis of COPD.403,404
The mTOR signaling pathway is highly activated in multiple cells in COPD lungs, which enhances the susceptibility to pulmonary inflammation405 and emphysema.406 Metformin-activated AMPK pathway ameliorates the COPD phenotype in mice,407 indicating that nutritional signals also affect the process of COPD.
Increased genomic instability and telomere dysfunction are associated with COPD. In COPD patients, the levels of DNA damage markers in lung tissue and peripheral blood cells are increased,408,409 telomeres are shortened,387,410 and the expression of DNA repair-related proteins is decreased,411 all of which are related to disease severity.412 By regulating the expression of telomere protective protein 1 (TPP1), telomerase reverse transcriptase and telomerase (TERC), we can improve cell senescence413 and COPD phenotypes in mice.414 In addition, aging cells release incremental proinflammatory cytokines through SASP reactions, resulting in chronic lung inflammation.415
Benign prostatic hyperplasia (BPH)
BPH is a common urogenital disease in middle-aged and elderly men. Previous studies have indicated that increasing age is an important risk factor for the development of BPH. At present, the pathogenesis of BPH is believed to include upregulation of androgen receptor expression, increased levels of inflammation-promoting growth factors, metabolic syndrome, endocrine and neurotransmitter changes caused by oxidative stress and epithelial–mesenchymal interactions, and lifestyle and dietary habits. All of these mechanisms can lead to the proliferation and apoptosis of epithelial cells and stromal cells in prostate tissue. These effects result in an increase in the number of cells and eventually lead to the development of BPH.416
Changes in androgen levels and tissue remodeling caused by aging are generally considered to be the major determinants of BPH. There is clinical evidence that taking a 5α-reductase inhibitor can reduce the concentration of dihydrotestosterone in prostate tissue, thereby preventing the further development of BPH. Compared with controls, androgen-depleted animals have lower bacterial counts and inflammation, reducing the risk of BPH development.417,418
Recent research also suggests that BPH may be an immune-inflammatory disease. Approximately 90% of prostate immune cells are T lymphocytes, which promote the release of cytokines and growth factors. These factors further lead to abnormal remodeling of the prostate structure characterized by tissue injury, chronic immune responses, and fibromuscular growth. Autoimmune responses associated with T cells may induce abnormal proliferation of epithelial and stromal cells involved in epithelial–mesenchymal transition (EMT). Both epithelial and stromal cells of the prostate can upregulate proinflammatory signals and trigger an inflammatory response following a bacterial challenge. As inflammation progresses, macrophages and MCP1, IL-8, IL-1, transforming growth factor-β2, and C-C motif chemokine 3 accumulate locally. This accumulation can increase the rate of cell growth, aggravate inflammation,419 and promote the development of BPH.420,421 In one study, compared with the patients in a nonstromal group, the BPH patients with inflammation in the stromal group had larger prostate volumes, a higher incidence of acute urinary retention, and lower maximum urine flow.
A key to the development of BPH in elderly individuals is metabolic syndrome, which is associated with low testosterone and hyperestrogenism. The characteristics of metabolic syndrome include T2DM, hypertension, obesity, high insulin levels, and low high-density lipoprotein-cholesterol levels. Metabolic syndrome components alone are risk factors for the development of BPH. IGF-1 has been shown to have strong mitogenic and antiapoptotic effects on prostate tissue. As the prostate volume increases, the patient’s serum insulin level increases, and IGFBP-3/PSA levels decrease. These effects suggest that early interventions that improve the insulin level may help control BPH.422,423
Aging-related macular degeneration (AMD)
AMD is a degenerative disease of the macula that leads to severe visual loss in the elderly population.424 Clinically, early AMD is characterized by the deposition of lipoproteinaceous drusen on the Bruch membrane accompanied by pigmentary abnormalities in the RPE and progresses into two late forms: dry (atrophic) and wet (neovascular) AMD.425
In the retina, senescence of the RPE, neurons, microglia, and endothelial cells accelerates AMD. Senescent RPE cells destroy the external blood‒retinal barrier (BRB) between the retina and the choroid, resulting in drusen deposition and progressive macular damage.426 Senescent resident neurons contribute to photosensitive system disorders.427 Senescent retinal vascular endothelial cells decrease the oxygen supply capacity,427,428 and senescent microglia lead to persistent inflammation and loss of tissue homeostasis.429
Aging of the immune system decreases inflammatory regulation ability and immune clearance. Senescent phagocytes (such as macrophages and neutrophils) show reduced phagocytic activity and clearance and induce the production of atypical lipid species in the retina. A variety of immune cells, such as monocytes, neutrophils and T cells, invade the retina.430,431 In addition to affecting cellular functions, senescent cells lead to retinal degeneration by releasing SASP components. Retina-derived SASP induces BRB matrix degradation,428 destroys the tight junction proteins432 of the retinal barrier, recruits and activates immune cells to increase inflammation or releases angiogenic growth factors and VEGF to participate in angiogenesis.433
Senescent cells also show abnormal metabolic regulation. For example, senescent macrophages show abnormal ABCA1-mediated cholesterol metabolism, which reduces drusen clearance and promotes retinal aging.432 In addition to macrophages, peripheral blood mononuclear cells have similar roles in lipid metabolism regulation.431,434,435 In RPE cells, NAD+, mediated by the AMPK/mTOR pathway, leads to photoreceptor degeneration436,437 by affecting the metabolism of glycophospholipids, lipids and proteins in the retina.438,439
Autophagy and protein homeostasis affects AMD progression. The expression of autophagy-related proteins and autophagy flux are decreased in the retinas of aged rats440 and AMD patients.441 Nrf2/p62-mediated autophagy regulate the formation and accumulation of drusen in RPE cells.442 The accumulation of drusen further drives lysosomal damage443 and mitochondrial autophagy dysfunction,444 resulting in a vicious cycle of damage.445 In addition, the UPR446 and sumoylation447 regulate the aging process of the retina and RPE by regulating the degradation of proteins involved in the cell cycle,447 autophagy,448 and cytokine processes.
Mitochondrial dysfunction is also associated with AMD progression. The RPE in elderly individuals and AMD patients shows a decrease in the number of mitochondria and impaired activity. In addition, the shift from OXPHOS to glycolysis causes RPE dysfunction and subsequent photoreceptor death.449 The humanin peptide encoded by the mitochondrial gene humanin plays a potential protective role in RPE cells by reducing oxidative stress. Humanin enhances mitochondrial function and biogenesis via increases in mtDNA mass, mitochondrial number, and the protein expression level of the mitochondrial transcription factor mtTFA, which is a key protein involved in mitochondrial biogenesis.450 The traditional drugs lutein451 and tudca452 and new lipid mediators called elovanoids453 can protect photoreceptors by ameliorating oxidative stress.
In addition, mtDNA and nuclear DNA damage,454,455 telomere dysfunction,412 methylation changes[46], and RPE stem cell senescence456 contribute to AMD progression.
Presbycusis
Presbycusis, also referred to as aging-related hearing loss (ARHL), is a progressive form of sensorineural hearing loss that occurs with aging and is a common condition in the elderly population.457
Accumulation of dysfunctional mitochondria might promote presbycusis progression.457,458 Decreased mitochondrial complex IV activity has been observed in the cochleae of elderly presbycusis patients459 and SAMP8 mice.460 Cdk5rap1 knockout in mice induces premature hearing loss by inducing mitochondrial dysfunction and mitochondrial tRNA modification dysregulation.458 In addition, lactate dehydrogenase B–knockout mice show hearing loss at high frequencies that is due to the regulation of mitochondrial membrane potential and ATP levels.461 Mitochondrial biogenesis is decreased in the spiral ganglion neurons (SGNs) of aged SAMP8 mice and increased in senescence-accelerated mouse resistant 1 strain mice.460 Mitochondrial-mediated apoptosis is also involved in the pathogenesis of presbycusis. The expression of Bax in the cochleae of SAMP8 mice is increased,460 and deletion of Bak prevents the aging-related loss of SGNs and hair cells in C57BL/6 J mice and the occurrence of presbycusis.462 In addition, oxidative stress damages hair cells and destroys the structure of the cochlea.457,463 Some studies have found significant associations between presbycusis and polymorphisms of antioxidant-related enzymes in the cochlea, including CYP1A1, UCP21, and MSR.464 SOD1465- and Gpx1460-knockout mice demonstrate accelerated aging-related cochlear hair cell loss and an increased susceptibility to noise-induced hearing loss.
Aging destroys protein homeostasis in the inner ear and leads to alterations in ion and water homeostasis that result in ARHL-specific dysfunction466 Autophagy alterations contribute to AMD. During the senescence of SGNs in SAMP8 mice, lc3-II is upregulated with lipofuscin accumulation.460 Mitophagy is essential for cell survival and cochlear functions. Recently, BCL-2 interacting protein 3-like (Bnip3) and NIX knockout in mice was shown to promote presbycusis by downregulating mitophagy.467 In addition, miR-34a promotes presbycusis in mice by inducing mitophagy impairment.468
The role of inflammation in ARHL has attracted much attention.469 In humans, there is a significant correlation between a reduction in the hearing threshold and the levels of four systemic markers of inflammation (leukocyte count, neutrophil count, IL-6, and C-reactive protein).470,471 There is also an association between systemic inflammation and ARHL,472,473 and the levels of IL-1β and TNF-α460 and the number and morphology of macrophages in the cochlea showed aging-related changes.474 Transcriptome data from young and old C57BL/6 mice show that the expression of genes related to the immune response and inflammatory pathways is increased in old cochleae.475
Increased genomic instability is associated with the onset of presbycusis. Mice expressing error-prone mtDNA polymerase gamma (PolgD257A) or with POLGD knockout show defective mtDNA replication fidelity and premature aging, which leads to early-onset ARHL.476 Increased levels of mtDNA oxidative damage markers have been observed in the cytoplasm of cells in the cochleae of SAMP8 mice.477 In addition, ROS-induced DNA damage drives the aging of cochlear cells in SAMP8 mice and helps to accelerate presbycusis.478 mtDNA deletion and mutation are increased in the cochleae of ARHL patients,479,480 and deletion of mtDNA4977 is closely related to the severity of presbycusis.481
Epigenetic factors are also involved in the process of presbycusis. Altered methylation modifications of connexin,482 amino acid transporter483 and signaling pathway-related proteins484 contribute to the risks of presbycusis in different populations.
Cancer
The link between aging and cancer is complex. Although there is clear evidence that cells entering a senescent state can act as a barrier to tumorigenesis, some studies have demonstrated that, in certain conditions, persistent senescent cells can acquire pro-tumorigenic properties.
Senescent cells can initiate both intrinsic and extrinsic mechanisms to inhibit tumorigenesis. Induction of stable growth arrest forms a natural barrier to tumorigenesis which works as a typical intrinsic antitumor manner. In Oncogene-induced senescence, aberrant activation of proto-oncogenes such as RAS fuels unscheduled DNA replication, causing malignant cellular growth. In turn, cells trigger firm proliferative arrest and senescence by initiating key cell cycle arrest genes such as TP53, p16INK4a, and p21, which counteract malignant growth.485 In PTEN loss-induced cellular senescence, cells activate mTOR-p53-mediated signaling to block the cell cycle, and induce a cellular senescence phenotype.486 In addition, cells undergoing senescence enhance tumor suppression by cell-extrinsic manner. Senescent cells promote senescence or death of neighboring cells through direct cell-cell interactions or inflammatory SASP factors release, causing increased production of ROS and a sustained DNA damage response, thereby also limiting the proliferation of neighboring precancerous or cancerous cells.485 In addition, SASP factors containing immune surveillance enhancers such as CCL2, IL-15, CXCL1, etc. that drive macrophages, lymphocytes and NK cells to recruit to tumor sites, which activate immune surveillance. Chromatin reader bromodomain-containing protein 4,487 P53-P21 axis, and NFκB signaling pathway488 regulate immune surveillance by regulating the expression and type of SASP.
However, studies in the past decade also proved established that the persistent senescent cells induce pro-tumorigenic effects by producing a proinflammatory and immunosuppressive microenvironment.485 Due to the diversity and highly dynamic nature of SASP factors, different SASP factors may have opposite functions on each other, or some SASP factors may have contradictory multiple functions on themselves, which results in SASP factors having both tumor-suppressive and tumor-promoting effects. SASP factors secreted by senescent cells include pro-proliferation factors such as IL-6 and IL-8,489 tissue angiogenic remodeling factors such as VEGF and CXCL5,490,491 and pro-cancer metastasis and invasion factors such as MMPs and GDF-15.492,493 These factors drive tumorigenesis by creating a chronic inflammatory microenvironment that supports cancer growth, disrupting the extracellular matrix and promoting tissue vascular remodeling effects. SASP factors-driven immunosuppression is also responsible for the pro-tumorigenic effects of senescent cells. For example, IL-6 causes the recruitment of myeloid-derived suppressor cells to the tumor microenvironment,494 and myeloid-derived suppressor cells not only blocks IL-1-mediated cellular senescence, but also reduces immune surveillance through the suppression of antitumor cells such as CD8+ T cells and NK cells.495 Hepatocytes with NrasG12V-induced senescence attract a subpopulation of CCR2+ myeloid cells by secreting CCL2, and these cells bind to NK cells, ultimately blocking tumor immunosurveillance.496
In the therapy-induced senescence model, cells undergoing senescence regain proliferative properties through reversibility of senescence, or acquire senescence-associated stemness by reprogramming mechanisms and turn into highly aggressive tumors, which drive tumor recurrence and progression.485 Therefore, there may be very complex mechanisms by which aging promotes tumorigenesis.
Senescence has dual roles in tumorigenesis. Whether senescent cells exert tumor-suppressive or tumor-promoting effects depends on numerous factors: the triggers of senescence, the number and persistence of senescent cells, the type and period of tumor tissue, the immune status of the body, and the status of key senescence proteins such as TP53. Despite recent advances in understanding the biology of senescence in cancer, the process of targeting senescence for cancer prevention and treatment still faces many challenges.
Summary
Overall, the existing evidence indicates that the hallmarks of aging are the common drivers of aging-related diseases. However, the diverse aging-related diseases in different organs and systems have their own combinations of molecular hallmarks of aging. Mitochondrial dysfunction (Fig. 5), defective autophagy, loss of proteostasis (Fig. 6), induction of cellular senescence and alteration of intercellular communication by the SASP (Fig. 7) are common causes of various aging-related diseases. Moreover, numerous aging-related diseases are associated with a chronic inflammatory status, which is frequently attributable to the long-term accumulation of senescent cells in various tissues. Other hallmarks of aging, such as genomic instability, epigenetic alteration, telomere attrition, deregulation of nutrient sensing, and dysbiosis of the gut microbiota, are also related to many major aging-related diseases, including cardiovascular diseases, neurodegenerative diseases, metabolic diseases, and chronic respiratory diseases. Therefore, intricate mechanisms underlie different aging-related diseases. Understanding the common and different mechanisms will provide new insights to aid in the development of therapeutic strategies against aging-related diseases.

Mitochondrial dysfunction contributes to diverse aging-related diseases. With aging, an increase in ROS production in mitochondria leads to oxidative stress, causing oxidative damage to DNA (especially mtDNA), lipids, and proteins. An increased mtDNA mutation rate causes increased frequencies of errors or mutations in mtDNA-encoded enzyme subunits, resulting in impaired OXPHOS. mtDNA is released into the cytoplasm or outside the cell and participates in SASP secretion by activating cGAS-STING pathways. Decreased mitophagy mediated by the PINK1/parkin ubiquitin pathway results in impaired clearance of damaged mitochondria. Reduced mitochondrial biogenesis mediated by PGC1 and NRF decreases the number of newborn mitochondria. During aging, mitochondria show altered quality control changes, Drp1/FIS1-mediated mitochondrial fission decreases, and MFN/OPA-mediated mitochondrial fusion increases, affecting mitochondrial shape and function. The mitophagy defects and mitochondrial dysfunction trigger Aβ and tau accumulation, leading to synaptic dysfunction and cognitive deficits during AD development. The metabolic transition from OXPHOS to glycolysis leads to altered metabolite generation. Mitochondrial pathway-mediated apoptosis is an important form of cell death. Mitochondrial dysfunction contributes to AD, HF, diabetes, OP, OA, presbycusis, NAFLD, COPD, AMD, and atherosclerosis by inducing oxidative stress, inflammation, apoptosis, and metabolic alterations. (Fig. 5 includes modified templates from Servier Medical Art (http://www.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.)

SASP related to various age-related diseases. Senescent cells that have a proinflammatory SASP can cause substantial pathogenic effects, resulting in various aging-related diseases. In the tissue microenvironment, the SASP involves chemokines, cytokines, proteases, and growth factors, which have a range of negative effects on neighboring cells, the surrounding extracellular matrix and other structural components. Senescent cells exhibit increased expression of chemokines, such as CCL2 and MCP1, which promotes the recruitment of monocytes, macrophages and lymphocytes in the vascular endothelium, islets, liver, synovium, and retinas. The accumulation of proinflammatory factors, such as IL-6, IL-1β, TNF-α, and IL-8, exacerbates the pathogenesis of various age-related diseases. Proteases destroy the external BRB and cartilage by inducing matrix degradation in AMD and OA. Growth factors, such as TGF-β and IGF-1, induce the abnormal proliferation of epithelial and stromal cells involved in EMT in BPH. The multifaceted SASP of senescent cells promotes the progression of various diseases and may be a therapeutic target. (Fig. 6 includes modified templates from Servier Medical Art (http://www.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.)

Molecular mechanisms for proteostasis in aging-related diseases. Aging, genetic mutations, environmental and lifestyle insults, and various other stresses cause increases in the amounts of unfolded, misfolded, and oxidized proteins, which lead to activation of the protein degradation system of the UPS and lysosomal proteolysis (including nonselective autophagy and selective autophagy, such as mitophagy and reticulophagy). Chaperones help refold unfolded proteins and assist in the formation of autophagosomes and ubiquitin-proteasomes. Balanced proteostasis leads to healthy aging and longevity. Disrupted proteostasis induces protein aggregation, cellular organelle function loss, increased ROS production and chronic inflammation, which lead to the development of many aging-related diseases
Interventions and treatments for aging-related diseases
Aging is an inevitable pathophysiological process caused by many factors that lead to a progressive reduction in the ability to resist stress and contribute to a variety of aging-related diseases. Healthy longevity without aging-related diseases has been pursued by humans since ancient times. The prevention and treatment of aging-related diseases is promising but challenging. However, there are many mechanisms of aging, and there are also differences in the development of different aging-related diseases. There are nondrug therapies for aging-related disease prevention and treatment, such as CR, nutrition, exercise, and intestinal microbiota transplantation, as well as drug therapies targeting different aging mechanisms and symptoms of aging-related diseases (Fig. 8). Next, this review will focus on the advances in laboratory and clinical studies on these interventions and treatments in the contexts of different aging-related diseases.

