
Abstract
Non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH) has become the leading cause of liver disease worldwide. NASH, an advanced form of NAFL, can be progressive and more susceptible to developing cirrhosis and hepatocellular carcinoma. Currently, lifestyle interventions are the most essential and effective strategies for preventing and controlling NAFL without the development of fibrosis. While there are still limited appropriate drugs specifically to treat NAFL/NASH, growing progress is being seen in elucidating the pathogenesis and identifying therapeutic targets. In this review, we discussed recent developments in etiology and prospective therapeutic targets, as well as pharmacological candidates in pre/clinical trials and patents, with a focus on diabetes, hepatic lipid metabolism, inflammation, and fibrosis. Importantly, growing evidence elucidates that the disruption of the gut–liver axis and microbe-derived metabolites drive the pathogenesis of NAFL/NASH. Extracellular vesicles (EVs) act as a signaling mediator, resulting in lipid accumulation, macrophage and hepatic stellate cell activation, further promoting inflammation and liver fibrosis progression during the development of NAFL/NASH. Targeting gut microbiota or EVs may serve as new strategies for the treatment of NAFL/NASH. Finally, other mechanisms, such as cell therapy and genetic approaches, also have enormous therapeutic potential. Incorporating drugs with different mechanisms and personalized medicine may improve the efficacy to better benefit patients with NAFL/NASH.
Similar content being viewed by others

Introduction
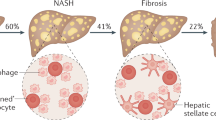
Hepatic steatosis (fatty liver) is one of the most prevalent chronic liver diseases worldwide, affecting approximately one quarter of the global population, and is predicted to become the leading indication for liver transplantation by 2030, posing a significant burden on global health.1,2,3,4,5,6 According to the history of alcohol intake, fatty liver is artificially categorized into two common forms: alcohol-associated liver disease (ALD) and non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH).4,7,8,9,10 While ALD is defined by the presence of hepatic steatosis associated with significant alcohol consumption, NAFL is a generic term that includes a series of liver diseases with different injury severities and consequent fibrosis.4,9 Among these, hepatic steatosis is referred to as NAFL, which is defined as the composition of fat that takes up 5–10% of the liver’s weight. NASH is associated with inflammation and fibrosis, which may progress to cirrhosis and hepatocellular carcinoma (HCC).11,12,13,14 About 20% of patients with NAFL develop NASH, and over 40% of patients with NASH progress to fibrosis.15,16 However, HCC can also develop in the absence of cirrhosis.17,18
Fatty degeneration of the liver, as a pathological change, was first proposed by William Bowman who found that fat accumulation in the liver through observing human liver specimens under the microscope in 1842.19 For the next hundred years, it was generally believed that long-term alcohol consumption was the major cause of the fatty liver; however, a considerable proportion of fatty liver was identified in obese and diabetic people without drinking history.20,21 In 1980, Dr Jurgen Ludwig first proposed the concept of NASH22 and Dr Fenton Schaffner suggested the concept of non-alcohol-associated fatty liver disease (NAFLD) in 1986.23 The term NAFLD has evolved throughout history with advances in the understanding of disease pathophysiology and diagnostic methods.14 NASH is the subtype of NAFLD that can culminate in cirrhosis, HCC, and even death.24 However, the molecular mechanisms underlying the transition from NAFL to NASH are complex and not yet fully understood.13 NAFL/NASH, as a multisystem metabolic disease, is also associated with extrahepatic organ diseases, such as cardiovascular disease (CVD),25,26 chronic kidney disease (CKD),27,28 dementia, and sleep apnea.29,30 Despite increasing liver-related mortality, CVD remains the primary cause of death in patients with NAFL/NASH.16
Clinical progression of NAFL/NASH
Although NAFL/NASH develops at different rates among individuals, it typically follows four stages.31 The first stage involves liver fat accumulation, also known as NAFL. The second stage is referred to as early NASH (F0 no fibrosis and F1 negligible fibrosis) and is characterized by fatty infiltration and liver inflammation. The diagnosis of NASH requires the presence of steatosis, ballooning, and lobular inflammation in liver biopsy. Other histological changes, including portal inflammation, polymorphonuclear infiltrates, Mallory–Denk bodies, apoptotic bodies, clear vacuolated nuclei, microvacuolar steatosis, and megamitochondria, can be seen in NASH, but are not necessary for the diagnosis.7 The third stage, known as fibrosis (F2 advanced fibrosis and F3 bridging fibrosis), is caused by chronic liver inflammation and injury, which results in the excessive accumulation of extracellular matrix (ECM) proteins, including collagen, in the liver. The fourth stage is liver cirrhosis (F4), a severe stage of NAFL/NASH that can be life threatening and develop into end-stage liver disease (ESLD), which is fatal without a transplant.4,7,15,32
NAFL/NASH-related epidemiology
The prevalence of NAFL/NASH is parallel with age, the development of obesity, and type 2 diabetes mellitus (T2DM), and it varies with country and ethnicity.15 Globally, it is estimated that NAFL/NASH accounts for approximately 25% of the general population. By 2030, this percentage is expected to increase, and the proportion of patients with NAFL/NASH affected by terminal diseases will be even higher.3,6,15 Notably, in T2DM patients, the global prevalence of NASL/NASH is two-fold higher than in the general population, amounting to 55.5%, and the highest prevalence was reported in Europe (68%).33 The global prevalence of NASH among patients with T2DM is 37.3%. Approximately 17% of patients with NAFL/NASH and T2DM have developed advanced fibrosis.33 Age affects the incidence of NAFL/NASH, with the mean age of 70–79 having the highest prevalence (33.99%), followed by 60–69 (28.9%), 50–59 (27.4%), and 40–49 (26.53), and 30–39 holding the lowest prevalence (22.43%).15 A study has revealed that lipid turnover, the balance between lipid storage and removal, in adipose tissue decreases with age, whether weight loss or gain.34 The decrease in lipid turnover rate was associated with insulin resistance (IR), dyslipidemia, and metabolic disorders that could increase the risk of obesity, NAFL/NASH, and other chronic diseases.35 Furthermore, the frequency and severity of NAFL/NASH vary by geographic region and ethnicity. Specifically, the Middle East was found to have the greatest frequency of NAFL/NASH (31.79%), followed by Asia (27.37%), South America (24.13%), North America (24.13%), and Europe (23.71%), while Africa had the lowest prevalence (13.48%).4,15 The prevalence in different regions is closely related to their genetic background, lifestyle, and economic status. Current estimates of direct medical costs for NAFLD exceed $100 billion annually in the United States, with the majority of that spent on NASH and its subsequent diseases.36 In addition to cirrhosis and HCC, NAFL/NASH significantly increases the incidence of multiple extrahepatic complications such as T2DM, CVD, CKD, and some extrahepatic malignancies.37 Patients with NAFL/NASH had a 64% increased risk of CVD, and the incidence of CVD is proportional to the severity of NAFL/NASH.38 Patients with NAFL/NASH also develop coronary atherosclerosis, myocardial alterations, and arrhythmias, all of which raise the risk of heart failure.39 NAFL/NASH also significantly increases the risk of extrahepatic cancers such as colorectal tumors,40 gastric cancer,41 pancreatic cancer,42 uterus cancer,43 and breast cancer.44 Hence, it is important to design effective treatment for metabolic syndrome and cancer screening programs for patients with NAFL/NASH. It is also needed to take effective interventions to prevent and control the prevalence of NAFL/NASH to reduce the economic and social burden.
