
Abstract
The outbreak of COVID-19 has become a global crisis, and brought severe disruptions to societies and economies. Until now, effective therapeutics against COVID-19 are in high demand. Along with our improved understanding of the structure, function, and pathogenic process of SARS-CoV-2, many small molecules with potential anti-COVID-19 effects have been developed. So far, several antiviral strategies were explored. Besides directly inhibition of viral proteins such as RdRp and Mpro, interference of host enzymes including ACE2 and proteases, and blocking relevant immunoregulatory pathways represented by JAK/STAT, BTK, NF-κB, and NLRP3 pathways, are regarded feasible in drug development. The development of small molecules to treat COVID-19 has been achieved by several strategies, including computer-aided lead compound design and screening, natural product discovery, drug repurposing, and combination therapy. Several small molecules representative by remdesivir and paxlovid have been proved or authorized emergency use in many countries. And many candidates have entered clinical-trial stage. Nevertheless, due to the epidemiological features and variability issues of SARS-CoV-2, it is necessary to continue exploring novel strategies against COVID-19. This review discusses the current findings in the development of small molecules for COVID-19 treatment. Moreover, their detailed mechanism of action, chemical structures, and preclinical and clinical efficacies are discussed.
Similar content being viewed by others

Introduction
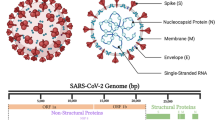
COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to more than 6 million deaths worldwide.1 SARS-CoV-2 is a betacoronavirus and possesses a positive-sense single-stranded RNA genome that contains 14 open reading frames (ORFs) (Fig. 1). Two ORFs encode polyproteins PP1a and PP1b.2 Four ORFs encode a series of structural proteins, including the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins. In the SARS-CoV-2 lifecycle, S protein, which recognizes the human ACE2 receptor and is cleaved by host proteases, is responsible for virus binding and entry into host cells.3,4 Subsequently, Mpro and PLpro are necessary for the production and function of non-structural proteins (NSPs). The key NSP RNA-dependent RNA polymerase (RdRp, also known as NSP12) catalyzes the synthesis of viral RNA and plays a central role in the lifecycle of SARS-CoV-2.5,6,7 Therefore, targeting these functional proteins is a rational strategy to inhibit infection and the replication of SARS-CoV-2. Infection with SARS-CoV-2 activates the host immune system, which may elicit a dysfunctional inflammatory response and cause organ damage.8,9,10 Therefore, therapeutic interventions targeting the immune system are also potential approaches for COVID-19 therapy.

Schematic illustration of the genome of SARS-CoV-2 and its structure. The size of SARS-CoV-2 genome is close to 30 kb; it contains 14 open reading frames (ORFs) and encodes 29 proteins. Two ORFs, comprising approximately two-thirds of the genome, encode two polyproteins, which are digested by M protease (Mpro) and Papain-like protease (PLpro) into 16 nonstructural proteins (nsps). Four ORFs encode a series of structural proteins, including the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins
Small molecules targeting specific signals and functions are widely applied in the treatment of diseases. Compared with biologics such as monoclonal antibodies and plasma products, small molecules are more flexible in binding with target molecules when acting as antagonist or agonist.11,12 Their lower production cost and higher stability also make them ideal therapeutic agents for both clinical and research applications. In parallel with the growing understanding of the pathogenic mechanisms of SARS-CoV-2 infection, small molecules from natural sources or those produced via chemical synthesis have demonstrated their immense therapeutic potential by intervening with various processes.13,14,15 The development of small molecules to treat COVID-19 has been achieved by several strategies, including computer-aided lead compound design and screening, natural product discovery, drug repurposing, and combination therapy. In this review, we present a comprehensive overview of the latest progress in the development of small molecule therapeutics for COVID-19 treatment. These therapeutic compounds are classified according to their chemical structures. The anti-COVID-19 molecular mechanisms are also discussed.
COVID-19 therapeutic targets for small molecules
RNA-dependent RNA polymerase (RdRp)
RdRp of SARS-CoV-2 is composed of NSP12 as the catalytic subunit and the NSP7–NSP8 complex as accessory subunits.16,17,18 RdRp is central to RNA transcription and viral replication, and may thus be an ideal target for anti-SARS-CoV-2 drugs (Fig. 2). The structural conformation of the SARS-CoV-2 RdRp complex is highly similar to that of SARS-CoV RdRp.17,19 NSP12 is classified into three domains: an N-terminal nidovirus RdRp-associated nucleotidyltransferase domain (residues 1–250), an interface region (residues 251–398), and the core RdRp domain (residues 399–932). NSP12 is formed by polymerase motifs A to G. These motifs are conserved in most RNA viruses.17 Studies of this RdRp domain have provided information on the role of these conserved motifs during RNA synthesis. Briefly, initial nucleotide recognition is mediated by positively charged Lys and Arg residues, which are located in motifs D and F of NSP12. The nucleotide flips into the active site through interaction with motifs A, B, and F to form a base pair with the template nucleotide, close to the active site. The incoming NTP forms a phosphodiester bond with the product RNA and after catalysis releases pyrophosphate. Then, the conformation of the active site immediately changes to an open state through a subtle rotation of motif A for the next nucleotide addition cycle.20,21,22 RdRp is the primary target of many existing antiviral nucleotide drugs. Based on its high conservation in diverse RNA viruses, repurposing of existing nucleotide drugs is an effective strategy that could shorten drug development time.18,19

Lifecycle of SARS-CoV-2. The SARS-CoV-2 S protein recognizes the ACE2 receptor while being cleaved by the host proteases and entering into the target cells. Then, the gRNA is released and translated into pp1a and pplb, thereby being digested into the NSPs necessary for viral replication. Under the catalyzation of RdRp, new gRNAs are produced and encode the structural proteins to assemble the progeny virus
The possible antiviral mechanism of nucleotide drugs is threefold; they can act as mutagens, as obligate chain terminators, and as non-obligate chain terminators (Fig. 3).23,24 Mutagens incorporated into RNA strands can cause permanent mutations.25,26 Obligate terminators lacking a 3-OH group will terminate RNA extensions immediately, while non-obligate chain termination usually proceeds when a drug contains both a natural base and a 3-OH on the sugar but has a modified ribose skeleton that disrupts translocation.27,28

Antiviral mechanisms of nucleotide drugs. The triphosphate form of remdesivir acts as a non-obligate chain terminator to exert an inhibition effect (Protein Data Bank entries 7VB219). The active form of molnupiravir can be directly incorporated into RNA as a substrate instead of cytidine triphosphate (C) or uridine triphosphate (U), thereby leading to mutated RNA products (Protein Data Bank entry 7OZU38). The triphosphate form of AT-527 (AT-9010) incorporates at the 3′ end of the RNA product, causing termination of RNA synthesis (Protein Data Bank entry 7ED551)
Remdesivir was first developed for the prevention of the Ebola virus infection.29,30,31 It is a non-obligate chain terminator of SARS-CoV-2.32 A study conducted by Yin et al. revealed that the triphosphate form of remdesivir (GS-441524) mimics a nucleotide and is covalently linked to the replicating RNA, thus blocking further synthesis of SARS-CoV-2 RNA.19 Kokic et al. reported that incorporation of remdesivir into the RNA product could stop RNA synthesis after the addition of three more nucleotides.33 They showed that the stalling is caused by the C1ʹ-cyano group in the remdesivir ribose moiety. Insight into this non-obligate chain termination mechanism may facilitate the search for compounds with potential to interfere with SARS-CoV-2 replication.16,34
Molnupiravir, an orally available antiviral drug, is a mutagen of SARS-CoV-2.35,36,37 According to research reported by Kabinger et al., the active form of molnupiravir, beta-D-N4-hydroxycytidine triphosphate, can be directly incorporated into RNA as a substrate instead of cytidine triphosphate or uridine triphosphate, leading to mutated RNA products.38 Structural analysis of RdRp-mutated RNA indicated that beta-D-N4-hydroxycytidine triphosphate formed a stable base pair with G or A in the RdRp active region, thus escaping proofreading and synthesizing mutated RNA. Like molnupiravir, ribavirin abrogates viral RNA synthesis by incorporation into nascent RNA strands.39,40,41,42 Cheung et al. confirmed it is a mutagen for influenza virus by increasing the G-to-A and C-to-T mutation rates in vitro.39 The molecular docking study of Bylehn et al. indicated that it binds strongly at the active site of SARS-CoV-2 RdRp.43 However, their results revealed that ribavirin does not bind the nucleotide on the complementary strand as effectively and seems to act by a different mechanism.
Favipiravir is another inhibitor of RdRp with two possible mechanisms of action.44,45,46,47 Shannon et al. demonstrated its active form could result in SARS-CoV-2 lethal mutagenesis by incorporation into the nascent viral RNA by error-prone SARS-CoV-2 RdRp, provoking C-to-U and G-to-A mutations in the SARS-CoV-2 genome.48 This mutagen mechanism of favipiravir was also reported by Peng et al.49. A study conducted by Naydenova et al. indicated that favipiravir could suppress the replication of SARS-CoV-2 RNA in the presence of natural nucleotides by weak incorporation into the RNA prime strand.50 They revealed that favipiravir–RTP represents an unusual, non-productive binding mode at the catalytic site of SARS-CoV-2 RdRp, thus inducing non-obligate chain termination.
The obligate chain terminator AT-527 is a guanosine nucleotide analog that serves as an orally available prodrug with inhibitory effects on hepatitis C virus (HCV) RdRp.51,52 Shannon et al. reported a 2.98 Å cryo-EM structure of the SARS-CoV-2 RdRp–RNA complex, showing the triphosphate form of AT-527 (AT-9010) bound at three sites of NSP12.51 Their results showed that after AT-9010 is incorporated at the end of the RNA product strand, its modified ribose group will prevent correct alignment of incoming NTP, thereby causing obligate chain termination.
Due to the conserved structure of RdRp, the effects of several molecules interfering with other viral RdRps against RdRp of SARS-CoV-2 were also studied.17,53 For example, sofosbuvir is an oral nucleoside that is used to treat chronic HCV infection.54,55,56,57 Appleby et al. indicated that the metabolized form of sofosbuvir could be recognized by HCV RdRp (NS5B) and incorporated into the growing chain. The presence of fluoro and methyl modifications at the 2′ position promotes non-obligate chain termination of HCV RNA.58 Enzymatic assays demonstrated that sofosbuvir acts as a competitive inhibitor of SARS-CoV-2 RdRp,59 revealing it might act as a non-obligate terminator. Another molecule, galidesivir, was initially designed to inhibit filovirus RNA polymerase activity indirectly through non-obligate RNA chain termination.60,61,62 It exhibited activity against numerous viruses, including yellow fever virus, dengue virus, Japanese encephalitis virus, West Nile virus, zika virus, and tick-borne encephalitis virus, in cell cultures and animal models.63 Molecular docking assays also revealed galidesivir is attached to the catalytic center of SARS-CoV-2 RdRp, and its binding mechanism needs to be further studied.61
Main protease (Mpro)
SARS-CoV-2 Mpro (also named NSP5 or 3C-like protease) is a key enzyme that plays a vital role in viral replication and transcription.64,65,66 After membrane fusion, genomic RNA (gRNA) of SARS-CoV-2 is released into the cytosol of the target cell (Fig. 2). The gRNA of SARS-CoV-2 contains two large replicase ORFs, ORF1a and ORF1b. These ORFs encode two N-terminal polyproteins, PP1a and PP1ab, respectively.67 Mpro mainly digests both polyproteins at more than 11 conserved sites, thus helping to release NSPs.68 These NSPs are involved in the production of subgenomic RNA, encoding four major structural proteins and other helper proteins.69,70,71 Since no human protease has a structure similar to that of Mpro, it is an attractive target for SARS-CoV-2 treatment.72 The SARS-CoV-2 Mpro crystal structure revealed it is a homodimer containing two protomers (promoters A and B), and each protomer is composed of three domains.68,73,74 The substrate binding site was located between (i) Domains I and II and (ii) Domain III. It regulates the dimerization of Mpro, which is necessary for its catalytic activity.72 The active sites of Mpro between Domains I and II are composed of four sites (S1′, S1, S2, and S4), which often accommodate four fragments (P1′, P1, P2, and P3, respectively) of inhibitors.68,73,74,75 Among them, covalent linkage with the Cys-145 residue in the S1′ site is beneficial for the activity of inhibitors.70,76 Non-covalent SARS-CoV-2 Mpro inhibitors binding with Mpro in different patterns have also became clinical candidates for treating SARS-CoV-2.77,78
Mpro always accommodates four fragments—P1′, P1, P2, and P3—which occupy the S1′, S1, S2, and S4 pockets of Mpro, respectively. Following this rule, novel molecules against SARS-CoV-2 Mpro were developed by structure-based design methods. For example, Dai et al. designed and synthesized two lead compounds (11a and 11b) targeting Mpro70 (Fig. 4). In their design, an aldehyde was selected as a new warhead along with an (S)-γ-lactam ring in order to form a covalent bond with cysteine. A cyclohexyl or 3-fluorophenyl was introduced in P2, while an indole group was introduced into P3. The resulting 11a and 11b were covalently bound to Cys-145 of Mpro according to the X-ray crystal structures of their complexes with SARS-CoV-2 Mpro. Qiao et al. designed new inhibitors by fixing P1 as an optimal fragment, using P2 that was derived from either boceprevir or telaprevir and allowing P3 to change.79 According to their results, one of the most potent compounds, MI-23, covalently bound to the catalytic residue Cys-145 of SARS-CoV-2 Mpro as expected. The binding pattern of the representative compound MI-23 with Mpro is consistent with its design concept. Based on the structure of ML188(R), a non-covalent inhibitor of SARS-CoV Mpro, Kitamura et al. proposed a strategy for designing the SARS-CoV-2 Mpro inhibitor and obtained a novel Mpro inhibitor 23R with high specificity to SARS-CoV-2 and SARS-CoV Mpro.77 Furthermore, they designed covalent SARS-CoV-2 Mpro inhibitors Jun9-62-2R and Jun9-57-3R using novel cysteine reactive warheads to improve the target specificity of aldehyde warhead.80 To optimize oral bioavailability of Mpro inhibitors, Quan et al. chose alpha-ketoamide as warhead P1’, and P1, P2, and P3 were fixed as pyridine, tert-butylbenzene, and tert-butyl, respectively, similar to the groups in ML188.81 The resulting compound Y180 showed high oral bioavailability in mice and efficiently protected transgene mice from SARS-CoV-2 and variant infection.

Different binding models of inhibitors in complex with SARS-CoV-2 Mpro. a Binding models of inhibitors 11a and 11b complexing with SARS-CoV-2 Mpro (the Protein Data Bank entries for SARS-CoV-2 Mpro complexing with 11a and 11b are 6LZE and 6M0K, respectively70) b Binding model of inhibitor nirmatrelvir in complex with SARS-CoV-2 Mpro (Protein Data Bank entry 7RFW).84 c Binding model of non-covalent inhibitor 23R in complex with SARS-CoV-2 Mpro (Protein Data Bank entry 7KX577)
Besides the rational design of novel compounds, several SARS-CoV-2 Mpro inhibitors were discovered by optimizing existing Mpro inhibitors through drug design.82,83 The drug PF-07321332, more commonly known as nirmatrelvir, was optimized from the SARS-CoV Mpro inhibitor PF-00835231.84 Meanwhile, Zhang et al. optimized the structure of the alpha-ketoamide Mpro inhibitor 11r to increase its half-life and solubility and reduce its interaction with plasma proteins.72 Then, the authors replaced the P2 cyclohexyl moiety with a small cyclopropyl to increase the antiviral activity by scarifying the broad-spectrum nature.65 The molecule 13b was located in the substrate binding cleft of Mpro and interacted with the Glu-166 residue, thus disturbing the correct shape of the S1 pocket and inactivating the enzyme.72,85 Kenller et al. presented the design and characterization of three hybrid reversible covalent SARS-CoV-2 Mpro inhibitors named BBH-1, BBH-2, and NBH-2 by splicing the SARS-CoV protease inhibitors boceprevir and narlaprevir.86 By substituting the ketoamide group of boceprevir with the keto-benzothiazole moiety or introducing the nitrile warhead, they directed the warhead into the oxyanion hole. Then, they substituted the P1 group of boceprevir and narlaprevir with a Gln-mimic γ-lactam, thereby synthesizing the hybrid reversible covalent inhibitors BBH-1, BBH-2, and NBH-2. A study by Amporndanai et al. indicated that ebselen and its derivative MR6-31-2 solely bind at the Mpro catalytic site by donating a selenium atom, forming a covalent bond and blocking the His-41 and Cys-145 catalytic dyad.87
The three-dimensional structure of SARS-CoV-2 Mpro is highly similar to that of SARS-CoV Mpro.72,88,89,90 Therefore, repurposing of drugs is a good strategy to develop drugs against SARS-CoV-2. Two SARS-CoV Mpro inhibitors, GRL-1720 and 5 h, have shown anti-SARS-CoV-2 activity.91,92,93 According to X-ray structural analysis, 5 h fully occupies all binding pockets and is stabilized by six direct hydrogen bonds with the residues inside the binding groove of SARS-CoV-2 Mpro, and covalent bonds are formed between 5 h and the Cys-145 residue.91 Su et al. reported that myricetin inhibits SARS-CoV-2 Mpro.94 According to a crystal structure of the SARS-CoV-2 Mpro–myricetin complex, an exact covalent bond can be observed between the sulfur atom of Cys-145 and the C6’ atom of the pyrogallol group of myricetin, revealing the potential of pyrogallol as an alternative warhead of an Mpro inhibitor. High-throughput screens were also applied to repurpose molecules with potential inhibitory effects on SARS-CoV-2 Mpro.95,96,97 For example, Günther et al. applied X-ray fragment screening experiments with approved drugs and drugs in clinical trials, and identified 37 compounds that bind to Mpro .88 Moreover, they obtained structural evidence for interaction of seven compounds at active and allosteric sites of Mpro, and identified two allosteric sites representing attractive targets for drug development. Another high-throughput screening study was conducted by Drayman et al. on a library of 1900 clinically safe drugs against OC43, which is also a betacoronavirus.98 As a result, they identified the most potent SARS-CoV-2 Mpro inhibitor, masitinib, and characterized the mechanism by X-ray crystallography. Virtual high-throughput screening methodology was also applied in identifying novel inhibitors from a large collection. Jin et al. assayed more than 10000 compounds through structure-based virtual screening and high-throughput screening, and identified ebselen as a promising inhibitor of SARS-CoV-2 Mpro.68
Papain-like protease (PLpro)
PLpro (NSP3) is an important coronavirus enzyme that digest polyproteins by recognizing the conserved sequence LXGG, thus generating a functional replicase complex which enables viral spread99,100,101 (Fig. 2). In addition, it is implicated in both the ubiquitination and inhibition of ISGylation on host proteins as an evasion mechanism against host antiviral immune responses.102,103,104 Shin et al. demonstrated that SARS-CoV-2 PLpro prefers to cleave the conserved LRGG motif at the C-terminus of interferon-stimulated gene 15 (ISG15), which attenuates type I interferon immune responses elicited by viral infection.105 This dual functionality of PLpro makes it an attractive antiviral target for SARS-CoV-2 treatment. PLpro has four subdomains: the ubiquitin-like domain, the Thumb domain, the Finger domain, and the Palm domain (Fig. 5).105 The substrate binding pockets are located at the interface of the Palm and Thumb domains, which include a conserved catalytic triad of Cys-111. The other two core residues, Phe-69 and Val-66, mediate interactions of PLpro with ISG15.105 Substrates accessing the active site are regulated by a flexible blocking loop 2 (BL2).99 The key Tyr-268 residue on BL2 is vital for regulating the function of the enzyme.102 In addition, the zinc finger domain comprises four cysteines which also contribute to the structural integrity and protease activity of PLpro.106,107,108 These sites are hotspots on PLpro, which have led to the discovery of drug leads with clinical potential for COVID-19 treatment.

GRL-0617 is a non-covalent inhibitor of SARS-CoV PLpro, and it exhibited inhibitory effects against SARS-CoV-2 in vitro.103,109,110 Gao et al. demonstrated that GRL0617 not only occupies the substrate pockets, but also induces closure of the BL2 loop and narrows the substrate binding cleft, thus preventing binding of the LXGG motif of the substrate.99 This BL2 conformational change was also observed by Ma et al. through X-ray co-crystal analysis of PLpro complexed with GRL0617 (Fig. 5).111 Further, Shin et al. reported that GRL-0617 treatment of SARS-CoV-2-infected cells led to a marked increase in IRF3 ISGylation and significantly rescued the expression of IFN-responsive genes.105 According to Fu et al., GRL0617 blocks the binding of the ISG15 LRGG C-terminus to PLpro, thus interfering with cleavage of ISG15.112 Moreover, through a high-throughput screening and subsequent lead optimization, they identified two PLpro inhibitors, Jun9-72-2 and Jun9-75-4. Both inhibitors demonstrated improved enzymatic inhibition and antiviral activity compared to GRL0617. In addition, Zhao et al. identified SARS-CoV-2 PLpro inhibitors by high-throughput screening.108 They found that YM155, an anticancer drug candidate, efficiently inhibited the activity of SARS-CoV-2 PLpro. By analyzing crystal structures of SARS-CoV-2 PLpro and its complex with YM155, they found that YM155 simultaneously targets the substrate binding pocket, the ISG15 binding site, and the zinc finger motif of enzyme.
Based on substrate specificity and the structure of SARS-CoV-2 PLpro, rational design of compounds would greatly facilitate the development of novel PLpro inhibitors.100 For instance, by using a Hybrid Combinatorial Substrate Library, Rut et al. revealed the molecular rules governing PLpro substrate specificity, and designed and biochemically characterized potent inhibitors (VIR250 and VIR251) with high selectivity for SARS-CoV-2 PLpro.100 Further, they found that both inhibitors could selectively inhibit the activities of PLpro in both SARS-CoV and SARS-CoV-2. This revealed a high level of sequence and structural similarity between these PLpro in the substrate binding pocket. The crystal structures of VIR250 and VIR251 in complex with SARS-CoV-2 PLpro reveal they inhibit the enzyme by forming a covalent link with the Cys-111 residue and provide a structural basis for the observed substrate specificity profiles. Osipiuk et al. synthesized six naphthalene-based compounds derived from GRL0617. Five of them are further amine-functionalized derivatives of GRL0617, and one is a simplified variant of GRL0617 without a chirality center.112,113 All these compounds exhibited inhibition activities of PLpro, and the crystal structure indicated these inhibitors bind to protease S4/S3 sites, thus blocking peptide recognition. Shan et al. also synthesized a series of reported ScoV PLpro inhibitors (11–13) that partially resemble GRL0617 with a shared naphthyl subunit.114 Co-crystal structure analysis of SARS-CoV-2 PLpro-12 revealed 12 occupies a pocket between the S1 position and the catalytic position of SARS-CoV-2 PLpro, and the three hydrophobic rings of 12 are engaged simultaneously with the phenyl ring of Tyr-268, thus closing the binding pocket.114
Spike glycoprotein (S protein) and angiotensin-converting enzyme 2 (ACE2)
SARS-CoV-2 virus entry into host cells depends on the viral S protein.115,116,117 In brief, the S protein recognizes the peptidase domain (PD) of the ACE2 receptor in host cells (Fig. 2). This initiates recognition of the virus and host cell receptor–viral membrane fusion.118,119,120 It was thought that targeting the virus entry process is more advantageous than targeting the subsequent stages of the SARS-CoV-2 lifecycle, thus many efforts have been made to find inhibitors blocking this process.121,122,123 Small molecules targeting the S protein, ACE2, and the S protein–ACE2 complex were found to potentially inhibit SARS-CoV-2 infection.124,125 The SARS-CoV-2 S protein consists of two subunits; S1 comprises the receptor binding domain (RBD) and S2 is responsible for viral membrane fusion.126,127,128,129,130 Previous studies revealed that the high affinity between the S protein RBD and the human ACE2 receptor could partially explain the efficient transmission of SARS-CoV-2 among humans.131,132,133 The structure of the SARS-CoV-2 RBD was found to have more ACE2-interacting residues than the SARS-CoV RBD.119 Compensating mutations in the S protein RBD of further variants (especially the Delta and Omicron variants) possibly account for their heightened transmissibility and immune evasion.134,135 Thus, interference with binding between them is beneficial for viral inhibition. A six-helical bundle (6-HB) structure of S2 conjuncts the viral and cell membranes for a fusion reaction.136 Blocking the 6-HB domain is considered effective for developing fusion inhibitors EK1 (Fig. 6).137,138 In human ACE2, Lys-31 and Lys-353 are sensitive to the RBD.139 Its glycosylation sites Asn-90 and Asn-322 also demonstrated the ability to interfere with S protein binding in a recent study.140 Glycosylation of asparagine residues within the RBD is an important mediator of ACE2 binding.141

S2 subunit of SARS-CoV-2 S protein involves the HR1 and HR2 trimers to form a 6-HB domain. The binding model of the EK1 inhibitor in complex with the HR1 motif is presented (Protein Data Bank entry 7C53)679
The effects of molecules binding with S protein against SARS-CoV-2 were investigated. A previous study revealed that the RBD of the S protein of SARS-CoV-2 recognizes oligosaccharides containing sialic acid.142 Based on this, Petitjean et al. investigated the biophysical properties of S1 subunit binding to sialic acids or 9-O-acetylated sialic acid (9-AcSA) using force–distance (FD) curve-based atomic force microscopy.143 Then, they designed novel blocking molecules with various topologies and carrying multiple salic acid or 9-AcSA residues. They reported that 9-AcSA-derived porphyrin has strong inhibitory effects on SARS-CoV-2. Yi et al. searched for S protein RBD inhibitors by screening compounds from the Chinese herbal medicine licorice.144 They found that glycyrrhetinic acid (GA) and licorice saponin A3 target the S protein RBD, and Tyr-453 is a key residue for the affinity of triterpenoids with the S protein RBD. Another strategy to inhibit SARS-CoV-2 S protein is to disrupt the disulfide pairs of RBD.145,146 Disulfide bond formation is central to the dynamic structure of many viral receptor binding and entry/fusion proteins.147 The SARS-CoV-2 S protein RBD contains four disulfide pairs, which may interact with thiol-based reducing agents.146,148 Shi et al. reported that the preclinical thiol-based reducing agents P2110 and P2165 target a conserved hydrophobic binding pocket in the RBD, thus inhibiting SARS-CoV-2 infection.146 In detail, proteomic and reactive cysteine mapping showed that the disulfide pairs Cys-379–Cys-432 and Cys-391–Cys-525 are redox-sensitive and can be reduced by P2110 and P2165. A significant conformational change of the RBD was observed after reduction of both disulfide pairs. They also indicated that P2110 and P2165 could modulate the extracellular redox poise required for SARS-CoV-2 entry into cells, which is beneficial for preventing viral infection.
Besides finding molecules with inhibitory effects on the S protein, studies focused on finding molecules which can inhibit the RBD–ACE2 interaction.149,150,151 For example, Pei et al. applied a computer-aided approach based on the RBD binding residues on ACE2 to design ultrashort peptide inhibitors against SARS-CoV-2.152 Based on the critical residues of ACE2, they initially obtained the peptide inhibitor SI1. Then, using a “docking–activity test–molecular simulation–sequence improvement” scheme, they successfully obtained ultrashort peptides SI5α and SI5α-b, which had significantly higher activity. By analyzing the binding sites of ultrashort peptides to RBD, the residues from Glu-484 to Tyr-505 on the RBD were determined as the “binding pocket” in this study, which may be helpful for the design of RBD inhibitors or antibodies. A similar computer-aided strategy for the identification of novel inhibitors disrupting the RBD–ACE2 interaction was reported by Gupta et al. In their study, machine learning classifiers were applied for the prediction of new small molecular modulators of the SARS-CoV-2 S protein RBD–ACE2 interaction. Using this RBD: hACE2 predictor, they identified more than 300 novel small molecule scaffolds that can be repurposed for SARS-CoV-2. Panda et al. took the structure-based drug design approach for screening inhibitors with an affinity against Mpro and S protein.153 Molecular docking simulations indicated that the obtained molecule, PC786, has a binding affinity toward the RBDs of all the chains in the trimeric S protein. Their protein–protein interaction analysis revealed that conformational changes occur when PC786 interacts with the RBD–ACE2 complex, revealing that the binding of PC786 with S protein substantially affects S protein binding to the ACE2 domain. Lee et al. showed that both Etravirine and Dolutegravir preferentially bind to primary ACE2-interacting residues on the RBD domain, implying that these two drugs may inhibit attachment of SARS-CoV-2.154 Xiong et al. showed that the novel inhibitors DC-RA016 and DC-RA052 have the ability to interfere with the SARS-CoV-2 S protein RBD–ACE2 interaction, thus playing an anti-SARS-CoV-2 role.155
Host proteases
After binding to the ACE2 receptor of host cells, S protein needs to be activated by host protease at the putative cleavage site located at the boundary of the S1 and S2 subunits, thus exposing the S2 subunit for viral entry (Fig. 2).128,156,157 This cleavage is performed by host cells proteases, including serine protease transmembrane protease, serine 2 (TMPRSS2), cysteine protease cathepsin L (CTSL), and the arginine protease furin.54,121,158 TMPRSS2 was thought to play an essential role in SARS-CoV-2 viral entry.159,160,161 It enables rapid endosome-independent virus entry of SARS-CoV-2 into the cells (within 10 min).162 CTSL also enhances SARS-CoV-2 infection in both human cells and human ACE2 transgenic mice.163,164,165 CTSL is critical for SARS-CoV-2 entry via endocytosis during infection.157 The furin cleavage site also has a critical role in SARS-CoV-2 infection,164,166,167,168 since a study has revealed that its cleavage site at the S1/S2 boundary is essential for S-protein-mediated cell–cell fusion and entry into human lung cells.168 Based on these observations, inhibitors of TMPRSS2, CTSL, and furin were identified as promising therapeutical agents for COVID-19 treatment.169
The structure of TMPRSS2 is characterized by an N-terminal cytoplasmic domain, a transmembrane domain, a class A LDL receptor domain, a scavenger receptor cysteine-rich domain, and an activation domain linked to a serine protease domain via a disulfide bond.54,159,170 Since no crystal structure of TMPRSS2 is available, repurposing or optimizing inhibitors against well-known serine proteases may facilitate the discovery of effective TMPRSS2 inhibitors against SARS-CoV-2.170,171,172 For example, Sun et al. identified structurally similar serine proteases using a structure-based phylogenetic computational tool to find potential inhibitors of TMPRSS2.173 According to their computational results, six serine peptidases, including kallikrein-related B1, had a high structural similarity to the TMPRSS2 S1 protease domain. The kallikrein-related B1 inhibitor avoralstat with high potential to be repurposed for COVID-19 therapy was identified. In addition, based on a previously designed peptidomimetic tetrapeptide with inhibitory activity against matriptase, Shapira et al. developed a small library of peptidomimetic compounds to screen for inhibitors of TMPRSS2.174 Through the screening process, they found that N-0385, containing a ketobenzothiazole warhead, inhibits TMPRSS2. Then, by building a homology model of TMPRSS2 using the crystal structure of matriptase, they found that the catalytic Ser-441 residue of the enzyme forms a covalent bond with the warhead of N-0385. This contributes to its inhibitory activity against TMPRSS2. Rational structure-based drug design was also applied to discover TMPRSS2 inhibitors by Mahoney et al..175 Based on molecular docking studies using a published homology model of TMPRSS2 and substrate specificity data from PS-SCL, a set of ketobenzothiazole inhibitors of HGF-activating serine proteases (including HGF activator [HGFA], matriptase, and hepsin) were developed. After further optimization, they identified multiple potent inhibitors of TMPRSS2. Four of these analogs displayed activity at subnanomolar concentrations, both in the enzyme assay and in blocking the entry of VSV-SARS-CoV-2 chimeras into human Clau-3 epithelial lung cells. Besides blocking the cleavage function of TMPRSS2, molecules with the ability to reduce TMPRSS2 expression on host cells also drew attention for anti-COVID-19 research. A high-throughput screening using a library of 2560 FDA-approved or currently investigated clinical compounds was carried out by Chen et al. to identify small molecules that reduce TMPRSS2 expression.176 They found that halofuginone modulates TMPRSS2 levels through proteasomal-mediated degradation that involves the E3 ubiquitin ligase component DDB1- and CUL4-associated factor 1.