Possible interventions and treatments against aging-related diseases. Proof-of-principle therapeutic strategies used in cell experiments, animal experiments, and clinical trials are shown together. Daily lifestyle changes, such as exercise, dietary interventions, and weight loss, can inhibit aging and reduce the occurrence and development of aging-related diseases, subsequently promoting healthy aging and longevity. Drug therapy is the main strategy targeting aging. Antiaging drugs exert their effects by reducing the number of senescent cells, alleviating the SASP, and exerting anti-inflammatory and antioxidant effects while affecting multiple signaling pathways. Altering the metabolism or composition of the gut microbiota with drugs or through microbiota transplantation can also inhibit aging and aging-related diseases. Moreover, cell replacement therapy, cell transplantation, gene therapy and immunotherapy can be used to promote healthy aging and longevity and to treat aging-related diseases. mTOR mammalian target of rapamycin, NAD+ nicotinamide adenine dinucleotide, SGLT2 sodium-glucose cotransporter 2, ER endoplasmic reticulum, BET bromo- and extraterminal
Alzheimer’s disease (AD)
Before the clinical signs and symptoms of AD appear, lifestyle interventions, including good nutrition and physical exercise, can improve the cognitive status of individuals and attenuate both the development and progression of AD.497,498 For example, the Mediterranean diet significantly reduces the risks of cognitive impairment and AD. Some studies have also found that adding olive oil and nuts to one’s diet can improve cognitive function. In addition, long-chain fatty acids, including omega-3 polyunsaturated fatty acids, eicosapentaenoic acid, and docosahexaenoic acid, has been found to be beneficial for cognitive and mental health. These fatty acids can delay aging-related cognitive potential decline as well as AD.499 The ketogenic diet is another specific diet studied for its effects on neurodegenerative diseases.500,501 In addition, some studies have shown that physical excise may also be a lifestyle intervention that can prevent AD in elderly people.501
In terms of drug targets, neurodegenerative diseases share common features of protein aggregation, such as neurofibrillary tangles and Lewy bodies. A common strategy for AD treatment is to clear Aβ through the application of the amyloid cascade hypothesis.502 The expression of Aβ is mainly reduced via immunotherapy or by inhibiting γ- and β-secretase.503 The aggregation of phosphorylated tau proteins in neurons leads to neuronal damage and AD; therefore, it is considered another potential drug target for AD. However, the long-term effects of these drugs still need to be verified, and results have differed among the existing clinical trials.
In recent years, there has been an increasing emphasis on strategies involving nonamyloid targets for the treatment of AD, including therapeutic approaches to combat inflammation and oxidative stress; to provide synaptic and neuronal protection; to affect vascular factors; to provide mitochondrial protection; and to intervene at the epigenetic level. There has also been an increase in research on “drug repurposing”. Two typical examples of repurposed drugs are escitalopram and metformin.504,505,506 Cognitive impairment caused by elevated homocysteine concentrations has been reported to be inhibited following the administration of B vitamin supplements and may be particularly beneficial in individuals with one or more conditions.507,508 Aducanumab is an antiamyloid monoclonal antibody and was the first disease-modifying therapy approved for AD. Several clinical studies on the use of aducanumab for mild cognitive impairment and mild AD in patients started in 2021. The EMERGE trial showed that high-dose aducanumab can slow the progression of clinical cognitive impairment. However, another clinical trial, ENGAGE, did not find a significant protective effect. Therefore, this clinical trial evidence is contradictory, and more phase III studies are needed to determine the efficacy of aducanumab in the future.509,510,511
At present, cholinesterase inhibitors, such as tacrine and donepezil, are primarily used clinically to relieve the symptoms of AD. However, with increasing age and the aggravation of symptoms, the number of active neurons decreases, which makes it difficult for drugs to effectively treat AD. The main currently approved drugs are cholinesterase inhibitors (acetylcholinesterase inhibitors) and N-methyl-D-aspartate (NMDA) receptor antagonists. Acetylcholinesterase inhibitors generally reduce the hydrolysis of acetylcholine released from presynaptic neurons into the synaptic cleft by inhibiting acetylcholinesterase in the synaptic cleft. These effects enhance the stimulation of cholinergic receptors and improve cognitive function in patients with mild to moderate disease.512,513 The representative NMDA receptor antagonist is memantine, which can antagonize NMDA receptors and regulate glutamate activity for the treatment of patients with advanced disease.514 Sodium oligomannate and aducanumab are indicated for the treatment of moderate stages of AD.515 Although these treatments can alleviate cognitive impairment and improve quality of life in patients with mild to moderate AD, clinical evidence in the past 20 years has demonstrated that they have no significant effect on disease onset or progression.516,517
A number of researchers have proposed mechanistic links among oxidative stress, inflammation, and neurodegeneration; therefore, the use of natural plant components and dietary antioxidants to prevent neuronal damage may be a therapeutic approach to reduce AD risk.518 Examples include drugs targeting Aβ and tau, natural anti-inflammatory antioxidants (such as the polyphenol derivatives curcumin, Ginkgo biloba extract, etc.), free radical scavengers, nerve growth factors, and anti-infective treatments. In traditional Chinese medicine, many natural herbs have been indicated to be effective alternative treatments for AD, such as Astragalus, Artemisia, Ginseng, Ginkgo polygonatum, Chuanxiong, and Lycium barbarum.519,520
Decreased removal of dysfunctional mitochondria is an important mechanism of AD. The development of strategies to treat this impairment of mitophagy would benefit from the screening and identification of new mitophagy regulators.521,522 Recently, a research team used a combination of unsupervised machine learning (including vector representation of molecular structure, pharmacophore fingerprints and conformational fingerprints) and cross-species methods to screen and validate new mitophagy-inducing natural compounds. They found two effective compounds, kaempferol and rhapontigenin, that increased glutamatergic and cholinergic neuron survival and function, eliminated Aβ and tau lesions, and improved memory in animals.523
Recent studies have found that gut microbes are key regulators of many diseases and that gut microbes are involved in neurodegeneration through the gut microbiota–brain axis. This finding supports the possibility of new microbiota-based therapeutic options. Although many studies have found a possible relationship between the gut microbiota and AD pathogenesis, the inconsistency and reproducibility of existing clinical results need to be addressed before clinical application.196
Parkinson’s disease (PD)
There is increasing evidence that the Mediterranean diet, which includes high amounts of fruits, vegetables, whole grains, fish, and unsaturated fatty acids, reduces the incidence of PD.524 Many antioxidants, such as flavonoids, carotenoids, quercetin, curcumin, and resveratrol, have been shown to reduce the risk of PD.525,526 NAD+ supplementation may delay the onset of premature aging and aging-related neurodegeneration.527 Some traditional Chinese herbal medicines also inhibit the progression of PD, such as dogwood, ginseng, Pueraria, and skullcap.528 Microglia are activated due to aging, exogenous or endogenous infection, oxidative stress, and genetic factors, which may lead to neuroinflammation and neurodegeneration. The prevention of microglial activation represents a potential therapeutic strategy. With the development of science and technology, cell transplantation and gene therapy have brought good prospects to the treatment of PD. However, both are in the experimental stage and have not been used in clinical practice. Notably, transplantation of human induced pluripotent stem cells into primate dopaminergic neurons has been found to lead to the formation of dense neurites in the striatum.529,530
Nondrug treatments for PD include surgical treatments that can relieve the clinical symptoms, such as deep brain stimulation and rehabilitation and exercise therapy.531 At present, the drugs for the treatment of PD are divided into two main categories: anticholinergic drugs (such as Antan) and drugs related to the action of dopamine in the brain.532,533 To improve PD pathology, attempts have been made to reduce α-syn expression. Targeting specific posttranslationally modified forms of α-syn, such as the phosphorylated, nitrated, oxidized or truncated forms, may also be a strategy for PD therapy.534 In terms of targeting neuroinflammation, aspirin and ibuprofen are associated with a 13% lower risk of PD development. Masitinib is a novel tyrosine kinase inhibitor that targets the proliferation and activation of mast cells and microglia, reducing the risk of motor nerve injury.535,536
Gastrointestinal dysfunction is also an important pathogenic mechanism during the development of PD. In PD, a highly complex relationship between the gut microbiota and the brain, including the vagus nerve and α-Syn in the enteric nervous system, alters gut permeability and inflammation. Additionally, it causes changes in gut microbes and their metabolic activity. However, the results from studies on the link between exposure to certain types of oral antibiotics and an increased risk of PD are inconsistent and still controversial. Moreover, there are also differences in the results of existing animal experiments and human experiments regarding the therapeutic efficacy of FMT.537
Heart failure (HF)
Aging can lead to decreased cardiac function and HF. Antiaging strategies can reduce the progression of HF. For example, drugs that delay cellular senescence and adoption of healthy lifestyles (smoking cessation, physical exercise,538 dietary interventions, and weight control) could be effective.539 These interventions can be presented directly to the patient along with education and lifestyle guidance, but low adherence hinders their efficacy. However, CR could exert an extensive and unfavorable influence on aging-associated diseases such as nervous system diseases, cardiac diseases and cancer. Additionally, recommendation of one specific diet over another is difficult because of the limited available evidence.540 Regarding drugs, treatment with the sodium-glucose cotransporter 2 inhibitor dapagliflozin for 12 weeks has been found to significantly improve cardiac function and exercise capacity in patients with HF with preserved ejection fraction. This treatment improves the quality of life and is well tolerated in patients with chronic HF with preserved ejection fraction.541 Navitoclax is a Bcl-2 inhibitor that targets cellular senescence and activates apoptosis in senescent cardiomyocytes. This inhibitor can improve left ventricular systolic function and conduction velocity, and suppress myocardial fibrosis and cardiac hypertrophy in mice with Ang II-induced HF.542
Cycloastragenol, a major natural compound in Astragalus membranaceus, has been shown to have multiple pharmacological effects, such as antiaging, anti-inflammatory and antifibrotic effects. Cycloastragenol is also the only telomerase activator discovered to date that can delay telomere shortening by inducing telomerase expression.543 In rats, cycloastragenol can ameliorate cardiac insufficiency and cardiac remodeling by increasing cardiomyocyte autophagy and inhibiting the expression of matrix metalloproteinase 2 (MMP-2) and MMP-9, indicating that cycloastragenol may be a therapeutic candidate for HF.544 Moreover, the dietary safety of cycloastragenol is very good, and its subchronic toxicity and genotoxicity have not been found.545 In spontaneously hypertensive rats, Eriobotrya japonica leaf extract has been found to effectively reduce cardiomyocyte apoptosis and myocardial fibrosis, and improve cardiac function.546 Furthermore, Alpinate Oxyphyllae Fructus has antiapoptotic and prosurvival effects in ROS-induced aging hearts.547 Aconitum carmichaelii Debeaux has various biological activities, such as anti-inflammatory, analgesia, antiaging and energy metabolism–altering effects, and is currently used for the treatment of cardiovascular diseases such as chronic HF and coronary heart disease.548
Cell replacement therapy is a new strategy for HF that is aimed at improving function by replacing dysfunctional cells with sufficient differentiated human stem cells. Li, J. et al. reported that stem cell antigen 1 (Sca1)+ cells isolated from the bone marrow of young mice were engrafted into aged mice and could inhibit aging and promote the regeneration of cardiac endothelial cells.549 Cell replacement-based therapeutic strategies are viable for designing therapies to enhance the repair and regeneration of injured hearts in humans. Moreover, MSC-based cardiac regenerative treatment may be a novel therapeutic approach. However, the potential cytotoxicity to host cells should be considered when choosing MSC-based cell interventions. These strategies to enhance cardiac cells may further stimulate paracrine effects, and this needs to be taken into account in cardiac regeneration medicine. Currently, there are multiple strategies for cardiac regeneration under active development, each with its own advantages and challenges. In the future, new developments will be necessary to achieve cardiac regeneration. For example, stimulating endogenous cardiac regeneration by mobilizing and modulating resident stem cells is actually the targeting step in most current strategies for cardiac regeneration. Future research and clinical application should also aim to enhance the regeneration capacity of endogenous resident cardiac endothelial cells to promote therapeutic neovasculogenesis in the injured heart. However, both intrinsic and extrinsic regulators need to be taken into account when designing therapeutic strategies to enhance the regeneration of the injured heart. For example, the immunological activity of cells can engage in complex interactions with resident heart cells and the extracellular matrix of tissue, ultimately leading to cell death in the heart.
Aging-induced alterations in gut microbial composition and metabolism are associated with the development of HF. Since the launch of the Human Microbiome Project, intestinal microecology research has developed rapidly, providing new directions for studying the mechanisms of drug action in HF. With regard to deregulated nutrient sensing, studies have shown that interventions affecting the gut microbiota or its related enzymes can regulate intestinal microbial metabolism and reduce circulating trimethylamine oxide (TMAO) levels. Thus, they can treat HF and improve outcomes in HF patients.550 In addition, the yellow wine polyphenolic compound prevents doxorubicin-induced HF by regulating the gut microbiota composition and metabolic functions. These findings support the use of dietary polyphenols for the treatment of HF through microbiota modulation.551 Furthermore, potassium alginate oligosaccharides alter the composition of the gut microbiota to increase microbial diversity, potentially preventing the development of hypertension and HF in spontaneously hypertensive rats.552 Therefore, exploring therapeutic strategies targeting the gut microbiota may enrich the therapeutic concepts of HF and improve therapeutic efficacy against this disease.
Atherosclerosis
Patients with atherosclerosis should change their lifestyle, control their total intake of calories, and reduce the proportion of fat, especially saturated fat, in their total caloric intake.553 Unlike other interventions, lifestyle adjustments have few side effects. In many cases, diet is the main driver, because dietary interventions can reduce ROS production, reduce nuclear DNA and mtDNA damage accumulation, and maintain mitochondrial homeostasis. Over the past few decades, epidemiological, clinical andbasic studies have demonstrated that dietary interventions are key strategies to inhibit aging, promote health and prevent atherosclerosis.554 In addition, the combination of a low-fat diet with drug therapy may yield enhanced therapeutic effects.
The Mediterranean diet can prevent atherosclerosis by interfering with multiple signaling pathways that promote the development of the disease. The Mediterranean diet has beneficial effects on blood lipids, lipoprotein particles, inflammation, oxidative stress and the expression of proatherosclerotic genes.555 This diet is rich in fruits, vegetables, red wine and olive oil, which contains flavones, polyphenols, and stilbenes that may activate SIRT1. Therefore, this diet can reduce the incidence of multiple aging-related diseases, such as cardiovascular disease, neurodegenerative disease and metabolic disease.556 For example, the Mediterranean diet may improve the function of circulating EPCs in elderly individuals, thereby improving endothelial function in cardiovascular disease.557
Senolytics are standard antiaging treatments that are designed to identify and target senescent cells. Common drugs in this class include dasatinib and quercetin. In ApoE−/− mice, quercetin increases SIRT1 expression and reduces lipid deposition.558 Combination treatment with dasatinib and quercetin reduces aging markers, improves vasomotor function and reduces aortic calcification in hypercholesterolemic mice.559 However, the oral bioavailability of quercetin is relatively poor. Moreover, the SASP of senescent cells promotes senescence of the secreting cells and other cells. Therefore, preventing the secretory behavior of senescent cells and eliminating the deleterious effects of intercellular communication can also combat cellular senescence. This idea has resulted in the development of novel antiaging drugs called senomorphics, which are represented by metformin. Metformin is commonly used to treat diabetes and affects longevity through AMPK activation. In ApoE-/- mice, metformin inhibits endothelial cell senescence and atherosclerosis.560 In addition, studies have shown that adding a high dose (1%) of metformin to the diet reduces the lifespan of mice.561 However, metformin has obvious side effects, such as abdominal distension and diarrhea.158
Traditional Chinese medicines such as Bazi Bushen have been shown to regulate aging and treat atherosclerosis through antilipid, anti-inflammatory and antiapoptotic mechanisms.562 Berberine (BBR) is an alkaloid derived from plants with anti-inflammatory and lipid-regulatory properties, making it a potential therapeutic candidate for atherosclerosis.563 BBR also promotes autophagy in macrophages by activating SIRT1-induced deacetylation of transcription factor EB (TFEB), thereby inhibiting atherosclerosis.564 In addition, BBR can downregulate the choline-TMA-TMAO metabolic pathway in intestinal bacteria with vitamin-like effects and treat atherosclerosis.565
Type 2 diabetes mellitus (T2DM)
Lifestyle intervention is an effective and feasible method for reducing diabetes risk.566 For example, weight loss resulting from dietary and physical activity interventions effectively reduces the incidence of T2DM.567 Pancreatic β cell and adipocyte senescence leads to insulin resistance and worsens metabolic homeostasis. Distinctive potent markers and molecular mechanisms of cellular senescence have been found in these two types of cells. Therefore, the senotherapeutic strategies for adipose tissue and β cells are different.568 Senolysis can specifically remove senescent cells without harming healthy cells. In a transgenic INK-ATTAC model, B/B homodimerizer treatment specifically kills p16-positive cells. In addition, in 12-week high-fat diet (HFD)-fed mice, B/B homodimerizer treatment improves β cell function and glucose tolerance.283 A senolytic drug, ABT263 (a Bcl-2 inhibitor), also improves glucose metabolism and β-cell function and decreases the expression of markers of aging, senescence, and the SASP in an HFD-fed mouse model.283 Unfortunately, ABT263 has possible adverse effects; specifically, it is toxic to platelets and induces transient thrombocytopenia and thrombopathy.569 Other senolytic drugs, such as ABT-199 (a Bcl-2 inhibitor) and ABT737 (a Bcl-2, Bcl-xL, and Bcl-w inhibitor), can also remove senescent β-cells.570 Dasatinib and quercetin have been found to eliminate both p16high and p21high senescent cells in the pancreas and adipose tissues in a T2DM animal model.571,572 Unexpectedly, it has been reported that treatment with dasatinib and quercetin worsens liver disease progression in a diethylnitrosamine (DEN)/HFD mouse model, slightly increases histological damage and tumorigenesis, and has no effect on senescent cell removal.573
Moreover, human senescent β cells also respond to senolysis, establishing the foundation for translation. These novel findings lay the framework to pursue the senolysis of β cells as a preventive and alleviating strategy for T2DM.283
In addition to promoting senolysis, the senomorphic drugs resveratrol and metformin can ameliorate β-cell senescence and improve metabolic balance in animal models.574,575 In diabetic individuals, metformin is suggested to improve glucose tolerance and insulin sensitivity by reducing telomere shortening, preventing inflammation and oxidation and shifting the gut macrobiotia composition.576,577,578,579 However, the use of metformin by older adults can cause deficiencies in folate-related B vitamins, which are associated with reduced cognitive performance.580
Additionally, impaired ER homeostasis has been found to induce the misfolding of proinsulin and the pathogenesis of diabetes. Numerous studies have found that administration of the ER stress modulators 4-PBA and TUDCA improves β-cell functions and the secretion of insulin.581 PERK inhibitors have also been shown to improve the secretion of insulin.582 Modulating the gut microbiota with probiotics, prebiotics, synbiotics and via fecal microbial transplantation is a promising approach to treat diabetes.287 The Chinese traditional medicine BBR is a natural alkaloid extracted from Berberis aristata and Coptis chinensis. It has been confirmed that BBR has hypoglycemic effects in Chinese participants by modulating the gut microbiota.583
Nonalcoholic fatty liver disease (NAFLD)