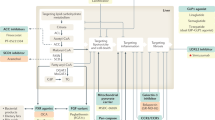
Currently, there are no approved treatments specific to NAFL/NASH despite the high incidence and growing global health impact. While steady progress has been made in the understanding of NAFL/NASH pathophysiology and identification of therapeutic targets, relatively slow progress was achieved in the treatment of all aspects of NAFL/NASH even after years of intense research,5,13,45 although several approved drugs for treating metabolic-related disorders and diseases showed promising outcomes in patients with NAFL/NASH, including Orlistat46 used for the treatment of obesity (Fig. 1). Appropriate therapeutic targets and potent drug candidates are urgently demanded. Herein, we highlight the current understanding of the pathogenesis of NAFL/NASH and outline potential therapeutic targets and corresponding drug candidates in preclinical/clinical trials or patents for treating NAFL/NASH. These emerging therapies namely target diabetes, hepatic lipid metabolism, inflammation, and fibrosis. In addition, advanced research on the signaling pathways that participate in NAFL/NASH pathogenesis has been recognized, including extracellular vesicles (EVs) and gut microbiota, which may provide more rationales and strategies for individualized approaches for future management of NAFL/NASH.

Timeline of NAFL/NASH-related drug development. Drugs at different clinical stages are indicated in different colors: phase 4 drugs are marked in red, phase 3 drugs are marked in orange, phase 2 drugs are marked in yellow, phase 1 drugs are marked in brown, and preclinical drugs are marked in cyan. All colors of drugs in the following figures are the same. Created with BioRender
Signaling pathways driving NAFL/NASH development and related therapeutic targets
The development of NAFL/NASH is considered to initiate from simple steatosis as the first hit that is not enough to induce inflammation and fibrosis; however, during disease progression, a following second hit, including oxidative stress, is necessary to aggravate liver damage.47 NASH is the result of multiple factors acting simultaneously, including genetic variants, abnormal lipid metabolism, oxidative stress, altered immune response, and imbalances in the gut microbiota.48 The “multiple hits” implies that liver inflammation, instead of steatosis, is the primary cause of NASH progression to fibrosis, therefore probably multiple mechanisms act in synergy to promote disease progression.49 The substrate-overload lipotoxic liver injury model of NAFL/NASH revealed that the liver is overwhelmed in dealing with the primary metabolic energy substrates, carbohydrates, and fatty acids, which subsequently lead to the accumulation of toxic lipid species.50,51,52 These metabolites can further induce hepatocellular stress, injury, and hepatic death, resulting in fibrogenesis and genomic instability that make patients susceptible to cirrhosis and HCC (Fig. 2).5,53 While numbers of the current drugs in clinical trials generally achieved the effect of improving NASH histopathology (hepatic steatosis, etc.) or without worsening fibrosis, future studies are needed to translate into appropriate clinical applications.54 On the other hand, patients may exhibit different NAFL/NASH phenotypes due to unique genetic predispositions and idiosyncrasies within the disease, a single treatment is unlikely to reverse NAFL/NASH across all patients, pharmacological combinations and personalized therapy will be favored in the future.53,55,56,57 Here we focus on the signaling pathways that drive NAFL/NASH pathogenesis and summarize the relevant agents and therapies.

Schematic summary of the pathogenesis and interorgan crosstalk of NAFL/NASH. Increased lipid synthesis and uptake in the liver exceeds lipid oxidation and excretion, leading to lipid accumulation and lipotoxicity, inflammatory response, cell death, and fibrosis. Besides the liver, insulin-sensitive organs, such as adipose tissue and muscle, produce adipokines and myokines, respectively, which promote inflammation and oxidative stress in the liver. The gut microbiota regulates the inflammatory response and hepatic lipid accumulation through the metabolism of PAMPs, bile acids, etc. Innate immune responses involved in NAFL/NASH include activation of resident Küpffer cells and recruitment of leukocytes (e.g., neutrophils, monocytes) to the liver. Lymphocyte-mediated adaptive immunity is an additional factor promoting liver inflammation. EVs act as drivers of inflammation in NAFL/NASH activating immune cells and HSC. In NAFL/NASH progression, lipotoxicity-induced hepatocyte death is an important driver including apoptosis, necroptosis, pyroptosis, and ferroptosis. Arrows (red) indicate upregulation and arrows (blue) indicate downregulation in NAFL/NASH. Produced with the assistance of Servier Medical Art (https://smart.servier.com). DNL de novo lipogenesis, FA fatty acid, FAO fatty acid oxidation, TG triglyceride, VLDL very-low-density lipoprotein, ER endoplasmic reticulum, MPO myeloperoxidase, NE neutrophil elastase, BAFF B cell-activating factor, TGF-β transforming growth factor beta, TNF-α tumor necrosis factor-alpha, IL interleukin, IFN interferon, CCL2 C-C motif ligand 2, TRAIL tumor necrosis factor-related apoptosis-inducing ligand, CHOP C/EBP homologous protein, RIP receptor-interacting serine-threonine kinase, MLKL mixed lineage kinase domain-like protein, NLPR3 NACHT, LRR, and PYD domains-containing protein 3, GSDMD gasdermin D, GSH glutathione, GSSG glutathione disulfide, GPX4 glutathione peroxidase 4, ROS reactive oxygen species, NAFL nonalcoholic fatty liver, EVs extracellular vesicles
Lifestyle interventions
Lifestyle interventions, including dietary change, exercise, and weight loss, are the major treatment strategies for NAFL patients without fibrosis development.58 So far, weight loss is the key to improve the histopathological features of NASH, with a clear dose-response association. It was reported that weight loss of at least 3–5% could improve hepatic steatosis, and 5–7% weight loss was necessary to reduce inflammatory activity. In addition, a weight loss of more than 10% indicated the regression of fibrosis.59