CTSL is a lysosomal cysteine protease. It contains an L domain of alpha-helices and an R domain of beta-sheets.177,178,179 Gallinamide A is a potent covalent inhibitor of several parasite-derived cysteine proteases, as well as human CTSL.180,181 Ashhurst et al. demonstrated that Gallinamide A and analogs could directly interact with CTSL and potently inhibit SARS-CoV-2 infection in vitro.182 Structure-based design of CTSL inhibitors was carried out by Phan et al. According to their report, good peptidyl substrates can be converted into CTSL inhibitors that are active at submicromolar concentrations by a single thioamide substitution in the peptide backbone.169 By designing and scanning several thioamide-stabilized peptide scaffolds, they found that the peptide RS1A inhibits CTSL activity with >25-fold higher specificity compared to the other cathepsins. According to computational modeling analysis, the P1 thioamide N–H group of the peptide interacts with the His-163 catalytic triad of CTSL. In a recent preprint reported by Frueh et al., an orally available CTSL inhibitor K777 exhibited anti-viral ability and efficiently reduced COVID-19-related pulmonary pathology in African green monkeys.183 Despite these achievements, the ubiquitous expression of CTSL raises concern about the side effects of CTSL inhibitors.184 Combined use of a CTSL inhibitor and other protease inhibitors or development of a CTSL inhibitor with multiple functions might be effective in preventing viral infection at a lower dose and in reducing side effects. Thus, Hu et al. found that calpain inhibitors II and XII, and GC-376 have a dual mechanism of action by inhibiting both viral Mpro and host CTSL in vitro.185 In addition, Sacco et al. found that Mpro inhibitors targeting the hydrophobic methionine side chain in the S1 pocket are also active against CTSL, which paved the way for the design of dual inhibitors that target both viral Mpro and host CTSL.186
Furin recognizes and cleaves a polybasic stretch of an RRAR motif in the S1/S2 boundary of S protein. It is worth noting that the cleavage site of furin was only identified in SARS-CoV-2, and not in other lineages of betacoronaviruses.187,188,189,190 Even Papa et al. indicated that knockout of furin significantly suppressed but not abolished SARS-CoV-2 S-protein-mediated cell–cell fusion.191 Johnson et al. revealed that RRAR cleavage site mutation attenuates SARS-CoV-2 pathogenesis in both hamster and K18-hACE2 transgenic mouse models.167 Peacock et al. found that SARS-CoV-2 virus lacking the S1/S2 furin cleavage site was shed to lower titers from infected ferrets and was not transmitted to cohoused sentinel animals, unlike the wild-type virus.168 Thus, Cheng et al. reported that two molecular inhibitors of furin, decanoyl-RVKR-chloromethylketone (CMK) and naphthofluorescein, significantly inhibited syncytium formation in S-protein-expressing cells and cytopathic effects (CPEs) in SARS-CoV-2-infected cells.187 According to their results, CMK abolished CPEs and decreased virus titer in the preinfection treatment experiments, while it did not decrease virus production and infectivity but only decreased CPEs in postinfection treatment. This revealed that CMK affects the viral entry stage of SARS-CoV-2, and that it likely ameliorates viral virulence and pathogenicity. In addition, another furin inhibitor, naphthofluorescein, showed affinity at the replication stage when the virus entered the cell downstream.192,193 Authors speculated CMK and naphthofluorescein might act differently for furin substrates located in different compartments. It remains to be clarified whether naphthofluorescein’s function depends on furin activity or other new targets. Paszti-Gere et al. revealed that another furin inhibitor, MI-1851, could exert anti-SARS-CoV-2 effects on cells by suppressing the cleavage of S protein.194
Immune regulation
SARS-CoV-2 infection activates both innate and adaptive immune responses, which may cause excessive inflammatory reactions and dysregulate the adaptive host immune response.9,195,196,197 Many studies have reported the influence of SARS-CoV-2 infection on the immune system of COVID-19 patients. In detail, lymphopenia was widely observed in patients with severe COVID-19.67,198 The proportion of lymphocytes is considered a reliable indicator of disease severity.199 In patients with severe COVID-19, the proportions of circulating CD4+ T cells, CD8+ T cells, B cells, and natural killer cells also decreased, while the proportions of immunosuppressive regulatory T cells were moderately increased in patients with mild COVID-19.200,201,202 Moreover, the levels of proinflammatory cytokines and chemokines (such as IL2, IL7, IL10, GSCF, IP10, MCP1, MIP1A, TNFα, and IL6) were significantly increased in severe patients.198,201,203 As a result of virus recognition, downstream immune-regulatory pathways such as nuclear factor κB (NF-κB), and Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathways are activated (Fig. 7). These pathways are crucial for the antiviral response.204,205,206 In fact, mortality of COVID-19 patients is often caused by acute respiratory distress syndrome (ARDS), and ARDS is the result of dysregulated hyperinflammation in response to viral infection.198,207,208 Thus, various immune regulators were developed or repurposed for COVID-19 treatment (Fig. 3). Most immune regulators, such as glucocorticoids, function as inflammatory extinguishers. Here, we present the immunomodulatory mechanism of these molecules against COVID-19.

Illustration of SARS-CoV-2-induced immune responses and pro-inflammatory signaling pathways
The JAK family consists of four non-receptor tyrosine protein kinases, JAK1, JAK2, JAK3, and TYK2.209,210 They are often activated when proinflammatory cytokines bind to their receptors, thus amplifying the inflammation caused by SARS-CoV-2 infection.211 So far, more than 50 cytokines that transmit their signals via JAK proteins have been identified.212,213,214 Based on this, it was recognized that JAK inhibitors could help to prevent the cytokine storm in severe COVID-19 patients.213,215 Baricitinib, a JAK1/JAK2 inhibitor, blocks the immune cascade and reduces SARS-CoV-2 replication in patients.216,217,218 According to a study conducted by Stebbing et al., type-1 interferons (IFNs), specifically IFN-α2, increased ACE2 expression in human liver cells could increase the viral load, and this induction is fully inhibited by the JAK inhibitor baricitinib.219 A study reported by Nystrom et al. indicated that baricitinib could block the cytokine-induced JAK/STAT/APOL1 signaling, which may rescue a severe kidney disease called COVID-19-associated nephropathy.220 Other JAK inhibitors, such as tofacitinib, ruxolitinib, and nezulcitinib, were also shown to exert effects against COVID-19 in clinical studies.221,222,223 According to a study of Yan et al., the JAK1/2 inhibitor ruxolitinib could normalize the SARS-CoV-2-induced complement hyperactivation in lung epithelial cells.224 Ruxolitinib was also clinically related to increased serum levels of inflammatory cytokines such as IL6 and the acute phase protein ferritin and cardiac improvement.225 Tofacitinib is a JAK1/JAK3 inhibitor known to be effective against cytokine signaling. It also inhibits JAK2 with a lower potency.226,227,228 Several studies indicated that it suppresses S-protein-potentiated STAT1 signaling and combats lung tissue-resident memory T cells which cause chronic inflammation and fibrosis when treating COVID-19.210,229,230
Bruton’s tyrosine kinase (BTK) is a cytoplasmic non-receptor tyrosine kinase (TK) expressed in all cells of the hematopoietic lineage, particularly B cells, mast cells, and macrophages.231,232 In addition, BTK-deficient macrophages are defective in expressing proinflammatory cytokines and preferentially polarize into anti-inflammatory M2 macrophages, even upon virus infection.233 A previous study indicated that inhibition of BTK attenuated neutrophil extracellular traps released into the lung with reduced levels of TNFα, IL1β, IL6, KC, and MCP-1 in mice after influenza A virus infection.233 Since cytokine release syndrome and resident macrophages may lead to pulmonary injury associated with COVID-19, Treon et al. reported that inhibitors of the BTK pathway may protect against pulmonary injury in COVID-19 patients.234 Chong et al. also suggested continuing BTK inhibitor treatment in patients who receive it for therapy of B cell malignancies with COVID-19, since the potential benefit of attenuation of M1 polarization to mitigate the immediate risk of COVID-19-related mortality outweighs the potential medium- to long-term risk of impaired humoral immunity.235 The BTK inhibitors ibrutinib, zanubrutinib, and acalabrutinib have been found to protect against pulmonary injury in a small group of participants infected with SARS-CoV-2.232,236,237
NF-κB is a proinflammatory transcription factor critically involved in both inflammatory and thrombotic responses.238,239 Its upregulation was widely observed in the development of SARS-CoV-2 infection.240,241,242,243 In addition, N protein and NSP5 of SARS-CoV-2 facilitate NF-κB hyperactivation, thus inducing inflammation.244,245,246 Therefore, NF-κB has become a potential immunotherapeutic target for COVID-19 treatment.247,248,249 Sharma et al. reported that curcumin could potently inhibit the inflammatory response elicited by SARS-CoV-2 S protein in cells by deactivating MAPK/NF-κB signaling.250 Lee et al. found that the NF-κB inhibitor pyrrolidine dithiocarbamate suppresses ACE2 protein expression in human lung cell lines, which indicates another potential mechanism by which NF-κB inhibitors may combat COVID-19.251
The Nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is activated when viral infection-associated pathogens are recognized by the innate immune system.252,253 Activation of the NLRP3 inflammasome pathway leads to release of the proinflammatory cytokines IL18 and IL1β, which mediate cytokine release and pyroptosis during lung injury and ARDS.254,255,256 Rodrigues et al. demonstrated that the NLRP3 inflammasome is activated in COVID-19 patients. Inflammasome-derived products such as IL18 in the serum were correlated with disease severity.257 A study of Pan et al. revealed that the N protein of SARS-CoV-2 promotes NLRP3 inflammasome activity and induces an excessive immune response.258 Therefore, inhibitors targeting the NLRP3 inflammasome might serve as drugs to treat COVID-19.259 A study conducted by Zeng et al. demonstrated that inhibition of the NLRP3 inflammasome by MCC950 alleviated excessive lung inflammation. Further, they showed that MCC950 could reduce COVID-19-like pathology in human ACE2 transgenic mice.260
Structures of small molecule drugs for COVID-19 therapy
Nucleoside/nucleotide analogs
Nucleoside/nucleotide analogs were investigated widely in the area of antiviral drugs (Fig. 8).261,262,263 Generally, nucleoside/nucleotide analogs resemble naturally occurring nucleosides, and act as normal nucleotides, being recognized by viral polymerases or cellular enzymes, and prevent virus replication.264,265 Various nucleoside/nucleotide analogs have been applied for clinical antiviral therapies. Besides the first anti-HSV drug, acyclovir,266 other nucleoside/nucleotide analogs such as zidovudine against HIV, telbivudine against HBV, and sofosbuvir against HCV also exhibited specific therapeutic effects.267,268,269 Although achievements have been made in the area of DNA virus application, these analogs are still facing challenges in the treatment of infections with RNA viruses with higher spread and mutation rates. In the area of SARS-CoV-2, looking for nucleoside/nucleotide analogs is the preferred strategy, as no homolog of RdRp has been found in human cells. Since the RdRp of SARS-CoV-2 is conserved, exploring the anti-SARS-CoV-2 effects of pre-existing antiviral nucleoside/nucleotide analogs against the virus has been shown to be an effective way.270,271 Nucleobase analogs and double-stranded RNA (dsRNA) compounds with anti-SARS-CoV-2 effects will also be discussed in this section. Although they do not function by imitating nucleosides, those analogs and compounds interfere with viral infection by various mechanisms.

Chemical structure of representative small molecules and their backbone (labeled in red)
As a constituent of ATP and cAMP, adenosine participates in numerous processes in the human body.272,273 Therefore, numerous adenosine analogs have been synthesized against various diseases, including COVID-19. Among the existing adenosine analogs against COVID-19, the most investigated one is remdesivir. It was developed by Gilead to combat the Ebola virus, and it bears the structure of an adenine c-nucleoside modified by monophosphoramide and cyano groups.274 As a nucleotide prodrug, remdesivir is metabolized by the host cell to the pharmacologically active triphosphate to inhibit the activation of RdRp.31 In a study reported by Pruijssers et al., remdesivir exhibited a potent in vitro inhibition ability against SARS-CoV-2 replication in human lung cells and primary human airway epithelial cells.275 Its in vivo effect was also confirmed in SARS-CoV-2-infected rhesus macaques. Remdesivir treatment in rhesus macaques with COVID-19 efficiently prevented progression to pneumonia. Holshue et al. first reported its clinical application, which described an immediate improvement in clinical symptoms in the first confirmed case of SARS-CoV-2 after receiving remdesivir administration.276,277 However, according to a recent study conducted by Stevens et al., remdesivir resistance was observed in SARS-CoV-2 after 13 passages of co-culturing with GS-441525.278 Although it is encouraging that natural variants did not propagate remdesivir resistance mutations, this study emphasized that the extended use of remdesivir might increase the possibility for SARS-CoV-2 to adapt to remdesivir. It is worth noting that remdesivir is a prodrug of GS-441524, which has also been proved to be effective against COVID-19.279,280 GS-441524 is also developed by Gilead, which is the dephosphoramidated ribonucleoside parent nucleus of remdesivir.281 Pharmacokinetic analysis showed that GS-441524 is the predominant metabolite of remdesivir reaching the lungs. Based on its easy synthesis and high lung loads, Yan et al. claimed it is superior to remdesivir for COVID-19 treatment.280 Li et al. reported that GS-441524 effectively inhibited SARS-CoV-2 in three cell lines (Vero E6, Calu-3, and Caco-2).282 In addition, remdesivir can only be given intravenously, and there is a pressing medical need for oral antivirals. Xie et al. performed an in vitro and in vivo drug metabolism and pharmacokinetics assessment to examine the potential of GS-441524 as an oral drug.283 In further in vivo studies in CD-1 mice, GS-441524 displayed a favorable oral bioavailability of 57%. Due to these advantages, the first study of orally administered GS-441524 for COVID-19 in humans was started on January 1, 2021, and conducted by Copycat Sciences. The clinical results suggested the high safety and low toxicity of orally administered GS-441524 in healthy people.284,285 Although further clinical studies of the compound remain to be implemented, GS-441524 has potential as an oral drug for treatment of COVID-19. Further, another prodrug of GS-441524 named VV116 was developed by the Shanghai Institute of Materia Medica. VV116 is derived from GS-441524 by esterification of all three hydroxyl groups and replacing a hydrogen atom on the basic group with a D atom.286 Wu et al. reported that VV116 is highly effective in inhibiting SARS-CoV-2 replication in cell-based and animal models.287 A clinical study of VV116 showed that it has good safety and efficacy.288 Moreover, studies have shown that VV116 exhibits antiviral activity against the Alpha, Beta, Delta, and Omicron variants with high oral bioavailability and good chemical stability.289 Two international phase II/III clinical trials of VV116 are underway. Besides remdesivir and its analogs, another adenosine analog, galidesivir, also is notable as an anti-SARS-CoV-2 drug. Galidesivir was developed by BioCryst Pharmaceuticals and was originally intended as a drug for HCV treatment.63 Unlike the pyrrolotriazine group in the abovementioned compounds, galidesivir bears a pyrrolopyrimidine group as its nucleobase. A molecular docking study conducted by Aftab et al. indicated that galidesivir binds effectively to SARS-CoV-2 RdRp, suggesting its potential use to treat COVID-19.290
Cytidine analogs have also been investigated for COVID-19 treatment. One of the cytidine analogs, molnupiravir, is the synthetic ribonucleoside derivative N4-hydroxycytidine developed by Merck and Ridgebace. It is a prodrug of β-D-N4-hydroxycytidine (EIDD-1931), which was originally developed for treating seasonal influenza.291 Unlike the abovementioned remdesivir, which terminates the elongation of viral genes, molnupiravir contains two forms of tautomers that can pair with A and T,35 thus causing large mutations in RNA products and preventing SARS-CoV-2 replication. According to the results reported by Sheahan et al., administration of molnupiravir improved pulmonary function and reduced virus titer and weight loss in mice infected with SARS-CoV-2.292 On November 4, 2021, it was first approved by the UK Medicines and Health Products Regulatory Agency (MHRA) for treating adults with mild to moderate COVID-19. Thus, molnupiravir was the word’s first orally administered anti-SARS-CoV-2 drug. A recent study revealed that the SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir.293 However, the potential side effect of molnupiravir of eliciting mutation in mammalian cells has raised concern.294 Azvudine is a cytosine analog which was also found to be efficient to treat SARS-CoV-2.295 It was previously approved for HIV inhibition.296 Recently, Zhang et al. observed that azvudine significantly inhibited viral load, promoted lymphocyte subsets, protected histological structures, and reduced inflammation caused by SARS-CoV-2 infection.297
Several guanosine analogs were reported to be efficient for inhibiting SARS-CoV-2.298 The most investigated one is ribavirin. It is a broad-spectrum antiviral drug with triazole structure, whose conformation is similar to that of guanosine.299 In 1970, it was first synthesized by Joseph T. Witkowski of ICN Pharmaceuticals.300 In 2013, it was approved by the FDA for the treatment of chronic HCV infection.61 Eslami et al. showed that combination therapy with ribavirin can effectively improve disease symptoms in severe COVID-19 patients.301 Later, results of an open-label randomized phase II trial showed that this triple therapy in hospitalized patients with COVID-19 pneumonia can effectively alleviate symptoms and shorten the duration of viral shedding and hospital stay in patients.302 Combination treatment with ribavirin, which is currently clinically available and cheap, with other antiviral drugs may become the treatment of choice in COVID-19 patients. In addition to ribavirin, the guanosine analog thioguanosine potentially inhibits SARS-CoV-2 by binding to Mpro.303 The guanine analog triazavirin was reported to be a promising agent to treat SARS-CoV-2.304 A pilot trial by Wu et al. indicated that triazavirin can inhibit the tendency to bind to ACE2, and triazavirin showed a significantly better therapeutic effect and higher safety in the treatment of COVID-19 compared with a placebo or standard therapy.305
The uridylate analog sofosbuvir was also believed to play an anti-SARS-CoV-2 role.306 It was discovered in 2007 by Pharmasset (Gilead) and approved for HCV treatment.307 Previous studies have also shown that it can inhibit Zika virus replication.308,309 Sofosbuvir needs to be triphosphorylated to its active form (2’-F, Me-UTP) to be recognized by HCV polymerase, thereby preventing viral replication.310 A study by Chien et al. showed that the activated triphosphate form of Sofosbuvir can bind to RdRp of SARS-CoV-2.311 Currently, several clinical trials studying the effects of sofosbuvir on SARS-CoV-2 are being carried out.301 According to a multicenter Egyptian study involving 174 patients with COVID-19, patients receiving combination treatment with sofosbuvir/daclatasvir demonstrated shorter hospital stay, faster PCR negativity, and possibly reduced mortality.312 However, according to a meta-analysis by Kow et al., sofosbuvir-based direct-acting antiviral agents have no protective effects against the development of severe illness in patients with COVID-19 with the current dosing regimen.313 In a previous study, sofosbuvir demonstrated higher anti-viral efficiency against West Nile virus in hepatic cells than in lung cells.314 This liver-targeting characteristic of sofosbuvir raises concerns for its use in treating SARS-CoV-2. In this regard, future studies should be conducted to improve sofosbuvir’s targeting of the SARS-CoV-2-attacked organs by structural optimization or formulation improvement.
Favipiravir, a pyrazine analog with no nucleoside-like structure, can also be phosphorylated and acts as a nucleotide analog that selectively inhibits viral RdRp.315 It is being developed and manufactured by Toyama Chemical (a subsidiary of Fujifilm) and was approved for influenza virus treatment in Japan in 2014. An in vitro study showed that favipiravir exerts beneficial effects in Vero E6 cells infected with SARS-CoV-2 with a half-maximal effective concentration (EC50) of 61.88 μM and a half-cytotoxic concentration (CC50) of >400 μM.276 Many clinical trials proposed to use favipiravir in the treatment of COVID-19. Cai et al. reported that after favipiravir treatment, a significant improvement in chest CT of COVID-19 patients was observed, indicating that favipiravir is associated with better therapeutic responses in COVID-19 patients in terms of disease progression and viral clearance.316 In a multicenter randomized study, Dabbous et al. discovered that the patients who received favipiravir had a lower mean duration of hospitalization than patients in the chloroquine group.317 Thus, favipiravir has been recommended by Thailand’s Department of Disease Control for mild to moderate COVID-19 cases in both adults and children, while recommendations from India include mild COVID-19 patients with or without comorbidities.45 Furthermore, Rabie discovered a derivative of favipiravir named cyanorona-20 as a promising anti-SARS-CoV-2 compound.318 Pyrazine derivatives may serve as guides for further discovery of anti-SARS-CoV-2 agents.
As the basis of nucleotides, pyrimidines widely participate in viral metabolism. Thus, nucleobase analogs were found to effectively inhibit SARS-CoV-2 by various pathways. Among them, baricitinib, a pyrrolopyrimidine analog, is widely applied for treatment of severe COVID-19 in combination with remdesivir.319 Baricitinib is an oral selective inhibitor of JAK1 and JAK2.320 It was initially predicted by artificial intelligence algorithms as a potential treatment strategy against SARS-CoV-2. According to a study by Bronte et al., baricitinib improved the clinical outcomes of SARS-CoV-2 infection, affected the immune landscape in participants with COVID-19, and modified immune-suppressive features of myeloid cells.321 A study by Marconi et al. suggested that baricitinib reduces 28-day and 60-day mortality when used in addition to the current standard of care.322 As such, baricitinib plus standard of care could be a treatment option to reduce overall deaths globally. Another pyrrolopyrimidine analog, abivertinib, was found to depress cytokine production in patients with COVID-19.323 Several pyrimidine analogs have also been found to combat SARS-CoV-2. For example, according to a recent study conducted by Huntington et al., GLPG-0187, which bears a pyrimidin ring, effectively blocked SARS-CoV-2 pseudovirus infection across multiple viral variants, especially the Omicron and Delta pseudovirus variants, in a dose-dependent manner.324 Indu et al. reported that raltegravir combats SARS-CoV-2, because it demonstrated the highest interaction energy with Mpro and had high bioavailability among 65 FDA-approved small molecule antiviral drugs.325 Fostamatinib might be used to treat severe COVID-19.326 Other pyrimidine analogs, including ambrisentan and apilimod, were also reported to be promising agents for SARS-CoV-2 treatment.327,328
Another compound class, dsRNA, was also found to inhibit SARS-CoV-2. Rintatolimod, a Toll-like receptor 3 (TLR3) agonist, was reported to exert antiviral effects in human pancreatic cancer cells by activating the innate immune system, suggesting it could be used in the treatment of cancer patients who suffer from SARS-CoV-2 infection.329 Poly-ICLC is a synthetic complex of carboxymethylcellulose, polyinosinic-polycytidylic acid, and poly-L-lysine dsRNA.330 A phase I trial to study the safety and immunogenicity of poly-ICIC in healthy vaccinated COVID-19 adults is in its recruitment stage.
Flavonoids
Flavonoids are a class of bioactive substances derived from plants. Chemically, flavonoids have a C6-C3-C6 skeleton structure, which consists of two phenyl rings and an oxygen heterocyclic ring.331 By regulating key enzymes participating in biological processes, flavonoids possess antioxidant, anticancer, anti-inflammatory, and antiviral properties.332 Due to their broad bioactivity, they may play complex roles to treat SARS-CoV-2 infection by blocking ACE2 receptor in host cells, directly inhibiting viral RdRp and Mpro, and affecting the activity of various inflammatory enzymes (such as phospholipase A2, cyclooxygenases [COXs], TK, and so on).333 These mechanisms make flavonoids an excellent supportive care strategy for patients suffering from chronic post-COVID-19 syndrome.
Flavonols, also named 3-hydroxyflavones, are the most abundant and widely distributed flavonoids in the nature. Chemically, these molecules differ from many other flavonoids due to the hydroxyl group at position 3 of the flavonol skeleton. Quercetin, the most abundant flavonoids in edible plants, is a flavonol with five hydroxy groups placed at the 3-, 3′-, 4′-, 5-, and 7-positions. It has broad-spectrum antiviral ability against a variety of viruses, including HIV, poliovirus, Sindbis virus, respiratory viruses, Mayarovirus, and Mengo virus.334,335 Pan et al. reported that quercetin may exert anti-SARS-CoV-2 effects by affecting the binding of viral S protein to the ACE2 receptor.336 Further, the anti-SARS-CoV-2 effect of quercetin was also thought to be achieved by (i) inhibiting Mpro and PLpro proteinase of SARS-CoV-2 and (ii) acting as a zinc ionophore.337 Currently, several clinical studies of quercetin are underway. A phase IV clinical study supported by the Ministry of Health of Saudi Arabia on quadruple therapy with quercetin, zinc, bromelain, and vitamin C for COVID-19 patients is in its recruitment stage (NCT04468139). Myricetin, a 7-hydroxyflavonol, has been isolated from the leaves of Myrica rubra and other plants. In research conducted by Su et al., myricetin inhibited Mpro at >90% at a concentration of 10 μM, and its EC50 value in Vero E6 cells infected with SARS-CoV-2 was 8.00 μM.94 The 3-hydroxyl group of flavonol can be glycosylated, thus forming flavonol glycosides, which are found in plants. As a quercetin O-glycoside, quercitrin is obtained by placing an alpha-L-rhamnosyl moiety at position 3 of quercetin via a glycosidic linkage. Several in silico studies have reported that quercitrin may be used against SARS-CoV-2 based on its affinity to the serine protease TMPRSS2, Mpro, and PLpro.338,339,340
There are a series of compounds whose backbone consists of a flavonol structure. They have also been found to be effective in combating COVID-19. For example, flavonolignans are a family of compounds containing a flavonol moiety linked together with coniferyl alcohol.341 Silymarin, extracted from the botanical source Silybum marianum, is a mixture of flavonolignans (silybin, isosilybin, silychristin, and siliandrin) and a flavonol (taxifolin).342 It is commonly known for its hepatoprotective potential.343 Its anti-SARS-CoV-2 effect was thought to be achieved by inhibiting the expression of the host cell surface receptor TMPRSS2.342 Hanafy et al. developed silymarin/curcumin dual-loaded BSA nanoparticles as an inhalable delivery system to treat pneumonia.344 According to their results, silymarin exhibited antiviral activity against SARS-CoV-2 at a concentration of 25 μg/mL in vitro. They reported that silymarin could protect the lungs during SARS-CoV-2 infection due to their anti-inflammatory and antioxidant effects, and it could inhibit the ACE2 receptor, thus preventing viral entry. As a natural-derived compound mixture, silymarin might be a good option for treating COVID-19 owing to its multifunction properties. A phase III clinical study of silymarin is in its recruitment stage, which is aimed at assessing the clinical outcome in adults with COVID-19 pneumonia under standard care plus placebo or oral silymarin (NCT04394208).
In addition to the abovementioned flavanols, the anti-SARS-CoV-2 effects of flavones, which have a 2-phenyl-1-benzopyran-4-one backbone, were also studied. Luteolin is the most investigated flavone compound. Luteolin is a flavone which bears four hydroxy groups located at the 3′-, 4′-, 5-, and 7-locations. It is obtained from the plant Reseda luteola. It was first isolated in pure form and named in 1829 by the French chemist Michel Eugène Chevreul.345 Results obtained from relaxed complex scheme analysis, classical molecular docking simulations, and metadynamics simulations suggest luteolin blocks SARS-CoV-2 entry into cells.346,347 A system pharmacology and bioinformatic analysis study conducted by Xie et al. indicated it has great potential to be used for treating COVID-19/asthma comorbidity due to its effects on viruses, regulating inflammation and immune responses, reducing oxidative stress, and regulating blood circulation.348 Luteolin was found to be safe for human use and showed good drug properties. Clinical results suggest that oral luteolin supplementation improves the recovery of olfactory function after COVID-19. Besides the above common flavones, amentoflavone, a hydroxyflavone and bioflavonoid, also has shown binding affinity with Mpro, RdRp, NSP13, NSP15, and ACE2 in several in silico surveys.349,350,351 Similar to flavanols, the hydroxy groups of flavones can be glycosylated, thus forming flavone glycosides. Baicalin, a 7-O-glucuronide of baicalein, is a biologically active flavonoid of natural origin obtained primarily from the roots of Scutellaria baicalensis Georgi. Zandi et al. have demonstrated that baicalein and its aglycon baicalein can directly inhibit the activity of SARS-CoV-2 RdRp and that it exhibits in vitro anti-SARS-CoV-2 activity with an EC50 of 4.5 µM and an EC90 of 7.6 µM.352 Su et al. also found its binding activity with Mpro and proved its anti-SARS-CoV-2 activity in vitro. Their further study revealed that baicalin and baicalein as two bioactive ingredients of Shuanghuanglian (a Chinese traditional medicine) provides supporting evidence for the antiviral activity of Shuanghuanglian. However, their exact antiviral ability has to be verified in animal models or clinical trials.
The effects of flavanols represented by epigallocatechin gallate (EGCG) against COVID-19 have also been studied. EGCG is a phenolic antioxidant found in a number of plants, including green and black tea, with reported antiviral effects against influenza virus, HIV, and HBV.353,354 Unlike other flavonoids with a chromone part, it bears a 2-phenyl-3,4-dihydro-2H-chromen-3-ol skeleton.355 EGCG exerts inhibitory effects on SARS-CoV-2 replication through its actions on ACE2, Mpro, and RdRp.356 Jang et al. demonstrated that EGCG inhibits SARS-CoV-2 Mpro activity in 293T cells in a dose-dependent manner without signs of cytotoxicity at any dose used.357 Chiou et al. conducted an in vitro study on the inhibitory effects of EGCG against SARS-CoV-2 Mpro. EGCG inhibited the activity of SARS-CoV-2 Mpro, thus suggesting its potential application in the treatment of SARS-CoV-2 infection.358 It is worth noting that a clinical phase II/III study of EGCG is underway to determine its chemoprophylactic effects on COVID-19 in healthy workers (NCT04446065). Other flavanols, including cianidanol,359 epicatechin gallate,360,361 and procyanidin,362 have also been found to have potential anti-SARS-CoV-2 effects in vitro.