Lifestyle modifications, including dietary changes, physical exercise, and weight loss, are the basic treatments for NAFLD. Weight loss is the key goal of lifestyle modifications. Studies have reported that a weight loss of 5% reduces hepatic fat content by approximately 30% and that a weight loss of 7–10% may be sufficient to reduce hepatic inflammation in obese subjects suffering from NAFLD.584
Polyphenols are antioxidants that provide beneficial effects for NAFLD patients.585 Vitamin C at a dose of 800 mg/day improves hepatic histological pathologies in NASH patients without T2DM.586
A meta-analysis has shown that supplementation with probiotics in NAFLD patients can decrease the expression of inflammatory factors in the liver.587 Other research has shown that FMT from healthy individuals to NAFLD patients can improve small intestinal permeability, but FMT has only minimal effects on liver dysfunction and metabolic syndrome.588
No drug is currently approved for the treatment of NAFLD. However, because the incidence of NAFLD is closely related to T2DM and obesity, various targeted antidiabetic and antiobesity drugs have been evaluated for NAFLD treatment. Metformin, a first-line antidiabetic drug, has been shown to reduce body weight, protect against oxidative stress, preserve mitochondrial function, and inhibit cellular senescence. However, a meta-analysis has reported that although metformin improves liver function and increases insulin sensitivity to some extent, it does not improve NASH-related outcomes.587 Liraglutide and semaglutide are two analogs of GLP-1 that have been tested in patients with NAFLD. Liraglutide promotes weight loss and can reduce hepatic steatosis. Additionally, semaglutide reduces hepatic fat accumulation in patients with NASH, but the effect on fibrosis has not been proven.589 PPARs agonizts play critical roles in regulating hepatic fatty acid β-oxidation. Additionally, they have been demonstrated to improve insulin sensitivity and to have anti-inflammatory properties. A phase 2 clinical trial has shown that saroglitazar, a double agonist of PPARα/γ, can significantly improve liver function, attenuate insulin resistance, ameliorate dyslipidemia and decrease hepatic steatosis in patients with NAFLD or NASH.590 In addition, elafibranor, a PPARα/β/δ agonist, was used in the GOLDEN-505 trial with 247 NASH patients. In this study, elafibranor could not achieve histological resolution in 162 patients with NASH but ameliorated insulin resistance, improved plasma lipid profiles and attenuated hepatic steatosis in a subgroup with significant inflammation.591
In addition, several traditional Chinese medicines have been identified as promising candidates for NAFLD treatment. BBR, an active single compound isolated from Rhizoma Coptidis, has been verified to significantly ameliorate hepatic steatosis in NAFLD patients in a randomized clinical trial and to directly regulate the expression of genes related to hepatic lipid metabolism. However, anorexia, upset stomach, diarrhea and constipation are common side effects in patients treated with BBR.348 Cordycepin, the major component of the fungus Cordyceps militaris, has been reported to protect against hepatic steatosis, inflammation, and fibrosis in mice through stimulation of the AMPK signaling pathway.592 Breviscapine, isolated from the traditional Chinese herb Erigeron breviscapus, attenuates hepatic lipid accumulation, inflammation, and fibrosis in mice via direct inhibition of TGF-β-activated kinase 1.593
Osteoarthritis (OA)
A senolytic cocktail of dasatinib and quercetin effectively has been found to eliminate senescent cells in joints and to alleviate OA symptoms in an older mouse model.370,594 UBX0101, an E3 ubiquitin-protein ligase, specifically degrades p53 by inhibiting the binding of p53 and MDM2. In one mouse model, injection of UBX0101 selectively eliminated senescent cells and reduced proteoglycan loss, which alleviated the cartilage degradation and pain caused by OA.339 Fenofibrate, a PPARα agonist, clears SA-β-gal-positive chondrocytes by promoting apoptosis.595 Fisetin, a flavonoid drug, alleviates inflammation and prolongs the lifespan of chondrocytes.596,597 Navitoclax (ABT263) eliminates p16-positive chondrocytes by promoting apoptosis.598
Senomorphics remove the SASP of senescent cells. Thus far, drugs targeting TNF or IL-1, such as lutikizumab (an inhibitor of IL-1α and IL-1β), etanercept (a TNF inhibitor), and canakinumab (an anti-IL-1β antibody), have achieved satisfactory therapeutic results in clinical trials.599,600,601 Specific knockout of MMP13 in chondrocytes alleviates OA-induced meniscal–ligamentous injury. Treatment of mice with CL821989 (a selective inhibitor of MMP13) relieves the symptoms of OA by increasing collagen content and inhibiting chondrocyte death.602 Metformin, quercetin, A-769662 and Protectin DX have been found to alleviate OA progression through the AMPK pathway in a mouse OA model.348,603,604 Rapamycin has been found to delay articular cartilage degeneration by activating autophagy in a murine model of osteoarthritis.605 Rapamycin has many side effects, such as hyperlipidemia, gastrointestinal disorders, and respiratory and urinary infections, but many of these effects are reversible.606 In addition, many drugs, such as wogonin, genistein, Sargassum serratifolium, apremilast and sinomenine, have been shown to alleviate the progression of OA by reducing inflammation.607,608,609,610,611
Osteoporosis (OP)
Elimination of senescent cells can alleviate the pathogenesis of OP. The INK-ATTAC “suicide” transgene encoding inducible caspase 8 is expressed specifically in senescent cells and specifically kills senescent cells in mice.284,559,612 In an old INK-ATTC mouse model, administration of AP20187 has been found to specifically eliminate p16-positive cells. AP20187 treatment reduces senescent bone cells, alleviates trabecular bone loss in the spine and bone cortex loss in the femur and improves bone strength.370 However, this is not the approach proposed for application in humans. Senolytics target the senescent cell antiapoptotic pathway and lead to the apoptosis of senescent cells without harming healthy cells.613,614 Injection of a senolytic combination (dasatinib and quercetin) into 20-month-old male mice reduces p16 expression and the proportion of senescent bone cells in bone.613 Senomorphic drugs, such as JAK inhibitors and mTOR inhibitors, do not kill senescent cells but inhibit the secretion of SASP components.615 The JAK inhibitor ruxolitinib reduces the size of the secretome derived from senescent cells.370,615 Ruxolitinib treatment reduces the levels of proinflammatory factors and improves spine trabecular bone microarchitecture and bone strength.370 It has been reported that other drugs can also inhibit bone loss and improve bone mass and microstructure in animal models, such as resveratrol, apocynin (an inhibitor of NADPH oxidases) and SRT3025 (a Sirt1 activator).366,616,617
MSCs are multipotent cells capable of differentiating into osteoblasts, adipocytes, or chondrocytes.596 MSC transplantation promises to increase osteoblast differentiation, block osteoclast activation and rebalance bone formation and resorption.618 Many ncRNAs and extracellular vesicles transplantation therapies are promising approaches to enhance the therapeutic effects and efficacy of MSCs.619,620,621,622,623,624
Targeted inhibition of cellular senescence is a novel therapeutic strategy for preventing or even reversing age-related OP and concurrently treating a variety of other aging-related diseases. This approach does not focus on the bones but on the basic aging mechanisms of the whole body. If the results of clinical studies are consistent with those of preclinical studies, it may be possible to improve the health of the aging population.
Chronic obstructive pulmonary disease (COPD)
Senolytics is an antiaging treatment that has attracted much attention recently, and dasatinib and quercetin have been used in clinical trials for idiopathic pulmonary fibrosis.625 However, to date, no convincing clinical trials have confirmed that senolytic therapies are effective and safe for COPD.626 MSCs have also been used as a potential treatment for COPD.627,628 In COPD rats, transplanted MSCs repair damaged tissues through anti-inflammatory effects, inflammation regulation and growth factors released by the MSCs. Thus far, some research teams have tried to implant functional lung tissue formed by lung stem cells to improve gas exchange capacity in COPD, but they have not achieved the desired effect.629 Antioxidant therapy is also a current strategy to combat the aging process and may protect against COPD. Studies have shown that atomized glutathione treatment can improve the respiratory function of COPD patients,630 and erdosteine thiol compounds have been found to decrease exacerbations and shorten hospital stays.631 Resveratrol has anti-inflammatory and antioxidant properties, making it a potentially useful treatment for aging-related COPD.632,633 In patients with COPD and diabetes, metformin treatment can reduce COPD-specific emergency treatment and hospitalization rates.634 However, the side effects of metformin are had been apparent in some other clinical trials, such as diarrhea and bloating.635 Furthermore, melatonin treatment improves dyspnea in COPD patients.636
Traditional Chinese medicines are also used for the treatment or adjuvant treatment of COPD. In a preclinical study, curcumin reduced pneumonitis manifestations in COPD patients by suppressing apoptotic signaling via regulation of oxidative stress.637
Benign prostatic hyperplasia (BPH)
Dietary prophylaxis can antagonize the hormonal regulation of BPH and reduce the development of BPH. The useful dietary compounds include polyphenols, isoflavones, and flavonoids that are present in fruits, broccoli, and green tea.638 Androgen-induced prostate cell growth can also be inhibited using drugs that modulate cellular metabolism. For example, the widely used antidiabetic drug metformin has hypoglycemic effects and antiproliferative and proapoptotic properties. Metformin inhibits the effects of testosterone on estrogen receptor alpha upregulation and estrogen receptor beta downregulation during BPH. Although metformin does not affect 5α-reductase, it decreases the mRNA levels of IGF-I and its receptor. IGF regulates cell proliferation and apoptosis through a complex system that is dependent on receptor and binding protein expression.639
In recent years, drug treatments for BPH have included α1-adrenoceptor blockers (α-blockers), 5α-reductase inhibitors, phosphodiesterase inhibitors, and gonadotropin-releasing hormone drugs. Treatment of BPH with the 5α-reductase type II inhibitor finasteride is a standard clinical procedure. Concomitant administration of dutasteride and testosterone inhibits the growth of BPH-1 cells.637 However, 5α-reductase type II inhibitors can cause side effects such as sexual dysfunction and allergic reactions, which should be monitored during drug application. Vanillic acid has been shown to inhibit AR, estrogen receptor alpha and steroid receptor coactivator-1.640 Treatment with vanillic acid (a known anti-inflammatory agent) results in decreased prostate weight and increased epithelial thickness. The neuropeptide 5-HT also attenuates AR expression, thereby preventing prostate branching.
Transurethral resection of the prostate is a standard surgical procedure and remains the most clinically effective long-term treatment. As stated in the BPH/lower urinary tract symptoms (LUTS) guidelines, the surgical approach should be individualized based on surgical risk (anesthesia and bleeding), prostate volume, and patient preference (preservation of sexual function).641
Aging-related macular degeneration (AMD)
The Bcl-2-inhibitory drugs UBX1325 and UBX1967 show promise for the treatment of AMD by promoting apoptosis in senescent cells. In addition, small compounds targeting prosurvival pathways, such as inhibitors of bromo- and extraterminal domain proteins, show prospective therapeutic value by ameliorating RPE cell loss in vitro and protecting ganglion cells in vivo.642,643
Several induced pluripotent stem cell (iPSC)-derived RPE cell therapies are in the clinical trial stage and are promising strategies for improving AMD treatment in the near future. Transplantation of RPE cell monolayers or biological patches consisting of RPE monolayers derived from human embryonic stem cells into elderly patients with dry AMD results in improved corrected visual acuity.644,645 In addition, treating AMD by transplanting RPE cells derived from skin fibroblast-generated pluripotent stem cells (iPSCs) improves the VFQ-25 score.646
Many clinical trials have analyzed the role of antioxidative stress in AMD therapy and have shown a certain beneficial effect on the clinical presentation of AMD. For example, antioxidant nutrient formulas;647 the Mediterranean diet;648 and some traditional medicines, such as resveratrol,649 coenzyme Q1050 and melatonin,650 have been used.
The ameliorative effects of traditional Chinese medicine formulas on AMD have been observed among small samples. Several compounds with potential therapeutic effects in Chinese medicines that reduce oxidation, inflammation and apoptosis have been identified by in vitro experiments. However, there is still a lack of multicenter, large-sample, randomized controlled studies to evaluate the effects of traditional Chinese medicines in the treatment of AMD.651
Presbycusis
Aspirin has anti-inflammatory and antioxidant properties that target the senescence-like phenotype and proinflammatory cytokine secretion. A randomized double-blind clinical trial is currently being conducted to evaluate the potential efficacy of low-dose aspirin in the treatment of presbycusis,652.653 However, aspirin has been reported to increase the risk of gastrointestinal bleeding in healthy elderly individuals.654
Stem cell therapy is also used in the treatment of presbycusis. Intracerebral neural stem cell transplantation in C57BL/6 J mice with presbycusis slightly restores auditory function.655 In animal models of GABAergic ouabain-induced deafness, such as gerbils, transplantation of otic neural progenitor cells improves hearing,656,657 but exogenous cell transplantation in the inner ear of humans is still a challenge.658
Antioxidants are also used in the treatment of ARHL. The enzyme cofactor α-lipoic acid,659 melatonin,660 antioxidant extracts from Ginkgo biloba leaves661 (EGb761), the synthetic superoxide dismutase/catalase simulant euk-207,478 coenzyme Q10, and the coenzyme Q10 analog coenzyme Q10-ter457 exert certain preventative effects against ARHL by improving mitochondrial function and ameliorating oxidative stress in animal models. A protective effect of dietary vitamin C intake against ARHL has been observed in elderly patients,662 but a protective effect against ARHL was not observed in a double-blind randomized clinical trial on EGb761.663
Cancer
Inducing cancer cell senescence is one of antitumor method by inhibiting cancer cell proliferation.664 SASP released by senescent cancer cells recruit immune cells and suppress the growth of adjacent tumor cells. However, chronic and long-term accumulation of SASP will promote tumor growth and metastasis.665 Therefore, an ideal method to eliminate tumor cells can include two steps: senescence-inducing therapy and senolytic therapy.486
There are many factors that can induce senescence of tumor cells, such as chemotherapy,666 CDK upregulation,667 telomere attrition,668 epigenetic modulation,669 and oncogenes induction.670 Chemotherapy drugs can trigger senescence in many types of cancer, such as topoisomerase I and II inhibitors (Doxorubicin, daunorubicin, mitoxantrone etoposide and camptothecin),671,672,673,674 Platinum-based compounds (cisplatin, carboplatin, oxaliplatin),675,676,677,678,679 alkylating agents (temozolomide, dacarbazine and busulfan),680 and microtubule inhibitors (paclitaxel, docetaxel and vinca alkaloids).681,682 Cell cycles of tumor cells can be arrested by using CDK inhibitors to inhibit tumor proliferation. For example, CDK4/6 inhibitors (Palbociclib, abemaciclib and ribociclib),683 CDK2/4/6 inhibitor (PF-06873600),684 CDK7 inhibitors (ICEC0942, SY-1365, SY-5609 and LY3405105),685 and CDK12 inhibitor (CDK12-IN-3)686 are potently used in treatment of various tumors. Telomerase inhibitions (BIBR15 and GRN163L) can induce senescence of tumor cells.687,688,689 DNA methyltransferases inhibitor (Decitabine)690 and histone deacetylase inhibitors (GCN5, p300/CBP, PCAF, and Tip 60)669 induce senescence in tumor cells via Epigenetic modulation.
The SASP factors derived from senescent tumor cells can mediate senescence of neighbor cells. Senescent tumor cells secrete the inflammatory cytokine IL-1α, IL-6, and IL-8. Proinflammatory cytokines and infiltrating immune cells can eliminate tumor cells and suppress tumor progression. However, persistently suffering SASP factors may promote tumor growth and metastasis. Senolytic agents can selectively eliminate senescent cells and minimize side effect of senescence-inducing therapy.486 Antiapoptotic BCL-2 family proteins (BCL-2, BCL-XL, and BCL-W) are therapeutic targets for eliminating senescent tumor cells. Navitoclax (ABT263 and ABT737) specifically eliminates various senescent cells via inhibiting BCL-2 family proteins and promoting cell apoptosis.598,691,692 And Dasatinib, quercetin, and Fisetin eliminate senescent cells via inhibiting PI3K-AKT pathway.693,694,695
Due to the heterogeneity of tumors, there is no drug that can induce senescence of all tumor cells. In addition, in existing treatments that induce tumor cell senescence, it is difficult to accurately assess the effect of drug-induced tumor cell senescence with current technology. Second, senescence-inducing drugs do not only induce tumor cell senescence but also cause the senescence of normal cells. In addition, although instantly accumulated SASP can promote tumor cell senescence and suppress tumors, sustained SASP can promote cancer progression, which is also an issue to be addressed. Therefore, the search for tumor-specific targeted senescence-inducing drugs or therapeutic approaches in senescence therapy is an important research direction for future tumor therapy.
Summary
Antiaging interventions and treatments for aging-related diseases face great challenges. If the function of aging-related signaling pathways in physiological processes is compromised, patients receiving antiaging therapy may experience side effects. Over the past decade, antiaging drugs have continually emerged and many drugs have entered the clinical research stage (Table 1). There are increasing cases of toxicity and side effects caused by high-dose and prolonged use of antiaging drugs. For example, metformin, known to lower blood sugar and delay aging, is the most widely used oral hypoglycemic drug for T2DM worldwide and can also reduce the incidence of human aging-related diseases such as neurodegenerative diseases. However, it also has many side effects, some of which may have severe consequences. Therefore, before metformin becomes the mainstay of antiaging therapy, a better understanding of the mechanisms underlying its beneficial effects on aging is required.635 The adverse effects of antiaging therapy need to be overcome. In addition, combining antiaging therapies with other established strategies for aging-related diseases may yield better therapeutic outcomes than individual strategies. Future opportunities and challenges will involve determining the optimal combination of synergistic treatment regimens for each patient.
Conclusion and future perspectives
For thousands of years, a healthy longevity has been the goals pursued by human beings. With the establishment and improvement of the public health system, human life expectancy has increased significantly, but also faces the challenge of increasing the number of aging-related diseases. Research on aging-related diseases has also become a major focus of scientific research and public health systems. New research methods, new drugs, and new clinical treatments are emerging, offering opportunities for revealing the mechanisms of aging, and the prevention, or treatment of aging-related diseases. The establishment and use of new methodologies such as genomics, proteomics, metabolomics, single-cell transcriptomics, and artificial intelligence applications can directly target aging and aging-related diseases for the study of mechanisms and exploration of prevention and treatment methods.
In the first part of this review, we summarize new developments in the onset of aging at three different levels: the molecular level, the cellular level, and systemic alterations. The molecular mechanisms underlying the onset of aging are summarized from multiple perspectives, at multiple levels and in terms of multi-organ interactions.
Research on the mechanisms of aging is a very active area in academia and a difficult area of research in the biomedical field. The study of aging mechanisms will be extremely important for delaying aging, reducing the occurrence of aging-related diseases, and maintaining a long and healthy life in human body. Especially, in recent years, research on epigenetic regulation, proteostasis, autophagy, cellular senescence, stem cell, has provided us with new directions for aging mechanism study. However, the causes of human aging are multifaceted, and the mechanisms of aging are extremely complex. So far, although a variety of theories on the mechanism of aging have been proposed by academics, all of which have their own experimental basis, but they all have their limitations to explain the complex mechanisms of aging. Therefore, we should adopt a comprehensive and multi-perspective approach when we explain the aging mechanisms. At present, there has been great progress in the research on the molecular mechanisms of aging, and there has been a breakthrough in the understanding of the biological and genetic mechanisms of the aging process, as well as a profound understanding of the pathogenesis of aging-related diseases. However, these findings are still far from being able to delay human aging and reduce the occurrence of aging-related diseases. Therefore, there is still a long way to go in the study of aging mechanisms.
In the second and third section, we summarize the characteristics of the mechanisms of aging-related disease development, and present the latest interventions and therapeutic approaches for aging-related diseases in response to aging. Recently, new research technologies, new drugs, and new clinical treatment methods are emerging, offering new opportunities for revealing the regulation mechanisms of aging and the treatment of aging-related diseases. Although some of the technologies and methods are still in the experimental stage and have not been directly used for the diagnosis and treatment of aging-related diseases, they have good future applications in the future.