A prospective study evaluated the impact on patients with histologically proven NASH of lifestyle changes through a hypocaloric diet (750 kcal/d, calorie deficit) combined with exercise (walk 200 min per week) to reduce weight over 52 weeks. Paired liver biopsies showed the greater weight loss caused by lifestyle changes was related to the improvement of NASH histological characteristics. Among the patients with weight loss ≥10%, the rates of NAFLD activity score (NAS) reduction, NASH, and fibrosis regression were the highest.60 In addition, a small randomized controlled trial using a combination of diet, exercise, and behavior modification for 48 weeks showed that histology of the lifestyle intervention group improved significantly at 48 weeks, of which 67% have improved NAS. Liver steatosis, lobular inflammation, and ballooning in the intervention group were also improved. However, no improvement was observed in fibrosis.61 In addition, the clinical practice guidelines published by the European Association for the Study of the Liver, European Association for the Study of Diabetes, and European Association for the Study of Obesity recommended that the Mediterranean diet (MD) pattern, which contains high amounts of whole grains and monounsaturated fatty acids (MUFAs), as the first-choice diet for patients with NAFL/NASH.62 MD showed improved hepatic steatosis and reduced visceral fat in patients with NAFL in both adults and adolescents,63,64 and persistent MD might reduce the prevalence of NAFL and improve IR in patients with NAFL.65 MD was also shown to reduce platelet activation and hepatic collagen deposition, reducing the risk of CVD in patients with NAFLD.66 The fiber and polyphenols in whole grains of MD reduced energy intake and increased Lactobacillus and Bifidobacterium in the gut that is beneficial for improving NAFLD.67,68 Some butyrate-producing bacteria also increased that is beneficial for improving NAFLD.68 Recent research showed that a green-MD, which contains more green plants and polyphenols and less red or processed meat, led to double intrahepatic fat loss in patients with NAFLD compared with traditional MD.69 However, it is important to note that existing studies have focused on early NAFLD or NAFLD prevention; whether MD is effective for individuals with NASH and advanced disease states requires further investigation.
Clinical trials showed the remission of steatosis occurs with weight reduction achieved by lifestyle interventions, which remains the cornerstone of treatment. However, the effectiveness of lifestyle modification is still limited by difficulties in implementing lifestyle changes, as patients with NAFL/NASH may lack preparation for changing and adopting a healthier lifestyle, particularly regarding physical activity.70 Moreover, the life quality of the patients could be persistently affected by advanced symptoms and diseases, such as hepatic fibrosis, cirrhosis, and HCC, special focus should be made on pharmacological treatment in addition to lifestyle-related interventions.58
Pharmacological interventions
Although lifestyle interventions have been shown to improve fatty liver in NAFL patients, advanced disease conditions, such as substantial fibrosis, are unlikely to be cured by simple lifestyle interventions; therefore, pharmacological interventions remain highly demanded. Here we summarize the progress of pharmacological intervention strategies and their respective pursuant signaling pathways, including targeting metabolism, cellular stress, inflammation, and fibrosis.
Glucose and lipid metabolisms
Metabolic disorders, such as steatosis, are considered essential steps in the pathogenesis of NAFL/NASH (Fig. 3), targeting the abnormal fatty acid and glucose metabolism to prevent liver fat accumulation and the production of a profibrotic environment appear to be promising therapeutic strategies.71,72 Here we review some of the most promising therapeutic targets for NASH, and describe compounds being evaluated against these targets in clinical or preclinical stages (Tables 1 and 2).

Glucose and lipid metabolisms and targeting drugs for NASH. Depiction of the drugs actions sites that are currently in preclinical and clinical trials, based on their primary locus of activity. Targets include those that regulate lipids and glucose homeostasis, such as GLP-1 signaling, mTOR signaling, PPAR signaling, BAs metabolism, DNL and NEFA metabolism, and gut microbiota targets in humans. Agonists are indicated with a green arrow and antagonists with a red inhibitor. Drugs at different clinical stages are as indicated. Created with BioRender. ACLY ATP-citrate lyase, ACC acetyl-coenzyme A carboxylase, FASN fatty acid synthase, SCD stearoyl-CoA desaturase, GLP glucagon-like peptide, FGF fibroblast growth factor, NEFA non-esterified fatty acid, FXR farnesoid X receptor, RXR retinoid X receptor, THR thyroid hormone receptor, mTOR mammalian target of rapamycin, PPARα/δ/γ peroxisome proliferator-activated receptors PPARα, PPARδ, and PPARγ, BAs bile acids, ChREBP carbohydrate response element-binding protein, SREBP sterol regulatory element-binding protein, TCA tricarboxylic acid, FMT fecal microbiota transplantation, OCA obeticholic acid, UDCA ursodeoxycholic acid
Targeting de novo lipogenesis (DNL) and hepatic lipid metabolism
DNL can be upregulated by 20–30% in patients with NAFL/NASH compared to healthy controls, and increased lipogenesis is the key feature associated with fatty liver.73 DNL refers to the endogenous synthesis of lipids from dietary sources (usually carbohydrates or stored energy depots), including fatty acid synthesis, fatty acid elongation/unsaturation, and assembly into triglycerides (TG).74 DNL is mainly regulated by two key transcription factors: sterol regulatory element-binding protein 1c (SREBP-1c, activated by insulin) and carbohydrate regulatory element-binding protein (ChREBP, activated by elevated glucose).75 There are also two crucial enzymes that regulate DNL: acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS). ACC introduces a carboxyl group into acetyl-CoA to produce malonyl-CoA, and FAS is responsible for converting malonyl-CoA to fatty acid chains. DNL is closely associated with excessive glucose intake and the development of IR, which further contributes to the development of NAFL/NASH.