Phenylpropanoids
Phenylpropanoids are a family of plant-derived compounds with a C6–C3 structure. In general, phenylpropanoids are derived from the shikimic acid pathway via phenylalanine and tyrosine. This phenylpropanoid metabolism pathway is a major anabolic pathway in plants, which plays a vital role in several processes, especially biotic and abiotic stress responses.363 Phenylpropanoids act as antioxidants and free radical scavengers. Their applications as antioxidant, anticancer, antiviral, anti-inflammatory, and antibacterial agents have attracted interest.364 Several phenylpropanoids were found to exert anti-SARS-CoV-2 effects. Some of them have demonstrated potential anti-SARS-CoV-2 effects in vitro or by computational analysis.
Hydroxycinnamic acid derivatives belong to the basic phenylpropanoids. Based on the C6-C3 structure, they also possess an aromatic carboxylic acid substituted by phenolic hydroxyl groups. As a common derivative of hydroxycinnamic acid, caffeic acid possesses a phenyl ring substituted by hydroxy groups at the 3- and 4-positions.365 It is an orally bioavailable small molecule mainly found in Pavetta indica and Eupatorium cannabinum. Further studies have shown its potential antiviral activity against HBV366 and HPIV3.367 Several in silico molecular docking studies have revealed it could specifically bind to SARS-CoV-2 Mpro368 and Membrane protein.369 Chlorogenic acid, the ester of caffeic acid and quinic acid, is often found in coffee and black tea. Several studies have pointed out that chlorogenic acid and its derivatives have good antiviral activity against various types of viruses, including HIV, influenza A virus, herpes simplex virus (HSV), and hepatitis B virus (HBV).370 Its anti-SARS-CoV-2 ability was first predicted by Yu et al., whose molecular docking study revealed that chlorogenic acid could stably bind with ACE2, indicating it may inhibit SARS-CoV-2 entry into cells.371 Another molecular docking simulation conducted by Gizawy et al. suggested that chlorogenic acid can interact with the Asn-142, His-164, Arg-188, and Met-165 residues of the active site in Mpro of SARS-CoV-2.372 According to their in vitro study on Vero E6 cells, chlorogenic acid had an IC50 of 360 μg/mL and a selective index (CC50/IC50) of 8 against SARS-CoV-2. Chen et al. found that chlorogenic acid and its isomers (chlorogenic acid, neochlorogenic acid, and cryptochlorogenic acid) all exhibited ACE2 inhibitory activities with IC50 values of about 40 μM.373 As a chlorogenic acid derivative, isochlorogenic acid A is the diester obtained by the condensation of the hydroxy groups at positions 3 and 5 of (−)-quinic acid with the carboxy group of trans-caffeic acid. Recent computational studies have predicted it to have binding ability with Mpro of SARS-CoV-2.374,375 Salvianolic acid B (Sal-B), one of the main active ingredients of Salvia miltiorrhiza, is a hydroxycinnamic acid with strong antioxidant effects.376 Hu et al. revealed that by binding to the RBD of S protein and ACE2, Sal-B can inhibit the entry of SARS-CoV-2 pseudovirus into cells that highly express ACE2.377 A similar anti-SARS-CoV-2 effect can be also achieved by Sal-A and Sal-C. According to a study by Wang et al., Sal-A dose-dependently alleviates the pathological alterations in mice with acute lung inflammation due to infection with SARS-CoV-2 S protein-pseudotyped virus in a dose-dependent manner.378 Sal-C has been reported to potently bind to the 6-HB core of S protein, thereby inhibiting SARS-CoV-2 infection.379 According to the in vitro results, Sal-C potently inhibits the membrane fusion of S protein–overexpressing HEK293T and Vero E6 cells with an IC50 of 1.71 μM.
Like other natural products, hydroxycinnamic acid derivatives often occur as glycosides in nature. For example, forsythoside A is a phenylethanol glycoside product isolated from the dried fruit of forsythia, of which it is the main active ingredient.380,381 Chen et al. demonstrated that forsythoside A acid could form suitable steric complementarities with the binding interface of ACE2 with SARS-CoV-2 S protein by ACE2 bio-chromatography screening.373 Fu et al. found it has strong docking affinities with S protein’s RBD of SARS-CoV-2 and its variants (Alpha [B.1.1.7], Beta [B.1.351], and Delta [B.1.617]), as well as NRP1 and Mpro .382 Moreover, biolayer interferometry binding (BLI) analysis results revealed that forsythoside A may block or interfere with the binding of the RBD to other receptors in the body (e.g., ACE2) by binding to the RBD.383
Besides hydroxycinnamic acid derivatives, coumarins and lignans also belong to the phenylpropanoids. Coumarins bear a 2H-chromen-2-one (2H-1-benzopyran-2-one or benzo-alpha-pyrone) ring.384 Bergamottin, a natural product found in bergamot, exhibits a structure similar to that of furanocoumarin. Zhou et al. have reported its inhibitory activity against SARS-CoV-2 both in vitro and in vivo.385 According to their results, bergamottin interferes with various stages of viral life cycle, including blocking the viral fusion and reducing the viral RNA replication, and effectively protects a golden Syrian hamster model from SARS-CoV-2 infection. It is worth noting that bergamottin inhibits CYP450 activity like ritonavir, which means that it may be promising to combine it with other anti–SARS-CoV-2 drugs. Lignans are constituted by the union of two phenylpropane units.386 As a lignan and glycoside, phillyrin is the main active ingredient of the traditional Chinese medicine Forsythia suspensa. Ma et al. reported that phillyrin could significantly inhibit SARS-CoV-2 and HCoV-229E replication in vitro.387 Further, Lai et al. discovered that phillyrin could be used to treat COVID-19 and influenza co-infection since it not only inhibits the replication of both viruses, but also possesses the ability to regulate hypoxia-cytokine storm based on bioinformatics network pharmacology analysis.388
Terpenoids
Terpenoids are a large class of natural compounds based on isoprene units. They display various biological activities and have diverse structures. Their lipophilicity was assumed to empower their tendency to enter into cellular membranes, thus affecting functions of membrane proteins or disrupting membrane integrity. Terpenoids exert multiple effects, including anti-inflammatory and antiviral effects.389 By enhancing the adaptive immune response and inverting the chronic inflammatory response elicited by the virus, terpenoids are thought to assist in the treatment of COVID-19 and associated symptoms.390,391 Many preclinical studies have found terpenoids with direct anti-SARS-CoV-2 effects by binding them to proteins or viral receptors.
Sesquiterpenoids are a class of enormously diverse natural products derived from a 15-carbon precursor. Artemisinin is the sesquiterpene endoperoxide lactone extracted from the herb Artemisia annua as the basis for the currently preferred treatment for malaria.392,393 Antiviral activities of artemisinin and its analogs against HSV type 1, Epstein–Barr virus, HBV, HCV, bovine viral diarrhea virus, and human cytomegalovirus have been reported.394,395,396 Molecular dynamics analysis revealed that artemisinin interacts with Lys-353 and Lys-31, which are binding hotspots of the SARS-CoV-2 S protein, in two patterns.397 Cao et al. conducted an in vitro study of artemisinin analogs, which revealed Arteannuin B exerts the strongest anti-SARS-CoV-2 effects, with an EC50 of 10.28 ± 1.12 μM.398 Artesunate and dihydroartemisinin showed similar EC50 values of 12.98 ± 5.30 μM and 13.31 ± 1.24 μM, respectively. Another in vitro study found that artemisinin alone showed an estimated IC50 of about 70 μM, and the clinically used artemisinin derivatives artesunate, artemether, and dihydroartemisinin were ineffective or cytotoxic at elevated micromolar concentrations.399 An open-label, non-randomized controlled trial by Li et al. revealed that the combination of artemisinin and piperaquine shortens the time SARS-CoV-2 remains in the body.400 Besides artemisinin analogs, other sesquiterpenoids, including beta-eudesmol,401 nootkatone,402,403 and lactupicrin,404 were all included in computational studies of SARS-CoV-2.
Diterpenoids are a chemically heterogenous group of compounds, all with a C20 carbon skeleton based on four isoprene units.405 Andrographolide is a diterpenoid extracted from traditional Chinese medicine. It is a well-known diterpenoid with broad therapeutical applications, including the inhibitory effect on HIV virus, influenza A virus H1N1, H3N2 and influenza B virus.406 In a molecular docking study of Rajagopal et al., andrographolide was predicted to bind to Mpro of SARS-CoV-2. Further, according to an in vitro anti-SARS-CoV-2 assay by Hu et al., andrographolide (EC50 = 11.12 µM, CC50 = 95.73 µM, SI = 8.61) showed excellent anti-SARS-CoV-2 activity.391 A phase III clinical study using andrographolide for COVID-19 treatment is in the recruitment stage in Thailand (NCT05019326). Another diterpenoid, paclitaxel, is a compound extracted from the Pacific yew tree Taxus brevifolia with antineoplastic activity. Paclitaxel binds to tubulin and inhibits the disassembly of microtubules, thereby resulting in the inhibition of cell division. This agent also induces apoptosis by blocking the function of the B-cell Leukemia 2 (Bcl-2) protein, which inhibits apoptosis. Using a network-based drug repurposing strategy, Adhami et al. found that paclitaxel has four interactions with genes associated with SARS-CoV-2 infection, which is the most remarkably identified candidate drug for COVID-19.407 Molecular docking and molecular dynamics simulation analyses by Pingali et al. indicated that paclitaxel has high affinity to RdRp of SARS-CoV-2.408 However, the impairment of the helper/suppressor T cell ratio and depletion of CD4 + T cells, CD8 + T cells, and natural killer cells during paclitaxel therapy can result in susceptibility to infection and pneumonia.409 Paclitaxel also increases the alveolar capillary membrane permeability, resulting in several adverse effects, including pulmonary diffusion dysfunction, which raises concerns for COVID-19 treatment.410 Other diterpenoids, including triptolide,411 oridonin,412,413 and carnosic acid,414,415 were also identified to have anti-SARS-CoV-2 activities by computational methods.
The beneficial effects of triterpenoids such as glycyrrhizic acid in COVID-19 were investigated. Glycyrrhizic acid is obtained from licorice, and it has been shown to inhibit various viruses, such as HIV, HBV, and herpes zoster virus.416 Molecular docking studies have identified its binding affinity with Mpro, S protein, and NSP15 of SARS-CoV-2. Zhao et al. developed highly biocompatible glycyrrhizic acid nanoparticles with the ability to inhibit murine coronavirus MHV-A59.417 Their results indicated that glycyrrhizic acid nanoparticles could reduce proinflammatory cytokine production caused by MHV-A59 or SARS-CoV-2 N protein, indicating their potential for COVID-19 treatment. Bardoxolone and bardoxolone methyl are oleanolic acid-derived synthetic triterpenoid compounds that activate the Nrf2 pathway and inhibit the NF-κB pathway, and they can be used to treat chronic kidney diseases.418 Sun et al. determined the binding activities of both compounds to the active site cysteine of SARS-CoV-2 Mpro with computational analyses.419 Further in vitro experiments showed that bardoxolone methyl and bardoxolone inhibit SARS-CoV-2 replication in Vero E6 cells with EC50 values of 0.29 μM (SI = 23.9) and 0.43 μM (SI = 56.6), respectively, and in human Calu-3 cells with EC50 values of 0.20 μM (SI = 5.8) and 0.42 μM (SI = 28.2), respectively. Alpha-hederin is a triterpenoid saponin that is produced by attaching a 2-O-(6-deoxy-alpha-L-mannopyranosyl)-alpha-L-arabinopyranosyl residue to hederagenin at position 3 via a glycosidic linkage. Studies have shown its potential to inhibit SARS-CoV-2 RdRp, Mpro, and the S protein RBD domain by molecular docking methods.420,421
Carotenoids are a group of compounds with a polyene chain backbone, mostly eight-isoprenoid building blocks (tetraterpenoids). They are biosynthesized by plants, bacteria, and fungi but not humans; therefore, humans need to obtain them from the diet.422 Carotenoids exhibit many health and pharmaceutical effects in the body, and they have been used to treat COVID-19 and related symptoms.423 The carotenoid crocetin is a 20-carbon natural carotenoid which is also a diterpenoid and a vitamin A analog.424 Kordzadeh et al. identified it as a candidate drug for COVID-19 treatment based on its high binding energies to S protein and Mpro of SARS-CoV-2 virus.425 Further, in a phase I/II clinical trial on COVID-19 patients suffering from severe respiratory complications, a single injection of LEAF-4L6715 (a liposomal nanocarrier encapsulating crocetin) enhances the oxygenation of vascular tissue and therefore has the potential to improve the clinical outcomes of ARDS and COVID-19 in severely impacted patients.426 The sodium salt of the trans-isomer of crocetin, trans sodium crocetinate, also has entered a phase I/II clinical trial for treatment of COVID-19 patients. Crocin is a glucoside derived from crocetin. As an antioxidant, crocin has been investigated for the treatment of hyperglycemia, metabolic syndrome, hypertriglyceridemia, and hypercholesterolemia.427,428 It was reported that crocin has the potential to limit the progression and severity of SARS-CoV-2 infection due to its antioxidant, anti-inflammatory, and immunomodulatory properties.429 By employing computational methods, Kordzadeh et al. and Aanouz et al. identified its binding affinity towards the Mpro of SARS-CoV-2.401,425 Stalin et al. also reported its distinctive strong interaction with the RBD of SARS-CoV-2 S protein.430 Beta-carotene is a vitamin A precursor composed of two retinyl groups. In critical COVID-19 patients, the concentration of beta-carotene is decreased compared to the reference range.431 In a study conducted by Xia et al., the binding affinity of beta-carotene to the AKT1 pocket was determined, suggesting potential therapeutic effects on COVID-19.432 Astaxanthin is derived from a hydride of beta-carotene. It is a carotenoid with no vitamin A activity but still has antioxidant and anti-inflammatory properties. Some studies have shown that it can be used to prevent and counteract the symptoms of COVID-19.433,434 An in -silico study also revealed that it can interact with SARS-CoV-2 proteins (Mpro, RdRp, NSP15, and S protein).435 In addition, the three main forms of vitamin A, retinol, retinal, and tretinoin, are also carotenoids with potential anti-COVID-19 effects. Vitamin A is the most evaluated nutrient due to its impact on immunity. It is a key regulator of immune function and augments the innate response to RNA viruses. The dsRNA formed within the cells by viral pathogens is primarily sensed by pattern recognition receptors including retinoic acid-inducible gene I (RIG-I) and RIG-I-like receptors (RLRs). Vitamin A has been demonstrated to decrease mortality due to measles and Ebola in clinical studies.436,437 Based on this, vitamin A is considered to hold benefits for COVID-19 patients as therapeutic agent or as adjuvant with vaccines. Although with unspecific antiviral mechanisms and effects, it is encouraging to study its benefit for COVID-19 patients due to its high safety, low cost, and availability in most of the developing countries. To verify its function, two phase II clinical trials have been registered to evaluate the effects of vitamin A supplementation on disease in children with COVID-19 (NCT04920760) or in patients with COVID-19-related olfactory dysfunction (NCT04900415).
Cannabinoids are a diverse group of compounds derived from Cannabis sativa. Most of them are terpenoids with complex anti-inflammatory and antiviral effects.438 The effects of cannabinoids on COVID-19 patients have been investigated.439 Cannabidiol is an orally available cannabinoid that is largely related to the human endocannabinoid system. It has been approved by the FDA and EMA for treatment of Dravet syndrome and Lennox–Gastaut syndrome.440,441,442 Raj et al. screened 32 cannabinoids with binding affinity to SARS-CoV-2 Mpro.443 Five cannabinoids were selected, and their antiviral abilities were tested in vitro. Cannabidiol (IC50 = 7.91 μM) was found to be exert more potent antiviral effects against SARS-CoV-2 in vitro compared to the reference drugs lopinavir, chloroquine, and remdesivir (IC50 ranges of 8.16–13.15 μM). Nguyen et al. reported that cannabidiol treatment could significantly inhibit SARS-CoV-2 replication in mice.444 Moreover, they found that patients with a medical record of cannabidiol for seizure-related conditions exhibited a lower SARS-CoV-2 infection rate than non-cannabidiol patients, which revealed cannabidiol is negatively associated with indications of SARS-CoV-2 infection. Currently, seven clinical trials on cannabidiol for the treatment of COVID-19 and related diseases are underway. Based on these studies, cannabidiol may be a promising drug for treating COVID-19. Dronabinol, also named delta-9-tetrahydrocannabinol (Δ9-THC), is the primary psychoactive component of cannabis (marijuana). Mohammed et al. demonstrated that Δ9-THC could lead to a 100% survival rate, decreased lung inflammation, and the suppression of cytokine storm in a mouse model of ARDS induced by staphylococcal enterotoxin B, suggesting Δ9-THC could be used to treat ARDS associated with COVID-19.445 Pitakbut et al. reported that Δ9-THC acts as an inhibitor against both Mpro and ACE2 with IC50 values of 16.23 ± 1.71 µM and 11.47 ± 3.60 µM, respectively.446
Steroids
Steroids are compounds that contain four cycloalkane rings with a perhydrocyclopentano[α]phenanthrene core structure. They are found in plants and animals. Synthetic steroids were developed to enhance their biological activities. Steroids play an important role in people’s lives.447 Various sex hormones, corticosteroids, vitamin D, cholesterol, and cardiac glycosides are natural steroid compounds with vital physiological activity. Steroids have been used by the pharmaceutical industry and have various applications, such as anticancer drugs, anti-inflammatory agents, anticonvulsants, contraceptives, anti-autoimmune disease drugs, and fertility stimulants.448,449 Steroids such as glucocorticoids exert effects on severe COVID-19 patients due to their anti-inflammatory effects. Other steroids, including vitamin D and sex hormones, are also beneficial for COVID-19 patients due to their immunomodulatory action.450,451,452
The effectiveness of two main classes of corticosteroids, glucocorticoids and mineralocorticoids, in COVID-19 treatment were explored in different observational studies.453 During the initial phase of the SARS-CoV-2 pandemic, 44.9% of hospitalized patients with COVID-19 pneumonia received glucocorticoid therapy.454 The clinical practice guideline of “The Infectious Diseases Society of America” recommends the use of glucocorticoids in severe COVID-19 patients.455 As a synthetic glucocorticoid, dexamethasone is derived from a hydride of a pregnane with anti-inflammatory function. In a preliminary report of a controlled, open-label trial comparing a range of possible treatments for hospitalized patients with COVID-19, dexamethasone treatment showed a reduction in 28-day mortality in patients with COVID-19 who received respiratory support.456 Another randomized clinical trial comparing intravenous dexamethasone plus standard care with standard care revealed a statistically significant increase in the number of days alive and the number of days free of mechanical ventilation over a 28-day period in the dexamethasone group.457 The glucocorticoid methylprednisolone is an FDA-approved anti-inflammatory and systemic immunosuppressive corticosteroid. A study proposed that high-dose methylprednisolone significantly decreased the recovery time compared with dexamethasone in COVID-19 patients.458 Through describing the clinical characteristics and outcomes in patients with COVID-19 pneumonia who developed ARDS or died, Wu et al. found that treatment with methylprednisolone decreased the risk of death among patients with ARDS.459 In another clinical trial on 46 severe COVID-19 patients, Wang et al. concluded that early, low-dose, and short-term application of methylprednisolone was associated with better clinical outcomes, which revealed it should be considered before the occurrence of ARDS.460 However, there are concerns since Li et al. reported that high-dose methylprednisolone potentially increased the mortality of patients with severe COVID-19.461 The possible reasons might be the delayed clearance of virus under high-dose glucocorticoid treatment. Thus, despite the effective anti-inflammatory effect, glucocorticoids should be applied carefully, giving due consideration to factors such as initiation of the therapy, dosage, and route of administration.
Vitamin D is a group of steroids that have an open ring structure. It is an essential metabolite clinically associated with infection, reproduction, the cardiovascular condition, and cancer.462,463,464,465,466 In a retrospective, observational study of Carpagnano et al., COVID-19 patients with severe vitamin D deficiency had a significantly higher mortality risk and poor prognosis rate, suggesting adjunctive treatment with vitamin D might improve disease outcomes.467 The proposed mechanisms whereby vitamin D reduces the risk of COVID-19 have been clearly summarized in a review of Barrea et al..468 For these reasons, numerous clinical studies have used vitamin D as supplementation in COVID-19 treatment. Cholecalciferol, also known as vitamin D3, is the endogenous form of vitamin D. According to a randomized clinical trial in patients with mild to moderate COVID-19, 5000 IU daily oral vitamin D3 supplementation for 2 weeks reduces the time to recovery for cough and gustatory sensory loss among patients.469 Calcifediol is the major circulating metabolite of vitamin D3, and it is the best indicator of the body’s vitamin D stores. In a pilot randomized clinical study in 76 patients hospitalized with COVID-19 infection, administration of a high dose of calcifediol significantly reduced the need for ICU admission of patients when all of them received the best available therapy and the same standard care.470 A retrospective, multicenter, open, non-randomized cohort study reported by Alcali-Diaz et al. evaluated if calcifediol supplementation influences in-hospital mortality of COVID-19 patients under standard care and the best available treatment.471 According to their results, treatment with calcifediol was significantly associated with lower in-hospital mortality during the first 30 days. Moreover, a multicenter, randomized, double-blind, placebo-controlled clinical trial revealed that calcifediol was able to improve the immune function of COVID-19 patients by increasing blood lymphocyte counts.472
Sex hormones are another class of steroids widely studied for COVID-19 treatment. Adverse outcomes are more common among elderly and male COVID-19 patients. The levels of sex hormones such as progesterone, which has been shown to modulate a more robust immune response, are low in these people.473,474 Based on these observations, it is rational to consider sex hormones for treatment to alleviate COVID-19 inflammatory and cytokine storm events, as they can influence immune system function against SARS-CoV-2 infection, thus reducing the adverse effects of COVID-19. Estradiol, a 17-β-hydroxy steroid, is a naturally occurring hormone in females. Studies revealed that it may combat COVID-19 by inhibiting the SARS-CoV-2 S-protein-induced ACE2-dependent activation of NOX2, MCP-1, and ROS production.475,476 Baristaite et al. reported that treatment of A549 human lung epithelial cells with 17-β-estradiol reduced the cellular mRNA levels of ACE2 and TMPRSS2.477 This outcome may contribute to reduced SARS-CoV-2 infection of lung epithelial cells. Estrogen is thought to inhibit initial viral responses and attenuate cytokine-storm-induced endothelial dysfunction, so it might serve as a novel therapy for COVID-19 patients.476 Progesterone is another sex hormone with immunomodulatory and anti-inflammatory functions. Su et al. revealed that higher levels of progesterone alleviate COVID-19 symptoms, since progesterone promotes the innate antiviral response both in vitro and in vivo.478 Yuan et al. indicated that treatment with progesterone ameliorated the severity of SARS-CoV-2-caused pneumonia in a Syrian hamster model.479 In addition, a randomized, controlled pilot trial suggested that supplementation with progesterone in hospitalized men with moderate to severe COVID-19 resulted in shorter periods of oxygen supplementation (median, 4.5 vs. 7.5 days) and shorter hospitalization periods (median, 7.0 vs. 9.5 days) as compared with control subjects.480
Azoles
Azoles are nitrogen-, sulfur-, and oxygen-containing compounds with a five-membered ring system. Azoles comprise various rings, including thiazole, oxazole, triazole, imidazole, and pyrazole. Most of them are known as antifungal agents, and other bioactivities such as antidiabetic, immunosuppressant, anti-inflammatory, anticancer, and antiviral effects also contribute to their pharmaceutical functions.481,482,483 Many synthetic small molecules bearing core structures of azoles have been studied in the fight against COVID-19.
Thiazoles are five-membered heterocyclic compounds containing sulfur and nitrogen. Nitazoxanide belongs to the class of thiazoles, and is also a synthetic benzamide.484 As a broad-spectrum antiviral drug, nitazoxanide inhibits a broad range of influenza A and B viruses including influenza A (pH1N1) and the avian A (H7N9) virus, as well as viruses that are resistant to neuraminidase inhibitors.485 Riccio et al. demonstrated that nitazoxanide could hamper the glycosylation of SARS-CoV-2 S protein, thus hindering infectivity of the virus.486 This study also revealed that nitazoxanide is equally effective against different variants of SARS-CoV-2, including the Delta variant. According to a preprint provided by Miorin et al., nitazoxanide exhibited an IC50 of 4.04 μM in Vero E6 cells against SARS-CoV-2, and a significant inhibitory effect was observed in different human cell lines including stem cell-derived human alveolar epithelial type 2 cells.487 This in vitro inhibitory effect was also confirmed against different SARS-CoV-2 variants (Beta, Gamma, and Delta). Moreover, this study also confirmed the antiviral activity of nitazoxanide by oral treatment in hamsters. The clinical application potential of nitazoxanide was also verified in placebo-controlled trial.488,489 The safety of high-dose nitazoxanide was also proved in a phase I clinical trial in healthy volunteers recently.