Antiaging interventions and treatments for aging-related diseases face great challenges. An important way to achieve healthy aging is through early intervention and prevention. Early lifestyle intervention can promote healthy longevity and reduce aging-related diseases. Through a variety of lifestyle interventions, it is hoped that the aging process can be slowed and the incidence of aging-related diseases may be reduced. In terms of pharmacological interventions and treatments for aging-related diseases, most of the drugs now applied clinically may focus more on symptom relief after the onset of disease and lack therapeutic approaches to address the causes of aging and aging-related diseases. Research on the latest therapeutic approaches, such as stem cell transplantation, elimination of senescent cells, promotion of antiaging factor expression and inhibition of pro-aging factor expression, and tissue or organ regeneration, provides new directions for treatments of aging-related diseases. In addition, there have been significant technological developments, mainly through gene therapy, nanomaterial drug carriers, therapeutic antibodies or small molecule drugs, which have also contributed to advances in the treatment of aging-related diseases. Some of these methods and technologies have been applied in the clinic, and some are undergoing model animal studies and small-scale clinical studies. The application of these state-of-the-art technological approaches and new targeted drugs will facilitate the treatment and clinical application of aging-related diseases.
Interestingly, various alternative medicine approaches including traditional Chinese medicine treatment, acupuncture, dietary supplementation, etc., have also shown certain efficacy in the antiaging and the treatment of aging-related diseases. However, some of these methods still lack systematic mechanistic studies and clinical evidence-based medical evaluations, which may be directions for future research. Despite many advances in the study of human aging-related mechanisms and interventions, many contributing factors remain unclear. Therefore, the effects of these interventions need to be validated through long-term studies. More basic and in-depth clinical research needs to be conducted to discover new methods to fight aging and aging-related diseases. Large-scale evidence-based medical studies are also needed to verify the feasibility and safety of these methods in order to avoid adverse reactions and maximize the benefit to elderly individuals.
The goal of aging medicine is gradually changing from disease treatment to prevention of the occurrence and development of aging-related diseases. In other words, geriatric medicine is moving away from focusing on postdisease treatment to targeting aging-related chronic disease risk factors for preintervention. Research on the mechanisms of aging and on intervention measures and methods has an important role in improving human health and prolonging lifespan. Due to the aging of the global population, antiaging and healthy aging pursuits are undoubtedly important tasks for public health organizations, scientific research departments and drug research and development departments. Although there are many challenges to the research of aging and aging-related diseases and many questions still need to be addressed, promoting healthy aging has important and far-reaching socioeconomic and public health implications. With the emergence of new technologies and methods of modern biology, and the development and utilization of new drug discovery methods, research on aging mechanisms will further facilitate the prevention, diagnosis, and treatment of aging-related diseases, thus promoting healthy longevity for humans.
References
Northrop, J. H. The influence of the intensity of light on the rate of growth and duration of life of Drosophila. J. Gen. Physiol. 9, 81–86 (1925).
McCay, C. M., Maynard, L. A., Sperling, G. & Barnes, L. L. The Journal of Nutrition. Volume 18 July-December, 1939. Pages 1–13. Retarded growth, life span, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. Nutr. Rev. 33, 241–243 (1975).
Klass, M. R. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech. Ageing Dev. 22, 279–286 (1983).
Niccoli, T. & Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 22, R741–R752 (2012).
Wilkerson, H. L. Problems of an aging population: public health aspects of diabetes. Am. J. Public Health Nations Health 37, 177–188 (1947).
Cortes-Canteli, M. & Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 75, 942–951 (2020).
Hoehn, M. M. & Yahr, M. D. Parkinsonism: onset, progression and mortality. Neurology 17, 427–442 (1967).
Kaufman, P. & Poliakoff, H. Studies on the aging heart; the pattern of rheumatic heart disease in old age (a clinical pathological study). Ann. Intern Med. 32, 889–904 (1950).
Yan, M. et al. Cardiac aging: from basic research to therapeutics. Oxid. Med. Cell Longev. 2021, 9570325 (2021).
Hernandez Cordero, A. I. et al. Airway aging and methylation disruptions in HIV-associated chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 206, 150–160 (2022).
Grunewald, M. et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 373, eabc8479 (2021).
Chen, X. et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 81, 87–99 (2022).
Ding, J. et al. The association between aging-related monocyte transcriptional networks and comorbidity burden: the multi-ethnic study of atherosclerosis (MESA). Geroscience https://doi.org/10.1007/s11357-022-00608-1 (2022).
Bettonte, S., Berton, M. & Marzolini, C. Magnitude of drug-drug interactions in special populations. Pharmaceutics 14, 789 (2022).
Zhao, Y., Seluanov, A. & Gorbunova, V. Revelations about aging and disease from unconventional vertebrate model organisms. Annu. Rev. Genet. 55, 135–159 (2021).
Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 (1956).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Li, Z. et al. Folic acid inhibits aging-induced telomere attrition and apoptosis in astrocytes in vivo and in vitro. Cereb. Cortex 32, 286–297 (2022).
Bharath, L. P. et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55 e46 (2020).
Tracy, T. E. et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 185, 712–728.e714 (2022).
Lei, J. et al. Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell 13, 220–226 (2022).
van der Laan, L. et al. Epigenetic aging biomarkers and occupational exposure to benzene, trichloroethylene and formaldehyde. Environ. Int. 158, 106871 (2022).
Mossad, O. et al. Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N(6)-carboxymethyllysine. Nat. Neurosci. 25, 295–305 (2022).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Aman, Y. et al. Autophagy in healthy aging and disease. Nat. Aging 1, 634–650 (2021).
Aguilera, A. & Garcia-Muse, T. Causes of genome instability. Annu. Rev. Genet. 47, 1–32 (2013).
Siametis, A., Niotis, G. & Garinis, G. A. DNA damage and the aging epigenome. J. Invest. Dermatol. 141, 961–967 (2021).
Williams, A. B. & Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Har. Perspect. Med. 6, a026070 (2016).
Regulski, M. J. Cellular senescence: what, why, and how. Wounds 29, 168–174 (2017).
Hou, J. et al. Aging with higher fractional exhaled nitric oxide levels are associated with increased urinary 8-oxo-7,8-dihydro-2’-deoxyguanosine concentrations in elder females. Environ. Sci. Pollut. Res. Int. 23, 23815–23824 (2016).
Gorini, F. et al. The genomic landscape of 8-oxodG reveals enrichment at specific inherently fragile promoters. Nucleic Acids Res. 48, 4309–4324 (2020).
Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020).
Sanchez-Roman, I. et al. Molecular markers of DNA repair and brain metabolism correlate with cognition in centenarians. GeroScience 44, 103–125 (2022).
Storci, G. et al. Genomic stability, anti-inflammatory phenotype, and up-regulation of the RNAseH2 in cells from centenarians. Cell Death Differ. 26, 1845–1858 (2019).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Dou, Z. et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Kauppila, T. E. S., Kauppila, J. H. K. & Larsson, N. G. Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71 (2017).
Son, J. M. & Lee, C. Mitochondria: multifaceted regulators of aging. BMB Rep. 52, 13–23 (2019).
Chiang, J. L. et al. Mitochondria in ovarian aging and reproductive longevity. Ageing Res. Rev. 63, 101168 (2020).
Quan, Y., Xin, Y., Tian, G., Zhou, J. & Liu, X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid. Med. Cell Longev. 2020, 9423593 (2020).
Yu, C. H. et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649.e618 (2020).
Loo, T. M., Miyata, K., Tanaka, Y. & Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 111, 304–311 (2020).
Son, J. M. & Lee, C. Aging: all roads lead to mitochondria. Semin. Cell Dev. Biol. 116, 160–168 (2021).
Jauhari, A. et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Invest. 130, 3124–3136 (2020).
Gonzalo, S., Kreienkamp, R. & Askjaer, P. Hutchinson-Gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 33, 18–29 (2017).
Bin Imtiaz, M. K. et al. Declining lamin B1 expression mediates age-dependent decreases of hippocampal stem cell activity. Cell Stem Cell 28, 967–977.e968 (2021).
Miller, K. N. et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506–5526 (2021).
Wang, Y. et al. Cytoplasmic DNA sensing by KU complex in aged CD4(+) T cell potentiates T cell activation and aging-related autoimmune inflammation. Immunity 54, 632–647.e639 (2021).
Zhao, J., Wang, J., Chen, D. J., Peterson, S. R. & Trewhella, J. The solution structure of the DNA double-stranded break repair protein Ku and its complex with DNA: a neutron contrast variation study. Biochemistry 38, 2152–2159 (1999).
Fagundes, N. J. R., Bisso-Machado, R., Figueiredo, P., Varal, M. & Zani, A. L. S. What we talk about when we talk about “Junk DNA”. Genome Biol. Evol. 14, evac055 (2022).
Xu, T. et al. Polymorphic tandem DNA repeats activate the human telomerase reverse transcriptase gene. Proc. Natl Acad. Sci. USA 118, e2019043118 (2021).
Zhu, Y., Liu, X., Ding, X., Wang, F. & Geng, X. Telomere and its role in the aging pathways: telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 20, 1–16 (2019).
Laberthonniere, C., Magdinier, F. & Robin, J. D. Bring it to an end: does telomeres size matter? Cells 8, 30 (2019).
Frenck, R. W. Jr., Blackburn, E. H. & Shannon, K. M. The rate of telomere sequence loss in human leukocytes varies with age. Proc. Natl Acad. Sci. USA 95, 5607–5610 (1998).
Zeichner, S. L. et al. Rapid telomere shortening in children. Blood 93, 2824–2830 (1999).
Takubo, K. et al. Telomere shortening with aging in human liver. J. Gerontol. A Biol. Sci. Med. Sci. 55, B533–B536 (2000).
Howard, J. T. et al. Telomere shortening and accelerated aging in US Military Veterans. Int. J. Environ. Res. Public Health 18, 1743 (2021).
Cordoba-Lanus, E. et al. Telomere shortening and accelerated aging in COPD: findings from the BODE cohort. Respir. Res. 18, 59 (2017).
Turner, K. J., Vasu, V. & Griffin, D. K. Telomere biology and human phenotype. Cells 8, 73 (2019).
Blackburn, E. H., Epel, E. S. & Lin, J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350, 1193–1198 (2015).
Sun, L. et al. Transient induction of telomerase expression mediates senescence and reduces tumorigenesis in primary fibroblasts. Proc. Natl Acad. Sci. USA 116, 18983–18993 (2019).
Mojiri, A. et al. Telomerase therapy reverses vascular senescence and extends lifespan in progeria mice. Eur. Heart J. 42, 4352–4369 (2021).
Ningarhari, M. et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J. Hepatol. 74, 1155–1166 (2021).
Cen, J. et al. Anti-aging effect of estrogen on telomerase activity in ovariectomised rats-animal model for menopause. Gynecol. Endocrinol. 31, 582–585 (2015).
Chakravarti, D., LaBella, K. A. & DePinho, R. A. Telomeres: history, health, and hallmarks of aging. Cell 184, 306–322 (2021).
Sen, P., Shah, P. P., Nativio, R. & Berger, S. L. Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016).
Lopez, V., Fernandez, A. F. & Fraga, M. F. The role of 5-hydroxymethylcytosine in development, aging and age-related diseases. Ageing Res. Rev. 37, 28–38 (2017).
Zarakowska, E. et al. Oxidation products of 5-methylcytosine are decreased in senescent cells and tissues of progeroid mice. J. Gerontol. A Biol. Sci. Med. Sci. 73, 1003–1009 (2018).
Cagan, A. et al. Somatic mutation rates scale with lifespan across mammals. Nature 604, 517–524 (2022).
Mendelsohn, A. R. & Larrick, J. W. Epigenetic drift is a determinant of mammalian lifespan. Rejuvenation Res. 20, 430–436 (2017).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018).
Shireby, G. L. et al. Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain 143, 3763–3775 (2020).
Loomba, R. et al. DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3, e96685 (2018).
Fiorito, G. et al. DNA methylation-based biomarkers of aging were slowed down in a two-year diet and physical activity intervention trial: the DAMA study. Aging Cell 20, e13439 (2021).
Demidenko, O. et al. Rejuvant(R), a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test. Aging 13, 24485–24499 (2021).
Belsky, D. W. et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife 9, e54870 (2020).
Paluvai, H., Di Giorgio, E. & Brancolini, C. The histone code of senescence. Cells 9, 466 (2020).
Yi, S. J. & Kim, K. New insights into the role of histone changes in aging. Int. J. Mol. Sci. 21, 8241 (2020).
Padeken, J., Methot, S. P. & Gasser, S. M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 23, 623–640 (2022).
Wang, Y., Yuan, Q. & Xie, L. Histone modifications in aging: the underlying mechanisms and implications. Curr. Stem Cell Res. Ther. 13, 125–135 (2018).
Zhang, W. et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015).
Kirfel, P., Vilcinskas, A. & Skaljac, M. Lysine acetyltransferase p300/CBP plays an important role in reproduction, embryogenesis and longevity of the pea aphid Acyrthosiphon pisum. Insects 11, 265 (2020).
Kane, A. E. & Sinclair, D. A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 54, 61–83 (2019).
Zhu, D. et al. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 6, eabb2529 (2020).
Liu, Z. et al. Large-scale chromatin reorganization reactivates placenta-specific genes that drive cellular aging. Dev. Cell 57, 1347–1368.e1312 (2022).
Liu, X., Li, M., Xia, X., Li, X. & Chen, Z. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440–445 (2017).
Coux, R. X., Owens, N. D. L. & Navarro, P. Chromatin accessibility and transcription factor binding through the perspective of mitosis. Transcription 11, 236–240 (2020).
Zhang, C. et al. ATF3 drives senescence by reconstructing accessible chromatin profiles. Aging Cell 20, e13315 (2021).
Wu, Q., Li, B., Liu, L., Sun, S. & Sun, S. Centrosome dysfunction: a link between senescence and tumor immunity. Signal Transduct. Target Ther. 5, 107 (2020).
Jackson-Cook, C. Constitutional and acquired autosomal aneuploidy. Clin. Lab Med. 31, 481–511 (2011).
Martin, J. M., Kellett, J. M. & Kahn, J. Aneuploidy in cultured human lymphocytes: I. Age and sex differences. Age Ageing 9, 147–153 (1980).
Nowinski, G. P. et al. The frequency of aneuploidy in cultured lymphocytes is correlated with age and gender but not with reproductive history. Am. J. Hum. Genet. 46, 1101–1111 (1990).
Azzalin, C. M. & Lingner, J. Cell biology. Telomere wedding ends in divorce. Science. 304, 60–62 (2004).
Dynek, J. N. & Smith, S. Resolution of sister telomere association is required for progression through mitosis. Science 304, 97–100 (2004).
Arendt, T., Ueberham, U. & Janitz, M. Non-coding transcriptome in brain aging. Aging 9, 1943–1944 (2017).
Wang, L. et al. PEGylated reduced-graphene oxide hybridized with Fe3O4 nanoparticles for cancer photothermal-immunotherapy. J. Mater. Chem. B 7, 7406–7414 (2019).
Nicholson, A. W. Structure, reactivity, and biology of double-stranded RNA. Prog. Nucleic Acid Res. Mol. Biol. 52, 1–65 (1996).
Tarallo, V. et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 (2012).
Saldi, T. K., Gonzales, P. K., LaRocca, T. J. & Link, C. D. Neurodegeneration, heterochromatin, and double-stranded RNA. J. Exp. Neurosci. 13, 1179069519830697 (2019).
Zhang, Y. et al. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548, 52–57 (2017).
Idda, M. L., Munk, R., Abdelmohsen, K. & Gorospe, M. Noncoding RNAs in Alzheimer’s disease. Wiley Interdiscip. Rev. RNA 9, 10.1002/wrna.1463 (2018).
Knupp, D. & Miura, P. CircRNA accumulation: a new hallmark of aging? Mech. Ageing Dev. 173, 71–79 (2018).
Hipp, M. S., Kasturi, P. & Hartl, F. U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019).
Sabath, N. et al. Cellular proteostasis decline in human senescence. Proc. Natl Acad. Sci. USA 117, 31902–31913 (2020).
Li, Y. et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 38, e98786 (2019).
Koyuncu, S. et al. Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature 596, 285–290 (2021).
Zhou, D., Borsa, M. & Simon, A. K. Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell 20, e13316 (2021).
Zhao, Q. et al. Activating transcription factor 3 involved in Pseudomonas aeruginosa PAO1-induced macrophage senescence. Mol. Immunol. 133, 122–127 (2021).
Sun, Y. et al. Loss of ATF4 leads to functional aging-like attrition of adult hematopoietic stem cells. Sci. Adv. 7, eabj6877 (2021).
Timmer, L. T., Hoogaars, W. M. H. & Jaspers, R. T. The role of IGF-1 signaling in skeletal muscle atrophy. Adv. Exp. Med. Biol. 1088, 109–137 (2018).
Ock, S. et al. Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157, 336–345 (2016).
Gong, Z. et al. Reductions in serum IGF-1 during aging impair health span. Aging Cell 13, 408–418 (2014).
Ascenzi, F. et al. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 18, e12954 (2019).
Yoshida, T. & Delafontaine, P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells 9, 1970 (2020).
Templeman, N. M. & Murphy, C. T. Regulation of reproduction and longevity by nutrient-sensing pathways. J. Cell Biol. 217, 93–106 (2018).
Gao, K., Li, Y., Hu, S. & Liu, Y. SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. eLife 8, e41792 (2019).
Princz, A. & Tavernarakis, N. SUMOylation in neurodegenerative diseases. Gerontology 66, 122–130 (2020).
Gong, L., Sun, Q. & Li, D. W. Sumoylation in cellular senescence and aging. Curr. Mol. Med. 16, 871–876 (2017).
Larrick, J. W., Larrick, J. W. & Mendelsohn, A. R. SUMO wrestles with mitophagy to extend lifespan. Rejuvenation Res. 23, 527–532 (2020).
Stein, K. C., Morales-Polanco, F., van der Lienden, J., Rainbolt, T. K. & Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 601, 637–642 (2022).
Fernandez, A. F. et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018).
Kitada, M. & Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 17, 647–661 (2021).
Hansen, M., Rubinsztein, D. C. & Walker, D. W. Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593 (2018).
Leidal, A. M., Levine, B. & Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 20, 1338–1348 (2018).
Zeidan, R. S., Han, S. M., Leeuwenburgh, C. & Xiao, R. Iron homeostasis and organismal aging. Ageing Res Rev. 72, 101510 (2021).
Guo, H. et al. Induction of autophagy via the ROS-dependent AMPK-mTOR pathway protects copper-induced spermatogenesis disorder. Redox Biol. 49, 102227 (2022).
Tang, D. & Kroemer, G. Ferroptosis. Curr. Biol. 30, R1292–R1297 (2020).
Aman, Y., Cao, S. & Fang, E. F. Iron out, mitophagy in! A way to slow down hepatocellular carcinoma. EMBO Rep. 21, e51652 (2020).
Pushkar, Y. et al. Aging results in copper accumulations in glial fibrillary acidic protein-positive cells in the subventricular zone. Aging Cell 12, 823–832 (2013).
Masaldan, S. et al. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 16, 322–331 (2018).
Fakouri, N. B. et al. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 286, 1058–1073 (2019).
Fouquerel, E. et al. Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell 75, 117–130.e116 (2019).
Meng, J., Lv, Z., Wang, Y. & Chen, C. Identification of the redox-stress signaling threshold (RST): increased RST helps to delay aging in C. elegans. Free Radic. Biol. Med. 178, 54–58 (2022).