ACLY inhibition: ATP-citrate lyase (ACLY) is a cytoplasmic enzyme responsible for the generation of acetyl-coenzyme A (acetyl-CoA) in DNL and cholesterol synthesis.76 The gene expression of ACLY increased both in patients with NAFLD77 and leptin receptor-deficient db/db mice.78 Bempedoic acid (ETC-1002), an ACLY inhibitor, alleviated high-fat diet (HFD)-induced NASH in male C57BL6/N mice, including decreased body weight gain, improved glycemic control, reduced hepatic TG and total cholesterol (TC), lowered mRNA expressions of inflammatory and fibrotic genes (Ccl2, Timp1, and Col1a1), and improvement in NAS score.79 In a phase 3 clinical trial (NCT02666664), Bempedoic acid significantly reduced low-density lipoprotein cholesterol (LDL-C) levels.80 Bempedoic acid was recently approved by US FDA for the treatment of heterozygous familial hypercholesterolemia (HeFH)81 and clinical atherosclerotic cardiovascular disease (ASCVD), due to the major risk factors for HeFH and ASCVD are the elevated LDL-C levels.79 In addition, hydroxy citric acid, another competitive inhibitor of ACLY, significantly reduced fatty acid synthesis and the levels of liver injury parameters, including alanine transaminase (ALT), aspartate transaminase (AST), gamma-glutamyltransferase (GGT), and lactate dehydrogenase (LDH) in rats fed HFD.82,83
ACC inhibition: ACC converts acetyl-CoA to malonyl-CoA and is a rate-limiting step in DNL. A preclinical study demonstrated that inhibition of ACC reduced liver fibrosis in a rat choline-deficient, HFD model.84 In a randomized and placebo-controlled trial of patients with NASH (NCT02856555), median relative decreases in magnetic resonance imaging-estimated proton density fat fraction (MRI-PDFF) were greater in patients treated with 20 mg of ACC inhibitor GS-0976 for 12 weeks (decrease of 29%) than those given placebo (decrease of 8%; p = 0.002).85 GS-0976 decreased hepatic steatosis, selected fibrosis markers, and liver biochemistry.85 The clinical study also indicated that after administration of GS-0976 for 12 weeks, the median hepatic DNL was decreased by 22% from baseline in patients with NASH (p = 0.004).86 MK-4074 is a small-molecule inhibitor specifically targeting liver ACC1/2. In preclinical animal models and clinical studies, administration of MK-4074 showed suppressed DNL and enhanced liver fatty acid oxidation (FAO), leading to significantly reduced hepatic TG content in preclinical studies.87 Based on the promising results from the pilot studies, phase 1 clinical studies have been conducted to assess changes in liver fat content (NCT01431521) in adult men and women with NAFL after multiple oral doses of MK-4074 and Pioglitazone hydrochloride. The results showed that the administration of MK-4074 for 1 month reduced liver TG by 36% in patients with hepatic steatosis. However, although liver TG content was reduced, plasma TG significantly increased by 200%.87 Similarly, dose-dependent reduction in liver fat reached 50–65% and a dose-dependent elevation in serum TG reached 8% with the ACC inhibitor PF-05221304 (NCT03248882). Notably, PF-05221304 combined with PF-06865571 (Diacylglycerol O-Acyltransferase 2 (DGAT2) inhibitor) has the potential to avoid some limitations of ACC inhibitor alone, including the ACC inhibitor-mediated elevation in serum TG (NCT03776175).88 In addition, an allosteric inhibitor of ACC1/2, NDI-010976, was well tolerated at doses up to 200 mg and resulted dose-dependently in inhibition of hepatic DNL in obese adult male subjects (NCT02876796).89
FAS inhibition: FAS is a rate-controlling enzyme that converts malonyl-CoA to palmitic acid during DNL. FAS mRNA expression in the liver is significantly higher in patients with NAFL/NASH than that in normal subjects.90 In a phase 2 clinical trial, administration of TVB-2640, a FAS inhibitor, showed promising results in adult patients with NASH with ≥8% liver fat and liver fibrosis (NCT03938246).91 TVB-2640 reduced liver fat by 9.6% in the 25 mg cohort and 28.1% in the 50 mg cohort from baseline compared to 4.5% increase in liver fat in the placebo cohort. A total of 23% of patients in 25 mg group and 61% in 50 mg group of TVB-2640 achieved a relative reduction of liver fat of ≥30% respectively, while only 11% of patients in the placebo group.91 A phase 2b (NCT04906421) trial is recruiting and subjects with liver fibrosis at stages F2–F3 will be enrolled to further evaluate the safety and efficacy of TVB-2640 in subjects with NASH. The FAS inhibitor (FT-4101) safely reduced hepatic DNL and steatosis of patients with NASH in a phase 1/2 clinical trial (NCT04004325).92 Another FAS inhibitor Orlistat, however, did not enhance weight loss or improve liver enzymes, measures of IR, and histopathology (NCT00160407).93
SCD1 inhibition: stearoyl coenzyme A desaturase 1 (SCD1) is an enzyme that catalyzes the rate-limiting step in the formation of MUFAs, specifically oleate and palmitoleate from stearoyl-CoA and palmitoyl-CoA.69,70 The expression of SCD1 in the liver was increased both in patients with NAFLD and ob/ob mice.94 Aramchol is a conjugate of cholic acid and arachidic acid that had an inhibitory effect on SCD1 activity to reduce liver fat content in patients with NASH (NCT01094158).95 In both isolated primary human hepatic stellate cells (HSCs) and a human hepatic stellate cell line (LX-2), Aramchol reduced fibrogenic gene expression by inhibiting SCD1 and inducing PPARγ.96 In a phase 2 clinical trial (NCT01094158) for NASH, liver fat content was decreased by 12.57% in patients treated with 300 mg/day of Aramchol compared with the increase of 6.39% in the placebo group.95 In a 52-week, double-blind, placebo-controlled, phase 2b trial (NCT02279524), Aramchol displayed a placebo-corrected decrease in liver TG, without reaching the prespecified significance (p = 0.066). NASH resolution without worsening fibrosis was achieved in 16.7% of Aramchol vs. 5% of the placebo, and fibrosis improvement by ≥1 stage without worsening NASH in 29.5% vs. 17.5%, respectively.97 Despite administration of 600 mg of Aramchol was unable to reduce liver fat, safety and changes in liver histology and enzyme improvements were observed; therefore, Aramchol is processing into a phase 3 trial (NCT04104321).97
SREBP inhibition: SREBP-1c is an insulin-sensitive transcription factor that plays a key role in the induction of lipogenic genes in the liver, which is transactivated by liver X receptor (LXR).98 The increased levels of nuclear SREBP-1c contributed to the hepatic steatosis99 and were regulated by insulin in mouse or rat T2DM models.100 Nuclear accumulation of mature forms of SREBP-1c and expression of its target genes is blocked by the mechanistic target of rapamycin kinase (mTOR) complex 1 (mTORC1) inhibitor rapamycin.101 The protein folliculin (FLCN) in adipocytes phosphorylates mTOR and retains TFE3 in the cytoplasm to inhibit WAT browning.102 This process is independent of canonical mTOR-S6K signaling.102 A recent study has shown that deletion of Flcn in the liver inhibited mTORC1 signaling to promote nuclear translocation of TFE3, which in turn activated lipid catabolism genes and suppressed DNL genes.103 This specific deletion of hepatic Flcn inhibited the activation of SREBP-1c and could prevent or reverse NASH in mice fed choline-deficient L-amino acid–defined and high-fat (CDAA-HF) diet.103 Previous reports have shown that hyperactivation of SREBP-1c promotes hepatic TG accumulation,104,105 suggesting that targeting SREBP-1c for regulating hepatic lipid metabolism might be an appropriate strategy for NASH treatment.105,106 Oltipraz (OPZ) is a synthetic dithiolethione with an antisteatotic effect by inhibiting the activity of LXR-α, thereby suppressing SREBP-1c activity.107 Administration of the thiol-reactive agent OPZ significantly attenuated the progression of histologic abnormalities, especially hepatic fibrosis in rats on a CDAA diet.108 The efficacy and safety of OPZ administration in patients with NAFLD were verified in the phase 2 clinical trials (NCT01373554 and NCT00956098). NCT01373554 revealed that 24-week treatment of OPZ significantly reduced the liver fat content in a dose-dependent manner in patients with NAFLD. Compared with the placebo group (–3.2%), absolute changes in the liver fat content were reduced by 7.7% and 13.9% for the low-dose and high-dose groups (p = 0.13 and p < 0.01), respectively.109,110 Clinical phase 3 trials (NCT04142749 and NCT02068339) are carried out to investigate the inhibitory role of OPZ on fatty acid synthesis in patients with NAFLD.