Oxazole is a five-membered heteroarene containing an oxygen in the 1-position and a nitrogen in the 3-position.490 Asapiprant, which contains an oxazole ring, is an antagonist of the prostaglandin D2 receptor (PTGDR).491 According to a study reported by Wong et al., treatment with asapiprant could protect aged mice from lethal SARS-CoV-2 infection, since elevated levels of prostaglandin D2 (PGD2) contribute to poor outcomes in SARS-CoV-2-infected aged mice.492 Proxalutamide is an androgen receptor antagonist, which also contains an oxazole ring, and it exhibited anti-SARS-CoV-2 potential in a clinical trial.493 Al-Wahaibi et al. synthesized novel oxazole-based macrocycles and evaluated their antiviral activities in vitro.494 Isopropyl triester-13 and triacid-14 exhibited IC50 values of 18.3 and 18.95 μM, respectively, on Vero E6 cells against SARS-CoV-2. Moreover, compound 13 exhibited a high inhibitory activity against Mpro of SARS-CoV-2 with an IC50 of 2.58 µM. Rivaroxaban, an orally bioavailable oxazolidine derivative, is an anticoagulant and a direct factor Xa inhibitor. Since COVID-19 can manifest with hypercoagulability, pulmonary intravascular coagulation, microangiopathy, and venous thromboembolism or arterial thrombosis, it is recommended to provide thromboprophylaxis with rivaroxaban in postdischarge patients.495
Triazole is a five-membered aromatic heterocyclic compound containing three nitrogen atoms. Itraconazole is a triazole antifungal agent used for treatment of systemic and superficial fungal infections.496 According to a study by Damme et al., itraconazole has antiviral activity in human Caco-2 cells with an EC50 of 2.3 μM against SARS-CoV-2.497 Yang et al. found that itraconazole could inhibit viral entry by targeting the 6-HB fusion core of SARS-CoV-2 S protein.498 In addition, Schloer et al. reported that traconazole–remdesivir combinations display synergistic effects and inhibit production of SARS-CoV-2 particles with >90% efficiency.499 According to their results, we can conclude that by interfering with different steps of the viral cycle, combination of drugs might be an effective and feasible way to combat fast-spreading SARS-CoV-2 variants. Selinexor contains structures of both triazole and hydrazine. It is a first-in-class small molecule inhibitor of chromosome region maintenance 1 protein (CRM1, also known as exportin 1 [XPO1]), with potential antineoplastic activity.500,501,502 By generating a series of transgenic fly lines for individual SARS-CoV-2 genes, Zhu et al. found that expression of ORF6 leads to reduced viability and tissue defects of flies, and selinexor could attenuate these phenotypes.503 Further experiments verified that ORF6 is a highly pathogenic protein encoded by the SARS-CoV-2 genome in human cell lines; thus, selinexor is a candidate drug for treatment of SARS-CoV-2-ORF6 protein-induced cellular damage.504 Kashyap et al. found that selinexor treatment reduced the viral load in the lungs and protected against tissue damage in the nasal turbinates and lungs in a ferret model of COVID-19.505 Bemcentinib is also a synthetic triazole with antifungal activity. It has an EC50 of 1.1 μM against SARS-CoV-2 in Vero E6 cells.506 Sitagliptin is a triazolopyrazine and a trifluorobenzene with multiple activities, including inhibitory effects on dipeptidyl peptidase-4 (DPP-4). DDP-4 is a target protein of the SARS-CoV-2 S protein; thus, sitagliptin is a candidate drug for COVID-19 treatment.507 Solertes et al. demonstrated that treatment with sitagliptin in hospitalized patients with type 2 diabetes and COVID-19 was associated with reduced mortality.508
The effects of other azoles, including pyrazole and selenzole, on COVID-19 were also studied. Ibrutinib is a pyrazolopyrimidine and a member of the acrylamides, and serves as an oral inhibitor of BTK that is used in the therapy of refractory chronic lymphocytic leukemia and mantle cell lymphoma.509,510 Treon et al. demonstrated that it may provide protection against lung injury and even improve pulmonary function in hypoxic patients with COVID-19.234 Five of the six COVID-19 patients receiving ibrutinib for Waldenstrom macroglobulinemia showed a steady improvement and resolution of COVID-19-related symptoms. Similar phenomena were observed in other reports of patients who have leukemia and COVID-19 at the same time. Ebselen is a benzoselenazole with anti-inflammatory, antioxidant, and cytoprotective activity.511 Jin et al. identified it as an antiviral agent targeting Mpro of SARS-CoV-2, and ebselen exerted inhibitory effects against SARS-CoV-2 with an EC50 of 4.67 μM.68 Two phase II clinical trials assaying ebselen’s effect in either moderate or severe COVID-19 patients are in the “enrolling by invitation” stage. Other compounds with an azole ring, such as zanubrutinib,512 acalabrutinib,237 and azilsartan,513 were also found to have affinity to SARS-CoV-2, and benzopyranylpyrazole-based hit compounds were demonstrated to inhibit SARS-CoV-2 replication in cells.514
Amides
Amides are compounds derived from oxoacids by replacement of an acidic hydroxy group with an amino group or a substituted amino group. The amide group plays a vital function in the composition of many bioactive compounds, including amino acids, peptides, and small molecule drugs. Due to their ability to form hydrogen bonds inside pockets of target proteins, amides have gained increasing attention in drug design and development.515,516 Amides, especially peptidomimetics and derivatives of amino acids with binding affinity to host proteases or SARS-CoV-2, have been designed or repurposed in many studies.41
Derivatives of amino acids are amides with broad medicinal values and development prospects. They were also widely investigated in recent COVID-19 research, especially as inhibitors of Mpro. Paxlovid is a co-packaged combination of nirmatrelvir and ritonavir.517,518 It is necessary to indicate that both compounds are derivatives of amino acids. Nirmatrelvir is a derivative of proline. It is an orally bioactive inhibitor of SARS-CoV-2 Mpro .519 Ritonavir is an L-valine derivative that has been applied as an HIV-1 protease inhibitor and as a cytochrome P450 (CYP3A) inhibitor.520 Ritonavir does not directly act on SARS-CoV-2 but is used to inhibit CYP3A-mediated metabolism of nirmatrelvir, resulting in increased plasma concentrations of nirmatrelvir.41 Owen et al. first developed Nirmatrelvir by optimizing PF-00835231, a potent inhibitor of SARS-CoV Mpro.84 According to their study, niramatrelvir exhibited good selectivity, safety, and protection against infection in a mouse-adapted SARS-CoV-2 model. Moreover, the results from a phase I single ascending dose study in healthy adult participants proved that nirmatrelvir was safe and well tolerated and exhibited a significant boost in plasma concentrations when co-administered with ritonavir.521 As Mpro is a highly conserved target protein, the antiviral potency of nirmatrelvir does not decrease when treating Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529) SARS-CoV-2 variants.522 However, recent preprints have reported that SARS-CoV-2 gains nirmatrelvir resistance after treatment with nirmatrelvir after in vitro culturing.523,524 There is also evidence that nirmatrelvir-resistant mutations have been acquired by the SARS-CoV-2 virus circulating in people.525,526,527 Boceprevir belongs to the imino acids and has potential activity against HCV genotype 1. Ma et al. first identified its anti-SARS-CoV-2 activity as an Mpro inhibitor and proved its in vitro activity.96 This anti-SARS-CoV-2 mechanism and effect was also proved by Fu et al. and Qiao et al..79,95 Based on the structure of boceprevir and another peptidomimetic compound, the HCV inhibitor telaprevir, Qiao et al. developed 32 new bicycloproline-containing Mpro inhibitors. As a result, two compounds (MI-09 and MI-30) showed excellent antiviral activity in cell-based assays, and significantly reduced lung viral loads and lung lesions in a transgenic mouse model of SARS-CoV-2 infection.79 Kneller et al. developed three hybrid peptidomimetic inhibitors, BBH-1, BBH-2, and NBH-2, by splicing components of boceprevir and narlaprevir, and proved their antiviral properties in vitro relative to nirmatrelvir.86 A study conducted by Xia et al. also showed the in vitro broad-spectrum coronavirus antiviral effect of two rationally designed inhibitors based on the peptidomimetic compounds GC-376, telaprevir, and boceprevir.528 To date, the development of peptidomimetics is the most used strategy in the search of anti-COVID-19 drugs.529 Other peptidomimetics, such as talaprevir, MG-132, and MDL-28170, were also found to have Mpro binding affinity. The cyclopeptide RTD-1, which has anti-SARS-CoV effects, was found to be safe to support its investigation for treatment of COVID-19.530
The effects of other amides, such as lopinavir, on SARS-CoV-2 were also studied. Lopinavir is a dicarboxylic acid diamide which is often used with ritonavir against HIV infections.531 Based on the structure of SARS-CoV-2 Mpro, Zhang et al. first reported it as a candidate drug against COVID-19.72 According to a study by Choy et al., lopinavir has in vitro activity against SARS-CoV-2 with an EC50 of 26.63 μM.532 Niclosamide is a secondary carboxamide resulting from the formal condensation of the carboxy group of 5-chlorosalicylic acid with the amino group of 2-chloro-4-nitroaniline.533 It has broad-spectrum antiviral activity, especially against the hepatitis virus, influenza virus, and rotavirus.534 It inhibits SARS-CoV-2 virus entry through TMEM16F inhibition and replication through autophagy induction.535,536 Weiss et al. showed that niclosamide potency is conserved against the Alpha, Beta, and Delta SARS-CoV-2 variants in Vero TMPRSS2 cells and the strong antiviral activity of niclosamide was validated in a human airway epithelial model.537 An inhaled niclosamide formulation was developed and tested in a murine infection model of SARS-CoV-2. Intranasal administration of niclosamide (0.24 mg·kg−1·day−1) to SARS-CoV-2-infected mice for 10 days improved survival and significantly reduced viral loads.538 Darunavir, with a similar anti-HIV effect, belongs to the carbamates.539 Computational evidence showed it may interact with the Mpro pocket.540 Besides, dalcetrapib, an anilide, is a cholesteryl ester transfer protein (CETP) inhibitor that can produce an increase in serum HDL-cholesterol levels and a decrease in serum LDL-cholesterol levels.541 Mancek-Keber et al. reported it can disrupt fusion within the RBD and the SARS-CoV-2 S protein.542 Niesor et al. claimed it can inhibit Mpro activity and viral replication in Vero E6 cells with IC50 values of 14.4 ± 3.3 μM and an EC50 value of 17.5 ± 3.5 μM.543
Alkaloids
Alkaloids are a complex class of compounds derived from plants with a basic character and bear at least one nitrogen atom, preferably in a heterocycle. Based on their core chemical structures, alkaloids can be classified into various subclasses, such as pyrrolidines, tropanes, quinolines, isoquinolines, and indoles.544 Many alkaloids possess biological activity, and have been applied in medicines. Since the COVID-19 outbreak, some alkaloids have been applied in clinical studies to verify their immune regulatory or antiviral effects. Moreover, many alkaloids with potential affinities to SARS-CoV-2 have been screened out by computational methods, and their therapeutic value against COVID-19 has been demonstrated.545,546
Quinoline alkaloids bear a common core structure of benzo-pyridine. Their antimalaria and immunomodulatory effects have been broadly investigated.547,548 Chloroquine is an aminoquinoline that is substituted at position 7 by chlorine. Since the 1940s, chloroquine has been investigated for the treatment of malaria.549 Chloroquine is also used off-label for the treatment of rheumatic diseases, as well as for the treatment and prophylaxis of Zika virus, HIV, dengue fever virus, and coronaviruses SARS-CoV and MERS-CoV.550,551 Previous studies revealed it has broad-spectrum antiviral activity by increasing the endosomal pH required for virus/cell fusion, as well as interfering with the glycosylation of cellular receptors of SARS-CoV.552 In the early stages of the COVID-19 pandemic, Wang et al. revealed that chloroquine functions both at the entry and at the post-entry stage of SARS-CoV-2 infection, and chloroquine exerts equal inhibitory effects on SARS-CoV-2 in Vero E6 cells infected by SARS-CoV-2 with an EC50 of 1.13 μM and an EC90 of 6.90 μM.553 Since chloroquine is a cheap and safe drug which has been approved for more than 70 years, it is a clinically applicable agent during the COVID-19 pandemic. A multicenter clinical trial conducted by Gao et al. showed that in China, it was effective and had an acceptable safety profile for COVID-19-related pneumonia.554 Hydroxychloroquine is similar to chloroquine, but its N-ethyl group at position 2 is hydroxylated. As a less toxic derivative of chloroquine, Liu et al. found that hydroxychloroquine is effective in inhibiting SARS-CoV-2 infection in vitro.555 In a pilot observational study, Gautret et al. provided evidence of a beneficial effect of co-administration of hydroxychloroquine with azithromycin in the treatment of COVID-19 and its potential effectiveness in the early reduction of contagiousness.556 On 28 March, 2020, the FDA authorized the emergency use of chloroquine and hydroxychloroquine to treat patients with COVID-19. However, further clinical studies provided no conclusive evidence supporting the use of chloroquine or hydroxychloroquine in the treatment of COVID-19. Thus, the FDA emergency use authorization (EUA) for hydroxychloroquine and chloroquine in the treatment of COVID-19 was revoked on 15 June, 2020. Based on the core structure of quinoline, quinazolines are synthetic molecules containing a benzene ring system fused to pyrimidine at two adjacent carbon atoms.557,558,559 Lapatinib, a member of the quinazoline class, has a role as an antineoplastic agent and as a TK inhibitor.560 Raymonda et al. showed that lapatinib has the potential to block SARS-CoV-2 infection by a high-throughput screening procedure.561 According to their in vitro results, lapatinib could inhibit SARS-CoV-2 RNA replication in pulmonary fibroblasts by over 50,000-fold. Apabetalone is another member of the quinazoline class with benefits in treating COVID-19. Gilham et al. demonstrated it could downregulate the cell surface receptors ACE2 and DPP-4, which are involved in SARS-CoV-2 entry.562 Moreover, their results revealed that the inhibitory effects of apabetalone on SARS-CoV-2 infection in vitro are comparable to those of antiviral agents.
Isoquinoline is a benzopyridine in which the nitrogen atom is not directly attached to the benzene ring. The isoquinoline structure occurs in a considerable number of alkaloids in widely separated plant families.563 Emetine is a pyridoisoquinoline comprising emetam with methoxy substituents at the 6′-, 7′-, 10-, and 11-positions.564 In a previous study, emetine was found to inhibit replication of buffalopox virus (BPXV), bovine herpesvirus 1 (BHV-1) and Newcastle disease virus (NDV).565 According to Wang et al., emetine has antiviral effects with an EC50 of 0.007 μM, suggesting it is >30-fold more effective than remdesivir (EC50: 0.24 μM) against SARS-CoV-2.566 Moreover, in vivo pharmacokinetics experiments revealed that emetine was enriched in the lung tissues to effective concentrations at 12 h posttreatment. Interestingly, molecule docking studies suggest that emetine has significant binding affinity toward RdRp (−9.5 kcal/mol), PLpro (−9.0 kcal/mol), the S protein RBD (−8.8 kcal/mol), and Mpro (−8.5 kcal/mol) of SARS-CoV-2.564 As a multitarget inhibitor of SARS-CoV-2, emetine was recognized to be a more potent drug.567 However, there are concerns that need further investigation since cardiovascular complications due to emetine have been reported.568,569 Hence, emetine can be used as a lead compound to design high-safety antiviral drugs in the future.570 Cepharanthine is a bisbenzylisoquinoline alkaloid from tubers of Stephania, which is used as an alopecia drug in Japan.571 Its antiviral ability has been verified in vitro against HIV, human T-lymphotropic virus type 1 (HTLV-1), HBV, SARS-CoV, and HCoV-OC43.572 The anti-SARS-CoV-2 effect of cepharanthine was verified by Ohashi et al. in vitro.573 According to their results, treatment with cepharanthine efficiently decreased the viral RNA concentration in infected cells, and the combination of cepharanthine with nelfinavir exhibited a synergistic effect.
The indole alkaloids with the 2,3-benzopyrrole core structure are important elements of many natural or synthetic molecules with significant biological activity. Melatonin is a therapeutic chemically synthesized form of the pineal indole melatonin with antioxidant properties. It is an effective anti-inflammatory agent and may inhibit SARS-CoV-2-induced cell damage by regulating mitochondrial physiology and enhancing the immune system.574,575,576 A study conducted by Zhai et al. revealed that melatonin could inhibit animal coronavirus infection in cells by reducing viral entry and replication.577 Cecon et al. demonstrated that administration of melatonin effectively attenuated severe symptoms and improved survival of human ACE2-expressing mice infected with SARS-CoV-2 by limiting the production of type I and type III interferons in the lungs.578 In addition, they demonstrated that melatonin could bind to an allosteric binding site of human ACE2, thus interfering with SARS-CoV-2 entry in endothelial cells.579 Indomethacin is a synthetic non-steroidal indole derivative with anti-inflammatory activity and chemopreventive properties.580 Amici et al. reported direct antiviral activity of indomethacin by inhibition of viral RNA synthesis against SARS-CoV and canine CoV, without being dependent on the COX inhibitory effect of indomethacin.581 Kiani et al. found an increase in percentage inhibition of SARS-CoV-2 to 93% in vitro when co-administered with 100 μM indomethacin compared with administration of ketotifen alone.582 An open-label randomized clinical trial of indomethacin for mild and moderate hospitalized COVID-19 patients indicated indomethacin use alongside standard treatment was associated with significant symptomatic relief and improved oxygen saturation levels.583 Lufotrelvir is an indolecarboxamide and its metabolic form PF-00835231 has strong and broad-spectrum inhibitory activity against numerous coronavirus 3CL proteases. Boras et al. provided ADME, safety, and in vitro and in vivo antiviral activity data that support lufotrelvir as a potential agent for COVID-19 treatment.584 The emvododstat bear core structure of tetrahydropyrido[3,4-b] indole is an orally available potent inhibitor of dihydroorotate dehydrogenase. Luban et al. found that treatment with emvododstat led to a dose-dependent reduction in the levels of SARS-CoV-2 nucleocapsid protein with an EC50 of 1.96 nM in infected Vero E6 cells.585 Lycorine is an indolizidine alkaloid found in Sternbergia clusiana and Pancratium trianthum, with inhibitory effects on RdRp activity of coronaviruses.586 The antiviral effect of lycorine was verified in Vero E6 cells infected with SARS-CoV-2, with an EC50 of 0.31 μM.587
Other small molecules
The anti-COVID-19 effects of various other naturally occurring molecules have also been investigated. Curcumin, a beta-diketone, is a natural dyestuff found in the root of Curcuma longa.588 As a broad-spectrum antiviral drug, curcumin can not only treat HIV virus, liver poison, and influenza A virus but has also been recognized as a therapeutic agent for COVID-19 as it affects cellular posttranscriptional and posttranslational modifications, thereby limiting viral multiplication.589,590 Bormann et al. demonstrated that curcumin effectively neutralizes SARS-CoV-2 at subtoxic concentrations in Vero E6 and human Calu-3 cells.591 Treatment significantly reduced SARS-CoV-2 RNA levels in cell culture supernatants. A clinical trial suggested that the use of nanomicelles containing curcumin in COVID-19 patients can accelerate recovery of the acute inflammatory phase, thus controlling the inflammatory response elicited by viral infection.592 Further, according to results from a randomized double-blind placebo-controlled trial, nanocurcumin can be effective in increasing oxygen saturation and reducing the severity of symptoms in COVID-19 patients; thus, it can be used as a complementary agent to accelerate the recovery of patients.593 Tamoxifen and clomiphene are derived from the natural product stilbene. They belong to the class of stilbenoids and are non-steroidal antiestrogens.594,595 Zu et al. showed that tamoxifen and clomiphene strongly antagonized SARS-CoV-2 infection, both in vitro and in vivo.596 They functioned by suppressing viral entry in the postbinding stage. In vivo experiments in a mouse model verified that tamoxifen and clomiphene effectively suppress infection of not only wild-type but also mutant SARS-CoV-2 variants such as P.1.351 and P.1.617.594 Ivermectin is a natural and broad-spectrum anti-infective agent found in Streptomyces avermitilis, and it can inhibit the replication of HIV-1, Newcastle disease virus and dengue virus.597 Research indicated it exerts inhibitory effects on SARS-CoV-2 replication in the early stages of infection. Ivermectin has recently been reported as a potent inhibitor of SARS-CoV-2 infection, with an excellent ability to reduce viral RNA levels in Vero-hSLAM cells.598 Carrageenan is a polysaccharide found in red algae with antiviral effects. Carrageenans, which are used in broadly used nasal and mouth sprays, have the potential to serve as first-line therapeutics to inhibit infection and transmission of SARS-CoV-2.599 Schutz et al. identified the mechanisms underlying the antiviral activity of one nasal and one mouth spray through in vitro assays.600 This antiviral effect was also observed by Froba et al. against several SARS-CoV-2 variants (Alpha, Beta, Gamma, and Delta).599 An aurothioglucose named auranofin exerted inhibitory effects on SARS-CoV-2 in Huh7 human liver cells for more than 24 h.601 It also suppressed the papain-like proteinase activity of SARS-CoV-2 in vitro with an IC50 of 0.75 ± 0.13 µM, and reduced the binding of the S protein of SARS-CoV-2 and human ACE2 in vitro with an IC50 of 22.2 ± 2.8 µM.602 Hypericin is an anthraquinone derivative that is naturally found in the yellow flower of Hypericum perforatum. It was identified as a candidate drug for COVID-19 therapy due to its inhibitory effects on SARS-CoV-2 PLpro in vitro.603,604
The effects of other synthetic small molecules on COVID-19 were also studied. For example, camostat, a benzoate ester, is a synthetic serine protease inhibitor.605 Hoffmann et al. demonstrated that camostat treatment significantly reduced Calu-3 infection with wild-type SARS-CoV-2 by blocking TMPRSS2 of target cells. According to their results, camostat reduced SARS-CoV-2 entry into cells with an EC50 of 1 µM and EC90 of 5 µM.606 In a retrospective analysis of 371 adult patients with COVID-19 pneumonia, Sakr et al. concluded that camostat treatment could be beneficial to critically ill COVID-19 patients.607 However, Chupp et al. claimed that camostat was not associated with a reduction in nasopharyngeal SARS-COV-2 viral load compared to placebo.608 Nafamostat, an analog of camostat and a member of the guanidines, also has potential anti-COVID-19 effects by blocking TMPRSS2 on target cells.609 Li et al. reported that nafamostat reduced SARS-CoV-2 infection in primary human airway epithelial cells and in the Calu-3 2B4 cell line, and exhibited greater antiviral efficiency compared with camostat.610 Moreover, they demonstrated that intranasal nafamostat treatment prior to or shortly after SARS-CoV-2 infection significantly reduced weight loss and lung tissue titers of mice infected by SARS-CoV-2. Jang et al. reported three cases of COVID-19 pneumonia who progressed while using antiviral drugs, needed supplementary oxygen therapy, and improved after treatment with nafamostat. However, according to the results of a phase Ib/IIa clinical study, no evidence of anti-inflammatory, anticoagulant, or antiviral activity of intravenous nafamostat in hospitalized COVID-19 patients was provided.611 The negative outcomes of the abovementioned TMPRSS2 inhibitors raise questions about the effectiveness of this target. It is worth noting that blocking TMPRSS2 might not function well when the virus has already infected the human body and caused symptoms. Thus, it is recommended to use TMPRSS2 inhibitors in the early stage of COVID-19 or to use them in combination with other anti-viral drugs. Amantadine is a synthetic amine with antiviral effects by interfering with the function of the transmembrane domain of the viral proteins.612 Its antiviral ability against SARS-CoV-2 has been tested in vitro in a study conducted by Fink et al., and was found to have an IC50 of around 100 μM.613 According to a case report, the use of amantadine may reduce the toxic effects of COVID-19, including ARDS, viral replication, and ventilator dependency.614,615 Currently, two phase III clinical trials determining if amantadine brings benefits in patients with COVID-19 are ongoing (NCT04952519; NCT04894617). Brilacidin, a non-peptidic small molecule mimetic of defensin, which is a type of host defense protein/peptide with antibacterial and antiviral activities, is also referred to as a SARS-CoV-2 inhibitor.616,617 Bakovic et al. demonstrated that brolacidin could impact viral entry and disrupt viral integrity, thus exerting inhibitory effects on SARS-CoV-2 infection in Calu-3 and Vero E6 cells.618 Other synthetic molecules, such as GLPG-0187 (a sulfonamide) and the cyclohexanone SIMR-2418, are also potential inhibitors of SARS-CoV-2 with proved in vitro antiviral effects.619,620
Small molecule drugs in clinical development
Approved/authorized products
So far, the FDA has approved two small molecular drugs, remdesivir (Veklury) and baricitinib (Olumiant), for the treatment of COVID-19.31,621 Remdesivir, which was developed by Gilead, is approved for the treatment of mild to moderate COVID-19 in adults and pediatric patients.32 Baricitinib, which was developed by Eli Lilly, is approved for the treatment of COVID-19 in hospitalized adults requiring supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).319 In addition, the FDA has granted EUA for the use of several unapproved drugs against COVID-19, which include two oral antiviral pills, nirmatrelvir/ritonavir (Paxlovid) and molnupiravir (Lagevrio).622,623 Four drugs, favipiravir (AVIFAVIR), proxalutamide, azvudine, and VV116 have been approved in Russia, Paraguay, China, and Uzbekistan, respectively (Table 1).45,288
Remdesivir was approved by the FDA on May 1, 2020 as the first treatment for COVID-19. On May 7, 2020, it was approved for emergency situations by the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan, and its use was authorized by the EMA on July 3, 2020. This approval is supported by the data from three randomized, controlled clinical trials that included patients hospitalized with mild to severe COVID-19. In detail, the first adaptive, randomized, double-blind, placebo-controlled trial to evaluate the safety and efficacy of remdesivir in hospitalized adults diagnosed with COVID-19 (ACTT-1) was supported by the National Institute of Allergy and Infectious Diseases (NIAID) (NCT04280705). The results of this trial were published by Beigel et al..277 In brief, 1062 patients (541 assigned to the remdesivir group and 521 to the placebo group) were included in this trial. Participants who received remdesivir had a shorter recovery time (10 days) compared with the placebo group (15 days). The second study to evaluate the antiviral activity of remdesivir in participants with moderate COVID-19 compared with standard care treatment was supported by Gilead Sciences (NCT04292730). According to the results presented by Spinner et al., patients who received 5-day remdesivir treatment had a significantly better clinical status than those who received standard care at 11 days after initiation of treatment.624 The third study to evaluate the safety and antiviral activity of remdesivir treatment in patients with severe COVID-19 was supported by Gilead Sciences (NCT04292899). According to the results, improvements in symptoms were similar in both groups of patients treated with 5-day remdesivir and 10-day remdesivir.625
Besides the above trials, a double-blind, randomized, placebo-controlled phase III trial conducted at 63 hospitals across five countries (Japan, Mexico, Singapore, South Korea, and the USA) by the NIAID (NCT04492475; EudraCT2020-003510-12) also revealed the anti-COVID-19 effects of remdesivir. Participants involving symptomatic, non-hospitalized patients with COVID-19 who are at high risk for disease progression (age ≥ 60 years, obesity, or certain co-existing medical conditions) were randomly assigned to the remdesivir group or the placebo group.626 A 3-day course of remdesivir had an acceptable safety profile. Compared with the placebo group, the remdesivir group had an 87% lower risk of COVID-19-related hospitalization or death and an 81% lower risk of COVID-19-related medically attended visits or death. However, according to another phase III, randomized, controlled, open-label trial (DisCoVeRy) conducted at 48 sites in Europe (France, Belgium, Austria, Portugal, and Luxembourg) (NCT04315948; EudraCT2020-000936-23), no clinical benefit from remdesivir treatment was observed in hospitalized COVID-19 patients with symptoms for more than 7 days who required oxygen support.627,628 The authors speculated that the discrepancy between their results and those from ATCC-1 might be explained by the differences in study populations.277 Among the patients without requirement of oxygen support in the DisCoVeRy trial, remdesivir significantly delayed the need for new mechanical ventilation or ECMO or death, consistent with what was reported in ACTT-1. In addition, a randomized, double-blind, placebo-controlled, multicenter trial of remdesivir carried out at ten hospitals in China (NCT04257656) also indicated that remdesivir was not associated with statistically significant clinical benefits in adult patients admitted to the hospital for severe COVID-19.629
Barcitinib was approved by the FDA on May 10, 2022 for treatment of COVID-19 in hospitalized adults requiring supplemental oxygen, non-invasive or invasive mechanical ventilation, or ECMO, and authorized under EUA for the same indication for pediatric patients (2–17 years old) in the USA.630 Barcitinib has also been approved by the PMDA of Japan on April 23, 2021 for treatment of pneumonia caused by COVID-19 (limited to patients requiring supplemental oxygen), and a marketing authorization application for barcitinib has been submitted in the European Union. The approval of barcitinib in both countries was supported by data from two clinical trials conducted by the NIAID.41 ACTT-2 (NCT04401579) is a randomized, double-blind, placebo-controlled trial evaluating if combination with baricitinib could improve the effects of remdesivir against COVID-19 in hospitalized adults. According to the results, combination treatment with the anti-inflammatory drug baricitinib and the antiviral drug remdesivir was safe and superior to remdesivir alone for the treatment of hospitalized patients with COVID-19 pneumonia.319 The clinical trial COV-BARRIER is a randomized, double-blind, placebo-controlled, parallel-group phase III study to verify if baricitinib is effective in hospitalized patients with COVID-19 (NCT04421027). First, the efficacy and safety results of baricitinib plus standard care (include systemic corticosteroids and remdesivir) in hospitalized adults with COVID-19 from 101 centers across 12 countries were presented.41 Baricitinib plus standard care lowered the absolute all-cause mortality risk to 5% at 28 days and 4.9% at 60 days. Another study reported the results from a critically ill cohort in COV-BARRIER not included in the main phase III trial.631 This study was conducted across 18 hospitals in Argentina, Brazil, Mexico, and the USA. According to the results, in critically ill hospitalized COVID-19 patients who received invasive mechanical ventilation or ECMO, combination treatment with baricitinib and standard care lowered the absolute all-cause mortality risk to 5% at 28 days and 17% at 60 days. This result is in line with the previously reported results of baricitinib in patients with less severe COVID-19.
Molnupiravir, which was developed by Merck, was first approved by the MHRA of the UK on November 4, 2021 for treatment of mild to moderate COVID-19 in adults with a positive SARS-COV-2 diagnostic test and who have at least one risk factor for developing severe illness.632 Further, it was authorized for the same indications by FDA on December 23, 2021. On the next day, it was approved for emergency use by the PMDA of Japan for treatment of diseases caused by SARS-CoV-2 infection.38,633 The approval and EUA of molnupiravir were mainly based on data from two clinical trials. The first one is a phase IIa double-blind, placebo-controlled, randomized trial evaluating the safety, tolerability, and antiviral efficacy of molnupiravir in patients with COVID-19 (NCT04405570). At the end of the 4-week study, the proportion of participants who achieved viral RNA clearance was higher in the 800-mg molnupiravir group (92.5%) than in the placebo group (80.3%).634 Moreover, the proportion of nasopharyngeal swabs containing infectious virus and the time to eliminate SARS-CoV-2 RNA were decreased. These results provide strong biological evidence indicating that molnupiravir can be used as an oral agent for COVID-19 treatment during the early stages of the disease. Another one is a phase II/III double-blind, placebo-controlled, randomized, multicenter clinical trial, MOVe-OUT, evaluating the safety, tolerability, and antiviral efficacy of molnupiravir in non-hospitalized adults with COVID-19 (NCT04575597). As shown by Bernal et al., data from the MOVe-OUT phase III trial indicate that initial treatment with molnupiravir within 5 days after the onset of symptoms reduces the risk of hospitalization for any cause or death through day 29.635
Nirmatrelvir/ritonavir, developed by Pfizer, is a co-packaged combination that is used to treat SARS-CoV-2 infection.636 The FDA issued an EUA for nirmatrelvir/ritonavir for the treatment of mild to moderate COVID-19 in adults and pediatric patients (≥12 years of age and weighing ≥40 kg) with SARS-CoV-2 infection and those patients who are at high risk of progression to severe COVID-19 on December 22, 2021. Nirmatrelvir/ritonavir was approved in Israel (December 26, 2021), Korea (December 27, 2021), UK (December 31, 2021), and the EU (January 28, 2022) for the treatment of COVID-19 in adults who do not require supplemental oxygen and are at increased risk of developing severe COVID-19. On February 10, 2022, the PMDA of Japan specially approved the use of Pfizer oral medicine, and signed a purchase agreement with Pfizer for 2 million people. On February 11, 2022, the National Medical Products Administration of the People’s Republic of China approved Paxlovid for emergency use in adults and adolescents with mild, common forms of COVID-19 within 5 days of onset and associated with severe risk factors for progression. The primary data supporting Paxlovid’s EUA came from EPIC-HR (NCT04960202), a randomized, double-blind, placebo-controlled clinical phase II/III trial of nirmatrelvir plus ritonavir in the treatment of non-hospitalized symptomatic adults with laboratory-confirmed SARS-CoV-2 infection. Results from this trial demonstrated the efficacy of oral administration of nirmatrelvir (300 mg) with ritonavir (100 mg) every 12 h for 5 days.519,637,638 Among non-hospitalized adults at high risk of progression to severe disease, treatment with nirmatrelvir plus ritonavir resulted in an 89.1% relative risk reduction of COVID-19-related hospitalization or death from any cause compared with the placebo group by day 28.518,519 Currently, the EPIC-HR trial is ongoing, and further data like the proportion of all-cause death will be published later. Besides, the phase II/III EPIC-SR trial which compares nirmatrelvir plus ritonavir and placebo for the treatment of non-hospitalized, symptomatic adults with COVID-19 who are at low risk of progressing to severe illness is also ongoing (NCT05011513). The phase II/III EPIC-PEP trial, which evaluates the efficacy and safety of two nirmatrelvir plus ritonavir regimens in preventing symptomatic SARS-COV-2 infection in adult household contacts of people infected with SARS-COV-2, is still recruiting participants (NCT05047601), and the EPIC-Pedstrial, a study of oral nirmatrelvir/ritonavir in non-hospitalized COVID-19 pediatric patients at risk for severe disease, is also recruiting participants. Further clinical trials are being conducted not only by Pfizer, but also in China, Japan, and other countries to further prove the safety and effectiveness of the drug.639
Favipiravir has been granted a conditional marketing authorization by the Russian Ministry of Health based on the interim results of a phase II/III clinical trial in May 2020640 (NCT04434248). In this adaptive, multicenter, open-label, randomized, phase II/III clinical trial, hospitalized patients with moderate COVID-19 were randomized at a 1:1:1 ratio to receive favipiravir with different dosages or standard care. According to their results, viral clearance was achieved in 62.5% of patients in the favipiravir groups and in 30% of patients in the standard care group on day 5, demonstrating a rapid antiviral response against SARS-CoV-2. However, according to the data from a randomized, double-blind, multicenter, and placebo-controlled trial in Saudi Arabia, favipiravir was not associated with faster viral clearance or a better clinical outcome when initiated within 5 days after onset of COVID-19 symptoms in adults with mild COVID-19.641 This conclusion was also drawn based on data from a prospective, randomized, open-label, multicenter trial of favipiravir for the treatment of COVID-19 at 25 hospitals across Japan642 (jRCTs041190120).
Proxalutamide, developed by Kintor Pharmaceuticals, has exhibited efficiency in preventing COVID-19 in a randomized, double-blind, placebo-controlled, multiregional clinical trial of Proxalutamide for hospitalized COVID-19 patients clinical trial conducted in Brazil. This trial was carried out with two different arms, the Northern Brazil arm (NCT04728802) and the Southern Brazil arm (NCT05126628). According to the combined results published by Cadegiani et al., the recovery rate was 121% higher in the proxalutamide group than in the placebo group at day 14 and 81% higher at day 28.493 Moreover, the all-cause mortality rate was 80% lower in the proxalutamide group than in the placebo group at day 14 and 78% lower at day 28. However, these results were suspicious since the fatality rate was as high as 49.4% in the placebo group and the trial was conducted very quickly. In reply to these suspicions, Cagegiani claimed the fatality rate was high due to the Gamma variant’s wide spread in Brazil at the time; about 43% of the hospitalized COVID-19 patients in the state of Amazonas were dying, according to official data. Recruitment was rapid because word got out that patients in the proxalutamide trial were recovering within days. Then, a randomized, double-blind, placebo-controlled clinical trial of proxalutamide was conducted in Brasilia, Brazil in men with COVID-19 in an outpatient setting (NCT04446429). Proxalutamide treatment reduced the rate of hospitalization by 91% in this trial.643 However, since tests of antiandrogens in COVID-19 patients were not encouraged except in the Brazilian trial,644 the anti-COVID-19 function of proxalutamine remains to be verified by clinical trials of other countries and organs (NCT04870606; NCT05009732). Also, its mechanism should be discussed in further studies.