Vyssokikh, M. Y. et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl Acad. Sci. USA 117, 6491–6501 (2020).
Ristow, M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 20, 709–711 (2014).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014).
Xiao, H. et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 180, 968–983 e924 (2020).
Smith, A. L. et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 1, 976–989 (2020).
Singh, B., Schoeb, T. R., Bajpai, P., Slominski, A. & Singh, K. K. Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 9, 735 (2018).
Fang, E. F. et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321 (2016).
Reiten, O. K., Wilvang, M. A., Mitchell, S. J., Hu, Z. & Fang, E. F. Preclinical and clinical evidence of NAD(+) precursors in health, disease, and ageing. Mech. Ageing Dev. 199, 111567 (2021).
Scheibye-Knudsen, M. et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855 (2014).
Fang, E. F. et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581 (2016).
Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 (2015).
Wiley, C. D. & Campisi, J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat. Metab. 3, 1290–1301 (2021).
Dollerup, O. L. et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 108, 343–353 (2018).
Dollerup, O. L. et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 598, 731–754 (2020).
Remie, C. M. E. et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am. J. Clin. Nutr. 112, 413–426 (2020).
Popov, L. D. Mitochondrial biogenesis: an update. J. Cell Mol. Med 24, 4892–4899 (2020).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705 (2021).
Konig, T. et al. MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23, 1271–1286 (2021).
Ryan, T. A. & Tumbarello, D. A. A central role for mitochondrial-derived vesicles in the innate immune response: implications for Parkinson’s disease. Neural Regen. Res. 16, 1779–1780 (2021).
Rhinn, M., Ritschka, B. & Keyes, W. M. Cellular senescence in development, regeneration and disease. Development 146, dev151837 (2019).
Kowald, A., Passos, J. F. & Kirkwood, T. B. L. On the evolution of cellular senescence. Aging Cell 19, e13270 (2020).
Birch, J. & Gil, J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 34, 1565–1576 (2020).
Palmer, A. K., Gustafson, B., Kirkland, J. L. & Smith, U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia 62, 1835–1841 (2019).
Cai, Y. et al. The landscape of aging. Sci. China Life Sci. 1–101 (2022).
Iram, T. et al. Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature 605, 509–515 (2022).
Prieto, L. I. & Baker, D. J. Cellular senescence and the immune system in cancer. Gerontology 65, 505–512 (2019).
Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 22, 687–698 (2021).
Desdin-Mico, G. et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376 (2020).
Deng, P. et al. Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 28, 1057–1073.e1057 (2021).
Revuelta, M. & Matheu, A. Autophagy in stem cell aging. Aging Cell 16, 912–915 (2017).
Zhang, D. et al. Autophagy inhibits the mesenchymal stem cell aging induced by D-galactose through ROS/JNK/p38 signalling. Clin. Exp. Pharm. Physiol. 47, 466–477 (2020).
Ho, T. T. et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 (2017).
Rodriguez-Ramirez, C. & Nor, J. E. p53 and cell fate: sensitizing head and neck cancer stem cells to chemotherapy. Crit. Rev. Oncog. 23, 173–187 (2018).
Manzano-Lopez, J. & Monje-Casas, F. Asymmetric cell division and replicative aging: a new perspective from the spindle poles. Curr. Genet. 66, 719–727 (2020).
Daynac, M. & Petritsch, C. K. Regulation of asymmetric cell division in mammalian neural stem and cancer precursor cells. Results Probl. Cell Differ. 61, 375–399 (2017).
Borsa, M. et al. Asymmetric cell division shapes naive and virtual memory T-cell immunity during ageing. Nat. Commun. 12, 2715 (2021).
Lengefeld, J. et al. Cell size is a determinant of stem cell potential during aging. Sci. Adv. 7, eabk0271 (2021).
Fafian-Labora, J. A. & O’Loghlen, A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 30, 628–639 (2020).
Xu, Q. et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 3, 1706–1726 (2021).
van Niel, G. et al. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 23, 369–382 (2022).
Kfoury, Y. S. et al. tiRNA signaling via stress-regulated vesicle transfer in the hematopoietic niche. Cell Stem Cell 28, 2090–2103.e2099 (2021).
Crewe, C. et al. An endothelial-to-adipocyte extracellular vesicle axis governed by metabolic state. Cell 175, 695–708 e613 (2018).
Narita, M. Juxtacrine regulation of cellular senescence. BMB Rep. 52, 3–4 (2019).
Hoare, M. et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992 (2016).
Giordano-Santini, R., Linton, C. & Hilliard, M. A. Cell-cell fusion in the nervous system: alternative mechanisms of development, injury, and repair. Semin Cell Dev. Biol. 60, 146–154 (2016).
Wang, Y., Welc, S. S., Wehling-Henricks, M. & Tidball, J. G. Myeloid cell-derived tumor necrosis factor-alpha promotes sarcopenia and regulates muscle cell fusion with aging muscle fibers. Aging Cell 17, e12828 (2018).
Chaigne, A. & Brunet, T. Incomplete abscission and cytoplasmic bridges in the evolution of eukaryotic multicellularity. Curr. Biol. 32, R385–R397 (2022).
Walters, H. E. & Cox, L. S. Intercellular transfer of mitochondria between senescent cells through cytoskeleton-supported intercellular bridges requires mTOR and CDC42 signalling. Oxid. Med. Cell Longev. 2021, 6697861 (2021).
Ma, S. et al. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus norvegicus aging. Cell 180, 984–1001.e1022 (2020).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Shannon, O. M. et al. Mediterranean diet and the hallmarks of ageing. Eur. J. Clin. Nutr. 75, 1176–1192 (2021).
Gonzalez, A. & Hall, M. N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 36, 397–408 (2017).
Lin, S. C. & Hardie, D. G. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313 (2018).
Gonzalez, A., Hall, M. N., Lin, S. C. & Hardie, D. G. AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 31, 472–492 (2020).
Fernandes, S. A. & Demetriades, C. The multifaceted role of nutrient sensing and mTORC1 signaling in physiology and aging. Front. Aging 2, 707372 (2021).
Johnson, S. C. Nutrient sensing, signaling and ageing: the role of IGF-1 and mTOR in ageing and age-related disease. Subcell. Biochem. 90, 49–97 (2018).
Kowalski, K. & Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 25, 48–60 (2019).
Wilmanski, T. et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 3, 274–286 (2021).
Kim, S. & Jazwinski, S. M. The gut microbiota and healthy aging: a mini-review. Gerontology 64, 513–520 (2018).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Wang, G. et al. Bridging intestinal immunity and gut microbiota by metabolites. Cell Mol. Life Sci. 76, 3917–3937 (2019).
Cryan, J. F. et al. The microbiota-gut-brain axis. Physiol. Rev. 99, 1877–2013 (2019).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Prim. 7, 33 (2021).
Jeppesen, D. K., Bohr, V. A. & Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 94, 166–200 (2011).
Scheibye-Knudsen, M. Neurodegeneration in accelerated aging. Dan. Med. J. 63, B5308 (2016).
Leandro, G. S., Sykora, P. & Bohr, V. A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. 776, 31–39 (2015).
Fielder, E., von Zglinicki, T. & Jurk, D. The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 60, S107–s131 (2017).
Gao, X. et al. Epigenetics in Alzheimer’s disease. Front. Aging Neurosci. 14, 911635 (2022).
De Jager, P. L., Srivastava, G., Lunnon, K., Burgess, J. & Schalkwyk, L. C. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163 (2014).
Ashrafian, H., Zadeh, E. H. & Khan, R. H. Review on Alzheimer’s disease: inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 167, 382–394 (2021).
Nisbet, R. M. & Götz, J. Amyloid-β and Tau in Alzheimer’s disease: novel pathomechanisms and non-pharmacological treatment strategies. J. Alzheimers Dis. 64, S517–s527 (2018).
Tong, B. C., Wu, A. J., Li, M. & Cheung, K. H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1745–1760 (2018).
Haass, C. & Selkoe, D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. 20, e3001694 (2022).
Lee, J. H. et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701 (2022).
Biundo, F. et al. Abolishing Tau cleavage by caspases at Aspartate(421) causes memory/synaptic plasticity deficits and pre-pathological Tau alterations. Transl. Psychiatry 7, e1198 (2017).
Roy, D. S. et al. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531, 508–512 (2016).
Perez Ortiz, J. M. & Swerdlow, R. H. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharm. 176, 3489–3507 (2019).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Miller, S. & Muqit, M. M. K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 705, 7–13 (2019).
Li, N., Shu, J., Yang, X., Wei, W. & Yan, A. Exosomes derived from M2 microglia cells attenuates neuronal impairment and mitochondrial dysfunction in Alzheimer’s disease through the PINK1/Parkin pathway. Front. Cell Neurosci. 16, 874102 (2022).
Gaikwad, S. et al. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. 36, 109419 (2021).
Han, X., Zhang, T., Liu, H., Mi, Y. & Gou, X. Astrocyte senescence and Alzheimer’s disease: a review. Front. Aging Neurosci. 12, 148 (2020).
Zhong, Y. et al. A review on pathology, mechanism, and therapy for cerebellum and tremor in Parkinson’s disease. NPJ Parkinsons Dis. 8, 82 (2022).
Shahnawaz, M. et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277 (2020).
Guo, W. et al. Association of LAG3 genetic variation with an increased risk of PD in Chinese female population. J. Neuroinflammation 16, 270 (2019).
Polymeropoulos, M. H. Revisiting the history of the 1997 alpha-synuclein discovery in Parkinson’s disease. J. Parkinsons Dis. 9, 443–444 (2019).
Blauwendraat, C. et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143, 234–248 (2020).
Sengupta, U. & Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 214, 102270 (2022).
Rocha, E. M., De Miranda, B. & Sanders, L. H. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257 (2018).
Savarese, G. et al. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc. Res. cvac013 https://doi.org/10.1093/cvr/cvac013 (2022).
Chen, K. et al. Klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ. Res. 128, 492–507 (2021).
Anderson, R. et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492 (2019).
Tang, X., Li, P. H. & Chen, H. Z. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front. Endocrinol. 11, 280 (2020).
Chen, M. S., Lee, R. T. & Garbern, J. C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 118, 1173–1187 (2022).
Weissman, D. & Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 171, 345–364 (2021).
Kowalczyk, P. et al. Mitochondrial oxidative stress-A causative factor and therapeutic target in many diseases. Int. J. Mol. Sci. 22, 13384 (2021).
Martini, H. et al. Selective cardiomyocyte oxidative stress leads to bystander senescence of cardiac stromal cells. Int. J. Mol. Sci. 22, 2245 (2021).
Zhang, X. et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation 137, 2052–2067 (2018).
Gu, J. et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 9, 82 (2018).
El-Nachef, D. et al. Repressive histone methylation regulates cardiac myocyte cell cycle exit. J. Mol. Cell Cardiol. 121, 1–12 (2018).
Berulava, T. et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 22, 54–66 (2020).
Chen, Y. et al. Fibroblast growth factor 20 attenuates pathological cardiac hypertrophy by activating the SIRT1 signaling pathway. Cell Death Dis. 13, 276 (2022).
Katsuumi, G., Shimizu, I., Yoshida, Y. & Minamino, T. Vascular senescence in cardiovascular and metabolic diseases. Front. Cardiovasc. Med. 5, 18 (2018).
Gevaert, A. B. et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ. Heart Fail. 10, e003806 (2017).
Jesel, L. et al. Atrial fibrillation progression is associated with cell senescence burden as determined by p53 and p16 expression. J. Clin. Med. 9, 36 (2019).
Sung, J. Y., Kim, S. G., Kim, J. R. & Choi, H. C. Prednisolone suppresses adriamycin-induced vascular smooth muscle cell senescence and inflammatory response via the SIRT1-AMPK signaling pathway. PLoS ONE 15, e0239976 (2020).
Xia, W., Chen, H., Xie, C. & Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 12, 8241–8260 (2020).
Jia, G., Aroor, A. R., Jia, C. & Sowers, J. R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1802–1809 (2019).
Howden, R. Nrf2 and cardiovascular defense. Oxid. Med. Cell Longev. 2013, 104308 (2013).
Xiao, X. et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct. Target Ther. 6, 354 (2021).
de Yebenes, V. G. et al. Aging-associated miR-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 40, 2408–2424 (2020).
Menghini, R. et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 120, 1524–1532 (2009).
Schiro, A. et al. Endothelial microparticles as conveyors of information in atherosclerotic disease. Atherosclerosis 234, 295–302 (2014).
Takahashi, T. & Kubo, H. The role of microparticles in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 9, 303–314 (2014).
Kugel, S. & Mostoslavsky, R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 39, 72–81 (2014).
Guarente, L. Franklin H. Epstein lecture: sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244 (2011).
Artal-Sanz, M. & Tavernarakis, N. Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 20, 394–401 (2009).
Schleicher, M. et al. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 180, 101–112 (2008).
Dermaku-Sopjani, M., Kolgeci, S., Abazi, S. & Sopjani, M. Significance of the anti-aging protein Klotho. Mol. Membr. Biol. 30, 369–385 (2013).
Wang, J. C. & Bennett, M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 111, 245–259 (2012).
Brozovich, F. V. et al. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharm. Rev. 68, 476–532 (2016).
Matthews, C. et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ. Res. 99, 156–164 (2006).
Liu, Y., Drozdov, I., Shroff, R., Beltran, L. E. & Shanahan, C. M. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ. Res. 112, e99–e109 (2013).
Grootaert, M. O. J., Finigan, A., Figg, N. L., Uryga, A. K. & Bennett, M. R. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ. Res. 128, 474–491 (2021).
Gosling, J. et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Invest. 103, 773–778 (1999).
Song, Y. et al. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 32, 103–109 (2012).
Gardner, S. E., Humphry, M., Bennett, M. R. & Clarke, M. C. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler. Thromb. Vasc. Biol. 35, 1963–1974 (2015).
Hamczyk, M. R. & Andres, V. Vascular smooth muscle cell loss underpins the accelerated atherosclerosis in Hutchinson-Gilford progeria syndrome. Nucleus 10, 28–34 (2019).
Potekhina, A. V. et al. Treg/Th17 balance in stable CAD patients with different stages of coronary atherosclerosis. Atherosclerosis 238, 17–21 (2015).
Fadini, G. P., Losordo, D. & Dimmeler, S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ. Res. 110, 624–637 (2012).
Lee, P. S. & Poh, K. K. Endothelial progenitor cells in cardiovascular diseases. World J. Stem Cells 6, 355–366 (2014).
Yiu, K. H. & Tse, H. F. Specific role of impaired glucose metabolism and diabetes mellitus in endothelial progenitor cell characteristics and function. Arterioscler. Thromb. Vasc. Biol. 34, 1136–1143 (2014).
Kalyani, R. R., Golden, S. H. & Cefalu, W. T. Diabetes and aging: unique considerations and goals of care. Diabetes Care 40, 440–443 (2017).
Khan, R. M. M. et al. From pre-diabetes to diabetes: diagnosis, treatments and translational research. Medicina 55, 546 (2019).
Lytrivi, M., Castell, A. L., Poitout, V. & Cnop, M. Recent insights into mechanisms of beta-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 432, 1514–1534 (2020).
de Zegher, F., Diaz, M. & Ibanez, L. Association between long telomere length and insulin sensitization in adolescent girls with hyperinsulinemic androgen excess. JAMA Pediatr. 169, 787–788 (2015).
Tamura, Y. et al. beta-cell telomere attrition in diabetes: inverse correlation between HbA1c and telomere length. J. Clin. Endocrinol. Metab. 99, 2771–2777 (2014).
Guo, N. et al. Short telomeres compromise beta-cell signaling and survival. PLoS ONE 6, e17858 (2011).
Tamura, Y., Takubo, K., Aida, J., Araki, A. & Ito, H. Telomere attrition and diabetes mellitus. Geriatr. Gerontol. Int. 16, 66–74 (2016).
Fonseca, S. G., Gromada, J. & Urano, F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol. Metab. 22, 266–274 (2011).
Zhang, I. X., Raghavan, M. & Satin, L. S. The endoplasmic reticulum and calcium homeostasis in pancreatic beta cells. Endocrinology 161, bqz028 (2020).
Fontana, L., Nehme, J. & Demaria, M. Caloric restriction and cellular senescence. Mech. Ageing Dev. 176, 19–23 (2018).
Eguchi, K. et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 15, 518–533 (2012).
Maedler, K. et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 127, 1589 (2017).
Chan, J. Y., Lee, K., Maxwell, E. L., Liang, C. & Laybutt, D. R. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia 62, 993–999 (2019).
Ehses, J. A. et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370 (2007).
Sone, H. & Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48, 58–67 (2005).
Aguayo-Mazzucato, C. et al. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 30, 129–142.e124 (2019).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Midha, A. et al. Unique human and mouse beta-cell senescence-associated secretory phenotype (SASP) reveal conserved signaling pathways and heterogeneous factors. Diabetes 70, 1098–1116 (2021).
Liu, Z., Wu, K. K. L., Jiang, X., Xu, A. & Cheng, K. K. Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci. 134, 315–330 (2020).
Iatcu, C. O., Steen, A. & Covasa, M. Gut microbiota and complications of type-2 diabetes. Nutrients 14, 166 (2021).
Gan, L., Chitturi, S. & Farrell, G. C. Mechanisms and implications of age-related changes in the liver: nonalcoholic fatty liver disease in the elderly. Curr. Gerontol. Geriatr. Res. 2011, 1–12 (2011).
Alqahtani, S. A. & Schattenberg, J. M. NAFLD in the elderly. Clin. Inter. Aging 16, 1633–1649 (2021).
Aravinthan, A. et al. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle 13, 1489–1494 (2014).
Palma, R. et al. The role of insulin resistance in fueling NAFLD pathogenesis: from molecular mechanisms to clinical implications. J. Clin. Med. 11, 3649 (2022).
Kim, J.-H., Jung, D. Y., Nagappan, A. & Jung, M. H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 8, 13734 (2018).
Rector, R. S. et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 52, 727–736 (2010).
Hunt, N. J., Kang, S. W., Lockwood, G. P., Le Couteur, D. G. & Cogger, V. C. Hallmarks of aging in the liver. Comput. Struct. Biotechnol. J. 17, 1151–1161 (2019).
Ramanathan, R., Ali, A. H. & Ibdah, J. A. Mitochondrial dysfunction plays central role in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 23, 7280 (2022).
Ipsen, D. H., Lykkesfeldt, J. & Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 75, 3313–3327 (2018).
Reinecke, F., Smeitink, J. A. M. & van der Westhuizen, F. H. OXPHOS gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta 1792, 1113–1121 (2009).
Dabravolski, S. A., Bezsonov, E. E. & Orekhov, A. N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 142, 112041 (2021).
Han, X. et al. Nicotinamide riboside exerts protective effect against aging-induced NAFLD-like hepatic dysfunction in mice. PeerJ 7, e7568 (2019).
Li, D.-J. et al. NAD+-boosting therapy alleviates nonalcoholic fatty liver disease via stimulating a novel exerkine Fndc5/irisin. Theranostics 11, 4381–4402 (2021).
Zhou, C.-C. et al. Hepatic NAD+deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharm. 173, 2352–2368 (2016).
Bozaykut, P., Sahin, A., Karademir, B. & Ozer, N. K. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech. Ageing Dev. 157, 17–29 (2016).
Volmer, R., van der Ploeg, K. & Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl Acad. Sci. USA 110, 4628–4633 (2013).