SREBP2 transcriptionally controls 3-hydroxy-3-methyglutaryl-coenzyme A (HMG-CoA) reductase, which is a key enzyme in cholesterol synthesis and ketogenesis that closely links to the development of NAFL/NASH.111,112 Statins (HMG-CoA reductase inhibitors) restrict cholesterol synthesis and are mostly used as hypolipidemic drugs. It has been shown that Statins increased the FAO capacity of the liver by inducing PPARα and prevented the development of MCD-induced NASH in mice; however, the authors did not claim that the effect of Statins on improving NASH may not be related to its cholesterol decreasing function.113 Simvastatin, first-generation statins, treatment in vivo or in vitro inhibited the activation of HSC in rats fed HFD.114 In addition, Atorvastatin, a third-generation synthetic statin that is more effective to reduce cholesterol and LDL-C, dissolved cholesterol crystals (the focal point of the coronal structure of activated Küpffer cells (KCs)) to improve fibrosis in obese and diabetic Alms1 mutant (foz/foz) mice fed high-fat (23%) diet containing 0.2% cholesterol.115 In a 6-year follow-up of more than 11 million subjects, Statins were observed to reduce the risk of NAFLD (adjusted odds ratio (AOR) 0.66; 95% confidence interval (CI) 0.65–0.67) and to reduce the risk of liver fibrosis (AOR 0.43; 95% CI 0.42–0.44).116 A case–control study showed a protective effect of Statins against NAFLD-associated HCC (OR = 0.20, 95% CI: 0.07–0.60, p = 0.004) in 102 subjects (patients with NAFLD-associated HCC, n = 34; NAFLD patients without HCC, n = 68).117 Probably due to the hepatotoxic effects of Statins, doctors may not prescribe statins to patients with high plasma aminotransferase levels.118 However, a post-hoc analysis of Statins use in a randomized controlled trial revealed that Statins therapy was safe for patients with prediabetes or T2DM and NASH, suggesting Statins may be a potential therapeutic strategy in those patients.119 Patients with NAFL/NASH have a higher risk of CVD.38 Further investigation is needed to fully demonstrate the safety and efficacy of statins, doctors may be able to try to use statins in patients with NASH to reduce the risk of CVD complications. Furthermore, in a phase 2, double-blind, randomized, placebo-controlled, multicenter study (NCT02633956) that evaluated the effect of Obeticholic acid (OCA, a synthetic bile acid (BA), and farnesoid X receptor (FXR) agonist), and the subsequent addition of Atorvastatin therapy, on lipoprotein metabolism in subjects with NASH (fibrosis stages 1–4), OCA-induced increases in LDL-C in patients were mitigated with Atorvastatin.120
Antidiabetic drugs for NASH treatment
NAFL/NASH is a metabolic-related liver disease with a bidirectional and significant relationship with obesity and T2DM.121 In patients with T2DM, the global prevalence of NAFL/NASH is more than 55%.33 T2DM has also been linked to a faster progression of NASH, cirrhosis, or HCC.122 Although there is no approved drug for the treatment of NAFL/NASH, various antidiabetic agents showed some efficacy. Here we discuss the putative molecular mechanisms that potentially link NAFLD and T2DM, as well as the current pharmacological treatments for NAFLD patients with a metabolic disorder.