VV116 was developed by Shanghai JunTop Biosciences Co., Ltd. According to data from an open, prospective cohort study of VV116 in Chinese participants infected with the SARS-CoV-2 Omicron variant (NCT05242042), participants who received VV116 within 5 days after the first positive PCR test of SARS-CoV-2 had a shorter viral shedding time than participants in the control group (8.56 vs. 11.13 days).289 VV116 exhibits a wide distribution in target organs of SARS-CoV-2 in rats and dogs.645 In this regard, VV116 might compensate for the liver-targeting limitation of remdesivir. VV116 has been approved for the treatment of COVID-19 in Uzbekistan and is being investigated in several phase III clinical trials in patients with COVID-19 (NCT05242042; NCT05279235; NCT05341609).
Azvudine was developed by Genuine Biotech Co., Ltd. According to data from a randomized, open-label, controlled clinical trial performed in China (ChiCTR2000029853), azvudine treatment plus standard care shortens the mean time of the first nucleic acid negative conversion in mild and common COVID-19 patients.282 Moreover, a randomized, single-arm clinical trial revealed that azvudine treatment cured COVID-19 patients, with the duration of nucleic acid negative conversion of 3.29 ± 2.22 days and hospital discharge at 9.00 ± 4.93 days.281 On July 25, 2022, azvudine was conditionally approved for the treatment of COVID-19 in China.
Candidates under phase III/IV clinical trials
Nitazoxanide is a broad-spectrum antiviral agent in vitro, so it was a logical choice to analyze its anti-SARS-CoV-2 effects (Table 2).534 According to data from a phase II multicenter, randomized, double-blind, placebo-controlled trial conducted in Brazil (NCT04552483), early nitazoxanide therapy in patients with mild COVID-19 reduced the viral load compared with the placebo group.488 A pilot proof-of-concept randomized double-blind clinical trial in patients hospitalized with moderate to severe COVID-19 also concluded that nitazoxanide is superior to placebo (NCT04348409), since patients in the nitazoxanide group had a lower mortality rate and faster hospital discharge compared to the placebo group.489 Based on these studies, several phase III trials were conducted in different countries. Among them, a randomized double-blind placebo-controlled clinical trial in 36 centers in the USA has given corresponding results (NCT04486313). According to the findings, nitazoxanide reduced the relative risk of progressing to severe illness in mild or moderate COVID-19 patients,646 suggesting it may reduce the progression to severe illness in high-risk participants. However, there is no significant difference in sustained clinical recovery time between the nitazoxanide and placebo treatment groups. The efficiency of nitazoxanide in patients should be tested by larger phase III trials with adequate statistical power (NCT04343248; NCT05157269; NCT05157243).
Camostat mesylate, an oral TMPRSS2 inhibitor, is used to treat chronic pancreatitis and reflux esophagitis. In a phase I clinical study (NCT04451083), it was shown to be safe and tolerable at a high dosage in healthy male subjects.647 A preprint article reported results of a phase II randomized, double-blind, placebo-controlled trial of camostat mesylate involving 70 COVID-19 outpatients (NCT04353284). In this trial, more rapid resolution of COVID-19 symptoms and amelioration of the loss of taste and smell was observed in the camostat group compared to the placebo group.608 However, treatment with camostat did not appear to be associated with a reduced nasopharyngeal SARS-COV-2 viral load in this trial. However, since camostat functions by inhibiting viral entry, it would possibly lead to similar viral load in the upper respiratory tract of patients in both groups. Thus, additional clinical trials are needed with a larger sample size to obtain more information about other symptomatic outcomes of camostat in early COVID-19. Currently, eight phase III clinical trials are registered at the ClinicalTrials.gov website, but none of their results have been reported.
Ciclesonide, a glucocorticoid, is applied for the treatment of obstructive airway diseases including asthma and chronic obstructive pulmonary disease.648 Based on its anti-inflammatory effect, it was hypothesized that it could decrease the symptom burden of COVID-19 in patients with prominent respiratory symptoms.649 In the phase II/III randomized, double-blind, placebo-controlled trial CONTAIN, it was analyzed if ciclesonide accelerates recovery from COVID-19 in outpatients (NCT04435795). However, the combination of inhaled and intranasal ciclesonide was not associated with an appreciable increase in symptom resolution among healthy young adults with COVID-19 who presented with cough, dyspnea, or fever compared with the placebo group according to the data of the phase II trial.650 The further phase III clinical trial was terminated since the researchers could not meet enrollment targets in Canada. Another phase III study evaluating the efficacy of inhaled ciclesonide was conducted in non-hospitalized participants with symptomatic COVID-19 (NCT04377711). In brief, 400 participants were enrolled and randomized in the ciclesonide arm or the placebo arm. The median time to alleviation of all COVID-19-related symptoms was 19.0 days in the ciclesonide arm and 19.0 days in the placebo arm, which suggested ciclesonide did not reduce the time to alleviate COVID-19-related symptoms.651 Furthermore, a meta-analysis evaluating the effect of inhaled ciclesonide in COVID-19 outpatients was conducted by Hsu et al..652 By searching and analyzing data from four randomized controlled trials, the authors concluded that inhaled ciclesonide could not relieve the symptoms for COVID-19 outpatients.
Rivaroxaban is a direct inhibitor of the coagulation factor Xa with anticoagulant activity.653 Since COVID-19 is associated with both venous and arterial thrombotic complications, prophylactic anticoagulation is widely recommended for hospitalized patients with COVID-19.654 Thus, several phase III/IV clinical trials have evaluated its anticoagulant ability in COVID-19 patients. Among them, ACTION is an academic-led, pragmatic, multicenter, open-label, randomized phase IV clinical trial conducted in Brazil (NCT04394377). It was designed to determine whether therapeutic anticoagulation with rivaroxaban improves clinical outcomes in hospitalized patients with COVID-19 and elevated D-dimer levels compared with standard prophylactic anticoagulation.655 As a result, in-hospital therapeutic anticoagulation with rivaroxaban or enoxaparin followed by rivaroxaban to day 30 did not improve clinical outcomes and increased bleeding compared with prophylactic anticoagulation,656 suggesting that a dosage of 20 mg rivaroxaban per day should be avoided as a routine anticoagulation strategy in hospitalized COVID-19 patients (NCT04662684). However, another phase III open-label, multicenter, randomized trial conducted at 14 centers in Brazil evaluated post-discharge thromboprophylaxis effects of rivaroxaban versus no anticoagulation in COVID-19 patients (NCT04662684).654 It was found that thromboprophylaxis with 10 mg/day rivaroxaban for 35 days improved clinical outcomes compared with no extended thromboprophylaxis in post-discharge patients with a high risk for venous thromboembolism. This study revealed that low-dose rivaroxaban at the time of hospital discharge and for another 35 days in the right patient population improves clinical outcomes. Despite these results, future trials with multiple study populations (such as COVID-19 patients at high risk of disease progression, or mild COVID-19 patients) are warranted to confirm the above findings, and the function of rivaroxaban when combined with different antiviral candidates. Thus, several related phase III/IV clinical studies are in active or recruiting status at present (NCT04351724; NCT04324463; NCT04715295).
Ivermectin is an anti-infective agent with activity against several parasitic nematodes and scabies and is the treatment of choice for onchocerciasis (river blindness). Since its anti-SARS-CoV-2 ability was observed in vitro and in animal models,598,657 ivermectin has attracted much attention in the fight against COVID-19. It has been widely promoted in some countries.658 Many phase III or IV clinical trials were conducted to understand the effect of ivermectin for the treatment of COVID-19. Among them, a double-blind, placebo-controlled, randomized trial involving 476 patients with mild COVID-19 was conducted in Colombia (NCT04405843). According to the data, a 5-day course of ivermectin initiated in the first 7 days after evidence of infection failed to significantly improve the time to resolution of symptoms compared with placebo.659 The researchers indicated that this may be due to the relatively healthy and young study population in this trial, highlighting the need to study the ability of ivermectin to prevent more severe COVID-19. Further, a phase III, multicenter, open-label, randomized clinical trial (I-TECH) evaluating the efficacy of ivermectin in 490 high-risk COVID-19 patients was conducted at 20 public hospitals and a COVID-19 quarantine center in Malaysia (NCT04920942). However, researchers concluded that ivermectin treatment during early illness of high-risk patients with mild to moderate COVID-19 did not prevent progression to severe disease.660 They also indicated that the open-label trial design might contribute to the underreporting of adverse events in the control group while overestimating the drug effects of ivermectin. Recently, data from a double-blind, randomized, placebo-controlled, adaptive platform trial involving a total of 3515 symptomatic SARS-CoV-2-positive adults recruited from 12 public health clinics in Brazil were published (NCT04727424). In line with previous reports, treatment with ivermectin did not result in a lower incidence of medical admission to a hospital or prolonged emergency department observation for COVID-19 among outpatients at high risk for serious illness.661
Fostamatinib is approved for treatment of immune thrombocytopenic purpura with potential anti-inflammatory and immunomodulating activities, and its metabolic active form is R406.662 Among healthy donor neutrophiles stimulated with COVID-19 patient plasma, treatment with R406 abrogated the release of neutrophil extracellular traps associating with mortality in COVID-19.663 Thus, fostamatinib was recognized to be a therapeutic regent for COVID-19, for which a phase II clinical trial was conducted in 60 hospitalized COVID-19 patients (NCT04579393). Results showed that fostamatinib with standard-care treatment decreased the all-cause mortality rate, days on supplemental oxygen, number of days in the ICU, and serious adverse event rate compared with the placebo group.664 However, larger randomized clinical trials should be conducted to reliably verify these findings and further investigate the full effects of fostamatinib on inflammation in patients. Thus, multicenter phase III studies evaluating the efficacy and safety of fostamatinib in COVID-19 subjects are underway (NCT04629703; NCT04924660).
Niclosamide is an oral anthelmintic drug approved for use against tapeworm infections.665 A phase II randomized, placebo-controlled clinical trial showed no significant difference in oropharyngeal clearance of SARS-CoV-2 at day 3 between the placebo and niclosamide groups666 (NCT04399356). However, due to the small enrollment pool and unavailable of drug blood levels, further studies should be considered in a wider range of patients. Currently, three phase III clinical studies are in the recruiting status to evaluate its efficacy in COVID-19 patients (NCT04558021; NCT04603924; NCT04870333). A phase IV open label, multi-arm, prospective, adaptive platform, randomized controlled trial involving niclosamide arm and niclosamide in combination with bromhexine arm was completed in June, 2022 (NCT05087381), and the results of the trial are awaiting publication. Since niclosamide is a historically well-tolerated and widely used anthelmintic drug, further escalation studies on this drug will be helpful in the fight against SARS-CoV-2.
Danoprevir boosted by ritonavir (Ganovo) is an HCV protease (NS3/4A) inhibitor, which was approved in China in 2018 to treat chronic HCV infection.667 According to the data from an open-label, single arm phase IV study in 11 COVID-19 patients (NCT04345276), Chen et al. concluded repurposing it for COVID-19 could be a promising therapeutic option.668 According to another study reported by Zhang et al., danoprevir/ritonavir-treated group exhibited shorter time to negative nucleic acid testing and a shorter hospital stay than lopinavir/ritonavir-treated group.669 However, given the lack of a placebo control group and the small sample size, further investigation should be conducted to verify this conclusion.
Candidates under phase I/II clinical trials
Phase I/II clinical trials often focus on drug safety, tolerance, pharmacokinetics, and the benefit/risk ratio in a small number of patients. Currently, many COVID-19 drugs are in these stages (Table 3), and some of them have demonstrated potential in entering a phase III clinical trial.41 For example, prostacyclin is a powerful vasodilator and inhibits platelet aggregation. Its sodium salt is used to treat primary pulmonary hypertension. Since endotheliopathy is a prominent feature of COVID-19 and associated with mortality in patients,670,671 prostacyclin, which has beneficial effects on the endothelium, might be useful adjunctive therapy for COVID-19 vaculopathy.672,673 To determine this effect, a multicenter, randomized phase II clinical trial was conducted in 80 adults with severe COVID-19 requiring mechanical ventilation and severe endotheliopathy (NCT04420741).674 No significant difference in the number of days alive without mechanical ventilation within 28 days was observed between the prostacyclin and placebo groups.675 Besides, two other phase II clinical trials investigating the potential benefits of prostacyclin in severe COVID-19 patients have been completed, but their results have not yet been published (NCT04445246; NCT04452669). Nezulcitinib (TD-0903) is an inhaled lung-selective inhibitor of JAKs with anti-inflammatory activities. The first study of nezulcitinib in human indicated good tolerance in healthy participants (NCT04402866).676 Further, a phase II study evaluating the efficiency, safety, pharmacodynamics, and pharmacokinetics of inhaled nezulcitinib in hospitalized patients with COVID-19-associated acute lung injury and impaired oxygenation was conducted in different countries (NCT04402866). This study was divided into two parts, with 25 and 110 participants, respectively,222 and the advanced dosage of 3 mg in Part 1 was applied for further investigation in Part 2. According to the data presented on the ClinicalTrials.gov website, ezulcitinib was associated with lower rates of all-cause mortality and serious adverse events compared to the placebo group.
Outlook
Small molecules have demonstrated their potential in the development of therapeutics against COVID-19. Viral proteins, host cell components, and immunoregulatory pathways have been identified as effective targets for COVID-19 treatment in regards to the pathogenic mechanisms of SARS-CoV-2. The diverse drug development strategies of small molecules contribute to their effectiveness. Because of global research efforts, some promising compounds, such as remdesivir, baricitinib, and nirmatrelvir/ritonavir, have already been approved or granted EUA in many countries. Moreover, there are more than 20 small molecule candidates in the phase III/IV clinical trial stages, which have the potential to further enrich the family of COVID-19 drugs.
Despite the above achievements, several issues need to be addressed. It is necessary to improve our understanding of SARS-CoV-2 and its lifecycle. The viral components involved in its pathological process must be characterized. The detailed mechanisms of viral replication and interaction with host cells must be elucidated in detail. It is also important to better understand the mechanisms by which the virus dysregulates the host immune system. This knowledge will contribute to the further development of anti-COVID-19 small molecules. SARS-CoV-2 variants are a critical issue. Several variants of concern, such as Alpha (B.1.1.7), Delta (B.1.617.2), and Omicron (B.1.1.529), have led to sustained challenges to the drug development industry. Drug resistance caused by viral mutations prompts us to continue searching for new compounds, targets, and drug combination strategies. Therefore, it is necessary to obtain up-to-date information regarding each variant to understand the structural influences induced by gene mutation. This would speed up and facilitate small molecule development and optimization. Furthermore, researchers should be encouraged to discover more compounds from natural products bearing multiple structural backbones with various activities. Enrichment of these natural backbones will inspire the structural design of potential small molecule drugs. Some findings of small molecules with promising anti-SARS-CoV-2 ability are still limited to the molecular docking simulation stage, while preclinical and clinical experimental evidence is needed to verify their therapeutic properties. Recently, some drugs have shown potential for use in combination therapy in clinical studies.677,678 Based on this, researchers should also develop drug combination strategies for existing small molecules to achieve synergistic therapeutic effects. The side effects of each candidate should also be addressed during drug development. In conclusion, the rapid progress in the development of anti-COVID-19 small molecule drugs has definitely strengthened global efforts to combat the SARS-CoV-2 pandemic.
References
Nalbandian, A. et al. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615 (2021).
Chen, Y., Liu, Q. & Guo, D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92, 418–423 (2020).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569 (2020).
Lan, T. C. T. et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 13, 1128 (2022).
Jiang, Y., Yin, W. & Xu, H. E. RNA-dependent RNA polymerase: structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 538, 47–53 (2021).
Jin, Z., Wang, H., Duan, Y. & Yang, H. The main protease and RNA-dependent RNA polymerase are two prime targets for SARS-CoV-2. Biochem. Biophys. Res. Commun. 538, 63–71 (2021).
Astuti, I. & Ysrafil. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab. Syndr. 14, 407–412 (2020).
Ovsyannikova, I. G., Haralambieva, I. H., Crooke, S. N., Poland, G. A. & Kennedy, R. B. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev. 296, 205–219 (2020).
Catanzaro, M. et al. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target Ther. 5, 84 (2020).
Madden, E. A. & Diamond, M. S. Host cell-intrinsic innate immune recognition of SARS-CoV-2. Curr. Opin. Virol. 52, 30–38 (2022).
Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 20, 669–688 (2021).
Tian, D. et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 137, 111313 (2021).
Billen, M., Schols, D. & Verwilst, P. Targeting chemokine receptors from the inside-out: discovery and development of small-molecule intracellular antagonists. Chem. Commun. 58, 4132–4148 (2022).
Warner, K. D., Hajdin, C. E. & Weeks, K. M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 17, 547–558 (2018).
Rubin, R. Baricitinib is first approved COVID-19 immunomodulatory treatment. J. Am. Med. Assoc. 327, 2281 (2022).
Hillen, H. S. et al. Structure of replicating SARS-CoV-2 polymerase. Nature 584, 154–156 (2020).
Gao, Y. et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368, 779–782 (2020).
Wang, Q. et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 182, 417–428e413 (2020).
Yin, W. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499–1504 (2020).
Malone, B., Urakova, N., Snijder, E. J. & Campbell, E. A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 23, 21–39 (2022).
Grellet, E., L’Hote, I., Goulet, A. & Imbert, I. Replication of the coronavirus genome: a paradox among positive-strand RNA viruses. J. Biol. Chem. 298, 101923 (2022).
Subissi, L. et al. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl Acad. Sci. USA 111, E3900–E3909 (2014).
Su, H. et al. Molecular insights into small-molecule drug discovery for SARS-CoV-2. Angew. Chem. Int. Ed. Engl. 60, 9789–9802 (2021).
Jeffreys, L. N. et al. Remdesivir-ivermectin combination displays synergistic interaction with improved in vitro activity against SARS-CoV-2. Int. J. Antimicrob. Agents 59, 106542 (2022).
Gordon, C. J., Tchesnokov, E. P., Schinazi, R. F. & Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 297, 100770 (2021).
Freistadt, M. S., Meades, G. D. & Cameron, C. E. Lethal mutagens: broad-spectrum antivirals with limited potential for development of resistance? Drug Resist. Updat. 7, 19–24 (2004).
Mitsuya, H., Yarchoan, R. & Broder, S. Molecular targets for AIDS therapy. Science 249, 1533–1544 (1990).
Hadj Hassine, I., Ben M’hadheb, M. & Menendez-Arias, L. Lethal mutagenesis of RNA viruses and approved drugs with antiviral mutagenic activity. Viruses. 14, 841 (2022).
Feldmann, H., Sprecher, A. & Geisbert, T. W. Ebola. N. Engl. J. Med. 382, 1832–1842 (2020).
Nili, A. et al. Remdesivir: a beacon of hope from Ebola virus disease to COVID-19. Rev. Med Virol. 30, 1–13 (2020).
Santoro, M. G. & Carafoli, E. Remdesivir: from Ebola to COVID-19. Biochem. Biophys. Res. Commun. 538, 145–150 (2021).
Lamb, Y. N. Remdesivir: first approval. Drugs 80, 1355–1363 (2020).
Kokic, G. et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 12, 279 (2021).
Potisopon, S., Ferron, F., Fattorini, V., Selisko, B. & Canard, B. Substrate selectivity of Dengue and Zika virus NS5 polymerase towards 2’-modified nucleotide analogues. Antivir. Res. 140, 25–36 (2017).
Tian, L. et al. Molnupiravir and its antiviral activity against COVID-19. Front. Immunol. 13, 855496 (2022).
Sendi, P., Razonable, R. R., Nelson, S. B., Soriano, A. & Gandhi, R. T. First-generation oral antivirals against SARS-CoV-2. Clin. Microbiol. Infect. 28, 1230–1235 (2022).
Masyeni, S. et al. Molnupiravir: a lethal mutagenic drug against rapidly mutating severe acute respiratory syndrome coronavirus 2-A narrative review. J. Med. Virol. 94, 3006–3016 (2022).
Kabinger, F. et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 28, 740–746 (2021).
Cheung, P. P. et al. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat. Commun. 5, 4794 (2014).
Sidwell, R. W., Robins, R. K. & Hillyard, I. W. Ribavirin: an antiviral agent. Pharm. Ther. 6, 123–146 (1979).
Drozdzal, S. et al. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updat. 59, 100794 (2021).
Jean, S. S., Lee, P. I. & Hsueh, P. R. Treatment options for COVID-19: the reality and challenges. J. Microbiol. Immunol. Infect. 53, 436–443 (2020).
Bylehn, F., Menendez, C. A., Perez-Lemus, G. R., Alvarado, W. & de Pablo, J. J. Modeling the binding mechanism of remdesivir, favilavir, and ribavirin to SARS-CoV-2 RNA-dependent RNA polymerase. ACS Cent. Sci. 7, 164–174 (2021).
Furuta, Y. et al. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 100, 446–454 (2013).
Joshi, S. et al. Role of favipiravir in the treatment of COVID-19. Int. J. Infect. Dis. 102, 501–508 (2021).
Lagocka, R., Dziedziejko, V., Klos, P. & Pawlik, A. Favipiravir in therapy of viral infections. J. Clin. Med. 10, 273 (2021).
Shiraki, K. & Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharm. Ther. 209, 107512 (2020).
Shannon, A. et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 11, 4682 (2020).
Peng, Q. et al. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovations. 2, 100080 (2021).
Naydenova, K. et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl Acad. Sci. USA 118, e2021946118 (2021).
Shannon, A. et al. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 13, 621 (2022).
Good, S. S. et al. AT-527, a double prodrug of a guanosine nucleotide analog, is a potent inhibitor of SARS-CoV-2 In vitro and a promising oral antiviral for treatment of COVID-19. Antimicrob. Agents Chemother. 65, e02479–20 (2021).
Melo-Filho, C. C. et al. Conserved coronavirus proteins as targets of broad-spectrum antivirals. Antivir. Res. 204, 105360 (2022).
Afdhal, N. et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N. Engl. J. Med. 370, 1889–1898 (2014).
Foster, G. R. et al. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. N. Engl. J. Med. 373, 2608–2617 (2015).
Keating, G. M. & Vaidya, A. Sofosbuvir: first global approval. Drugs 74, 273–282 (2014).
Lawitz, E. et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 368, 1878–1887 (2013).
Appleby, T. C. et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 (2015).
Sacramento, C. Q. et al. In vitro antiviral activity of the anti-HCV drugs daclatasvir and sofosbuvir against SARS-CoV-2, the aetiological agent of COVID-19. J. Antimicrob. Chemother. 76, 1874–1885 (2021).
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014).
Elfiky, A. A. Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 253, 117592 (2020).
Silva Arouche, T. D. et al. Interactions between remdesivir, ribavirin, favipiravir, galidesivir, hydroxychloroquine and chloroquine with fragment molecular of the COVID-19 main protease with inhibitor N3 complex (PDB ID:6LU7) using molecular docking. J. Nanosci. Nanotechnol. 20, 7311–7323 (2020).
Lim, S. Y. et al. A direct-acting antiviral drug abrogates viremia in Zika virus-infected rhesus macaques. Sci. Transl. Med. 12 (2020).
Holgersen, E. M. et al. Transcriptome-wide off-target effects of steric-blocking oligonucleotides. Nucleic Acid Ther. 31, 392–403 (2021).
Mengist, H. M., Fan, X. & Jin, T. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease M(pro). Signal Transduct. Target Ther. 5, 67 (2020).
Rut, W. et al. SARS-CoV-2 M(pro) inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 17, 222–228 (2021).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Jin, Z. et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Chen, Y., Wang, G. & Ouyang, L. Promising inhibitors targeting M(pro): an ideal strategy for anti-SARS-CoV-2 drug discovery. Signal Transduct. Target Ther. 5, 173 (2020).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Elseginy, S. A. Virtual screening and structure-based 3D pharmacophore approach to identify small-molecule inhibitors of SARS-CoV-2 M(pro). J. Biomol. Struct. Dyn. 1–17 (2021).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 (2020).
Faheem et al. Druggable targets of SARS-CoV-2 and treatment opportunities for COVID-19. Bioorg. Chem. 104, 104269 (2020).
Komatsu, T. S. et al. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Sci. Rep. 10, 16986 (2020).
Frecer, V. & Miertus, S. Antiviral agents against COVID-19: structure-based design of specific peptidomimetic inhibitors of SARS-CoV-2 main protease. RSC Adv. 10, 40244–40263 (2020).
Shi, T. H. et al. Andrographolide and its fluorescent derivative inhibit the main proteases of 2019-nCoV and SARS-CoV through covalent linkage. Biochem. Biophys. Res. Commun. 533, 467–473 (2020).
Kitamura, N. et al. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 65, 2848–2865 (2022).
Unoh, Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Qiao, J. et al. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378 (2021).
Ma, C. et al. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Quan, B. X. et al. An orally available M(pro) inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 7, 716–725 (2022).
Amin, S. A., Banerjee, S., Ghosh, K., Gayen, S. & Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 29, 115860 (2021).
Sharma, P. et al. Identification of potential drug candidates to combat COVID-19: a structural study using the main protease (mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 39, 6649–6659 (2021).
Owen, D. R. et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Liang, J. et al. Interaction of the prototypical alpha-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 87, 107292 (2020).
Kneller, D. W. et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 13, 2268 (2022).
Amporndanai, K. et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 12, 3061 (2021).
Gunther, S. et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372, 642–646 (2021).
Gupta, A. et al. Structure-based virtual screening and biochemical validation to discover a potential inhibitor of the SARS-CoV-2 main protease. ACS Omega 5, 33151–33161 (2020).
Khodadadi, E. et al. Study of combining virtual screening and antiviral treatments of the Sars-CoV-2 (Covid-19). Microb. Pathog. 146, 104241 (2020).
Hattori, S. I. et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 12, 668 (2021).
Narayanan, A., Toner, S. A. & Jose, J. Structure-based inhibitor design and repurposing clinical drugs to target SARS-CoV-2 proteases. Biochem. Soc. Trans. 50, 151–165 (2022).
Yang, H. & Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol 19, 685–700 (2021).
Su, H. et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 12, 3623 (2021).
Fu, L. et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 11, 4417 (2020).
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 30, 678–692 (2020).
Vuong, W. et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11, 4282 (2020).
Drayman, N. et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 373, 931–936 (2021).
Gao, X. et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 11, 237–245 (2021).
Rut, W. et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci. Adv. 6, eabd4596 (2020).
Weglarz-Tomczak, E. et al. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 11, 3640 (2021).
Klemm, T. et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 39, e106275 (2020).
McClain, C. B. & Vabret, N. SARS-CoV-2: the many pros of targeting PLpro. Signal Transduct. Target Ther. 5, 223 (2020).
Patchett, S. et al. A molecular sensor determines the ubiquitin substrate specificity of SARS-CoV-2 papain-like protease. Cell Rep. 36, 109754 (2021).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Esposito, S. et al. Host and viral zinc-finger proteins in COVID-19. Int. J. Mol. Sci. 23 (2022).
Tan, H., Hu, Y., Jadhav, P., Tan, B. & Wang, J. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 65, 7561–7580 (2022).
Zhao, Y. et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 12, 877–888 (2021).
Franko, N., Teixeira, A. P., Xue, S., Charpin-El Hamri, G. & Fussenegger, M. Design of modular autoproteolytic gene switches responsive to anti-coronavirus drug candidates. Nat. Commun. 12, 6786 (2021).
Ratia, K. et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl Acad. Sci. USA 105, 16119–16124 (2008).
Ma, C. et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a FlipGFP-based reporter assay. ACS Cent. Sci. 7, 1245–1260 (2021).
Fu, Z. et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 12, 488 (2021).
Osipiuk, J. et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 12, 743 (2021).
Shan, H. et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 28, 855–865e859 (2021).
Henderson, R. et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 27, 925–933 (2020).
Jackson, C. B., Farzan, M., Chen, B. & Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23, 3–20 (2022).
Zhang, J. et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 372, 525−530 (2020).
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Yang, J. et al. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 11, 4541 (2020).
Ahmad, I., Pawara, R., Surana, S. & Patel, H. The repurposed ACE2 inhibitors: SARS-CoV-2 entry blockers of Covid-19. Top. Curr. Chem. 379, 40 (2021).
Day, C. J. et al. Multidisciplinary approaches identify compounds that bind to human ACE2 or SARS-CoV-2 spike protein as candidates to block SARS-CoV-2-ACE2 receptor interactions. mBio. 12, e03681-20 (2021).
Xiang, Y., Wang, M., Chen, H. & Chen, L. Potential therapeutic approaches for the early entry of SARS-CoV-2 by interrupting the interaction between the spike protein on SARS-CoV-2 and angiotensin-converting enzyme 2 (ACE2). Biochem. Pharm. 192, 114724 (2021).
Lin, C. et al. Ceftazidime is a potential drug to inhibit SARS-CoV-2 infection in vitro by blocking spike protein-ACE2 interaction. Signal Transduct. Target Ther. 6, 198 (2021).
Wang, L. et al. Discovery of potential small molecular SARS-CoV-2 entry blockers targeting the spike protein. Acta Pharm. Sin. 43, 788–796 (2022).
Malik, Y. S. et al. Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet. Q 40, 68–76 (2020).
Chan, J. F. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236 (2020).
Huang, Y., Yang, C., Xu, X. F., Xu, W. & Liu, S. W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharm. Sin. 41, 1141–1149 (2020).
Xia, S. et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020).
Yang, J. et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 586, 572–577 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 183, 1735 (2020).
Ou, J. et al. V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J. Virol. 95, e0061721 (2021).
Tai, W. et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol. Immunol. 17, 613–620 (2020).
Yin, W. et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 375, 1048–1053 (2022).
Zhang, J. et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 374, 1353–1360 (2021).
Zhu, Y., Yu, D., Yan, H., Chong, H. & He, Y. Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. J. Virol. 94, e00635-20 (2020).
Schmitz, K. S. et al. Potency of fusion-inhibitory lipopeptides against SARS-CoV-2 variants of concern. mBio 13, e0124922 (2022).
Xue, S. et al. A novel cyclic gamma-AApeptide-based long-acting pan-coronavirus fusion inhibitor with potential oral bioavailability by targeting two sites in spike protein. Cell Discov. 8, 88 (2022).
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127-20 (2020).
Mehdipour, A. R. & Hummer, G. Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. Proc. Natl Acad. Sci. USA 118, e2100425118 (2021).
Azad, T. et al. Nanoluciferase complementation-based bioreporter reveals the importance of N-linked glycosylation of SARS-CoV-2 S for viral entry. Mol. Ther. 29, 1984–2000 (2021).
Nguyen, L. et al. Sialic acid-containing glycolipids mediate binding and viral entry of SARS-CoV-2. Nat. Chem. Biol. 18, 81–90 (2022).