Lebeaupin, C. et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 69, 927–947 (2018).
Dasgupta, D. et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology 159, 1487–1503.e1417 (2020).
Kern, P. et al. Creld2 function during unfolded protein response is essential for liver metabolism homeostasis. FASEB J. 35, e21939 (2021).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Carotti, S. et al. Lipophagy impairment is associated with disease progression in NAFLD. Front. Physiol. 11, 850 (2020).
Ferrara-Romeo, I. et al. The mTOR pathway is necessary for survival of mice with short telomeres. Nat. Commun. 11, 1168 (2020).
Gong, Z., Tas, E., Yakar, S. & Muzumdar, R. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol. Cell Endocrinol. 455, 115–130 (2017).
Amir, M. & Czaja, M. J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 5, 159–166 (2014).
Ogrodnik, M. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 15691 (2017).
Zhang, J. et al. The p66shc-mediated regulation of hepatocyte senescence influences hepatic steatosis in nonalcoholic fatty liver disease. Med. Sci. Monit. 26, e921887 (2020).
Jin, J. et al. Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Rep. 16, 744–756 (2016).
Nguyen, P. et al. Elimination of age-associated hepatic steatosis and correction of aging phenotype by inhibition of cdk4-C/EBPα-p300 axis. Cell Rep. 24, 1597–1609 (2018).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Disco. 16, 718–735 (2017).
Ohtani, N. & Hara, E. Gut‐liver axis‐mediated mechanism of liver cancer: a special focus on the role of gut microbiota. Cancer Sci. 112, 4433–4443 (2021).
Arab, J. P., Arrese, M. & Shah, V. H. Gut microbiota in non‐alcoholic fatty liver disease and alcohol‐related liver disease: Current concepts and perspectives. Hepatol. Res. 50, 407–418 (2020).
Fanta Barrow, S. K. et al. Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology 74, 704–722 (2021).
Geng, Y., Faber, K. N., de Meijer, V. E., Blokzijl, H. & Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 15, 21–35 (2021).
Nicklas, B. J. et al. Effects of caloric restriction on cardiorespiratory fitness, fatigue, and disability responses to aerobic exercise in older adults with obesity: a randomized controlled trial. J. Gerontol. A Biol. Sci. Med. Sci. 74, 1084–1090 (2019).
Linden, A. G. et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 59, 475–487 (2018).
Barazzoni, R., Gortan Cappellari, G., Ragni, M. & Nisoli, E. Insulin resistance in obesity: an overview of fundamental alterations. Eat. Weight Disord. 23, 149–157 (2018).
Huby, T. & Gautier, E. L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 22, 429–443 (2021).
Thibaut, R. et al. Liver macrophages and inflammation in physiology and physiopathology of non‐alcoholic fatty liver disease. FEBS J. 289, 3024–3057 (2021).
Strnad, P. et al. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLos ONE 11, e0159524 (2016).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Tran, S. et al. Impaired Kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity 53, 627–640.e625 (2020).
Remmerie, A. et al. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity 53, 641–657.e614 (2020).
Seidman, J. S. et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 52, 1057–1074.e1057 (2020).
Takatsu, M. et al. Calorie restriction attenuates cardiac remodeling and diastolic dysfunction in a rat model of metabolic syndrome. Hypertension 62, 957–965 (2013).
Jeon, O. H., David, N., Campisi, J. & Elisseeff, J. H. Senescent cells and osteoarthritis: a painful connection. J. Clin. Invest. 128, 1229–1237 (2018).
Wieland, H. A., Michaelis, M., Kirschbaum, B. J. & Rudolphi, K. A. Osteoarthritis - an untreatable disease? Nat. Rev. Drug Disco. 4, 331–344 (2005).
Barbour, K. E., Helmick, C. G., Boring, M. & Brady, T. J. Vital signs: prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation - United States, 2013-2015. MMWR Morb. Mortal. Wkly. Rep. 66, 246–253 (2017).
Goldring, M. B. & Marcu, K. B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 11, 224 (2009).
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P. & Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 (2011).
Sandell, L. J. Etiology of osteoarthritis: genetics and synovial joint development. Nat. Rev. Rheumatol. 8, 77–89 (2012).
Rim, Y. A., Nam, Y. & Ju, J. H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 21, 2358 (2020).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Martin, J. A., Brown, T. D., Heiner, A. D. & Buckwalter, J. A. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Relat. Res. S96–S103 (2004).
Price, J. S. et al. The role of chondrocyte senescence in osteoarthritis. Aging cell 1, 57–65 (2002).
Zhou, H. W., Lou, S. Q. & Zhang, K. Recovery of function in osteoarthritic chondrocytes induced by p16INK4a-specific siRNA in vitro. Rheumatology 43, 555–568 (2004).
Harbo, M. et al. The distribution pattern of critically short telomeres in human osteoarthritic knees. Arthritis Res. Ther. 14, R12 (2012).
Harbo, M. et al. The relationship between ultra-short telomeres, aging of articular cartilage and the development of human hip osteoarthritis. Mech. Ageing Dev. 134, 367–372 (2013).
Loeser, R. F. The role of aging in the development of osteoarthritis. Trans. Am. Clin. Climatol. Assoc. 128, 44–54 (2017).
Loeser, R. F., Gandhi, U., Long, D. L., Yin, W. & Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 66, 2201–2209 (2014).
Dai, S. M. et al. Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum. 54, 818–831 (2006).
Yan, H. M. et al. Efficacy of berberine in patients with non-alcoholic fatty liver disease. PLoS ONE 10, e0134172 (2015).
Hui, W. et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 75, 449–458 (2016).
Rego-Perez, I., Duran-Sotuela, A., Ramos-Louro, P. & Blanco, F. J. Mitochondrial genetics and epigenetics in osteoarthritis. Front. Genet. 10, 1335 (2019).
Zheng, L., Zhang, Z., Sheng, P. & Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 66, 101249 (2021).
Mobasheri, A. et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13, 302–311 (2017).
Peansukmanee, S. et al. Effects of hypoxia on glucose transport in primary equine chondrocytes in vitro and evidence of reduced GLUT1 gene expression in pathologic cartilage in vivo. J. Orthop. Res. 27, 529–535 (2009).
Bustamante, M. F. et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 77, 1636–1643 (2018).
Yang, X. et al. Pyruvate kinase M2 modulates the glycolysis of chondrocyte and extracellular matrix in osteoarthritis. DNA Cell Biol. 37, 271–277 (2018).
Liu-Bryan, R. Inflammation and intracellular metabolism: new targets in OA. Osteoarthr. Cartil. 23, 1835–1842 (2015).
Terkeltaub, R., Yang, B., Lotz, M. & Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 63, 1928–1937 (2011).
Matsuzaki, T. et al. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 73, 1397–1404 (2014).
Tsuchida, A. I. et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 16, 441 (2014).
Parfitt, A. M. Age-related structural changes in trabecular and cortical bone: cellular mechanisms and biomechanical consequences. Calcif. Tissue Int. 36, S123–S128 (1984).
Chen, Q. et al. DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J. Bone Min. Res. 28, 1214–1228 (2013).
Pignolo, R. J. et al. Defects in telomere maintenance molecules impair osteoblast differentiation and promote osteoporosis. Aging Cell 7, 23–31 (2008).
Saeed, H. et al. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J. Bone Min. Res. 26, 1494–1505 (2011).
Brennan, T. A. et al. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model Mech. 7, 583–592 (2014).
Saeed, H. et al. Telomerase activity promotes osteoblast differentiation by modulating IGF-signaling pathway. Biogerontology 16, 733–745 (2015).
Zeng, J., Xiao, Q., Li, X. & Chen, J. Advanced oxidation protein products aggravate agerelated bone loss by increasing sclerostin expression in osteocytes via ROSdependent downregulation of Sirt1. Int. J. Mol. Med. 47, 108 (2021).
Zeng, J. H. et al. Advanced oxidation protein products accelerate bone deterioration in aged rats. Exp. Gerontol. 50, 64–71 (2014).
Shah, S. et al. P53 regulation of osteoblast differentiation is mediated through specific microRNAs. Biochem. Biophys. Rep. 25, 100920 (2021).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Min. Res. 31, 1920–1929 (2016).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Idris, A. I. et al. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab. 10, 139–147 (2009).
Hu, L. et al. Mesenchymal stem cells: cell fate decision to osteoblast or adipocyte and application in osteoporosis treatment. Int. J. Mol. Sci. 19, 360 (2018).
Stenderup, K., Justesen, J., Clausen, C. & Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 33, 919–926 (2003).
Wang, H. et al. Impairment of osteoblast differentiation due to proliferation-independent telomere dysfunction in mouse models of accelerated aging. Aging Cell 11, 704–713 (2012).
Mattiucci, D., Maurizi, G., Leoni, P. & Poloni, A. Aging- and senescence-associated changes of mesenchymal stromal cells in myelodysplastic syndromes. Cell Transpl. 27, 754–764 (2018).
Guo, Y. et al. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 41, 101915 (2021).
Fathi, E., Charoudeh, H. N., Sanaat, Z. & Farahzadi, R. Telomere shortening as a hallmark of stem cell senescence. Stem Cell Investig. 6, 7 (2019).
Galderisi, U. et al. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 18, 1033–1042 (2009).
Vono, R., Jover Garcia, E., Spinetti, G. & Madeddu, P. Oxidative stress in mesenchymal stem cell senescence: regulation by coding and noncoding RNAs. Antioxid. Redox Signal 29, 864–879 (2018).
Moerman, E. J., Teng, K., Lipschitz, D. A. & Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 3, 379–389 (2004).
Christenson, S. A., Smith, B. M., Bafadhel, M. & Putcha, N. Chronic obstructive pulmonary disease. Lancet 399, 2227–2242 (2022).
Cho, S. J. & Stout-Delgado, H. W. Aging and lung disease. Annu Rev. Physiol. 82, 433–459 (2020).
Schneider, J. L. et al. The aging lung: physiology, disease, and immunity. Cell 184, 1990–2019 (2021).
Kurozumi, M., Matsushita, T., Hosokawa, M. & Takeda, T. Age-related changes in lung structure and function in the senescence-accelerated mouse (SAM): SAM-P/1 as a new murine model of senile hyperinflation of lung. Am. J. Respir. Crit. Care Med. 149, 776–782 (1994).
Ishii, M., Yamaguchi, Y., Yamamoto, H., Hanaoka, Y. & Ouchi, Y. Airspace enlargement with airway cell apoptosis in klotho mice: a model of aging lung. J. Gerontol. A Biol. Sci. Med. Sci. 63, 1289–1298 (2008).
Proenca de Oliveira-Maul, J. et al. Aging, diabetes, and hypertension are associated with decreased nasal mucociliary clearance. Chest 143, 1091–1097 (2013).
Tsuji, T., Aoshiba, K. & Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 174, 886–893 (2006).
Martinez de Toda, I., Mate, I., Vida, C., Cruces, J. & De la Fuente, M. Immune function parameters as markers of biological age and predictors of longevity. Aging 8, 3110–3119 (2016).
Kuwano, K. et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 54, 397–406 (2016).
Bodas, M., Patel, N., Silverberg, D., Walworth, K. & Vij, N. Master autophagy regulator transcription factor EB eegulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid. Redox Signal. 27, 150–167 (2017).
Yamada, Y. et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Lab Invest. 95, 625–634 (2015).
Barnes, P. J., Baker, J. & Donnelly, L. E. Autophagy in asthma and chronic obstructive pulmonary disease. Clin. Sci. 136, 733–746 (2022).
Mumby, S. & Adcock, I. M. Recent evidence from omic analysis for redox signalling and mitochondrial oxidative stress in COPD. J. Inflamm. 19, 10 (2022).
Wiegman, C. H. et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 136, 769–780 (2015).
Araya, J. et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 15, 510–526 (2019).
Tsubouchi, K., Araya, J. & Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 38, 18 (2018).
Vucic, E. A. et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 50, 912–922 (2014).
Qiu, W. et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 185, 373–381 (2012).
Rajendrasozhan, S., Yang, S. R., Kinnula, V. L. & Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177, 861–870 (2008).
Hodge, G. et al. Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes. Respir. Res. 16, 130 (2015).
Mercado, N. et al. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 406, 292–298 (2011).
Yao, H. et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 122, 2032–2045 (2012).
Baker, J. R. et al. MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. FASEB J. 33, 1605–1616 (2019).
Gu, C. et al. LncRNAmediated SIRT1/FoxO3a and SIRT1/p53 signaling pathways regulate type II alveolar epithelial cell senescence in patients with chronic obstructive pulmonary disease. Mol. Med. Rep. 15, 3129–3134 (2017).
Ganesan, S. et al. Aberrantly activated EGFR contributes to enhanced IL-8 expression in COPD airways epithelial cells via regulation of nuclear FoxO3A. Thorax 68, 131–141 (2013).
Hwang, J. W. et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 187, 987–998 (2011).
Cheng, X. Y., Li, Y. Y., Huang, C., Li, J. & Yao, H. W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 8, 22513–22523 (2017).
Caramori, G. et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax 66, 521–527 (2011).
Ceylan, E., Kocyigit, A., Gencer, M., Aksoy, N. & Selek, S. Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir. Med. 100, 1270–1276 (2006).
Albrecht, E. et al. Telomere length in circulating leukocytes is associated with lung function and disease. Eur. Respir. J. 43, 983–992 (2014).
Nyunoya, T. et al. Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am. J. Respir. Crit. Care Med. 179, 279–287 (2009).
Rossiello, F., Jurk, D., Passos, J. F. & d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 24, 135–147 (2022).
Ahmad, T. et al. Shelterin telomere protection protein 1 reduction causes telomere attrition and cellular senescence via sirtuin 1 deacetylase in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 56, 38–49 (2017).
Chen, R. et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 290, 30813–30829 (2015).
Kuilman, T., Michaloglou, C., Mooi, W. J. & Peeper, D. S. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Welén, K. & Damber, J. E. Androgens, aging, and prostate health. Rev. Endocr. Metab. Disord. 6, 62–77 (2022).
Shukla, G. C., Plaga, A. R., Shankar, E. & Gupta, S. Androgen receptor-related diseases: what do we know? Andrology 4, 366–381 (2016).
Jin, R. et al. Glucocorticoids are induced while dihydrotestosterone levels are suppressed in 5-alpha reductase inhibitor treated human benign prostate hyperplasia patients. Prostate 82, 1378–1388 (2022).
Tsunemori, H. & Sugimoto, M. Effects of inflammatory prostatitis on the development and progression of benign prostatic hyperplasia: a literature review. Int. J. Urol. 28, 1086–1092 (2021).
Kramer, G., Mitteregger, D. & Marberger, M. Is benign prostatic hyperplasia (BPH) an immune inflammatory disease? Eur. Urol. 51, 1202–1216 (2007).
Penna, G. et al. Human benign prostatic hyperplasia stromal cells as inducers and targets of chronic immuno-mediated inflammation. J. Immunol. 182, 4056–4064 (2009).
Xiong, Y., Zhang, Y., Tan, J., Qin, F. & Yuan, J. The association between metabolic syndrome and lower urinary tract symptoms suggestive of benign prostatic hyperplasia in aging males: evidence based on propensity score matching. Transl. Androl. Urol. 10, 384–396 (2021).
Xia, B. W. et al. The underlying mechanism of metabolic syndrome on benign prostatic hyperplasia and prostate volume. Prostate 80, 481–490 (2020).
Garcia-Garcia, J. et al. Pathophysiology of age-related macular degeneration. Implications for treatment. Ophthalmic Res. https://doi.org/10.1159/000524942 (2022).
Deng, Y. et al. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 9, 62–79 (2022).
Chaum, E., Winborn, C. S. & Bhattacharya, S. Genomic regulation of senescence and innate immunity signaling in the retinal pigment epithelium. Mamm. Genome 26, 210–221 (2015).
Lopez-Luppo, M. et al. Cellular senescence is associated with human retinal microaneurysm formation during aging. Invest. Ophthalmol. Vis. Sci. 58, 2832–2842 (2017).
Cabrera, A. P. et al. Senescence increases choroidal endothelial stiffness and susceptibility to complement injury: implications for choriocapillaris loss in AMD. Invest. Ophthalmol. Vis. Sci. 57, 5910–5918 (2016).
Damani, M. R. et al. Age-related alterations in the dynamic behavior of microglia. Aging cell 10, 263–276 (2011).
Lee, K. S., Lin, S., Copland, D. A., Dick, A. D. & Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflammation 18, 32 (2021).
Copland, D. A., Theodoropoulou, S., Liu, J. & Dick, A. D. A perspective of AMD through the eyes of immunology. Invest. Ophthalmol. Vis. Sci. 59, AMD83–AMD92 (2018).
Cao, L. et al. Abeta-induced senescent retinal pigment epithelial cells create a proinflammatory microenvironment in AMD. Invest. Ophthalmol. Vis. Sci. 54, 3738–3750 (2013).
Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell Mol. Life Sci. 77, 789–805 (2020).
Lin, J. B. et al. Macrophage microRNA-150 promotes pathological angiogenesis as seen in age-related macular degeneration. JCI insight 3, e120157 (2018).
Sene, A. et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 17, 549–561 (2013).
Zhao, C. et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Invest. 121, 369–383 (2011).
Jadeja, R. N. et al. Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging 10, 1306–1323 (2018).
Zhang, M. et al. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 10, 2464 (2020).
Chen, Y., Wang, J., Cai, J. & Sternberg, P. Altered mTOR signaling in senescent retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 51, 5314–5319 (2010).
Kozhevnikova, O. S., Telegina, D. V., Devyatkin, V. A. & Kolosova, N. G. Involvement of the autophagic pathway in the progression of AMD-like retinopathy in senescence-accelerated OXYS rats. Biogerontology 19, 223–235 (2018).
Golestaneh, N., Chu, Y., Xiao, Y. Y., Stoleru, G. L. & Theos, A. C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 8, e2537 (2017).
Ferrington, D. A., Sinha, D. & Kaarniranta, K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 51, 69–89 (2016).
Plotegher, N. & Duchen, M. R. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front. Cell Dev. Biol. 5, 110 (2017).
Konig, J. et al. Mitochondrial contribution to lipofuscin formation. Redox Biol. 11, 673–681 (2017).
Hohn, A., Jung, T., Grimm, S. & Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic. Biol. Med. 48, 1100–1108 (2010).
McLaughlin, T. et al. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 13, 16 (2018).
Sun, Q. et al. Inhibition of sumoylation alleviates oxidative stress-induced retinal pigment epithelial cell senescence and represses proinflammatory gene expression. Curr. Mol. Med. 18, 575–583 (2018).
Campello, L., Esteve-Rudd, J., Cuenca, N. & Martin-Nieto, J. The ubiquitin-proteasome system in retinal health and disease. Mol. Neurobiol. 47, 790–810 (2013).
Rohrer, B., Bandyopadhyay, M. & Beeson, C. Reduced metabolic capacity in aged primary retinal pigment epithelium (RPE) is correlated with increased susceptibility to oxidative stress. Adv. Exp. Med. Biol. 854, 793–798 (2016).
Minasyan, L., Sreekumar, P. G., Hinton, D. R. & Kannan, R. Protective mechanisms of the mitochondrial-derived peptide humanin in oxidative and endoplasmic reticulum stress in RPE cells. Oxid. Med. Cell Longev. 2017, 1675230 (2017).
Chae, S. Y., Park, S. Y. & Park, G. Lutein protects human retinal pigment epithelial cells from oxidative stressinduced cellular senescence. Mol. Med. Rep. 18, 5182–5190 (2018).