PPAR signaling: peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily. PPARs have three identified isotypes (α, β/δ, and γ), all of which are involved in lipid metabolism and glucose homeostasis in NAFL/NASH. PPARα gene expression in the liver of obese patients negatively correlated with steatosis, NASH severity, and IR.123 PPARβ/δ mRNA expression level was reduced in liver biopsies of patients with moderate or severe steatosis.124
PPARα is the primary regulator of hepatic fat catabolism during fasting.125 It has been demonstrated that deletion of PPARα promoted NAFL/NASH and hepatic inflammation in mice.126 It was observed that lipid accumulated massively in the liver of global PPARα knockout (Ppara−/−) mice fed with HFD.127 Hepatic steatosis was reduced in hepatocyte-specific PPARα deficient mice compared to Ppara−/− mice, probably due to increased FAO in other extrahepatic tissues like brown adipose tissue, muscle, and heart.128
Diabetic mice with hepatocyte-specific deletion of PPARγ had improved hepatic steatosis yet more severe IR, probably due to reduced insulin sensitivity in muscle and adipose tissues.129 PPARγ agonists reduced hepatic steatosis in patients with NAFLD possibly due to effects in adipose tissue, where PPARγ activation promoted adipogenesis in adipose tissue to decrease the fatty acids entering the liver.130 In addition, deletion of PPARγ in non-parenchymal liver cells including KCs and HSCs exacerbated liver damage and fibrogenic response to carbon tetrachloride (CCl4) challenge.131
The expression levels of PPARβ/δ are generally higher than PPARα and PPARγ. PPARβ/δ plays a critical role in the liver, skeletal muscle, adipose tissue and immune system.132 Transcriptional profiling of liver tissue revealed that PPARβ/δ deletion downregulated pathways including lipoprotein metabolism and glucose utilization, and upregulated genes connected to innate immunity and inflammation, which collectively correlated with increased plasma glucose and TG.133 PPARβ/δ-deficient mice with HFD were more prone to obesity.134 Activated PPARβ/δ suppresses hepatic glucose output and promotes beta-oxidation in muscle to regulate metabolic homeostasis.135 Meanwhile, fatty acid uptake by skeletal muscle appears to be influenced by hepatic PPARβ/δ, with hepatocyte-specific PPARβ/δ deficiency reducing muscle fatty acids uptake to avoid lipotoxicity in hepatocytes.136 In addition, PPARs are involved in anti-inflammatory effects through a mechanism known as transrepression that inhibits nuclear factor-κB (NF-κB), activator protein-1 (AP-1), signal transducer and activator of transcription (STAT), or nuclear factor of activated T cells.137
In general, PPARs are involved in glucose and lipid metabolism in multiple organs and contribute to the anti-inflammatory response in NAFL/NASH. Currently, there are several drug candidates for PPAR activations, which will be described later. In addition, compared to selective PPAR agonists, targeting two isotypes or pan-PPAR agonists combines the beneficial effects of selective PPAR agonists and improves NAFL/NASH more effectively in multiple ways.138,139
PPAR agonists: a variety of agents targeting different subtypes of PPAR are currently under preclinical and clinical studies. Pioglitazone, a promising PPARγ agonist, is currently under a phase 4 clinical trial (NCT00994682). The study included 44% of patients with type 2 diabetes who have NASH regression and 26% of non-diabetic patients who have NASH regression. A significant regression in fibrosis was observed only in patients with type 2 diabetes (p = 0.035). Compared with non-diabetic patients, Pioglitazone significantly improved insulin sensitivity in adipose tissue of diabetic patients (p < 0.001).140 However, compared to patients with prediabetes, Pioglitazone reduced liver fibrosis and adipose tissue insulin sensitivity at significantly higher levels in patients with type 2 diabetes.140,141 It is demonstrated that Elafibranor (GFT505), a dual PPARα/δ agonist, had liver-protective effects on steatosis, inflammation, and fibrosis in several animal models of NAFL/NASH.142,143 In a phase 2b clinical trial (NCT01694849), compared with the placebo group, NASH resolved without fibrosis worsening in a higher proportion of patients in the 120-mg Elafibranor group vs. the placebo group (19% vs. 12%; p = 0.045).143 However, Elafibranor exhibited poor anti-NASH effects in a phase 3 clinical trial (NCT02704403), probably due to its weak PPARα/δ agonistic activity and poor metabolic stability. Novel, structurally stable PPARα/δ agonists are still under investigation, such as new Triazolone derivatives.144 Other promising PPAR agonists were evaluated in randomized controlled phase 2/3 trials of NASH patients including the dual PPARα/γ agonist Saroglitazar (NCT03061721)145 and a pan-PPAR agonist Lanifibranor (NCT03008070).146 In a clinical 2b trial (NCT03008070), Lanifibranor achieved optimal results that patients treated with the 1200-mg dose of Lanifibranor had a decrease of at least two points in the SAF-A score (the activity part of the Steatosis, Activity, Fibrosis (SAF) scoring system) without worsening of fibrosis and reached the primary endpoint, which will ultimately determine the therapeutic potential of pan-PPAR agonist targeting inflammation and fibrosis to support further evaluation of Lanifibranor in the phase 3 trial.146 In addition, the study also reached several primary secondary endpoints, including NASH remission, no deterioration of NASH, and improved liver fibrosis. Furthermore, in preclinical studies, multiple PPAR agonists have achieved antifibrotic results in both animal and in vitro models, such as PPARα agonists (Bezafibrate147 and GW7647148).149 PPARα agonist Gemcabene prevented steatosis, inflammation, and hepatocyte ballooning, and inhibited fibrosis progression in a high-fat/high-calorie diet-fed murine model of NASH.150 PPAR δ agonist Seladelpar (MBX-8025) reduced steatosis and liver inflammation, and improved liver fibrosis in diabetic obse mice.151
While preclinical/clinical studies suggest that the dual/pan-PPAR agonists have a more significant effect in the treatment of disease when compared with PPARα or PPARγ agonists alone, improvement of the low agonistic activity and low metabolic stability of multiple agonists may still need to be improved. In addition, it is important to note that side effects occur frequently, such as diarrhea, nausea, peripheral edema, anemia, and weight gain.