Petitjean, S. J. L. et al. Multivalent 9-O-Acetylated-sialic acid glycoclusters as potent inhibitors for SARS-CoV-2 infection. Nat. Commun. 13, 2564 (2022).
Yi, Y. et al. Natural triterpenoids from licorice potently inhibit SARS-CoV-2 infection. J. Adv. Res. 36, 201–210 (2022).
Yao, H. et al. A high-affinity RBD-targeting nanobody improves fusion partner’s potency against SARS-CoV-2. PLoS Pathog. 17, e1009328 (2021).
Shi, Y. et al. Thiol-based chemical probes exhibit antiviral activity against SARS-CoV-2 via allosteric disulfide disruption in the spike glycoprotein. Proc. Natl Acad. Sci. USA 119, e2120419119 (2022).
Opstelten, D. J., de Groote, P., Horzinek, M. C., Vennema, H. & Rottier, P. J. Disulfide bonds in folding and transport of mouse hepatitis coronavirus glycoproteins. J. Virol. 67, 7394–7401 (1993).
Hati, S. & Bhattacharyya, S. Impact of thiol-disulfide balance on the binding of covid-19 spike protein with angiotensin-converting enzyme 2 receptor. ACS Omega 5, 16292–16298 (2020).
Nayak, S. K. Inhibition of S-protein RBD and hACE2 interaction for control of SARSCoV- 2 infection (COVID-19). Mini Rev. Med. Chem. 21, 689–703 (2021).
Shin, Y. H. et al. Inhibition of ACE2-spike interaction by an ACE2 binder suppresses SARS-CoV-2 entry. Angew. Chem. Int. Ed. Engl. 61, e202115695 (2022).
Junker, D. et al. COVID-19 patient serum less potently inhibits ACE2-RBD binding for various SARS-CoV-2 RBD mutants. Sci. Rep. 12, 7168 (2022).
Pei, P. et al. Computational design of ultrashort peptide inhibitors of the receptor-binding domain of the SARS-CoV-2 S protein. Brief Bioinform. 22, bbab298 (2021).
Panda, P. K. et al. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 6, eabb8097 (2020).
Lee, R. K. et al. Identification of entry inhibitors against delta and omicron variants of SARS-CoV-2. Int. J. Mol. Sci. 23, 4050 (2022).
Xiong, J. et al. Structure-based virtual screening and identification of potential inhibitors of SARS-CoV-2 S-RBD and ACE2 interaction. Front. Chem. 9, 740702 (2021).
Ling, R. et al. In silico design of antiviral peptides targeting the spike protein of SARS-CoV-2. Peptides 130, 170328 (2020).
Ou, X. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620 (2020).
Hoffmann, M., Kleine-Weber, H. & Pohlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779–784e775 (2020).
Abbasi, A. Z. et al. Spiking dependence of SARS-CoV-2 pathogenicity on TMPRSS2. J. Med. Virol. 93, 4205–4218 (2021).
de Loyola, M. B. et al. Alpha-1-antitrypsin: a possible host protective factor against Covid-19. Rev. Med. Virol. 31, e2157 (2021).
Matsuyama, S. et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl Acad. Sci. USA 117, 7001–7003 (2020).
Koch, J. et al. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 40, e107821 (2021).
Muus, C. et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 27, 546–559 (2021).
Zhao, M. M. et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target Ther. 6, 134 (2021).
Zhao, M. M. et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. 8, 53 (2022).
Cheng, J. et al. The furin-S2’ site in avian coronavirus plays a key role in central nervous system damage progression. J. Virol. 95, e02447–20 (2021).
Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293–299 (2021).
Peacock, T. P. et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 6, 899–909 (2021).
Phan, H. A. T., Giannakoulias, S. G., Barrett, T. M., Liu, C. & Petersson, E. J. Rational design of thioamide peptides as selective inhibitors of cysteine protease cathepsin L. Chem. Sci. 12, 10825–10835 (2021).
Mantzourani, C., Vasilakaki, S., Gerogianni, V. E. & Kokotos, G. The discovery and development of transmembrane serine protease 2 (TMPRSS2) inhibitors as candidate drugs for the treatment of COVID-19. Expert Opin. Drug Discov. 17, 231–246 (2022).
Breining, P. et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin. Pharm. Toxicol. 128, 204–212 (2021).
Stenke, L., Hast, R. & Reizenstein, P. Treatment of poor prognosis acute myeloid leukemia with aggressive and non-aggressive chemotherapy. Chemioterapia 6, 723–724 (1987).
Sun, Y. J. et al. Structure-based phylogeny identifies avoralstat as a TMPRSS2 inhibitor that prevents SARS-CoV-2 infection in mice. J. Clin. Invest. 131, e147973 (2021).
Shapira, T. et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 605, 340–348 (2022).
Mahoney, M. et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc Natl Acad Sci USA. 118, e2108728118 (2021).
Chen, Y. et al. A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nat. Commun. 12, 3907 (2021).
Chowdhury, S. F. et al. Exploring inhibitor binding at the S’ subsites of cathepsin L. J. Med. Chem. 51, 1361–1368 (2008).
Fujishima, A. et al. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 407, 47–50 (1997).
Wang, S. Q. et al. Virtual screening for finding natural inhibitor against cathepsin-L for SARS therapy. Amino Acids 33, 129–135 (2007).
Ashhurst, A. S. et al. Potent Anti-SARS-CoV-2 Activity by the Natural Product Gallinamide A and Analogues via Inhibition of Cathepsin L. J Med Chem. 65, 2956-2970 (2022).
Miller, B. et al. The marine cyanobacterial metabolite gallinamide A is a potent and selective inhibitor of human cathepsin L. J. Nat. Prod. 77, 92–99 (2014).
Ashhurst, A. S. et al. Potent anti-SARS-CoV-2 activity by the natural product gallinamide A and analogues via inhibition of cathepsin L. J. Med. Chem. 65, 2956–2970 (2022).
Frueh, F. W. et al. An orally available cathepsin L inhibitor protects lungs against SARS-CoV-2-induced diffuse alveolar damage in african green monkeys. Preprint at bioRxiv https://doi.org/10.1101/2021.07.20.453127 (2021).
Liu, T., Luo, S., Libby, P. & Shi, G. P. Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharm. Ther. 213, 107587 (2020).
Hu, Y. et al. Boceprevir, calpain inhibitors II and XII, and GC-376 have broad-spectrum antiviral activity against coronaviruses. ACS Infect. Dis. 7, 586–597 (2021).
Sacco, M. D. et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 6, eabe0751 (2020).
Cheng, Y. W. et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 33, 108254 (2020).
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020).
Osman, E. E. A., Rehemtulla, A. & Neamati, N. Why all the fury over furin? J. Med. Chem. 65, 2747–2784 (2022).
Zhang, L. et al. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl Acad. Sci. USA 118, e2109905118 (2021).
Papa, G. et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 17, e1009246 (2021).
Shang, J. et al. Cell entry mechanisms of SARS-CoV-2. Proc. Natl Acad. Sci. USA 117, 11727–11734 (2020).
Zhang, Y. & Tang, L. V. Overview of targets and potential drugs of SARS-CoV-2 according to the viral replication. J. Proteome Res. 20, 49–59 (2021).
Paszti-Gere, E. et al. In vitro characterization of the furin inhibitor MI-1851: albumin binding, interaction with cytochrome P450 enzymes and cytotoxicity. Biomed. Pharmacother. 151, 113124 (2022).
Azkur, A. K. et al. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 75, 1564–1581 (2020).
Bartleson, J. M. et al. SARS-CoV-2, COVID-19 and the ageing immune system. Nat. Aging 1, 769–782 (2021).
di Mauro, G., Scavone, C., Rafaniello, C., Rossi, F. & Capuano, A. SARS-Cov-2 infection: response of human immune system and possible implications for the rapid test and treatment. Int. Immunopharmacol. 84, 106519 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Tan, L. et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct. Target Ther. 5, 33 (2020).
Kempuraj, D. et al. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist 26, 402–414 (2020).
Tan, M. et al. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology 160, 261–268 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Coperchini, F., Chiovato, L., Croce, L., Magri, F. & Rotondi, M. The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 53, 25–32 (2020).
Goker Bagca, B. & Biray Avci, C. The potential of JAK/STAT pathway inhibition by ruxolitinib in the treatment of COVID-19. Cytokine Growth Factor Rev. 54, 51–62 (2020).
Hu, B., Huang, S. & Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 93, 250–256 (2021).
Lee, J. S. & Shin, E. C. The type I interferon response in COVID-19: implications for treatment. Nat. Rev. Immunol. 20, 585–586 (2020).
Vabret, N. et al. Advancing scientific knowledge in times of pandemics. Nat. Rev. Immunol. 20, 338 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Hu, X., Li, J., Fu, M., Zhao, X. & Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target Ther. 6, 402 (2021).
Zarrin, A. A., Bao, K., Lupardus, P. & Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 20, 39–63 (2021).
Levy, G., Guglielmelli, P., Langmuir, P. & Constantinescu, S. JAK inhibitors and COVID-19. J. Immunother. Cancer 10 (2022).
Cao, X. ISG15 secretion exacerbates inflammation in SARS-CoV-2 infection. Nat. Immunol. 22, 1360–1362 (2021).
Peterson, D., Damsky, W. & King, B. The use of Janus kinase inhibitors in the time of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J. Am. Acad. Dermatol. 82, e223–e226 (2020).
Song, P., Li, W., Xie, J., Hou, Y. & You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 509, 280–287 (2020).
Solimani, F., Meier, K. & Ghoreschi, K. Janus kinase signaling as risk factor and therapeutic target for severe SARS-CoV-2 infection. Eur. J. Immunol. 51, 1071–1075 (2021).
Goletti, D. & Cantini, F. Baricitinib therapy in covid-19 pneumonia—an unmet need fulfilled. N. Engl. J. Med. 384, 867–869 (2021).
Jorgensen, S. C. J., Tse, C. L. Y., Burry, L. & Dresser, L. D. Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy 40, 843–856 (2020).
Kay, J. et al. Changes in selected haematological parameters associated with JAK1/JAK2 inhibition observed in patients with rheumatoid arthritis treated with baricitinib. RMD Open. 6, e001370 (2020).
Stebbing, J. et al. JAK inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci Adv. 7, eabe4724 (2021).
Nystrom, S. E. et al. JAK inhibitor blocks COVID-19 cytokine-induced JAK/STAT/APOL1 signaling in glomerular cells and podocytopathy in human kidney organoids. JCI Insight. 7, e157432 (2022).
Chen, C. X. et al. JAK-inhibitors for coronavirus disease-2019 (COVID-19): a meta-analysis. Leukemia 35, 2616–2620 (2021).
Singh, D. et al. A phase 2 multiple ascending dose study of the inhaled pan-JAK inhibitor nezulcitinib (TD-0903) in severe COVID-19. Eur. Respir. J. 58, 2100673 (2021).
Tong, S. Y. C. & Petersiel, N. Tofacitinib reduced death or respiratory failure at 28 d in patients hospitalized with COVID-19 pneumonia. Ann. Intern. Med. 174, JC111 (2021).
Yan, B. et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci. Immunol. 6, eabg0833 (2021).
Neubauer, A. et al. Ruxolitinib for the treatment of SARS-CoV-2 induced acute respiratory distress syndrome (ARDS). Leukemia 34, 2276–2278 (2020).
Boyle, D. L. et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 74, 1311–1316 (2015).
Clark, J. D., Flanagan, M. E. & Telliez, J. B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 57, 5023–5038 (2014).
Govaerts, I., Jacobs, K., Vandepoel, R. & Cools, J. JAK/STAT pathway mutations in T-ALL, including the STAT5B N642H mutation, are sensitive to JAK1/JAK3 inhibitors. Hemasphere 3, e313 (2019).
Palasiewicz, K., Umar, S., Romay, B., Zomorrodi, R. K. & Shahrara, S. Tofacitinib therapy intercepts macrophage metabolic reprogramming instigated by SARS-CoV-2 Spike protein. Eur. J. Immunol. 51, 2330–2340 (2021).
Zununi Vahed, S., Hosseiniyan Khatibi, S. M., Ahmadian, E. & Ardalan, M. Targeting chronic COVID-19 lung injury; Tofacitinib can be used against tissue-resident memory T cells. Biomed. Pharmacother. 147, 112614 (2022).
Byrd, J. C. et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 369, 32–42 (2013).
Roschewski, M. et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 5, eabd0110 (2020).
Ni Gabhann, J. et al. Btk regulates macrophage polarization in response to lipopolysaccharide. PLoS ONE 9, e85834 (2014).
Treon, S. P. et al. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19-infected patients. Blood 135, 1912–1915 (2020).
Chong, E. A. et al. BTK inhibitors in cancer patients with COVID-19: “The Winner Will be the One Who Controls That Chaos” (Napoleon Bonaparte). Clin. Cancer Res. 26, 3514–3516 (2020).
Benner, B. & Carson, W. E. Observations on the use of Bruton’s tyrosine kinase inhibitors in SAR-CoV-2 and cancer. J. Hematol. Oncol. 14, 15 (2021).
Kaliamurthi, S. et al. Structure-based virtual screening reveals Ibrutinib and Zanubrutinib as potential repurposed drugs against COVID-19. Int. J. Mol. Sci. 22, 7071 (2021).
Aggarwal, B. B. Nuclear factor-kappaB: the enemy within. Cancer Cell 6, 203–208 (2004).
Mussbacher, M. et al. Cell type-specific roles of NF-kappaB linking inflammation and thrombosis. Front. Immunol. 10, 85 (2019).
Farahani, M. et al. Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 145, 112420 (2022).
Hariharan, A., Hakeem, A. R., Radhakrishnan, S., Reddy, M. S. & Rela, M. The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology 29, 91–100 (2021).
Nilsson-Payant, B. E. et al. The NF-kappaB transcriptional footprint is essential for SARS-CoV-2 replication. J. Virol. 95, e0125721 (2021).
Oh, H. & Ghosh, S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 252, 41–51 (2013).
Li, W. et al. SARS-CoV-2 Nsp5 activates NF-kappaB pathway by upregulating SUMOylation of MAVS. Front. Immunol. 12, 750969 (2021).
Wu, Y. et al. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-kappaB hyper-activation and inflammation. Signal Transduct. Target Ther. 6, 167 (2021).
Xia, J. et al. SARS-CoV-2 N protein induces acute lung injury in mice via NF-kB activation. Front. Immunol. 12, 791753 (2021).
Gudowska-Sawczuk, M. & Mroczko, B. The role of nuclear factor kappa B (NF-kappaB) in development and treatment of COVID-19: review. Int. J. Mol. Sci. 23, 5283 (2022).
Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651 (2009).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2, 17023 (2017).
Sharma, V. K. et al. Nanocurcumin potently inhibits SARS-CoV-2 spike protein-induced cytokine storm by deactivation of MAPK/NF-kappaB signaling in epithelial. Cells ACS Appl. Bio Mater. 5, 483–491 (2022).
Lee, M. C., Chen, Y. K., Tsai-Wu, J. J., Hsu, Y. J. & Lin, B. R. Zinc supplementation augments the suppressive effects of repurposed NF-kappaB inhibitors on ACE2 expression in human lung cell lines. Life Sci. 280, 119752 (2021).
Freeman, T. L. & Swartz, T. H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 11, 1518 (2020).
van den Berg, D. F. & Te Velde, A. A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 11, 1580 (2020).
Fung, S. Y., Yuen, K. S., Ye, Z. W., Chan, C. P. & Jin, D. Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg. Microbes Infect. 9, 558–570 (2020).
He, Y., Hara, H. & Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021 (2016).
Kolb, M., Margetts, P. J., Anthony, D. C., Pitossi, F. & Gauldie, J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107, 1529–1536 (2001).
Rodrigues, T. S. et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 218, e20201707 (2021).
Pan, P. et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 12, 4664 (2021).
Shah, A. Novel Coronavirus-induced NLRP3 inflammasome activation: a potential drug target in the treatment of COVID-19. Front Immunol. 11, 1021 (2020).
Zeng, J. et al. Specific inhibition of the NLRP3 inflammasome suppresses immune overactivation and alleviates COVID-19 like pathology in mice. EBioMedicine 75, 103803 (2022).
Deville-Bonne, D. et al. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: structural and catalytic properties. Antivir. Res. 86, 101–120 (2010).
Fung, J., Lai, C. L., Seto, W. K. & Yuen, M. F. Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J. Antimicrob. Chemother. 66, 2715–2725 (2011).
Garcia-Trejo, J. J., Ortega, R. & Zarco-Zavala, M. Putative repurposing of lamivudine, a nucleoside/nucleotide analogue and antiretroviral to improve the outcome of cancer and COVID-19 patients. Front. Oncol. 11, 664794 (2021).
Keeffe, E. B., Dieterich, D. T., Pawlotsky, J. M. & Benhamou, Y. Chronic hepatitis B: preventing, detecting, and managing viral resistance. Clin. Gastroenterol. Hepatol. 6, 268–274 (2008).
Leung, G. P. Iatrogenic mitochondriopathies: a recent lesson from nucleoside/nucleotide reverse transcriptase inhibitors. Adv. Exp. Med. Biol. 942, 347–369 (2012).
Luyt, C. E. et al. Acyclovir for mechanically ventilated patients with herpes simplex virus oropharyngeal reactivation: a randomized clinical trial. JAMA Intern. Med. 180, 263–272 (2020).
Chow, W. A., Jiang, C. & Guan, M. Anti-HIV drugs for cancer therapeutics: back to the future? Lancet Oncol. 10, 61–71 (2009).
Kowdley, K. V. et al. On-treatment HCV RNA as a predictor of sustained virological response in HCV genotype 3-infected patients treated with daclatasvir and sofosbuvir. Liver Int. 36, 1611–1618 (2016).
Wohlfarth, C. & Efferth, T. Natural products as promising drug candidates for the treatment of hepatitis B and C. Acta Pharm. Sin. 30, 25–30 (2009).
Vicenti, I., Zazzi, M. & Saladini, F. SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opin. Ther. Pat. 31, 325–337 (2021).
Wang, Y., Anirudhan, V., Du, R., Cui, Q. & Rong, L. RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J. Med. Virol. 93, 300–310 (2021).
Kazemzadeh-Narbat, M. et al. Adenosine-associated delivery systems. J. Drug Target 23, 580–596 (2015).
Vallon, V., Muhlbauer, B. & Osswald, H. Adenosine and kidney function. Physiol. Rev. 86, 901–940 (2006).
Santoro, M. G. & Carafoli, E. Remdesivir: From Ebola to COVID-19. Biochem. Biophys. Res. Commun. 538, 145–150 (2021).
Pruijssers, A. J. et al. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. Cell Rep. 32, 107940 (2020).
Holshue, M. L. et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 382, 929–936 (2020).
Beigel, J. H. et al. Remdesivir for the treatment of Covid-19—final report. N. Engl. J. Med. 383, 1813–1826 (2020).
Stevens, L. J. et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 14, eabo0718 (2022).
Cox, R. M. et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat. Commun. 12, 6415 (2021).
Yan, V. C. & Muller, F. L. Advantages of the parent nucleoside GS-441524 over remdesivir for covid-19 treatment. ACS Med. Chem. Lett. 11, 1361–1366 (2020).
Al-Tawfiq, J. A., Al-Homoud, A. H. & Memish, Z. A. Remdesivir as a possible therapeutic option for the COVID-19. Travel Med. Infect. Dis. 34, 101615 (2020).
Li, Y. et al. Remdesivir metabolite GS-441524 effectively inhibits SARS-CoV-2 infection in mouse models. J. Med. Chem. 65, 2785–2793 (2022).
Xie, J. & Wang, Z. Can remdesivir and its parent nucleoside GS-441524 be potential oral drugs? An in vitro and in vivo DMPK assessment. Acta Pharm. Sin. B 11, 1607–1616 (2021).
Rasmussen, H. B., Thomsen, R. & Hansen, P. R. Nucleoside analog GS-441524: pharmacokinetics in different species, safety, and potential effectiveness against Covid-19. Pharm. Res. Perspect. 10, e00945 (2022).
Tempestilli, M. et al. Pharmacokinetics of remdesivir and GS-441524 in two critically ill patients who recovered from COVID-19. J. Antimicrob. Chemother. 75, 2977–2980 (2020).
Zhang, R. et al. Oral remdesivir derivative VV116 is a potent inhibitor of respiratory syncytial virus with efficacy in mouse model. Signal Transduct. Target Ther. 7, 123 (2022).
Wu, C. R., Yin, W. C., Jiang, Y. & Xu, H. E. Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol. Sin. 1–13 (2022).
Qian, H. J. et al. Safety, tolerability, and pharmacokinetics of VV116, an oral nucleoside analog against SARS-CoV-2, in Chinese healthy subjects. Acta Pharmacol Sin. 1–9 (2022).
Shen, Y. et al. An open, prospective cohort study of VV116 in Chinese participants infected with SARS-CoV-2 omicron variants. Emerg. Microbes Infect. 11, 1518–1523 (2022).
Aftab, S. O. et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 18, 275 (2020).
Lee, C. C., Hsieh, C. C. & Ko, W. C. Molnupiravir—a novel oral anti-SARS-CoV-2 agent. Antibiotics (Basel) 10, 1294 (2021).
Sheahan, T. P. et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 12, eabb5883 (2020).
Li, P. et al. SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 32, 322–324 (2022).
Zhou, S. et al. beta-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 224, 415–419 (2021).
Fayzullina, D. et al. FNC: an advanced anticancer therapeutic or just an underdog? Front. Oncol. 12, 820647 (2022).
Chang, J. 4’-Modified nucleosides for antiviral drug discovery: achievements and perspectives. Acc. Chem. Res. 55, 565–578 (2022).
Zhang, J. L. et al. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal Transduct. Target Ther. 6, 414 (2021).
Su, S., Wang, Q. & Jiang, S. Facing the challenge of viral mutations in the age of pandemic: developing highly potent, broad-spectrum, and safe COVID-19 vaccines and therapeutics. Clin. Transl. Med. 11, e284 (2021).
Krajczyk, A. et al. Antivirally active ribavirin analogues–4,5-disubstituted 1,2,3-triazole nucleosides: biological evaluation against certain respiratory viruses and computational modelling. Antivir. Chem. Chemother. 23, 161–171 (2014).
Lau, J. Y., Tam, R. C., Liang, T. J. & Hong, Z. Mechanism of action of ribavirin in the combination treatment of chronic HCV infection. Hepatology 35, 1002–1009 (2002).
Eslami, G. et al. The impact of sofosbuvir/daclatasvir or ribavirin in patients with severe COVID-19. J. Antimicrob. Chemother. 75, 3366–3372 (2020).
Khamis, F. et al. Randomized controlled open label trial on the use of favipiravir combined with inhaled interferon beta-1b in hospitalized patients with moderate to severe COVID-19 pneumonia. Int J. Infect. Dis. 102, 538–543 (2021).
Kuzikov, M. et al. Identification of inhibitors of SARS-CoV-2 3CL-pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharm. Transl. Sci. 4, 1096–1110 (2021).
Abubakar, A. R. et al. Systematic review on the therapeutic options for COVID-19: clinical evidence of drug efficacy and implications. Infect. Drug Resist. 13, 4673–4695 (2020).
Wu, X. et al. Efficacy and safety of triazavirin therapy for coronavirus disease 2019: a pilot randomized controlled trial. Engineering 6, 1185–1191 (2020).
Hong, S. et al. Epigallocatechin gallate inhibits the uridylate-specific endoribonuclease Nsp15 and efficiently neutralizes the SARS-CoV-2 strain. J. Agric. Food Chem. 69, 5948–5954 (2021).
Sulkowski, M. S. et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N. Engl. J. Med. 370, 211–221 (2014).
Gardinali, N. R. et al. Sofosbuvir shows a protective effect against vertical transmission of Zika virus and the associated congenital syndrome in rhesus monkeys. Antivir. Res. 182, 104859 (2020).
Lin, Y. et al. Identification and characterization of Zika virus NS5 RNA-dependent RNA polymerase inhibitors. Int. J. Antimicrob. Agents 54, 502–506 (2019).
Leumi, S. et al. Identification of a novel replication-competent hepatitis C virus variant that confers the sofosbuvir resistance. Antivir. Res. 197, 105224 (2022).
Chien, M. et al. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 19, 4690–4697 (2020).
El-Bendary, M. et al. Efficacy of combined Sofosbuvir and Daclatasvir in the treatment of COVID-19 patients with pneumonia: a multicenter Egyptian study. Expert Rev. Anti Infect. Ther. 20, 291–295 (2022).
Kow, C. S., Javed, A., Ramachandram, D. & Hasan, S. S. Clinical outcomes of sofosbuvir-based antivirals in patients with COVID-19: a systematic review and meta-analysis of randomized trials. Expert Rev. Anti Infect. Ther. 20, 567–575 (2022).
Dragoni, F. et al. Evaluation of sofosbuvir activity and resistance profile against West Nile virus in vitro. Antivir. Res. 175, 104708 (2020).
Furuta, Y., Komeno, T. & Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 93, 449–463 (2017).
Cai, Q. et al. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering 6, 1192–1198 (2020).
Dabbous, H. M. et al. Safety and efficacy of favipiravir versus hydroxychloroquine in management of COVID-19: A randomised controlled trial. Sci. Rep. 11, 7282 (2021).
Rabie, A. M. Cyanorona-20: the first potent anti-SARS-CoV-2 agent. Int. Immunopharmacol. 98, 107831 (2021).
Kalil, A. C. et al. Baricitinib plus Remdesivir for hospitalized adults with covid-19. N. Engl. J. Med. 384, 795–807 (2021).
Patel, N. M. et al. Inhibition of the JAK/STAT pathway with baricitinib reduces the multiple organ dysfunction caused by hemorrhagic shock in rats. Ann. Surg. (2022).
Bronte, V. et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Invest. 130, 6409–6416 (2020).
Marconi, V. C. et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 9, 1407–1418 (2021).
Alizadehmohajer, N. et al. Screening of potential inhibitors of COVID-19 with repurposing approach via molecular docking. Netw. Model Anal. Health Inf. Bioinform. 11, 11 (2022).
Huntington, K. E. et al. Integrin/TGF-beta1 inhibitor GLPG-0187 blocks SARS-CoV-2 delta and omicron pseudovirus infection of airway epithelial cells in vitro, which could attenuate disease severity. Pharmaceuticals (Basel) 15, 618 (2022).
Indu, P. et al. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: a molecular docking and drug repurposing approach. J. Infect. Public Health 13, 1856–1861 (2020).
Tabassum, N., Zhang, H. & Stebbing, J. Repurposing fostamatinib to combat SARS-CoV-2-induced acute lung injury. Cell Rep. Med. 1, 100145 (2020).
Baranov, M. V., Bianchi, F. & van den Bogaart, G. The PIKfyve inhibitor apilimod: a double-edged sword against COVID-19. Cells 10, 30 (2020).
Maguire, J. J., Kuc, R. E. & Davenport, A. P. Defining the affinity and receptor sub-type selectivity of four classes of endothelin antagonists in clinically relevant human cardiovascular tissues. Life Sci. 91, 681–686 (2012).
Seya, T., Takeda, Y. & Matsumoto, M. A Toll-like receptor 3 (TLR3) agonist ARNAX for therapeutic immunotherapy. Adv. Drug Deliv. Rev. 147, 37–43 (2019).
Giantonio, B. J. et al. Toxicity and response evaluation of the interferon inducer poly ICLC administered at low dose in advanced renal carcinoma and relapsed or refractory lymphoma: a report of two clinical trials of the Eastern Cooperative Oncology Group. Invest. N. Drugs 19, 89–92 (2001).
Ko, M. J., Cheigh, C. I. & Chung, M. S. Relationship analysis between flavonoids structure and subcritical water extraction (SWE). Food Chem. 143, 147–155 (2014).
Panche, A. N., Diwan, A. D. & Chandra, S. R. Flavonoids: an overview. J. Nutr. Sci. 5, e47 (2016).
Liskova, A. et al. Flavonoids against the SARS-CoV-2 induced inflammatory storm. Biomed. Pharmacother. 138, 111430 (2021).
Cushnie, T. P. & Lamb, A. J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 26, 343–356 (2005).
Nguyen, T. L. A. & Bhattacharya, D. Antimicrobial activity of quercetin: an approach to its mechanistic principle. Molecules 27, 2494 (2022).
Pan, B. et al. Chinese herbal compounds against SARS-CoV-2: Puerarin and quercetin impair the binding of viral S-protein to ACE2 receptor. Comput. Struct. Biotechnol. J. 18, 3518–3527 (2020).
Derosa, G., Maffioli, P., D’Angelo, A. & Di Pierro, F. A role for quercetin in coronavirus disease 2019 (COVID-19). Phytother. Res. 35, 1230–1236 (2021).
Chikhale, R. V. et al. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 1–16 (2020).
Kandeel, M. et al. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struct. Dyn. 39, 5129–5136 (2021).
Rakshit, M., Muduli, S., Srivastav, P. P. & Mishra, S. Pomegranate peel polyphenols prophylaxis against SARS-CoV-2 main protease by in-silico docking and molecular dynamics study. J. Biomol. Struct. Dyn. 1–15 (2021).
Drouet, S., Tungmunnithum, D., Laine, E. & Hano, C. Gene expression analysis and metabolite profiling of silymarin biosynthesis during milk thistle (Silybum marianum (L.) Gaertn.) fruit ripening. Int. J. Mol. Sci. 21, 4730 (2020).
Palit, P., Mukhopadhyay, A. & Chattopadhyay, D. Phyto-pharmacological perspective of Silymarin: A potential prophylactic or therapeutic agent for COVID-19, based on its promising immunomodulatory, anti-coagulant and anti-viral property. Phytother. Res. 35, 4246–4257 (2021).
Gillessen, A. & Schmidt, H. H. Silymarin as supportive treatment in liver diseases: a narrative review. Adv. Ther. 37, 1279–1301 (2020).
Hanafy, N. A. N. & El-Kemary, M. A. Silymarin/curcumin loaded albumin nanoparticles coated by chitosan as muco-inhalable delivery system observing anti-inflammatory and anti COVID-19 characterizations in oleic acid triggered lung injury and in vitro COVID-19 experiment. Int. J. Biol. Macromol. 198, 101–110 (2022).
Imran, M. et al. Luteolin, a flavonoid, as an anticancer agent: a review. Biomed. Pharmacother. 112, 108612 (2019).
Maurya, V. K., Kumar, S., Prasad, A. K., Bhatt, M. L. B. & Saxena, S. K. Structure-based drug designing for potential antiviral activity of selected natural products from Ayurveda against SARS-CoV-2 spike glycoprotein and its cellular receptor. Virus Dis. 31, 179–193 (2020).
Shawky, E., Nada, A. A. & Ibrahim, R. S. Potential role of medicinal plants and their constituents in the mitigation of SARS-CoV-2: identifying related therapeutic targets using network pharmacology and molecular docking analyses. RSC Adv. 10, 27961–27983 (2020).