Alhasani, R. H. et al. Tauroursodeoxycholic acid protects retinal pigment epithelial cells from oxidative injury and endoplasmic reticulum stress in vitro. Biomedicines 8, 367 (2020).
Jun, B. et al. Elovanoids are novel cell-specific lipid mediators necessary for neuroprotective signaling for photoreceptor cell integrity. Sci. Rep. 7, 5279 (2017).
Kaarniranta, K., Pawlowska, E., Szczepanska, J., Jablkowska, A. & Blasiak, J. Role of mitochondrial DNA damage in ROS-mediated pathogenesis of age-related macular degeneration (AMD). Int. J. Mol. Sci. 20, 2374 (2019).
Ferrington, D. A. et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 145, 269–277 (2016).
Salero, E. et al. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell 10, 88–95 (2012).
Wang, J. & Puel, J. L. Presbycusis: an update on cochlear mechanisms and therapies. J. Clin. Med. 9, 218 (2020).
Miwa, T., Wei, F. Y. & Tomizawa, K. Cdk5 regulatory subunit-associated protein 1 knockout mice show hearing loss phenotypically similar to age-related hearing loss. Mol. Brain 14, 82 (2021).
Markaryan, A., Nelson, E. G. & Hinojosa, R. Major arc mitochondrial DNA deletions in cytochrome c oxidase-deficient human cochlear spiral ganglion cells. Acta Otolaryngol. 130, 780–787 (2010).
Menardo, J. et al. Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse Cochlea. Antioxid. Redox Signal 16, 263–274 (2012).
Tian, C. et al. Suppressed expression of LDHB promotes age-related hearing loss via aerobic glycolysis. Cell Death Dis. 11, 375 (2020).
Someya, S. et al. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl Acad. Sci. USA 106, 19432–19437 (2009).
Prasad, K. N. & Bondy, S. C. Increased oxidative stress, inflammation, and glutamate: potential preventive and therapeutic targets for hearing disorders. Mech. Ageing Dev. 185, 111191 (2020).
Kwon, T. J. et al. Methionine sulfoxide reductase A, B1 and B2 are likely to be involved in the protection against oxidative stress in the inner ear. Cells Tissues Organs 199, 294–300 (2014).
Keithley, E. M. et al. Cu/Zn superoxide dismutase and age-related hearing loss. Hear. Res. 209, 76–85 (2005).
Peixoto Pinheiro, B. et al. Age-related hearing loss pertaining to potassium ion channels in the cochlea and auditory pathway. Pflug. Arch. 473, 823–840 (2021).
Kim, Y. J. et al. BCL2 interacting protein 3-like/NIX-mediated mitophagy plays an important role in the process of age-related hearing loss. Neuroscience 455, 39–51 (2021).
Xiong, H. et al. Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis. Neurobiol. Aging 79, 30–42 (2019).
Kociszewska, D. & Vlajkovic, S. Age-related hearing loss: the link between inflammaging, immunosenescence, and gut dysbiosis. Int. J. Mol. Sci. 23, 7348 (2022).
Watson, N., Ding, B., Zhu, X. & Frisina, R. D. Chronic inflammation - inflammaging - in the ageing cochlea: A novel target for future presbycusis therapy. Ageing Res. Rev. 40, 142–148 (2017).
Verschuur, C., Agyemang-Prempeh, A. & Newman, T. A. Inflammation is associated with a worsening of presbycusis: evidence from the MRC national study of hearing. Int. J. Audio. 53, 469–475 (2014).
Lasisi, A. O. & Fehintola, F. A. Correlation between plasma levels of radical scavengers and hearing threshold among elderly subjects with age-related hearing loss. Acta Otolaryngol. 131, 1160–1164 (2011).
Nash, S. D. et al. Long-term assessment of systemic inflammation and the cumulative incidence of age-related hearing impairment in the epidemiology of hearing loss study. Gerontol. A Biol. Sci. Med. Sci. 69, 207–214 (2014).
Noble, K. V., Liu, T., Matthews, L. J., Schulte, B. A. & Lang, H. Age-related changes in immune cells of the human cochlea. Front. Neurol. 10, 895 (2019).
Su, Z. et al. Transcriptomic analysis highlights cochlear inflammation associated with age-related hearing loss in C57BL/6 mice using next generation sequencing. PeerJ 8, e9737 (2020).
Trifunovic, A. et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl Acad. Sci. USA 102, 17993–17998 (2005).
Valavanidis, A., Vlachogianni, T. & Fiotakis, C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C. Environ. Carcinog. Ecotoxicol. Rev. 27, 120–139 (2009).
Benkafadar, N. et al. ROS-induced activation of DNA damage responses drives senescence-like state in postmitotic cochlear cells: implication for hearing preservation. Mol. Neurobiol. 56, 5950–5969 (2019).
Falah, M. et al. The potential role for use of mitochondrial DNA copy number as predictive biomarker in presbycusis. Ther. Clin. Risk Manag. 12, 1573–1578 (2016).
Bai, U., Seidman, M. D., Hinojosa, R. & Quirk, W. S. Mitochondrial DNA deletions associated with aging and possibly presbycusis: a human archival temporal bone study. Am. J. Otol. 18, 449–453 (1997).
Markaryan, A., Nelson, E. G. & Hinojosa, R. Quantification of the mitochondrial DNA common deletion in presbycusis. Laryngoscope 119, 1184–1189 (2009).
Wu, X. et al. Reduced expression of Connexin26 and its DNA promoter hypermethylation in the inner ear of mimetic aging rats induced by d-galactose. Biochem. Biophys. Res. Commun. 452, 340–346 (2014).
Xu, J. et al. Elevated SLC26A4 gene promoter methylation is associated with the risk of presbycusis in men. Mol. Med. Rep. 16, 347–352 (2017).
Bouzid, A. et al. Down-expression of P2RX2, KCNQ5, ERBB3 and SOCS3 through DNA hypermethylation in elderly women with presbycusis. Biomarkers 23, 347–356 (2018).
Schmitt, C. A., Wang, B. & Demaria, M. Senescence and cancer - role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 19, 619–636 (2022).
Wang, L., Lankhorst, L. & Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 22, 340–355 (2022).
Tasdemir, N. et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Disco. 6, 612–629 (2016).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Ortiz-Montero, P., Londono-Vallejo, A. & Vernot, J. P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 15, 17 (2017).
Coppe, J. P., Kauser, K., Campisi, J. & Beausejour, C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574 (2006).
Chen, C. et al. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-kappaB pathway in colorectal cancer. Cell Death Dis. 10, 178 (2019).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Guccini, I. et al. Senescence reprogramming by TIMP1 deficiency promotes prostate cancer metastasis. Cancer Cell 39, 68–82.e69 (2021).
Ruhland, M. K. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7, 11762 (2016).
Di Mitri, D. et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 515, 134–137 (2014).
Eggert, T. et al. Distinct functions of senescence-associated immune responses in liver tumor Surveillance and tumor progression. Cancer Cell 30, 533–547 (2016).
Kivipelto, M. et al. The Finnish Geriatric Intervention Study to prevent cognitive impairment and disability (FINGER): study design and progress. Alzheimers Dement. 9, 657–665 (2013).
Lehtisalo, J. et al. Dietary changes and cognition over 2 years within a multidomain intervention trial-The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). Alzheimers Dement. 15, 410–417 (2019).
Kivipelto, M., Mangialasche, F. & Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 14, 653–666 (2018).
Vinciguerra, F. & Graziano, M. Influence of the Mediterranean and Ketogenic diets on cognitive status and decline: a narrative review. Nutrients 12, 1019 (2020).
De la Rosa, A. et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J. Sport Health Sci. 9, 394–404 (2020).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016).
Miziak, B., Błaszczyk, B. & Czuczwar, S. J. Some candidate drugs for pharmacotherapy of Alzheimer’s disease. Pharmaceuticals 14, 458 (2021).
Luchsinger, J. A. et al. Metformin, lifestyle intervention, and cognition in the diabetes prevention program outcomes study. Diabetes Care 40, 958–965 (2017).
Koenig, A. M. et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 31, 107–113 (2017).
Mitchell, E. S., Conus, N. & Kaput, J. B vitamin polymorphisms and behavior: evidence of associations with neurodevelopment, depression, schizophrenia, bipolar disorder and cognitive decline. Neurosci. Biobehav. Rev. 47, 307–320 (2014).
Román, G. C. & Mancera-Páez, O. Epigenetic factors in late-onset Alzheimer’s disease: MTHFR and CTH gene polymorphisms, metabolic transsulfuration and methylation pathways, and B vitamins. Int. J. Mol. Sci. 20, 319 (2019).
Yuksel, J. M., Noviasky, J. & Britton, S. Aducanumab for Alzheimer’s disease: summarized data from EMERGE, ENGAGE, and PRIME studies. Sr. Care Pharm. 37, 329–334 (2022).
Knopman, D. S., Jones, D. T. & Greicius, M. D. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 17, 696–701 (2021).
Kuller, L. H. & Lopez, O. L. ENGAGE and EMERGE: truth and consequences? Alzheimers Dement. 17, 692–695 (2021).
Remya, C. et al. Neuroprotective derivatives of tacrine that target NMDA receptor and acetyl cholinesterase - Design, synthesis and biological evaluation. Comput. Struct. Biotechnol. J. 19, 4517–4537 (2021).
Korabecny, J. et al. A systematic review on donepezil-based derivatives as potential cholinesterase inhibitors for Alzheimer’s disease. Curr. Med. Chem. 26, 5625–5648 (2019).
Uddin, M. S. et al. Multi-target drug candidates for multifactorial Alzheimer’s disease: AChE and NMDAR as molecular targets. Mol. Neurobiol. 58, 281–303 (2021).
Davies, P. & Maloney, A. J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2, 1403 (1976).
Bortolami, M. et al. Acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease - a patent review (2016-present). Expert Opin. Ther. Pat. 31, 399–420 (2021).
Saeedi, M. & Mehranfar, F. Challenges and approaches of drugs such as memantine, donepezil, rivastigmine, and aducanumab in the treatment, control and management of Alzheimer’s disease. Recent Pat. Biotechnol. 16, 102–121 (2022).
Román, G. C., Jackson, R. E., Gadhia, R., Román, A. N. & Reis, J. Mediterranean diet: the role of long-chain ω-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev. Neurol. 175, 724–741 (2019).
Howes, M. R., Fang, R. & Houghton, P. J. Effect of Chinese herbal medicine on Alzheimer’s disease. Int. Rev. Neurobiol. 135, 29–56 (2017).
Li, Q. et al. Current progress on neuroprotection induced by Artemisia, Ginseng, Astragalus, and Ginkgo traditional Chinese medicines for the therapy of Alzheimer’s disease. Oxid. Med. Cell Longev. 2022, 1–21 (2022).
Kerr, J. S. et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 40, 151–166 (2017).
Sun, N., Youle, R. J. & Finkel, T. The mitochondrial basis of aging. Mol. Cell 61, 654–666 (2016).
Xie, C. et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 6, 76–93 (2022).
de Carvalho, T. S. Calorie restriction or dietary restriction: how far they can protect the brain against neurodegenerative diseases? Neural Regen. Res. 17, 1640–1644 (2022).
Ciulla, M., Marinelli, L. & Cacciatore, I. Role of dietary supplements in the management of Parkinson’s disease. Biomolecules 9, 271 (2019).
Abdul-Latif, R., Stupans, I., Allahham, A., Adhikari, B. & Thrimawithana, T. Natural antioxidants in the management of Parkinson’s disease: review of evidence from cell line and animal models. J. Integr. Med. 19, 300–310 (2021).
Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
Zeng, B. Y. Effect and mechanism of Chinese herbal medicine on Parkinson’s disease. Int. Rev. Neurobiol. 135, 57–76 (2017).
Baker, D. J. & Petersen, R. C. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest. 128, 1208–1216 (2018).
Hou, Y., Dan, X., Babbar, M., Wei, Y. & Hasselbalch, S. G. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Wamelen, D. J. V., Rukavina, K., Podlewska, A. M. & Chaudhuri, K. R. Advances in the pharmacological and non-pharmacological management of non-motor symptoms in Parkinson’s disease: an update since 2017. Curr. Neuropharmacol. https://doi.org/10.2174/1570159X20666220315163856 (2022).
Hvingelby, V. S. et al. Interventions to improve gait in Parkinson’s disease: a systematic review of randomized controlled trials and network meta-analysis. J. Neurol. 269, 4068–4079 (2022).
Paz, R. M. & Murer, M. G. Mechanisms of antiparkinsonian anticholinergic therapy revisited. Neuroscience 467, 201–217 (2021).
Vaikath, N. N. et al. Antibodies against alpha-synuclein: tools and therapies. J. Neurochem. 150, 612–625 (2019).
Vermersch, P. et al. Efficacy and safety of masitinib in progressive forms of multiple sclerosis: a randomized, phase 3, clinical trial. Neurol. Neuroimmunol. Neuroinflamm. 9, e1148 (2022).
Vermersch, P., Brieva-Ruiz, L. & Fox, R. J. Efficacy and safety of masitinib in progressive forms of multiple sclerosis: a randomized, phase 3, clinical trial. Neurol. Neuroimmunol. Neuroinflamm. 9, e114 (2022).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e1412 (2016).
Palmer, K., Bowles, K. A., Paton, M., Jepson, M. & Lane, R. Chronic heart failure and exercise rehabilitation: a systematic review and meta-analysis. Arch. Phys. Med. Rehabil. 99, 2570–2582 (2018).
Tsao, C. W. et al. Heart disease and stroke statistics-2022 update: a report from the American Heart Association. Circulation 145, e153–e639 (2022).
Casaletto, K. B. et al. Microglial correlates of late life physical activity: relationship with synaptic and cognitive aging in older adults. J. Neurosci. 42, 288–298 (2022).
Nassif, M. E. et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat. Med. 27, 1954–1960 (2021).
Jia, K. et al. Senolytic agent navitoclax inhibits angiotensin II-induced heart failure in mice. J. Cardiovasc. Pharm. 76, 452–460 (2020).
Bernardes de Jesus, B. et al. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 10, 604–621 (2011).
Wang, J. et al. Cycloastragenol ameliorates experimental heart damage in rats by promoting myocardial autophagy via inhibition of AKT1-RPS6KB1 signaling. Biomed. Pharmacother. 107, 1074–1081 (2018).
Szabo, N. J. Dietary safety of cycloastragenol from Astragalus spp.: subchronic toxicity and genotoxicity studies. Food Chem. Toxicol. 64, 322–334 (2014).
Chiang, J. T. et al. Anti-apoptosis and anti-fibrosis effects of Eriobotrya japonica in spontaneously hypertensive rat hearts. Int. J. Mol. Sci. 19, 1638 (2018).
Chang, Y. M. et al. Anti-apoptotic and pro-survival effect of alpinate oxyphyllae fructus (AOF) in a d-galactose-induced aging heart. Int. J. Mol. Sci. 17, 466 (2016).
Zhou, G. et al. A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii Debeaux. J. Ethnopharmacol. 160, 173–193 (2015).
Li, J. et al. Long-term repopulation of aged bone marrow stem cells using young Sca-1 cells promotes aged heart rejuvenation. Aging Cell 18, e13026 (2019).
Lv, S., Wang, Y., Zhang, W. & Shang, H. Trimethylamine oxide: a potential target for heart failure therapy. Heart 108, 917–922 (2022).
Lin, H. et al. Yellow wine polyphenolic compound protects against doxorubicin-induced cardiotoxicity by modulating the composition and metabolic function of the gut microbiota. Circ. Heart Fail. 14, e008220 (2021).
Han, Z. L. et al. Potassium alginate oligosaccharides alter gut microbiota, and have potential to prevent the development of hypertension and heart failure in spontaneously hypertensive rats. Int. J. Mol.Sci. 22, 9823 (2021).
Visseren, F. L. J. et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 29, 5–115 (2022).
Torres, N., Guevara-Cruz, M., Velazquez-Villegas, L. A. & Tovar, A. R. Nutrition and atherosclerosis. Arch. Med. Res. 46, 408–426 (2015).
Ros, E. et al. Mediterranean diet and cardiovascular health: teachings of the PREDIMED study. Adv. Nutr. 5, 330S–336S (2014).
Perez-Lopez, F. R., Chedraui, P., Haya, J. & Cuadros, J. L. Effects of the Mediterranean diet on longevity and age-related morbid conditions. Maturitas 64, 67–79 (2009).
Marin, C., Yubero-Serrano, E. M., Lopez-Miranda, J. & Perez-Jimenez, F. Endothelial aging associated with oxidative stress can be modulated by a healthy mediterranean diet. Int. J. Mol. Sci. 14, 8869–8889 (2013).
Jiang, Y. H., Jiang, L. Y., Wang, Y. C., Ma, D. F. & Li, X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front. Pharm. 11, 512 (2020).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Karnewar, S. et al. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1115–1128 (2018).
Martin-Montalvo, A. et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192 (2013).
Huang, D. et al. Bazi Bushen capsule alleviates post-menopausal atherosclerosis via GPER1-dependent anti-inflammatory and anti-apoptotic effects. Front. Pharm. 12, 658998 (2021).
Zimetti, F. et al. The natural compound berberine positively affects macrophage functions involved in atherogenesis. Nutr. Metab. Cardiovasc Dis. 25, 195–201 (2015).
Zheng, Y. et al. Berberine-induced TFEB deacetylation by SIRT1 promotes autophagy in peritoneal macrophages. Aging 13, 7096–7119 (2021).
Ma, S. R. et al. Berberine treats atherosclerosis via a vitamine-like effect down-regulating Choline-TMA-TMAO production pathway in gut microbiota. Signal Transduct. Target Ther. 7, 207 (2022).
Weber, M. B., Gujral, U. P., Jagannathan, R. & Shah, M. Lifestyle interventions for diabetes prevention in South Asians: current evidence and opportunities. Curr. Diab. Rep. 21, 23 (2021).
Ho, M. et al. Effects of dietary and physical activity interventions on generic and cancer-specific health-related quality of life, anxiety, and depression in colorectal cancer survivors: a randomized controlled trial. J. Cancer Surviv. 14, 424–433 (2020).
Murakami, T., Inagaki, N. & Kondoh, H. Cellular senescence in diabetes mellitus: distinct senotherapeutic strategies for adipose tissue and pancreatic beta cells. Front. Endocrinol. 13, 869414 (2022).
Schoenwaelder, S. M. et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 118, 1663–1674 (2011).
Thompson, P. J. et al. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. 29, 1045–1060.e1010 (2019).
Novais, E. J. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213 (2021).
Zoico, E. et al. Senolytic effects of quercetin in an in vitro model of pre-adipocytes and adipocytes induced senescence. Sci. Rep. 11, 23237 (2021).
Raffaele, M. et al. Mild exacerbation of obesity- and age-dependent liver disease progression by senolytic cocktail dasatinib + quercetin. Cell Commun. Signal 19, 44 (2021).
Luo, G. et al. Resveratrol attenuates excessive ethanol exposure-induced beta-cell senescence in rats: a critical role for the NAD(+)/SIRT1-p38MAPK/p16 pathway. J. Nutr. Biochem. 89, 108568 (2021).
Kulkarni, A. S., Gubbi, S. & Barzilai, N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30 (2020).
Liu, J. et al. Association between antidiabetic agents use and leukocyte telomere shortening rates in patients with type 2 diabetes. Aging 11, 741–755 (2019).
Saisho, Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets 15, 196–205 (2015).