GLP-1 agonists: glucagon-like peptide-1 (GLP-1), a secreted peptide from enteroendocrine L cells, promotes insulin secretion and β-cell proliferation in the pancreas and regulates blood glucose levels.152 Interestingly, GLP-1 levels are decreased in NAFL/NASH patients.153 Liraglutide, a GLP-1 analog, is used as an antidiabetic agent by induction of insulin secretion. In a double-blind, randomized, placebo-controlled phase 2 study (NCT01237119), 39% of patients with NASH treated with Liraglutide showed NASH regression compared with 9% of patients in the placebo group. In addition, 9% of patients in the Liraglutide group developed fibrosis, while 36% of patients in the placebo group developed fibrosis. These observations indicate that Liraglutide is a well-tolerated disease-modifying intervention leading to histological resolution of NASH,154,155 reducing metabolic dysfunction, IR, and lipotoxicity in the pathogenesis of NASH.156 Another phase 2 study (NCT01399645) was conducted to test the effects of insulin vs. Liraglutide therapy on hepatic fat in patients with T2DM inadequately controlled with Metformin therapy. However, Liraglutide treatment did not significantly alter the liver mean proton density fat fraction (PDFF) (p = 0.15), magnetic resonance spectroscopy-PDFF (p = 0.80), liver volume (p = 0.30), or the total liver fat index (p = 0.39).157 Semaglutide, a GLP-1 receptor agonists (GLP-1-Ra) developed based on the extensive research behind the development of Liraglutide, has been used to treat T2DM. In a randomized, double-blind, placebo and active controlled phase 2 trial (NCT02453711), Semaglutide showed clinically relevant weight loss compared with placebo at all doses.158,159 Patients treated with 0.4 mg of Semaglutide had improvements in fibrosis stage compared with patients in the placebo group (43% vs. 33%) in a phase 2 clinical trial (NCT02970942).160,161,162 Given the potent effects of Semaglutide, several clinical trials are underway to determine whether Semaglutide alone or in combination with other drugs could better benefit patients with NASH (NCT04822181, NCT05016882, NCT04971785, NCT05195944, NCT04639414, and NCT04944992). Recently, a dual glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 receptor agonist Tirzepatide (LY3298176) significantly reduced NASH and fibrosis biomarkers in patients with T2DM.163,164 Another dual GIP/GLP-1 receptor agonist NNC0090-2746 improved glycemic control and reduced body weight and TC.165 In addition, a balanced glucagon-GLP-1 receptor agonist (Cotadutide) was observed to improve lipid profile, hepatic function indexes, and NAFLD fibrosis markers in type 2 diabetes patients controlled with Metformin.166
The most common adverse events of GLP agonists for NASH are mild to moderate gastrointestinal side effects, including nausea, diarrhea, indigestion, and vomiting, which show in a dose-dependent manner and are often transient.167 In addition to the most frequently observed gastrointestinal side effects, Liraglutide raised serum lipase and amylase levels. The absolute risk of Liraglutide-induced acute pancreatitis was higher when compared to placebo. Liraglutide may also contribute to an increased risk of acute gallbladder or biliary disease.168,169
SGLT2 inhibition: sodium-glucose cotransport protein 2 (SGLT2) inhibitors are a relatively new class of antidiabetic agents that lower blood glucose by inhibiting glucose reabsorption by SGLT2 in the proximal renal tubules.170 A growing number of studies have shown that most SGLT2 inhibitors are effective in improving steatosis and fibrosis in patients with NAFL/NASH and T2DM.171,172,173,174 In addition, SGLT2 inhibitors can also block KCs activation and associated inflammatory processes.175 Results of clinical trials (UMIN000015727 and jRCTs071180069) indicated that long-term Ipragliflozin treatment (IPR group) ameliorated hepatic fibrosis in patients with NAFL/NASH. It was reported that 67% of the IPR group (50 mg/day for 72 weeks) were relieved from NASH compared to 27.3% in the control group. In addition, none of the participants in the IPR group developed NASH, whereas 33.3% of the control group developed NASH. Compared to baseline measurements in patients with NASH, body weight, hemoglobin A1c (HbA1c), hepatic function indexes (AST, ALT, and GGT), body fat mass, and steatosis were significantly decreased after Ipragliflozin oral administration (50 mg/day) for 24 weeks.176,177 Empagliflozin, another SGLT2 inhibitor was analyzed in several clinical trials (Institutional Review Board of NAMS (approval number: 547-077/078),178 NCT02964715, IRCT20190122042450N3).179 After Empagliflozin administration for 6 months, there was a significant reduction in the mean controlled attenuation parameter (CAP) value from 282.07 ± 47.29 to 263.07 ± 49.93 dB/m and liver stiffness (LS) from 5.89 ± 4.23 to 5.04 ± 1.49 kPa.178 Empagliflozin (25 mg daily for 24 weeks) improved steatosis (67% vs. 26%, p = 0.025), ballooning (78% vs. 34%, p = 0.024), and fibrosis (44% vs. 6%, p = 0.008) significantly compared with historical placebo.180 Study also showed that CAP score significantly decreased in borderline with Empagliflozin (10 mg for 24 weeks) compared to placebo.179 These data suggest that long-term Empagliflozin treatment has improved liver steatosis and fibrosis in patients with NAFL/NASH and T2DM, leading to beneficial effects, such as weight loss and reduction in hepatic fat, transaminases, and GGT content. Canagliflozin, an SGLT2 inhibitor, significantly improved several hepatic functions or fibrosis markers (AST, fibrosis-4 index, and FM-fibro index), and metabolic parameters (HbA1c and body weight).181 Canagliflozin may be useful for the treatment of T2DM patients with NASH, especially those patients in hepatic fibrosis stages 1–3 (UMIN000023044).181 There is a reduction in visceral fat and an improvement in liver tests, including serum concentrations of AST, ALT, ferritin, and type IV collagen 7S, after treatment with Dapagliflozin by inhibiting SGLT2 (UMIN000022155 and UMIN000023574).182,183 However, administration of Dapagliflozin for 12 weeks did not improve hepatic steatosis in patients without T2DM (NCT02696941).184
Bile acids (BAs) therapeutics
BAs promote the intestinal absorption of lipid substances and improve lipid hydrolysis metabolism through regulating various lipid metabolism enzymes and enhance the lipid metabolism of the pancreas.185 The level of total fecal BAs was elevated in patients with NAFL/NASH, suggesting that the progression of NAFL/NASH might be associated with altered BAs homeostasis.186 Administration of cholic acid, a primary BA, changed the bacterial composition of the intestinal microbiome,187 and the increase of circulating BAs led to the toxic accumulation of BAs in hepatocytes, which propagates inflammation, oxidative stress, and the worsening of NAFL/NASH.188,189,190 FXR signaling is activated by BAs and the most potent of which is chenodeoxycholic acid (CDCA).191,192 FXR signaling was inhibited in patients with NAFLD and rats fed an HFD, probably due to deoxycholic acid (DCA), an FXR antagonistic secondary BA, increased while the agonistic CDCA was decreased.193 In the light of the potential hepatotoxic effects of BAs and BAs-induced FXR signaling to regulate insulin sensitivity and glycolipid metabolism, raising attention was attracted in the role of BAs in the treatment of NAFL/NASH (Tables 1 and 2). For instance, ursodeoxycholic acid (UDCA) is a hydrophilic, non-toxic, secondary BA in humans. In a phase 4 clinical trial (NCT04977661), UDCA improved hepatic aminotransferases and serum cytokine and chemokine (41%, 35%, 47%, and 37% for ALT, AST, IL-6, and CCL2/MCP-1, respectively).189
FXR agonists: FXR is found mainly in the liver and intestine, a major intercellular BA receptor activated during the fed state to regulate metabolism and inflammation.185,194,195 The interaction of BAs and intracellular FXR not only inversely regulates BA synthesis, but inhibits hepatic adipogenesis and steatosis, reduces hepatic gluconeogenesis, and increases peripheral insulin sensitivity through transcription of GLUT4.185,196,197,198 Deficiency of FXR leads to increased BA synthesis, which further contributes to liver fibrosis and inflammation and even to HCC.199,200,201