Xie, Y. Z. et al. A practical strategy for exploring the pharmacological mechanism of luteolin against COVID-19/asthma comorbidity: findings of system pharmacology and bioinformatics analysis. Front. Immunol. 12, 769011 (2021).
da Cunha, L. et al. Natural products with tandem anti-inflammatory, immunomodulatory and anti-sars-cov/2 effects: a drug discovery perspective against SARS-CoV-2. Curr. Med. Chem. 29, 2530–2564 (2022).
Hossain, R. et al. In silico screening of natural products as potential inhibitors of SARS-CoV-2 using molecular docking simulation. Chin. J. Integr. Med. 28, 249–256 (2022).
Ristovski, J. T., Matin, M. M., Kong, R., Kusturica, M. P. & Zhang, H. In vitro testing and computational analysis of specific phytochemicals with antiviral activities considering their possible applications against COVID-19. S. Afr. J. Bot. (2022).
Zandi, K. et al. Baicalein and baicalin inhibit SARS-CoV-2 RNA-dependent-RNA polymerase. Microorganisms 9, 893 (2021).
Bachar, S. C., Mazumder, K., Bachar, R., Aktar, A. & Al Mahtab, M. A review of medicinal plants with antiviral activity available in Bangladesh and mechanistic insight into their bioactive metabolites on SARS-CoV-2, HIV and HBV. Front. Pharm. 12, 732891 (2021).
da Silva-Junior, E. F. & Silva, L. R. Multi-target approaches of epigallocatechin-3-O-gallate (EGCG) and its derivatives against influenza viruses. Curr. Top. Med. Chem. 22, 1485–1500 (2022).
Wang, Y. Q., Li, Q. S., Zheng, X. Q., Lu, J. L. & Liang, Y. R. Antiviral effects of green tea EGCG and its potential application against COVID-19. Molecules 26, 3962 (2021).
Zhang, Z. et al. Potential protective mechanisms of green tea polyphenol EGCG against COVID-19. Trends Food Sci. Technol. 114, 11–24 (2021).
Jang, M. et al. Tea polyphenols EGCG and theaflavin inhibit the activity of SARS-CoV-2 3CL-protease in vitro. Evid. Based Complement Altern. Med. 2020, 5630838 (2020).
Chiou, W. C. et al. The inhibitory effects of PGG and EGCG against the SARS-CoV-2 3C-like protease. Biochem. Biophys. Res. Commun. 591, 130–136 (2022).
Arif, M. N. Catechin derivatives as inhibitor of COVID-19 main protease (Mpro): molecular docking studies unveil an opportunity against CORONA. Comb. Chem. High. Throughput Screen 25, 197–203 (2022).
Mahmud, S. et al. Plant-based phytochemical screening by targeting main protease of SARS-CoV-2 to design effective potent inhibitors. Biology (Basel) 10, 589 (2021).
Montone, C. M. et al. Characterization of the trans-epithelial transport of green tea (C. sinensis) catechin extracts with in vitro inhibitory effect against the SARS-CoV-2 papain-like protease activity. Molecules 26, 6744 (2021).
Jin, Y. H. et al. Natural polyphenols, 1,2,3,4,6-O-pentagalloyglucose and proanthocyanidins, as broad-spectrum anticoronaviral inhibitors targeting Mpro and RdRp of SARS-CoV-2. Biomedicines 10, 1170 (2022).
Dong, N. Q. & Lin, H. X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 63, 180–209 (2021).
Fang, C. Y. et al. Natural products: potential treatments for cisplatin-induced nephrotoxicity. Acta Pharm. Sin. 42, 1951–1969 (2021).
Xiang, J., Zhang, M., Apea-Bah, F. B. & Beta, T. Hydroxycinnamic acid amide (HCAA) derivatives, flavonoid C-glycosides, phenolic acids and antioxidant properties of foxtail millet. Food Chem. 295, 214–223 (2019).
Wang, G. F. et al. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antivir. Res. 83, 186–190 (2009).
Ozcelik, B., Kartal, M. & Orhan, I. Cytotoxicity, antiviral and antimicrobial activities of alkaloids, flavonoids, and phenolic acids. Pharm. Biol. 49, 396–402 (2011).
Youssef, F. S., Altyar, A. E., Omar, A. M. & Ashour, M. L. Phytoconstituents, in vitro anti-infective activity of Buddleja indica Lam., and in silico evaluation of its SARS-CoV-2 inhibitory potential. Front. Pharm. 12, 619373 (2021).
Bhowmik, D. et al. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 84, 104451 (2020).
Gamaleldin Elsadig Karar, M., Matei, M. F., Jaiswal, R., Illenberger, S. & Kuhnert, N. Neuraminidase inhibition of Dietary chlorogenic acids and derivatives—potential antivirals from dietary sources. Food Funct. 7, 2052–2059 (2016).
Yu, J. W., Wang, L. & Bao, L. D. Exploring the active compounds of traditional Mongolian medicine in intervention of novel coronavirus (COVID-19) based on molecular docking method. J. Funct. Foods 71, 104016 (2020).
El Gizawy, H. A. et al. Pimenta dioica (L.) Merr. bioactive constituents exert anti-SARS-CoV-2 and anti-inflammatory activities: molecular docking and dynamics, in vitro, and in vivo studies. Molecules 26, 5844 (2021).
Chen, X. et al. Identifying potential anti-COVID-19 pharmacological components of traditional Chinese medicine Lianhuaqingwen capsule based on human exposure and ACE2 biochromatography screening. Acta Pharm. Sin. B 11, 222–236 (2021).
Akdad, M., Moujane, S., Bouadid, I., Benlyas, M. & Eddouks, M. Phytocompounds from Anvillea radiata as promising anti-Covid-19 drugs: in silico studies and in vivo safety assessment. J. Environ. Sci. Health A Tox Hazard Subst. Environ. Eng. 56, 1512–1523 (2021).
Shah, S. et al. Prospecting for Cressa cretica to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2. J. Biomol. Struct. Dyn. 40, 5643–5652 (2022).
Xiao, Z. et al. Pharmacological effects of salvianolic acid B against oxidative damage. Front. Pharm. 11, 572373 (2020).
Hu, S. et al. Three salvianolic acids inhibit 2019-nCoV spike pseudovirus viropexis by binding to both its RBD and receptor ACE2. J. Med. Virol. 93, 3143–3151 (2021).
Wang, W. et al. Danshensu alleviates pseudo-typed SARS-CoV-2 induced mouse acute lung inflammation. Acta Pharm. Sin. 43, 771–780 (2022).
Yang, C. et al. Salvianolic acid C potently inhibits SARS-CoV-2 infection by blocking the formation of six-helix bundle core of spike protein. Signal Transduct. Target Ther. 5, 220 (2020).
Gong, L. et al. A review of pharmacological and pharmacokinetic properties of Forsythiaside A. Pharm. Res. 169, 105690 (2021).
Wang, Z. et al. Phytochemistry, pharmacology, quality control and future research of Forsythia suspensa (Thunb.) Vahl: a review. J. Ethnopharmacol. 210, 318–339 (2018).
Fu, K. et al. Forsythiaside A alleviated carbon tetrachloride-induced liver fibrosis by modulating gut microbiota composition to increase short-chain fatty acids and restoring bile acids metabolism disorder. Biomed. Pharmacother. 151, 113185 (2022).
Fu, Y. et al. Interfering effects on the bioactivities of several key proteins of COVID-19/variants in diabetes by compounds from Lianqiao leaves: In silico and in vitro analyses. Int J. Biol. Macromol. 207, 715–729 (2022).
Rostom, B., Karaky, R., Kassab, I. & Sylla-Iyarreta Veitia, M. Coumarins derivatives and inflammation: review of their effects on the inflammatory signaling pathways. Eur. J. Pharm. 922, 174867 (2022).
Zhou, M. et al. Bergamottin, a bioactive component of bergamot, inhibits SARS-CoV-2 infection in golden Syrian hamsters. Antivir. Res. 204, 105365 (2022).
Xu, X. Y., Wang, D. Y., Li, Y. P., Deyrup, S. T. & Zhang, H. J. Plant-derived lignans as potential antiviral agents: a systematic review. Phytochem. Rev. 21, 239–289 (2022).
Ma, Q. et al. Phillyrin (KD-1) exerts anti-viral and anti-inflammatory activities against novel coronavirus (SARS-CoV-2) and human coronavirus 229E (HCoV-229E) by suppressing the nuclear factor kappa B (NF-kappaB) signaling pathway. Phytomedicine 78, 153296 (2020).
Lai, Y. et al. Phillyrin for COVID-19 anD Influenza Co-infection: A Potential Therapeutic Strategy Targeting Host Based on Bioinformatics analysis. Front. Pharm. 12, 754241 (2021).
Xiao, S. et al. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med. Res. Rev. 38, 951–976 (2018).
Ge, J. et al. Natural terpenoids with anti-inflammatory activities: potential leads for anti-inflammatory drug discovery. Bioorg. Chem. 124, 105817 (2022).
Hu, Y. et al. Artemether, artesunate, arteannuin B, echinatin, licochalcone B and andrographolide effectively inhibit SARS-CoV-2 and related viruses in vitro. Front. Cell Infect. Microbiol. 11, 680127 (2021).
Ashley, E. A., Pyae Phyo, A. & Woodrow, C. J. Malaria. Lancet 391, 1608–1621 (2018).
Ma, N., Zhang, Z., Liao, F., Jiang, T. & Tu, Y. The birth of artemisinin. Pharm. Ther. 216, 107658 (2020).
Efferth, T. Beyond malaria: the inhibition of viruses by artemisinin-type compounds. Biotechnol. Adv. 36, 1730–1737 (2018).
Efferth, T. et al. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 47, 804–811 (2008).
Tu, Y. Artemisinin-A Gift from Traditional Chinese Medicine to the World (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 55, 10210–10226 (2016).
Rolta, R. et al. Phytocompounds of Rheum emodi, Thymus serpyllum, and Artemisia annua inhibit spike protein of SARS-CoV-2 binding to ACE2 receptor: in silico approach. Curr. Pharm. Rep. 7, 135–149 (2021).
Cao, R. et al. Anti-SARS-CoV-2 potential of artemisinins in vitro. ACS Infect. Dis. 6, 2524–2531 (2020).
Nair, M. S. et al. Artemisia annua L. extracts inhibit the in vitro replication of SARS-CoV-2 and two of its variants. J. Ethnopharmacol. 274, 114016 (2021).
Li, G. et al. Safety and efficacy of artemisinin-piperaquine for treatment of COVID-19: an open-label, non-randomised and controlled trial. Int. J. Antimicrob. Agents 57, 106216 (2021).
Aanouz, I. et al. Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: computational investigations. J. Biomol. Struct. Dyn. 39, 2971–2979 (2021).
Vincent, S., Arokiyaraj, S., Saravanan, M. & Dhanraj, M. Molecular docking studies on the anti-viral effects of compounds from Kabasura Kudineer on SARS-CoV-2 3CL(pro). Front. Mol. Biosci. 7, 613401 (2020).
Zubair, M. S. et al. GC-MS, LC-MS/MS, docking and molecular dynamics approaches to identify potential SARS-CoV-2 3-chymotrypsin-like protease inhibitors from Zingiber officinale Roscoe. Molecules 26, 5230 (2021).
Dey, D. et al. Molecular optimization, docking, and dynamic simulation profiling of selective aromatic phytochemical ligands in blocking the SARS-CoV-2 S protein attachment to ACE2 receptor: an in silico approach of targeted drug designing. J. Adv. Vet. Anim. Res 8, 24–35 (2021).
Pattanaik, B. & Lindberg, P. Terpenoids and their biosynthesis in cyanobacteria. Life (Basel) 5, 269–293 (2015).
Dai, Y. et al. Overview of pharmacological activities of Andrographis paniculata and its major compound andrographolide. Crit. Rev. Food Sci. Nutr. 59, S17–S29 (2019).
Adhami, M., Sadeghi, B., Rezapour, A., Haghdoost, A. A. & MotieGhader, H. Repurposing novel therapeutic candidate drugs for coronavirus disease-19 based on protein-protein interaction network analysis. BMC Biotechnol. 21, 22 (2021).
Pingali, M. S. et al. Docking and molecular dynamics simulation for therapeutic repurposing in small cell lung cancer (SCLC) patients infected with COVID-19. J. Biomol. Struct. Dyn. 1–10 (2021).
Fujimori, K., Yokoyama, A., Kurita, Y., Uno, K. & Saijo, N. Paclitaxel-induced cell-mediated hypersensitivity pneumonitis. Diagnosis using leukocyte migration test, bronchoalveolar lavage and transbronchial lung biopsy. Oncology 55, 340–344 (1998).
Nishino, M., Sholl, L. M., Hodi, F. S., Hatabu, H. & Ramaiya, N. H. Anti-PD-1-related pneumonitis during cancer immunotherapy. N. Engl. J. Med. 373, 288–290 (2015).
Chen, W. Y. et al. Prediction of potential therapeutic drugs against SARS-CoV-2 by using Connectivity Map based on transcriptome data. Eur. Rev. Med. Pharm. Sci. 25, 3122–3131 (2021).
Zhong, B. et al. Oridonin inhibits SARS-CoV-2 by targeting Its 3C-like protease. Small Sci. 2, 2100124 (2022).
Zhou, Y. F. et al. Harnessing natural products by a pharmacophore-oriented semisynthesis approach for the discovery of potential anti-SARS-CoV-2 agents. Angew. Chem. Int. Ed. Engl. 61, e202201684 (2022).
McCord, J. M., Hybertson, B. M., Cota-Gomez, A. & Gao, B. Nrf2 activator PB125(R) as a carnosic acid-based therapeutic agent against respiratory viral diseases, including COVID-19. Free Radic. Biol. Med. 175, 56–64 (2021).
Satoh, T., Trudler, D., Oh, C. K. & Lipton, S. A. Potential therapeutic use of the rosemary diterpene carnosic acid for Alzheimer’s Disease, Parkinson’s Disease, and long-COVID through NRF2 activation to counteract the NLRP3 inflammasome. Antioxidants (Basel). 11, 124 (2022).
Sun, Z. G., Zhao, T. T., Lu, N., Yang, Y. A. & Zhu, H. L. Research progress of glycyrrhizic acid on antiviral activity. Mini Rev. Med. Chem. 19, 826–832 (2019).
Zhao, Z. et al. Glycyrrhizic acid nanoparticles as antiviral and anti-inflammatory agents for COVID-19 treatment. ACS Appl. Mater. Interfaces 13, 20995–21006 (2021).
Cuadrado, A. et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharm. Sci. 41, 598–610 (2020).
Sun, Q. et al. Bardoxolone and bardoxolone methyl, two Nrf2 activators in clinical trials, inhibit SARS-CoV-2 replication and its 3C-like protease. Signal Transduct. Target Ther. 6, 212 (2021).
Baig, A. & Srinivasan, H. SARS-CoV-2 inhibitors from Nigella sativa. Appl. Biochem. Biotechnol. 194, 1051–1090 (2022).
Mir, S. A. et al. Identification of SARS-CoV-2 RNA-dependent RNA polymerase inhibitors from the major phytochemicals of Nigella sativa: an in silico approach. Saudi J. Biol. Sci. 29, 394–401 (2022).
Rodriguez-Concepcion, M. et al. A global perspective on carotenoids: metabolism, biotechnology, and benefits for nutrition and health. Prog. Lipid Res. 70, 62–93 (2018).
Saini, R. K. et al. Carotenoids: dietary sources, extraction, encapsulation, bioavailability, and health benefits—a review of recent advancements. Antioxidants (Basel). 11, 795 (2022).
Guo, Z. L. et al. Crocetin: a systematic review. Front. Pharm. 12, 745683 (2021).
Kordzadeh, A., Ramazani Saadatabadi, A. & Hadi, A. Investigation on penetration of saffron components through lipid bilayer bound to spike protein of SARS-CoV-2 using steered molecular dynamics simulation. Heliyon 6, e05681 (2020).
Mertes, P. M. et al. Liposomal encapsulation of trans-crocetin enhances oxygenation in patients with COVID-19-related ARDS receiving mechanical ventilation. J. Control Release 336, 252–261 (2021).
Korani, S., Korani, M., Sathyapalan, T. & Sahebkar, A. Therapeutic effects of Crocin in autoimmune diseases: a review. Biofactors 45, 835–843 (2019).
Liu, T. et al. Prospects and progress on crocin biosynthetic pathway and metabolic engineering. Comput. Struct. Biotechnol. J. 18, 3278–3286 (2020).
Ghasemnejad-Berenji, M. Immunomodulatory and anti-inflammatory potential of crocin in COVID-19 treatment. J. Food Biochem. 45, e13718 (2021).
Stalin, A. et al. An in-silico approach to identify the potential hot spots in SARS-CoV-2 spike RBD to block the interaction with ACE2 receptor. J. Biomol. Struct. Dyn. 1–16 (2021).
Pincemail, J. et al. Oxidative stress status in COVID-19 patients hospitalized in intensive care unit for severe pneumonia. a pilot study. Antioxidants (Basel). 10, 257 (2021).
Xia, Q. D. et al. Network pharmacology and molecular docking analyses on Lianhua Qingwen capsule indicate Akt1 is a potential target to treat and prevent COVID-19. Cell Prolif. 53, e12949 (2020).
Ahmadi, A. R. & Ayazi-Nasrabadi, R. Astaxanthin protective barrier and its ability to improve the health in patients with COVID-19. Iran. J. Microbiol. 13, 434–441 (2021).
Alesci, A., Aragona, M., Cicero, N. & Lauriano, E. R. Can nutraceuticals assist treatment and improve covid-19 symptoms? Nat. Prod. Res. 36, 2672–2691 (2022).
Mohammed Ali, H. S. H. et al. In silico screening of the effectiveness of natural compounds from algae as SARS-CoV-2 inhibitors: molecular docking, ADMT profile and molecular dynamic studies. J. Biomol. Struct. Dyn. 1–16 (2022).
Aluisio, A. R. et al. Vitamin A supplementation was associated with reduced mortality in patients with Ebola virus disease during the West African outbreak. J. Nutr. 149, 1757–1765 (2019).
Imdad, A. et al. Vitamin A supplementation for preventing morbidity and mortality in children from six months to five years of age. Cochrane Database Syst. Rev. 3, CD008524 (2022).
Fraguas-Sanchez, A. I. & Torres-Suarez, A. I. Medical use of cannabinoids. Drugs 78, 1665–1703 (2018).
Malinowska, B., Baranowska-Kuczko, M., Kicman, A. & Schlicker, E. Opportunities, challenges and pitfalls of using cannabidiol as an adjuvant drug in COVID-19. Int. J. Mol. Sci. 22, 1986 (2021).
Cristino, L., Bisogno, T. & Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 16, 9–29 (2020).
Devinsky, O. et al. Trial of Cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 376, 2011–2020 (2017).
Lattanzi, S. et al. Efficacy and safety of cannabidiol in epilepsy: a systematic review and meta-analysis. Drugs 78, 1791–1804 (2018).
Raj, V. et al. Assessment of antiviral potencies of cannabinoids against SARS-CoV-2 using computational and in vitro approaches. Int. J. Biol. Macromol. 168, 474–485 (2021).
Nguyen, L. C. et al. Cannabidiol inhibits SARS-CoV-2 replication through induction of the host ER stress and innate immune responses. Sci. Adv. 8, eabi6110 (2022).
Mohammed, A. et al. Delta9-tetrahydrocannabinol prevents mortality from acute respiratory distress syndrome through the induction of apoptosis in immune cells, leading to cytokine storm suppression. Int. J. Mol. Sci. 21, 6244 (2020).
Pitakbut, T., Nguyen, G. N. & Kayser, O. Activity of THC, CBD, and CBN on Human ACE2 and SARS-CoV1/2 main protease to understand antiviral defense mechanism. Planta Med. 88, 1047–1059 (2021).
Cole, T. J., Short, K. L. & Hooper, S. B. The science of steroids. Semin. Fetal Neonatal Med. 24, 170–175 (2019).
Abutaleb, A. R. A. et al. Myocarditis in duchenne muscular dystrophy after changing steroids. JAMA Cardiol. 3, 1006–1010 (2018).
Petrelli, F. et al. Association of steroids use with survival in patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Cancers (Basel). 12, 546 (2020).
Al-Lami, R. A., Urban, R. J., Volpi, E., Algburi, A. M. A. & Baillargeon, J. Sex hormones and novel corona virus infectious disease (COVID-19). Mayo Clin. Proc. 95, 1710–1714 (2020).
van de Veerdonk, F. L. et al. A guide to immunotherapy for COVID-19. Nat. Med. 28, 39–50 (2022).
Xu, Y. et al. The importance of vitamin d metabolism as a potential prophylactic, immunoregulatory and neuroprotective treatment for COVID-19. J. Transl. Med. 18, 322 (2020).
Fadel, R. et al. Early short-course corticosteroids in hospitalized patients With COVID-19. Clin. Infect. Dis. 71, 2114–2120 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J. Am. Med. Assoc. 323, 1061–1069 (2020).
Alexaki, V. I. & Henneicke, H. The role of glucocorticoids in the management of COVID-19. Horm. Metab. Res. 53, 9–15 (2021).
Group, R. C. et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 384, 693–704 (2021).
Tomazini, B. M. et al. Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19: the CoDEX randomized clinical trial. J. Am. Med. Assoc. 324, 1307–1316 (2020).
Pinzon, M. A. et al. Dexamethasone vs methylprednisolone high dose for Covid-19 pneumonia. PLoS ONE 16, e0252057 (2021).
Wu, C. et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan. China JAMA Intern. Med. 180, 934–943 (2020).
Wang, Y. et al. A retrospective cohort study of methylprednisolone therapy in severe patients with COVID-19 pneumonia. Signal Transduct. Target Ther. 5, 57 (2020).
Li, S., Hu, Z. & Song, X. High-dose But Not Low-dose Corticosteroids Potentially Delay Viral Shedding of Patients with COVID-19. Clin. Infect. Dis. 72, 1297–1298 (2021).
Charoenngam, N. & Holick, M. F. Immunologic effects of vitamin D on human health and disease. Nutrients. 12, 2097 (2020).
de la Guia-Galipienso, F. et al. Vitamin D and cardiovascular health. Clin. Nutr. 40, 2946–2957 (2021).
Jeon, S. M. & Shin, E. A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 50, 1–14 (2018).
Klimczak, A. M. & Franasiak, J. M. Vitamin D in human reproduction: some answers and many more questions. Fertil. Steril. 115, 590–591 (2021).
Martineau, A. R. et al. Vitamin D supplementation to prevent acute respiratory tract infections: systematic review and meta-analysis of individual participant data. Br. Med. J. 356, i6583 (2017).
Carpagnano, G. E. et al. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J. Endocrinol. Invest. 44, 765–771 (2021).
Barrea, L. et al. Vitamin D: a role also in long COVID-19? Nutrients. 14, 1625 (2022).
Sabico, S. et al. Effects of a 2-Week 5000 IU versus 1000 IU Vitamin D3 supplementation on recovery of symptoms in patients with mild to moderate Covid-19: a randomized clinical trial. Nutrients. 13, 2170 (2021).
Entrenas Castillo, M. et al. Effect of calcifediol treatment and best available therapy versus best available therapy on intensive care unit admission and mortality among patients hospitalized for COVID-19: a pilot randomized clinical study. J. Steroid Biochem. Mol. Biol. 203, 105751 (2020).
Alcala-Diaz, J. F. et al. Calcifediol treatment and hospital mortality due to COVID-19: a cohort study. Nutrients 13, 1760 (2021).
Maghbooli, Z. et al. Treatment with 25-hydroxyvitamin D3 (Calcifediol) is associated with a reduction in the blood neutrophil-to-lymphocyte ratio marker of disease severity in hospitalized patients with COVID-19: a pilot multicenter, randomized, placebo-controlled, double-blinded clinical trial. Endocr. Pract. 27, 1242–1251 (2021).
Khan, N. Possible protective role of 17beta-estradiol against COVID-19. J. Allergy Infect. Dis. 1, 38–48 (2020).
Peckham, H. et al. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun. 11, 6317 (2020).
Mauvais-Jarvis, F., Klein, S. L. & Levin, E. R. Estradiol, Progesterone, Immunomodulation, and COVID-19 Outcomes. Endocrinology. 161, bqaa127 (2020).
Youn, J. Y., Zhang, Y., Wu, Y., Cannesson, M. & Cai, H. Therapeutic application of estrogen for COVID-19: Attenuation of SARS-CoV-2 spike protein and IL-6 stimulated, ACE2-dependent NOX2 activation, ROS production and MCP-1 upregulation in endothelial cells. Redox Biol. 46, 102099 (2021).
Baristaite, G. & Gurwitz, D. Estradiol reduces ACE2 and TMPRSS2 mRNA levels in A549 human lung epithelial cells. Drug Dev. Res. 83, 961–966 (2022).
Su, S. et al. Modulation of innate immune response to viruses including SARS-CoV-2 by progesterone. Signal Transduct. Target Ther. 7, 137 (2022).
Yuan, L. et al. Female sex hormone, progesterone, ameliorates the severity of SARS-CoV-2-caused pneumonia in the Syrian hamster model. Signal Transduct. Target Ther. 7, 47 (2022).
Ghandehari, S. et al. Progesterone in addition to standard of care vs standard of care alone in the treatment of men hospitalized with moderate to severe COVID-19: a randomized, controlled pilot trial. Chest 160, 74–84 (2021).
Lindsay, J., Teh, B. W., Micklethwaite, K. & Slavin, M. Azole antifungals and new targeted therapies for hematological malignancy. Curr. Opin. Infect. Dis. 32, 538–545 (2019).
Nanjan, M. J., Mohammed, M., Prashantha Kumar, B. R. & Chandrasekar, M. J. N. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg. Chem. 77, 548–567 (2018).
Shafiei, M., Peyton, L., Hashemzadeh, M. & Foroumadi, A. History of the development of antifungal azoles: a review on structures, SAR, and mechanism of action. Bioorg. Chem. 104, 104240 (2020).
Stachulski, A. V. et al. Therapeutic potential of nitazoxanide: an appropriate choice for repurposing versus SARS-CoV-2? ACS Infect. Dis. 7, 1317–1331 (2021).
Rossignol, J. F. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antivir. Res. 110, 94–103 (2014).
Riccio, A. et al. Impairment of SARS-CoV-2 spike glycoprotein maturation and fusion activity by nitazoxanide: an effect independent of spike variants emergence. Cell Mol. Life Sci. 79, 227 (2022).
Miorin, L. et al. The oral drug nitazoxanide restricts SARS-CoV-2 infection and attenuates disease pathogenesis in Syrian hamsters. Preprint at bioRxiv https://doi.org/10.1101/2022.02.08.479634 (2022).
Rocco, P. R. M. et al. Early use of nitazoxanide in mild COVID-19 disease: randomised, placebo-controlled trial. Eur. Respir. J. 58, 2003725 (2021).
Blum, V. F. et al. Nitazoxanide superiority to placebo to treat moderate COVID-19—a pilot prove of concept randomized double-blind clinical trial. EClinicalMedicine 37, 100981 (2021).
Zhang, H. Z., Zhao, Z. L. & Zhou, C. H. Recent advance in oxazole-based medicinal chemistry. Eur. J. Med. Chem. 144, 444–492 (2018).
Santini, G. et al. Investigational prostaglandin D2 receptor antagonists for airway inflammation. Expert Opin. Investig. Drugs 25, 639–652 (2016).
Wong, L. R. et al. Eicosanoid signalling blockade protects middle-aged mice from severe COVID-19. Nature 605, 146–151 (2022).
Cadegiani, F. A. et al. Final results of a randomized, placebo-controlled, two-arm, parallel clinical trial of proxalutamide for hospitalized COVID-19 patients: a multiregional, joint analysis of the proxa-rescue AndroCoV trial. Cureus 13, e20691 (2021).
Al-Wahaibi, L. H. et al. Discovery of novel oxazole-based macrocycles as anti-coronaviral agents targeting SARS-CoV-2 main protease. Bioorg. Chem. 116, 105363 (2021).
Gerotziafas, G. T. et al. Guidance for the management of patients with vascular disease or cardiovascular risk factors and COVID-19: position paper from VAS-European independent foundation in angiology/vascular medicine. Thromb. Haemost. 120, 1597–1628 (2020).
De Beule, K. & Van Gestel, J. Pharmacology of itraconazole. Drugs 61, 27–37 (2001).
Van Damme, E. et al. In vitro activity of itraconazole against SARS-CoV-2. J. Med. Virol. 93, 4454–4460 (2021).
Yang, C. et al. Drug repurposing of itraconazole and estradiol benzoate against COVID-19 by blocking SARS-CoV-2 spike protein-mediated membrane fusion. Adv. Ther. 4, 2000224 (2021).
Schloer, S. et al. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-CoV-2 infection in vitro. Br. J. Pharm. 178, 2339–2350 (2021).
Azizian, N. G. & Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 13, 61 (2020).
Bader, J. C., Abdul Razak, A. R., Shacham, S. & Xu, H. Pharmacokinetics of selinexor: the first-in-class selective inhibitor of nuclear export. Clin. Pharmacokinet. 60, 957–969 (2021).
Syed, Y. Y. Selinexor: first global approval. Drugs 79, 1485–1494 (2019).
Zhu, J. Y., Lee, J. G., van de Leemput, J., Lee, H. & Han, Z. Functional analysis of SARS-CoV-2 proteins in Drosophila identifies Orf6-induced pathogenic effects with Selinexor as an effective treatment. Cell Biosci. 11, 59 (2021).
Lee, J. G. et al. Characterization of SARS-CoV-2 proteins reveals Orf6 pathogenicity, subcellular localization, host interactions and attenuation by Selinexor. Cell Biosci. 11, 58 (2021).
Kashyap, T. et al. Selinexor, a novel selective inhibitor of nuclear export, reduces SARS-CoV-2 infection and protects the respiratory system in vivo. Antivir. Res. 192, 105115 (2021).
Banerjee, S. et al. Drug repurposing to identify nilotinib as a potential SARS-CoV-2 main protease inhibitor: insights from a computational and in vitro study. J. Chem. Inf. Model. 61, 5469–5483 (2021).
Memis, H., Cakir, A., Durmus, M., Gok, S. & Bahcecioglu, O. F. Is sitagliptin effective for the treatment of COVID-19? Eur. J. Hosp. Pharm. 29, e6 (2021).
Solerte, S. B. et al. Sitagliptin treatment at the time of hospitalization was associated with reduced mortality in patients with type 2 diabetes and COVID-19: a multicenter, case-control, retrospective, observational study. Diabetes Care 43, 2999–3006 (2020).
Langerbeins, P. et al. The CLL12 trial: ibrutinib vs placebo in treatment-naive, early-stage chronic lymphocytic leukemia. Blood 139, 177–187 (2022).
Wang, M. L. et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 369, 507–516 (2013).
Haritha, C. V., Sharun, K. & Jose, B. Ebselen, a new candidate therapeutic against SARS-CoV-2. Int. J. Surg. 84, 53–56 (2020).
Kumar, A., Loharch, S., Kumar, S., Ringe, R. P. & Parkesh, R. Exploiting cheminformatic and machine learning to navigate the available chemical space of potential small molecule inhibitors of SARS-CoV-2. Comput. Struct. Biotechnol. J. 19, 424–438 (2021).