Dogan Turacli, I. et al. Potential effects of metformin in DNA BER system based on oxidative status in type 2 diabetes. Biochimie 154, 62–68 (2018).
de la Cuesta-Zuluaga, J. et al. Metformin is associated with higher relative abundance of mucin-degrading Akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care 40, 54–62 (2017).
Smith, A. D. et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS ONE 5, e12244 (2010).
Tang, C. et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia 55, 1366–1379 (2012).
Kim, M. J. et al. Specific PERK inhibitors enhanced glucose-stimulated insulin secretion in a mouse model of type 2 diabetes. Metabolism 97, 87–91 (2019).
Zhang, Y. et al. Gut microbiome-related effects of berberine and probiotics on type 2 diabetes (the PREMOTE study). Nat. Commun. 11, 5015 (2020).
Gepner, Y. et al. Effect of distinct lifestyle interventions on mobilization of fat storage pools: CENTRAL magnetic resonance imaging randomized controlled trial. Circulation 137, 1143–1157 (2018).
Abenavoli, L. et al. Dietary polyphenols and non-alcoholic fatty liver disease. Nutrients 13, 494 (2021).
An, H. S. et al. Caloric restriction reverses left ventricular hypertrophy through the regulation of cardiac iron homeostasis in impaired leptin signaling mice. Sci. Rep. 10, 7176 (2020).
Branković, M. et al. Lipotoxicity as the leading cause of non-alcoholic steatohepatitis. Int. J. Mol. Sci. 23, 5146 (2022).
Craven, L. et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am. J. Gastroenterol. 115, 1055–1065 (2020).
Armstrong, M. J. et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690 (2016).
Gawrieh, S. et al. Saroglitazar, a PPAR-alpha/gamma agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology 74, 1809–1824 (2021).
Ratziu, V. et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e1145 (2016).
Lan, T. et al. Cordycepin ameliorates nonalcoholic steatohepatitis by activation of the AMP-activated protein kinase signaling pathway. Hepatology 74, 686–703 (2021).
Lan, T. et al. Breviscapine alleviates NASH by inhibiting TGF-β-activated kinase 1-dependent signaling. Hepatology 76, 155–171 (2022).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Nogueira-Recalde, U. et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605 (2019).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Zheng, W. et al. Fisetin inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int. Immunopharmacol. 45, 135–147 (2017).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Fleischmann, R. M. et al. A phase II trial of lutikizumab, an anti-interleukin-1alpha/beta dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol. 71, 1056–1069 (2019).
Kloppenburg, M. et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 77, 1757–1764 (2018).
Schieker, M. et al. Effects of interleukin-1beta inhibition on incident hip and knee replacement: exploratory analyses from a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 173, 509–515 (2020).
Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 (2013).
Feng, K., Chen, Z., Pengcheng, L., Zhang, S. & Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell Physiol. 234, 18192–18205 (2019).
Piao, S. et al. Protectin DX attenuates IL-1beta-induced inflammation via the AMPK/NF-kappaB pathway in chondrocytes and ameliorates osteoarthritis progression in a rat model. Int. Immunopharmacol. 78, 106043 (2020).
Ye, J. et al. Interleukin-12p35 deficiency enhances mitochondrial dysfunction and aggravates cardiac remodeling in aging mice. Aging 12, 193–203 (2020).
Arriola Apelo, S. I. & Lamming, D. W. Rapamycin: an InhibiTOR of Aging Emerges From the Soil of Easter Island. J. Gerontol. A Biol. Sci. Med. Sci. 71, 841–849 (2016).
Wang, B., Sun, W., Bi, K., Li, Y. & Li, F. Apremilast prevents IL17induced cellular senescence in ATDC5 chondrocytes mediated by SIRT1. Int. J. Mol. Med. 47, 12 (2021).
Khan, N. M. et al. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic. Biol. Med. 106, 288–301 (2017).
Liu, F. C. et al. Chondroprotective effects of genistein against osteoarthritis induced joint inflammation. Nutrients 11, 1180 (2019).
Park, C. et al. Sargassum serratifolium extract attenuates interleukin-1beta-induced oxidative stress and inflammatory response in chondrocytes by suppressing the activation of NF-kappaB, p38 MAPK, and PI3K/Akt. Int. J. Mol. Sci. 19, 2308 (2018).
Wu, Y. et al. Sinomenine contributes to the inhibition of the inflammatory response and the improvement of osteoarthritis in mouse-cartilage cells by acting on the Nrf2/HO-1 and NF-kappaB signaling pathways. Int. Immunopharmacol. 75, 105715 (2019).
Xu, M. et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4, e12997 (2015).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Wong, S. K., Chin, K. Y. & Ima-Nirwana, S. Quercetin as an agent for protecting the bone: a review of the current evidence. Int. J. Mol. Sci. 21, 6448 (2020).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Lv, Y. J. et al. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics 8, 2387–2406 (2018).
Chen, X. et al. Nrf2 epigenetic derepression induced by running exercise protects against osteoporosis. Bone Res. 9, 15 (2021).
Jiang, Y., Zhang, P., Zhang, X., Lv, L. & Zhou, Y. Advances in mesenchymal stem cell transplantation for the treatment of osteoporosis. Cell Prolif. 54, e12956 (2021).
Qi, X. et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells repair critical-sized bone defects through enhanced angiogenesis and osteogenesis in osteoporotic rats. Int. J. Biol. Sci. 12, 836–849 (2016).
Zuo, R. et al. BM-MSC-derived exosomes alleviate radiation-induced bone loss by restoring the function of recipient BM-MSCs and activating Wnt/beta-catenin signaling. Stem Cell Res. Ther. 10, 30 (2019).
Li, L. et al. Exosomal miR-186 derived from BMSCs promote osteogenesis through hippo signaling pathway in postmenopausal osteoporosis. J. Orthop. Surg. Res. 16, 23 (2021).
Zhang, Y. et al. microRNA-935-modified bone marrow mesenchymal stem cells-derived exosomes enhance osteoblast proliferation and differentiation in osteoporotic rats. Life Sci. 272, 119204 (2021).
Xun, J. et al. Serum exosomes from young rats improve the reduced osteogenic differentiation of BMSCs in aged rats with osteoporosis after fatigue loading in vivo. Stem Cell Res. Ther. 12, 424 (2021).
Wang, Y. et al. Bone-targeted extracellular vesicles from mesenchymal stem cells for osteoporosis therapy. Int. J. Nanomed. 15, 7967–7977 (2020).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Baker, J. R., Donnelly, L. E. & Barnes, P. J. Senotherapy: a new horizon for COPD therapy. Chest 158, 562–570 (2020).
Zhang, W. G. et al. Regulation of transplanted mesenchymal stem cells by the lung progenitor niche in rats with chronic obstructive pulmonary disease. Respir. Res. 15, 33 (2014).
Liu, X., Fang, Q. & Kim, H. Preclinical studies of mesenchymal stem cell (MSC) administration in chronic obstructive pulmonary disease (COPD): a systematic review and meta-analysis. PLoS ONE 11, e0157099 (2016).
Horvath, L. et al. Engineering an in vitro air-blood barrier by 3D bioprinting. Sci. Rep. 5, 7974 (2015).
Dal Negro, R. W. et al. Effect of erdosteine on the rate and duration of COPD exacerbations: the RESTORE study. Eur. Respir. J. 50, PA675 (2017).
Moretti, M. et al. The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE Study. Drugs Exp. Clin. Res. 30, 143–152 (2004).
Culpitt, S. V. et al. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax 58, 942–946 (2003).
Beijers, R., Gosker, H. R. & Schols, A. Resveratrol for patients with chronic obstructive pulmonary disease: hype or hope? Curr. Opin. Clin. Nutr. Metab. Care 21, 138–144 (2018).
Bishwakarma, R. et al. Metformin use and health care utilization in patients with coexisting chronic obstructive pulmonary disease and diabetes mellitus. Int J. Chron. Obstruct. Pulmon. Dis. 13, 793–800 (2018).
Soukas, A. A., Hao, H. & Wu, L. Metformin as anti-aging therapy: is it for everyone? Trends Endocrinol. Metab. 30, 745–755 (2019).
de Matos Cavalcante, A. G. et al. Melatonin reduces lung oxidative stress in patients with chronic obstructive pulmonary disease: a randomized, double-blind, placebo-controlled study. J. Pineal Res. 53, 238–244 (2012).
Hee Jo, E. et al. Sensitization of GSH synthesis by curcumin curtails acrolein-induced alveolar epithelial apoptosis via Keap1 cysteine conjugation: a randomized controlled trial and experimental animal model of pneumonitis. J. Adv. Res. S2090-1232(22)00152-7 (2022).
Eleazu, C., Eleazu, K. & Kalu, W. Management of benign prostatic hyperplasia: could dietary polyphenols be an alternative to existing therapies? Front. Pharm. 8, 234 (2017).
Nørgaard, M., Darvalics, B. & Thomsen, R. W. Metformin use and long-term risk of benign prostatic hyperplasia: a population-based cohort study. BMJ Open 10, e041875 (2020).
Jung, Y. et al. Vanillic acid attenuates testosterone-induced benign prostatic hyperplasia in rats and inhibits proliferation of prostatic epithelial cells. Oncotarget 8, 87194–87208 (2017).
Enikeev, D. et al. EAU, AUA and NICE guidelines on surgical and minimally invasive treatment of benign prostate hyperplasia: a critical appraisal of the guidelines using the AGREE-II tool. Urol. Int. 106, 1–10 (2022).
Wakita, M. et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 11, 1935 (2020).
Li, J., Zhao, L., Urabe, G., Fu, Y. & Guo, L. W. Epigenetic intervention with a BET inhibitor ameliorates acute retinal ganglion cell death in mice. Mol. Vis. 23, 149–159 (2017).
Schwartz, S. D. et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379, 713–720 (2012).
da Cruz, L. et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 36, 328–337 (2018).
Mandai, M. et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 376, 1038–1046 (2017).
Carneiro, A. & Andrade, J. P. Nutritional and lifestyle interventions for age-related macular degeneration: a review. Oxid. Med. Cell Longev. 2017, 6469138 (2017).
Merle, B. M. J. et al. Mediterranean diet and incidence of advanced age-related macular degeneration: the EYE-RISK Consortium. Ophthalmology 126, 381–390 (2019).
Richer, S. et al. Resveratrol based oral nutritional supplement produces long-term beneficial effects on structure and visual function in human patients. Nutrients 6, 4404–4420 (2014).
Yi, C., Pan, X., Yan, H., Guo, M. & Pierpaoli, W. Effects of melatonin in age-related macular degeneration. Ann. N. Y Acad. Sci. 1057, 384–392 (2005).
Cao, Y. et al. Bioinformatical and biochemical analyses on the protective role of traditional Chinese medicine against age-related macular degeneration. Curr. Eye Res. 47, 1450–1463 (2022).
Lowthian, J. A. et al. Slowing the progression of age-related hearing loss: rationale and study design of the ASPIRIN in HEARING, retinal vessels imaging and neurocognition in older generations (ASPREE-HEARING) trial. Contemp. Clin. Trials 46, 60–66 (2016).
Robman, L. D. et al. Baseline characteristics and age-related macular degeneration in participants of the “ASPirin in Reducing Events in the Elderly” (ASPREE)-AMD trial. Contemp. Clin. Trials Commun. 20, 100667 (2020).
McNeil, J. J. et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N. Engl. J. Med. 379, 1509–1518 (2018).
Ren, H., Chen, J., Wang, Y., Zhang, S. & Zhang, B. Intracerebral neural stem cell transplantation improved the auditory of mice with presbycusis. Int. J. Clin. Exp. Pathol. 6, 230–241 (2013).
Corrales, C. E. et al. Engraftment and differentiation of embryonic stem cell-derived neural progenitor cells in the cochlear nerve trunk: growth of processes into the organ of Corti. J. Neurobiol. 66, 1489–1500 (2006).
Chen, W. et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 490, 278–282 (2012).
Wang, J. & Puel, J. L. Toward cochlear therapies. Physiol. Rev. 98, 2477–2522 (2018).
Seidman, M. D., Khan, M. J., Bai, U., Shirwany, N. & Quirk, W. S. Biologic activity of mitochondrial metabolites on aging and age-related hearing loss. Am. J. Otol. 21, 161–167 (2000).
Serra, L. S. M. et al. Role of melatonin in prevention of age-related hearing loss. PLoS ONE 15, e0228943 (2020).
Nevado, J. et al. Ginkgo biloba extract (EGb761) protects against aging-related caspase-mediated apoptosis in rat cochlea. Acta Otolaryngol. 130, 1101–1112 (2010).
Kang, J. W., Choi, H. S., Kim, K. & Choi, J. Y. Dietary vitamin intake correlates with hearing thresholds in the older population: the Korean National Health and Nutrition Examination Survey. Am. J. Clin. Nutr. 99, 1407–1413 (2014).
Polanski, J. F. & Cruz, O. L. Evaluation of antioxidant treatment in presbyacusis: prospective, placebo-controlled, double-blind, randomised trial. J. Laryngol. Otol. 127, 134–141 (2013).
Wyld, L. et al. Senescence and cancer: a review of clinical implications of senescence and senotherapies. Cancers 12, 2134 (2020).
Wang, B., Kohli, J. & Demaria, M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857 (2020).
Prasanna, P. G. et al. Therapy-induced senescence: opportunities to improve anticancer therapy. J. Natl Cancer Inst. 113, 1285–1298 (2021).
Bury, M., Le Calve, B., Ferbeyre, G., Blank, V. & Lessard, F. New insights into CDK regulators: novel opportunities for cancer therapy. Trends Cell Biol. 31, 331–344 (2021).
Gomez, D. L., Armando, R. G., Cerrudo, C. S., Ghiringhelli, P. D. & Gomez, D. E. Telomerase as a cancer target. Development of new molecules. Curr. Top. Med. Chem. 16, 2432–2440 (2016).
Ramaiah, M. J., Tangutur, A. D. & Manyam, R. R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 277, 119504 (2021).
Zhu, H. et al. Oncogene-induced senescence: from biology to therapy. Mech. Ageing Dev. 187, 111229 (2020).
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 (2009).
Cattrini, C., Capaia, M., Boccardo, F. & Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: data from a translational study. Cancer Treat. Res. Commun. 25, 100221 (2020).
Pommier, Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 (2006).
Skok, Z., Zidar, N., Kikelj, D. & Ilas, J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 63, 884–904 (2020).
Rottenberg, S., Disler, C. & Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 21, 37–50 (2021).
Dasari, S. & Tchounwou, P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharm. 740, 364–378 (2014).
de Vries, G., Rosas-Plaza, X., van Vugt, M., Gietema, J. A. & de Jong, S. Testicular cancer: determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 88, 102054 (2020).
Xiang, M., Colevas, A. D., Holsinger, F. C., Le, Q. X. & Beadle, B. M. Survival after definitive chemoradiotherapy with concurrent cisplatin or carboplatin for head and neck cancer. J. Natl Compr. Canc. Netw. 17, 1065–1073 (2019).
Rogers, B. B. et al. Oxaliplatin: detection and management of hypersensitivity reactions. Clin. J. Oncol. Nurs. 23, 68–75 (2019).
Hall, A. G. & Tilby, M. J. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 6, 163–173 (1992).
Perez, E. A. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 8, 2086–2095 (2009).
Klein, L. E., Freeze, B. S., Smith, A. B. 3rd & Horwitz, S. B. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle 4, 501–507 (2005).
O’Leary, B., Finn, R. S. & Turner, N. C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 13, 417–430 (2016).
Kumarasamy, V., Vail, P., Nambiar, R., Witkiewicz, A. K. & Knudsen, E. S. Functional determinants of cell cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res. 81, 1347–1360 (2021).
Sava, G. P., Fan, H., Coombes, R. C., Buluwela, L. & Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 39, 805–823 (2020).
Liu, H., Liu, K. & Dong, Z. Targeting CDK12 for cancer therapy: function, mechanism, and drug discovery. Cancer Res. 81, 18–26 (2021).
Phatak, P. & Burger, A. M. Telomerase and its potential for therapeutic intervention. Br. J. Pharm. 152, 1003–1011 (2007).
Pascolo, E. et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 277, 15566–15572 (2002).
Burchett, K. M., Yan, Y. & Ouellette, M. M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE 9, e85155 (2014).
Dhillon, S. Decitabine/cedazuridine: first approval. Drugs 80, 1373–1378 (2020).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016).
Dong, D. et al. Bcl2 inhibitor ABT737 reverses the Warburg effect via the Sirt3-HIF1alpha axis to promote oxidative stress-induced apoptosis in ovarian cancer cells. Life Sci. 255, 117846 (2020).
Coleman, R. L. et al. Dasatinib, paclitaxel, and carboplatin in women with advanced-stage or recurrent endometrial cancer: a pilot clinical and translational study. Gynecol. Oncol. 161, 104–112 (2021).
Tang, S. M. et al. Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed. Pharmacother. 121, 109604 (2020).
Jia, S. et al. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 10, 142 (2019).
Acknowledgements
This work was supported by funds from the National Key R&D Program of China (2018YFC2000100 and 2021YFE0114200), the National Natural Science Foundation of China (81770228, 81770858, 81600618, and 82271597), the Beijing Natural Science Foundation (7212086), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-1-050). We apologize to the authors of articles that could not be cited due to space constraints. We are very grateful to Professor Jin Liu of Beijing Normal University and to Kun Xu, Jiameng Yin, and Mingjun Wang from Beijing Hospital and Capital Medical University for their assistance with illustration. Figures 4–6 includes modified templates from Servier Medical Art (http://www.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the article. J.L. was responsible for the overall conceptualization, supervision, and writing (reviewing and editing) of the review. J.G. was responsible for conceptualization, writing of the original drafts of the “Theories and mechanisms of aging”, the “Conclusion and future perspectives” section, and preparation of Figs. 2, 3; X.H. was responsible for conceptualization, writing of the original draft for the sections on respiratory diseases, diseases of the visual system and hearing disease, and preparation of Fig. 5; L.D. was responsible for conceptualization, writing of the original draft for the sections on diseases of the locomotor system and endocrine system, and preparation of Fig. 6; M.Y. was responsible for conceptualization, writing of the original draft for the introduction and cardiovascular disease sections, and the summary in the “Interventions and treatments for aging-related diseases” section, and preparation of Figs. 1, 8; T.S. was responsible for conceptualization, writing of the original draft for the sections on nervous system and genitourinary system diseases, and the “Conclusion and future perspectives” section and preparation of Fig. 7; and W.T. was responsible for conceptualization, writing of the original draft for the digestive system disease sections and the summary in the “Pathogenic and regulatory mechanisms of aging-related diseases” section and preparation of Fig. 4. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, J., Huang, X., Dou, L. et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Sig Transduct Target Ther 7, 391 (2022). https://doi.org/10.1038/s41392-022-01251-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01251-0
This article is cited by
-
Bazi Bushen ameliorates age-related energy metabolism dysregulation by targeting the IL-17/TNF inflammatory pathway associated with SASP
Chinese Medicine (2024)
-
Exosomes in skin photoaging: biological functions and therapeutic opportunity
Cell Communication and Signaling (2024)
-
Nonlinear DNA methylation trajectories in aging male mice
Nature Communications (2024)
-
Cellular reprogramming as a tool to model human aging in a dish
Nature Communications (2024)
-
Insights into vaccines for elderly individuals: from the impacts of immunosenescence to delivery strategies
npj Vaccines (2024)