Ramirez-Salinas, G. L., Martinez-Archundia, M., Correa-Basurto, J. & Garcia-Machorro, J. Repositioning of Ligands that target the spike glycoprotein as potential drugs for SARS-CoV-2 in an in silico study. Molecules. 25, 5615 (2020).
Byun, W. G., Lee, J., Kim, S. & Park, S. B. Harnessing stress granule formation by small molecules to inhibit the cellular replication of SARS-CoV-2. Chem. Commun. 57, 12476–12479 (2021).
Xu, Y. et al. Design, synthesis, bioactivity evaluation, crystal structures, and in silico studies of new alpha-amino amide derivatives as potential histone deacetylase 6 inhibitors. Molecules. 27, 3335 (2022).
Zhu, J. et al. Progress on SARS-CoV-2 3CLpro inhibitors: inspiration from SARS-CoV 3CLpro peptidomimetics and small-molecule anti-inflammatory compounds. Drug Des. Dev. Ther. 16, 1067–1082 (2022).
Extance, A. Covid-19: what is the evidence for the antiviral Paxlovid? Br. Med. J. 377, o1037 (2022).
Mahase, E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. Br. Med. J. 375, n2713 (2021).
Hammond, J. et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 386, 1397–1408 (2022).
Molina, J. M. et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 96-week results of a randomised, double-blind, non-inferiority, phase 3 trial. Lancet HIV 7, e16–e26 (2020).
Singh, R. S. P. et al. Innovative randomized phase I study and dosing regimen selection to accelerate and inform pivotal COVID-19 trial of nirmatrelvir. Clin. Pharm. Ther. 112, 101–111 (2022).
Vangeel, L. et al. Remdesivir, molnupiravir and nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 198, 105252 (2022).
Jochmans, D. et al. The substitutions L50F, E166A and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. Preprint at bioRxiv https://doi.org/10.1101/2022.06.07.495116 (2022).
Zhou, Y. et al. Nirmatrelvir resistant SARS-CoV-2 variants with high fitness in vitro. Preprint at BioRxiv https://doi.org/10.1101/2022.06.06.494921 (2022).
Service, R. F. Bad news for Paxlovid? Resistance may be coming. Science 377, 138–139 (2022).
de Oliveira, V. M., Ibrahim, M. F., Sun, X., Hilgenfeld, R. & Shen, J. H172Y mutation perturbs the S1 pocket and nirmatrelvir binding of SARS-CoV-2 main protease through a nonnative hydrogen bond. Preprint at bioRxiv https://doi.org/10.1101/2022.07.31.502215 (2022).
Moghadasi, S. A. et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Preprint at bioRxiv https://doi.org/10.1101/2022.08.07.503099 (2022).
Xia, Z. et al. Rational design of hybrid SARS-CoV-2 main protease inhibitors guided by the superimposed cocrystal structures with the peptidomimetic inhibitors GC-376, telaprevir, and boceprevir. ACS Pharm. Transl. Sci. 4, 1408–1421 (2021).
Citarella, A., Scala, A., Piperno, A. & Micale, N. SARS-CoV-2 M(pro): a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules. 11, 607 (2021).
Park, A. Y. J. et al. Preclinical pharmacokinetics and safety of intravenous RTD-1. Antimicrob. Agents Chemother. 66, e0212521 (2022).
Magro, P., Zanella, I., Pescarolo, M., Castelli, F. & Quiros-Roldan, E. Lopinavir/ritonavir: repurposing an old drug for HIV infection in COVID-19 treatment. Biomed. J. 44, 43–53 (2021).
Choy, K. T. et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res 178, 104786 (2020).
Singh, S. et al. Niclosamide—a promising treatment for COVID-19. Br. J. Pharm. 179, 3250–3267 (2022).
Lokhande, A. S. & Devarajan, P. V. A review on possible mechanistic insights of Nitazoxanide for repurposing in COVID-19. Eur. J. Pharm. 891, 173748 (2021).
Braga, L. et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 594, 88–93 (2021).
Juang, Y. P. et al. Design, synthesis and biological evaluations of niclosamide analogues against SARS-CoV-2. Eur. J. Med. Chem. 235, 114295 (2022).
Weiss, A. et al. Topical niclosamide (ATx201) reduces Staphylococcus aureus colonization and increases Shannon diversity of the skin microbiome in atopic dermatitis patients in a randomized, double-blind, placebo-controlled Phase 2 trial. Clin. Transl. Med. 12, e790 (2022).
Jara, M. O. et al. Niclosamide inhalation powder made by thin-film freezing: multi-dose tolerability and exposure in rats and pharmacokinetics in hamsters. Int J. Pharm. 603, 120701 (2021).
Deeks, E. D. Darunavir/cobicistat/emtricitabine/tenofovir alafenamide: a review in HIV-1 infection. Drugs 78, 1013–1024 (2018).
Ngo, S. T., Quynh Anh Pham, N., Thi Le, L., Pham, D. H. & Vu, V. V. Computational determination of potential inhibitors of SARS-CoV-2 main protease. J. Chem. Inf. Model 60, 5771–5780 (2020).
Armitage, J., Holmes, M. V. & Preiss, D. Cholesteryl ester transfer protein inhibition for preventing cardiovascular events: JACC review topic of the week. J. Am. Coll. Cardiol. 73, 477–487 (2019).
Mancek-Keber, M. et al. Disruption of disulfides within RBD of SARS-CoV-2 spike protein prevents fusion and represents a target for viral entry inhibition by registered drugs. FASEB J. 35, e21651 (2021).
Niesor, E. J. et al. Inhibition of the 3CL protease and SARS-CoV-2 replication by dalcetrapib. ACS Omega 6, 16584–16591 (2021).
Bhambhani, S., Kondhare, K. R. & Giri, A. P. Diversity in chemical structures and biological properties of plant alkaloids. Molecules. 26, 3374 (2021).
Antonio, A. D. S., Wiedemann, L. S. M. & Veiga-Junior, V. F. Natural products’ role against COVID-19. RSC Adv. 10, 23379–23393 (2020).
Boozari, M. & Hosseinzadeh, H. Natural products for COVID-19 prevention and treatment regarding to previous coronavirus infections and novel studies. Phytother. Res. 35, 864–876 (2021).
Chu, X. M. et al. Quinoline and quinolone dimers and their biological activities: an overview. Eur. J. Med. Chem. 161, 101–117 (2019).
Moor, L. F. E., Vasconcelos, T. R. A., da, R. R. R., Pinto, L. S. S. & da Costa, T. M. Quinoline: an attractive scaffold in drug design. Mini Rev. Med. Chem. 21, 2209–2226 (2021).
White, N. J. et al. Malaria. Lancet 383, 723–735 (2014).
Kaur, R. & Kumar, K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem. 215, 113220 (2021).
Schrezenmeier, E. & Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 16, 155–166 (2020).
Weston, S. et al. Broad anti-coronavirus activity of food and drug administration-approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo. J. Virol. 94, e01218–20 (2020).
Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020).
Gao, J., Tian, Z. & Yang, X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 14, 72–73 (2020).
Liu, J. et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 6, 16 (2020).
Gautret, P. et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: a pilot observational study. Travel Med. Infect. Dis. 34, 101663 (2020).
Matada, B. S., Pattanashettar, R. & Yernale, N. G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 32, 115973 (2021).
Shang, X. F. et al. Biologically active quinoline and quinazoline alkaloids part I. Med. Res. Rev. 38, 775–828 (2018).
Shang, X. F. et al. Biologically active quinoline and quinazoline alkaloids part II. Med. Res. Rev. 38, 1614–1660 (2018).
Cha, M. Y. et al. Synthesis and biological evaluation of pyrimidine-based dual inhibitors of human epidermal growth factor receptor 1 (HER-1) and HER-2 tyrosine kinases. J. Med. Chem. 55, 2846–2857 (2012).
Raymonda, M. H. et al. Pharmacologic profiling reveals lapatinib as a novel antiviral against SARS-CoV-2 in vitro. Virology 566, 60–68 (2022).
Gilham, D. et al. Bromodomain and extraterminal protein inhibitor, apabetalone (RVX-208), reduces ACE2 expression and attenuates SARS-Cov-2 infection in vitro. Biomedicines. 9, 437 (2021).
Plazas, E., Avila, M. M., Munoz, D. R. & Cuca, S. L. Natural isoquinoline alkaloids: pharmacological features and multi-target potential for complex diseases. Pharm. Res. 177, 106126 (2022).
Snoussi, M. et al. Emetine, a potent alkaloid for the treatment of SARS-CoV-2 targeting papain-like protease and non-structural proteins: pharmacokinetics, molecular docking and dynamic studies. J. Biomol. Struct. Dyn. 1–14 (2021).
Khandelwal, N. et al. Emetine inhibits replication of RNA and DNA viruses without generating drug-resistant virus variants. Antivir. Res. 144, 196–204 (2017).
Wang, A. et al. Low dose of emetine as potential anti-SARS-CoV-2 virus therapy: preclinical in vitro inhibition and in vivo pharmacokinetic evidences. Mol. Biomed. 1, 14 (2020).
Ren, P. X. et al. A multi-targeting drug design strategy for identifying potent anti-SARS-CoV-2 inhibitors. Acta Pharm. Sin. 43, 483–493 (2022).
Brem, T. H. & Konwaler, B. E. Fatal myocarditis due to emetine hydrochloride. Am. Heart J. 50, 476–481 (1955).
Lemmens-Gruber, R., Karkhaneh, A., Studenik, C. & Heistracher, P. Cardiotoxicity of emetine dihydrochloride by calcium channel blockade in isolated preparations and ventricular myocytes of guinea-pig hearts. Br. J. Pharm. 117, 377–383 (1996).
Valipour, M. Different aspects of Emetine’s capabilities as a highly potent SARS-CoV-2 inhibitor against COVID-19. ACS Pharm. Transl. Sci. 5, 387–399 (2022).
Rogosnitzky, M., Okediji, P. & Koman, I. Cepharanthine: a review of the antiviral potential of a Japanese-approved alopecia drug in COVID-19. Pharm. Rep. 72, 1509–1516 (2020).
Bailly, C. Cepharanthine: an update of its mode of action, pharmacological properties and medical applications. Phytomedicine 62, 152956 (2019).
Ohashi, H. et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience 24, 102367 (2021).
Anderson, G. & Reiter, R. J. Melatonin: roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 30, e2109 (2020).
Mehrzadi, S., Karimi, M. Y., Fatemi, A., Reiter, R. J. & Hosseinzadeh, A. SARS-CoV-2 and other coronaviruses negatively influence mitochondrial quality control: beneficial effects of melatonin. Pharm. Ther. 224, 107825 (2021).
Reynolds, J. L. & Dubocovich, M. L. Melatonin multifaceted pharmacological actions on melatonin receptors converging to abrogate COVID-19. J. Pineal Res. 71, e12732 (2021).
Zhai, X. et al. Melatonin and other indoles show antiviral activities against swine coronaviruses in vitro at pharmacological concentrations. J. Pineal Res. 71, e12754 (2021).
Cecon, E. et al. Therapeutic potential of melatonin and melatonergic drugs on K18-hACE2 mice infected with SARS-CoV-2. J. Pineal Res. 72, e12772 (2022).
Feitosa, E. L. et al. COVID-19: rational discovery of the therapeutic potential of melatonin as a SARS-CoV-2 main Protease Inhibitor. Int J. Med Sci. 17, 2133–2146 (2020).
Amanullah, A. et al. Indomethacin elicits proteasomal dysfunctions develops apoptosis through mitochondrial abnormalities. J. Cell Physiol. 233, 1685–1699 (2018).
Amici, C. et al. Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: role of eIF2alpha kinase PKR. Cell Microbiol. 17, 1391–1404 (2015).
Kiani, P. et al. In vitro assessment of the antiviral activity of ketotifen, indomethacin and naproxen, alone and in combination, against SARS-CoV-2. Viruses. 13, 558 (2021).
Ravichandran, R. et al. An open label randomized clinical trial of Indomethacin for mild and moderate hospitalised Covid-19 patients. Sci. Rep. 12, 6413 (2022).
Boras, B. et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 12, 6055 (2021).
Luban, J. et al. The DHODH inhibitor PTC299 arrests SARS-CoV-2 replication and suppresses induction of inflammatory cytokines. Virus Res. 292, 198246 (2021).
Jin, Y. H. et al. Lycorine, a non-nucleoside RNA dependent RNA polymerase inhibitor, as potential treatment for emerging coronavirus infections. Phytomedicine 86, 153440 (2021).
Zhang, Y. N. et al. Gemcitabine, lycorine and oxysophoridine inhibit novel coronavirus (SARS-CoV-2) in cell culture. Emerg. Microbes Infect. 9, 1170–1173 (2020).
Nelson, K. M. et al. The essential medicinal chemistry of curcumin. J. Med. Chem. 60, 1620–1637 (2017).
Zahedipour, F. et al. Potential effects of curcumin in the treatment of COVID-19 infection. Phytother. Res. 34, 2911–2920 (2020).
Moghadamtousi, S. Z. et al. A review on antibacterial, antiviral, and antifungal activity of curcumin. Biomed. Res. Int. 2014, 186864 (2014).
Bormann, M. et al. Turmeric root and its bioactive ingredient curcumin effectively neutralize SARS-CoV-2 in vitro. Viruses. 13, 1914 (2021).
Hassaniazad, M. et al. A triple-blind, placebo-controlled, randomized clinical trial to evaluate the effect of curcumin-containing nanomicelles on cellular immune responses subtypes and clinical outcome in COVID-19 patients. Phytother. Res. 35, 6417–6427 (2021).
Hassaniazad, M. et al. The clinical effect of Nano micelles containing curcumin as a therapeutic supplement in patients with COVID-19 and the immune responses balance changes following treatment: a structured summary of a study protocol for a randomised controlled trial. Trials 21, 876 (2020).
Ghasemnejad-Berenji, M., Pashapour, S. & Ghasemnejad-Berenji, H. Therapeutic potential for clomiphene, a selective estrogen receptor modulator, in the treatment of COVID-19. Med Hypotheses 145, 110354 (2020).
Shagufta & Ahmad, I. Tamoxifen a pioneering drug: an update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 143, 515–531 (2018).
Zu, S. et al. Tamoxifen and clomiphene inhibit SARS-CoV-2 infection by suppressing viral entry. Signal Transduct. Target Ther. 6, 435 (2021).
Bryant, A. et al. Ivermectin for prevention and treatment of COVID-19 infection: a systematic review, meta-analysis, and trial sequential analysis to inform clinical guidelines. Am. J. Ther. 28, e434–e460 (2021).
Caly, L., Druce, J. D., Catton, M. G., Jans, D. A. & Wagstaff, K. M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 178, 104787 (2020).
Froba, M. et al. Iota-carrageenan inhibits replication of SARS-CoV-2 and the respective variants of concern alpha, beta, gamma and delta. Int. J. Mol. Sci. 22, 13202 (2021).
Schutz, D. et al. Carrageenan-containing over-the-counter nasal and oral sprays inhibit SARS-CoV-2 infection of airway epithelial cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 320, L750–L756 (2021).
Rothan, H. A. et al. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-COV-2) replication and attenuates inflammation in human cells. Virology 547, 7–11 (2020).
Gil-Moles, M. et al. Gold metallodrugs to target coronavirus proteins: inhibitory effects on the spike-ACE2 interaction and on PLpro protease activity by auranofin and gold organometallics*. Chemistry 26, 15140–15144 (2020).
Liang, J. J. et al. Investigation of small molecule inhibitors of the SARS-CoV-2 papain-like protease by all-atom microsecond modelling, PELE Monte Carlo simulations, and in vitro activity inhibition. Chem. Phys. Lett. 139294 (2021).
Pitsillou, E., Liang, J., Ververis, K., Hung, A. & Karagiannis, T. C. Interaction of small molecules with the SARS-CoV-2 papain-like protease: In silico studies and in vitro validation of protease activity inhibition using an enzymatic inhibition assay. J. Mol. Graph Model 104, 107851 (2021).
Sun, G. et al. Structural basis of covalent inhibitory mechanism of TMPRSS2-related serine proteases by camostat. J. Virol. 95, e0086121 (2021).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and Is Blocked by A Clinically Proven Protease Inhibitor. Cell 181, 271–280e278 (2020).
Sakr, Y. et al. Camostat mesylate therapy in critically ill patients with COVID-19 pneumonia. Intensive Care Med. 47, 707–709 (2021).
Chupp, G. et al. A Phase 2 Randomized, Double-Blind, Placebo-controlled Trial of Oral Camostat Mesylate for Early Treatment of COVID-19 Outpatients Showed Shorter Illness Course and Attenuation of Loss of Smell and Taste. medRxiv (2022).
Asakura, H. & Ogawa, H. Potential of heparin and nafamostat combination therapy for COVID-19. J. Thromb. Haemost. 18, 1521–1522 (2020).
Li, K., Meyerholz, D. K., Bartlett, J. A. & McCray, P. B. Jr The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. mBio 12, e0097021 (2021).
Quinn, T. M. et al. Randomised controlled trial of intravenous nafamostat mesylate in COVID pneumonitis: phase 1b/2a experimental study to investigate safety, pharmacokinetics and pharmacodynamics. EBioMedicine 76, 103856 (2022).
Smieszek, S. P., Przychodzen, B. P. & Polymeropoulos, M. H. Amantadine disrupts lysosomal gene expression: a hypothesis for COVID19 treatment. Int J. Antimicrob. Agents 55, 106004 (2020).
Fink, K. et al. Amantadine inhibits SARS-CoV-2 in vitro. Viruses. 13, 539 (2021).
Araujo, R., Aranda-Martinez, J. D. & Aranda-Abreu, G. E. Amantadine treatment for people with COVID-19. Arch. Med. Res. 51, 739–740 (2020).
Pahwa, R. Amantadine: an old drug reborn. Lancet Neurol. 20, 975–977 (2021).
Hu, Y., Jo, H., DeGrado, W. F. & Wang, J. Brilacidin, a COVID-19 drug candidate, demonstrates broad-spectrum antiviral activity against human coronaviruses OC43, 229E, and NL63 through targeting both the virus and the host cell. J. Med Virol. 94, 2188–2200 (2022).
Xu, C. et al. Brilacidin, a non-peptide defensin-mimetic molecule, inhibits SARS-CoV-2 infection by blocking viral entry. EC Microbiol 18, 1–12 (2022).
Bakovic, A. et al. Brilacidin demonstrates inhibition of SARS-CoV-2 in cell culture. Viruses. 13, 271 (2021).
Huntington, K. E. et al. Integrin/TGF-beta1 inhibitor GLPG-0187 blocks SARS-CoV-2 Delta and Omicron pseudovirus infection of airway epithelial cells which could attenuate disease severity. Pharmaceuticals (Basel). 15, 618 (2022).
Manandhar, A. et al. Discovery of novel small-molecule inhibitors of SARS-CoV-2 main protease as potential leads for COVID-19 treatment. J. Chem. Inf. Model 61, 4745–4757 (2021).
Mouffak, S., Shubbar, Q., Saleh, E. & El-Awady, R. Recent advances in management of COVID-19: a review. Biomed. Pharmacother. 143, 112107 (2021).
Tanne, J. H. Covid-19: FDA authorises pharmacists to prescribe Paxlovid. Br. Med. J. 378, o1695 (2022).
Gold, J. A. W. et al. Dispensing of Oral Antiviral Drugs for Treatment of COVID-19 by Zip Code-Level Social Vulnerability—United States, December 23, 2021-May 21, 2022. MMWR Morb. Mortal. Wkly Rep. 71, 825–829 (2022).
Spinner, C. D. et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. J. Am. Med. Assoc. 324, 1048–1057 (2020).
Goldman, J. D. et al. Remdesivir for 5 or 10 days in patients with severe covid-19. N. Engl. J. Med. 383, 1827–1837 (2020).
Gottlieb, R. L. et al. Early remdesivir to prevent progression to severe Covid-19 in outpatients. N. Engl. J. Med. 386, 305–315 (2022).
Ader, F. et al. Remdesivir plus standard of care versus standard of care alone for the treatment of patients admitted to hospital with COVID-19 (DisCoVeRy): a phase 3, randomised, controlled, open-label trial. Lancet Infect. Dis. 22, 209–221 (2022).
Ader, F. et al. Final results of the DisCoVeRy trial of remdesivir for patients admitted to hospital with COVID-19. Lancet Infect. Dis. 22, 764–765 (2022).
Wang, Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395, 1569–1578 (2020).
Favalli, E. G., Biggioggero, M., Maioli, G. & Caporali, R. Baricitinib for COVID-19: a suitable treatment? Lancet Infect. Dis. 20, 1012–1013 (2020).
Ely, E. W. et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir. Med. 10, 327–336 (2022).
Saravolatz, L. D., Depcinski, S. & Sharma, M. Molnupiravir anD Nirmatrelvir-ritonavir: Oral COVID antiviral drugs. Clin. Infect. Dis. ciac180 (2022).
Menendez-Arias, L. Decoding molnupiravir-induced mutagenesis in SARS-CoV-2. J. Biol. Chem. 297, 100867 (2021).
Fischer, W. A. 2nd et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 14, eabl7430 (2022).
Jayk Bernal, A. et al. Molnupiravir for oral treatment of covid-19 in nonhospitalized patients. N. Engl. J. Med. 386, 509–520 (2022).
Wang, Z. & Yang, L. In the age of Omicron variant: paxlovid raises new hopes of COVID-19 recovery. J. Med. Virol. 94, 1766–1767 (2022).
Burki, T. The future of Paxlovid for COVID-19. Lancet Respir. Med. 10, e68 (2022).
Najjar-Debbiny, R. et al. Effectiveness of paxlovid in reducing severe COVID-19 and mortality in high risk patients. Clin. Infect. Dis. ciac443 (2022).
Halford, B. The path to paxlovid. ACS Cent. Sci. 8, 405–407 (2022).
Ivashchenko, A. A. et al. AVIFAVIR for treatment of patients with moderate coronavirus disease 2019 (COVID-19): Interim results of a phase II/III multicenter randomized clinical trial. Clin. Infect. Dis. 73, 531–534 (2021).
Bosaeed, M. et al. Efficacy of favipiravir in adults with mild COVID-19: a randomized, double-blind, multicentre, placebo-controlled clinical trial. Clin. Microbiol Infect. 28, 602–608 (2022).
Doi, Y. et al. A prospective, randomized, open-label trial of early versus late favipiravir therapy in hospitalized patients with COVID-19. Antimicrob. Agents Chemother. 64, e01897–20 (2020).
McCoy, J. et al. Proxalutamide reduces the rate of hospitalization for COVID-19 male outpatients: a randomized double-blinded placebo-controlled trial. Front. Med. 8, 668698 (2021).
Welen, K. et al. A phase 2 trial of the effect of antiandrogen therapy on COVID-19 outcome: no evidence of benefit, supported by epidemiology and in vitro data. Eur. Urol. 81, 285–293 (2022).
Xie, Y. et al. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res. 31, 1212–1214 (2021).
Rossignol, J. F., Bardin, M. C., Fulgencio, J., Mogelnicki, D. & Brechot, C. A randomized double-blind placebo-controlled clinical trial of nitazoxanide for treatment of mild or moderate COVID-19. EClinicalMedicine 45, 101310 (2022).
Kitagawa, J. et al. A phase I study of high dose camostat mesylate in healthy adults provides a rationale to repurpose the TMPRSS2 inhibitor for the treatment of COVID-19. Clin. Transl. Sci. 14, 1967–1976 (2021).
Sonnappa, S. et al. Extrafine versus fine inhaled corticosteroids in relation to asthma control: a systematic review and meta-analysis of observational real-life studies. J. Allergy Clin. Immunol. Pract. 6, 907–915e907 (2018).
Zhang, X. Y. et al. Biological, clinical and epidemiological features of COVID-19, SARS and MERS and AutoDock simulation of ACE2. Infect. Dis. Poverty 9, 99 (2020).
Ezer, N. et al. Inhaled and intranasal ciclesonide for the treatment of covid-19 in adult outpatients: CONTAIN phase II randomised controlled trial. Br. Med. J. 375, e068060 (2021).
Clemency, B. M. et al. Efficacy of inhaled ciclesonide for outpatient treatment of adolescents and adults with symptomatic COVID-19: a randomized clinical trial. JAMA Intern. Med. 182, 42–49 (2022).
Hsu, C. K., Chao, C. M. & Lai, C. C. Inhaled ciclesonide for outpatients with COVID-19: a meta-analysis. J. Microbiol. Immunol. Infect. S1684-1182(22)00112-8 (2022).
Chan, N., Sobieraj-Teague, M. & Eikelboom, J. W. Direct oral anticoagulants: evidence and unresolved issues. Lancet 396, 1767–1776 (2020).
Ramacciotti, E. et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): an open-label, multicentre, randomised, controlled trial. Lancet 399, 50–59 (2022).
Lopes, R. D. et al. Randomized clinical trial to evaluate a routine full anticoagulation Strategy in Patients with Coronavirus Infection (SARS-CoV2) admitted to hospital: rationale and design of the ACTION (AntiCoagulaTlon cOroNavirus)-Coalition IV trial. Am. Heart J. 238, 1–11 (2021).
Lopes, R. D. et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet 397, 2253–2263 (2021).
de Melo, G. D. et al. Attenuation of clinical and immunological outcomes during SARS-CoV-2 infection by ivermectin. EMBO Mol. Med. 13, e14122 (2021).
Mega, E. R. Latin America’s embrace of an unproven COVID treatment is hindering drug trials. Nature 586, 481–482 (2020).
Lopez-Medina, E. et al. Effect of ivermectin on time to resolution of symptoms among adults with mild COVID-19: a randomized clinical trial. J. Am. Med. Assoc. 325, 1426–1435 (2021).
Lim, S. C. L. et al. Efficacy of ivermectin treatment on disease progression among adults with mild to moderate COVID-19 and comorbidities: the I-TECH randomized clinical trial. JAMA Intern. Med. 182, 426–435 (2022).
Reis, G. et al. Effect of early treatment with ivermectin among patients with Covid-19. N. Engl. J. Med. 386, 1721–1731 (2022).
Connell, N. T. & Berliner, N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood 133, 2027–2030 (2019).
Strich, J. R. et al. Fostamatinib inhibits neutrophils extracellular traps induced by COVID-19 patient plasma: a potential therapeutic. J. Infect. Dis. 223, 981–984 (2021).
Strich, J. R. et al. Fostamatinib for the treatment of hospitalized adults with coronavirus disease 2019: a randomized trial. Clin. Infect. Dis. 75, e491–e498 (2022).
Pearson, R. D. & Hewlett, E. L. Niclosamide therapy for tapeworm infections. Ann. Intern. Med. 102, 550–551 (1985).
Cairns, D. M. et al. Efficacy of niclosamide vs placebo in SARS-CoV-2 respiratory viral clearance, viral shedding, and duration of symptoms among patients with mild to moderate COVID-19: a phase 2 randomized clinical trial. JAMA Netw. Open 5, e2144942 (2022).
Ellis, S. Chinese approval for Ascletis’ HCV drug is first homegrown success. Nat. Biotechnol. 36, 675–676 (2018).
Chen, H. et al. First clinical study using HCV protease inhibitor danoprevir to treat COVID-19 patients. Medicines 99, e23357 (2020).
Zhang, Z. et al. A comparative study on the time to achieve negative nucleic acid testing and hospital stays between danoprevir and lopinavir/ritonavir in the treatment of patients with COVID-19. J. Med. Virol. 92, 2631–2636 (2020).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Matthay, M. A., Leligdowicz, A. & Liu, K. D. Biological mechanisms of COVID-19 acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 202, 1489–1491 (2020).
Bruggeman, L. A., Pellicoro, J. A., Horigan, E. A. & Klotman, P. E. Thromboxane and prostacyclin differentially regulate murine extracellular matrix gene expression. Kidney Int. 43, 1219–1225 (1993).
Kawabe, J. et al. Prostaglandin I2 promotes recruitment of endothelial progenitor cells and limits vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 30, 464–470 (2010).
Johansson, P. I. et al. The effect of prostacyclin (Iloprost) infusion at a dose of 1 ng/kg/min for 72 hours compared to placebo in mechanically ventilated patients with COVID-19: a structured summary of a study protocol for a randomized controlled trial. Trials 21, 746 (2020).
Johansson, P. I. et al. Prostacyclin in intubated patients with COVID-19 and severe endotheliopathy: a multicenter, randomized clinical trial. Am. J. Respir. Crit. Care Med. 205, 324–329 (2022).
Pfeifer, N. D., Lo, A., Bourdet, D. L., Colley, K. & Singh, D. Phase I study in healthy participants to evaluate safety, tolerability, and pharmacokinetics of inhaled nezulcitinib, a potential treatment for COVID-19. Clin. Transl. Sci. 14, 2556–2565 (2021).
Guzman-Esquivel, J. et al. Efficacy of the use of mefenamic acid combined with standard medical care vs. standard medical care alone for the treatment of COVID19: a randomized doubleblind placebocontrolled trial. Int. J. Mol. Med. 49, 29 (2022).
Marrone, A. et al. Remdesivir plus dexamethasone versus dexamethasone alone for the treatment of coronavirus disease 2019 (Covid-19) patients requiring supplemental O2 therapy: a prospective controlled nonrandomized study. Clin. Infect. Dis. 75, e403–e409 (2022).
Xia, S. et al. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct. Target Ther. 6, 288 (2021).
Acknowledgements
This research was funded by the National Natural Science Foundation (82003258), the Science Foundation of Chengdu (2022-YF05-01793-SN), and the Medico-Engineering Cooperation Funds from university of Electronic Science and Technology of China (No. ZYGX2021YGLH225). All Protein structure figures were made using PyMOL (Schrödinger, LLC). All chemical structures were made using Marvin Sketch (ChemAxon).
Author information
Authors and Affiliations
Contributions
S.B.L., X.H.C., and J.P.W. collected references and wrote the paper and tables. S.B.L. drew the figures. K.M. and X.M.D. provided valuable guidance and revised the paper. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, S., Chen, X., Wu, J. et al. Small molecules in the treatment of COVID-19. Sig Transduct Target Ther 7, 387 (2022). https://doi.org/10.1038/s41392-022-01249-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01249-8
This article is cited by
-
Gold nanoparticles combat enveloped RNA virus by affecting organelle dynamics
Signal Transduction and Targeted Therapy (2023)
-
Targetable elements in SARS-CoV-2 S2 subunit for the design of pan-coronavirus fusion inhibitors and vaccines
Signal Transduction and Targeted Therapy (2023)
-
Design of hACE2-based small peptide inhibitors against spike protein of SARS-CoV-2: a computational approach
Structural Chemistry (2023)
-
The Study of the Effect of Dimethylsulfoxide (or Diethylsulfoxide) on Quinine Sulfate-DNA Binding by UV–Vis and Steady-State Fluorescence Spectroscopies
Journal of Fluorescence (2023)
-
Engineered Therapeutic Antibody Against SARS-CoV-2
Current Clinical Microbiology Reports (2023)