OCA, an FXR agonist, regulates the expression of transcription factors that reduce BA synthesis and liver steatosis.202 In a clinical phase 2 trial (NCT01265498), OCA improved the histological features of NASH. A total of 45% of patients in the OCA group had improved liver histology compared with 21% of patients in the placebo group (p = 0.0002).203 Furthermore, in an 18-month clinical phase 3 trial (NCT02548351), 23% of the patient cohort who received OCA achieved a reduction of NAS by at least one score without worsening fibrosis compared to 12% (37/311) in the placebo group, indicating that OCA improved inflammation and fibrosis in patients with NASH.202 In addition, several other FXR agonists are currently under phase 2/3 trials, including Cilofexor, Tropifexor, and Nidufexor.204 In a recent phase 2b study (NCT02854605), Cilofexor (GS-9674) improved hepatic steatosis and liver transaminase in NASH patients. The relative decrease of MRI-PDFF in patients treated with 100 mg of Cilofexor for 24 weeks was 22.7%, while that of patients treated with placebo increased by 1.9% (p = 0.003). A total of 39% of the patients with Cilofexor treatment (p = 0.011) and 13% of the patients in the placebo group showed ≥30% reduction in MRI-PDFF (NCT02854605).205 A double-blind phase 2 study proved that EDP-305, a non-BA FXR agonist, mildly reduced ALT levels and liver fat content (NCT03421431). The mean reductions from baseline in ALT for patients receiving 2.5 and 1 mg of EDP-305 for 12 weeks were 27.9 U/L (p = 0.049) and 21.7 U/L (p = 0.304), respectively, compared to a decrease of 15.4 U/L for those receiving placebo. Absolute liver fat reduction was 7.1% with 2.5 mg EDP-305, 3.3% with EDP-305 1 mg, and 2.4% with placebo.206 While FXR agonists displayed promising efficacy in treating patients with NASH, almost all FXR agonists caused side reactions, such as pruritus and deterioration of the high-density lipoprotein (HDL-C)/LDL-C ratio.207
There is a strong association between impaired fibroblast growth factor 19 (FGF19) signaling and elevated levels of BAs in circulation.208,209,210 FGF19 modulates hepatic fat metabolism via multiple mechanisms, including accelerating lipid oxidation and repressing hepatic DNL, subsequently protecting the liver from steatosis.211 Aldafermin (NGM282), an engineered analog of the gut hormone FGF19, showed a tendency towards reducing liver fat and improving fibrosis yet with adverse events, including diarrhea, abdominal pain, and nausea in NASH patients.212,213,214
THR-β agonists: thyroid hormone receptor beta (THR-β) is the main thyroxine receptor in the liver and mediates cholesterol metabolism and excretion through BAs.215,216 THR-β agonists have been observed to reduce lipotoxicity, improve liver function and subsequently reduce liver fat by promoting fatty acid breakdown and stimulating mitochondrial biogenesis.217 Resmetirom (MGL-3196) is a selective THR-β agonist and is currently under clinical phase 2/3 trials (NCT02912260 and NCT03900429). In a 36-week paired liver biopsy study (NCT02912260), markers of fibrosis were reduced significantly by Resmetirom treatment, including the reductions in LS (p = 0.015) and the ratio of PRO-C3 (N-terminal type III collagen pro-peptide)/C3M (metalloproteinase-degraded collagen III) (p = 0.0004), a proposed measure of net fibrosis formation, in adult patients with NASH.218,219 Furthermore, the effective and safe daily doses of Resmetirom at 80 and 100 mg were used in the ongoing phase 3 NASH study (NCT03900429).218 GC-1 (Sobetirome) and VK2809 are NASH treatment candidates based on THR-β-agonism.220 In the human hepatocyte-derived Huh-7 cell line, treatment with GC-1 upregulated the transcription of mitochondrial carnitine palmitoyl transferase 1a (CPT1a), which is part of a mitochondrial outer membrane fatty acid transfer complex, with a dose-response comparable to that of the native THR ligand, triiodothyronine (T3).220,221 GC-1 also reduced fat accumulation and improved steatohepatitis induced in rats by a choline-methionine-deficient (CMD) diet.222 VK2809 has been shown to reduce the liver fat content in patients with NAFLD after 12 weeks of treatment.223
Other metabolic pathway targets
Fibroblast growth factor 21 (FGF21) was shown to participate in lipid oxidation and TG clearance in the liver.224,225 FGF21 agonists displayed promising effects in preclinical models of NAFL/NASH as well as in short-term clinical trials in patients with NASH.226 In a phase 2a study (NCT02413372), 16-week Pegbelfermin (BMS-986036, an FGF21 agonist) administration in patients with NASH and stage 1–3 fibrosis was associated with a significant reduction in hepatic steatosis measured by MRI-PDFF and improvement in lipid profiles, adiponectin concentration, and biomarkers of fibrosis and hepatic injury.227 To further evaluate the efficacy of Pegbelfermin, multicenter, double-blind, placebo-controlled, randomized trials (NCT03486899 and NCT03486912) are currently underway to focus on NASH patients with bridging fibrosis and cirrhosis.228 Efruxifermin is a long-acting Fc-FGF21 fusion protein designed to mimic the biological activity of FGF21. In a phase 2a clinical trial (NCT03976401), treatment of Efruxifermin in NASH patients (F1-F3 stage) indicated that the absolute changes from baseline in hepatic fat fraction were decreased in a dose-dependent manner, namely –12.3% (28 mg), –13.4% (50 mg), and –14.1% (70 mg) compared to 0.3% in the placebo group.229
Anti-cellular stress
Chronic disorders of lipid metabolism are closely associated with changes in the redox balance that affect metabolic-associated organelles, resulting in cell lipotoxicity, lipid peroxidation, chronic endoplasmic reticulum (ER) stress, and mitochondrial dysfunction (Figs. 2 and 3).230 Excessive accumulation of lipids leads to overproduction of reactive oxygen species (ROS) in different sources, including mitochondria, ER, and NADPH oxidase. Although there is no direct clinical evidence of a clear mechanism of action by which oxidative stress affects NAFLD, oxidative stress markers such as nitric oxide, thiobarbituric acid-reactive species,231 and malondialdehyde (MDA)232 may be measured clinically to determine the progression of NAFLD. Here we focus on the role of stress in mitochondria and ER in the development of NAFL/NASH.
Mitochondrial dysfunction
Energy homeostasis in hepatocytes is mainly mediated by oxidative mitochondrial metabolism, including β-oxidation of free fatty acids (FFAs), tricarboxylic acid (TCA) cycle, ATP synthesis, and ROS production.233,234,235 Wild-type mice exhibited a marked reduction in FAO in liver mitochondria after 4 weeks of HFD feeding, and this effect was restored after 8 weeks,236 suggesting a resilient mitochondrial functional change in obesity-induced metabolic disorder. Inefficient β-oxidation of fatty acids leads to the accumulation of toxic lipids such as hepatic diacylglycerols, ceramides, and long-chain acylcarnitines, accelerating inflammation and the NASH process.237 During IR, the hepatic TCA cycle decreases mitochondrial respiratory efficiency by increasing electron deposition into inefficient respiratory chains that are prone to generate ROS.238 During the development of NAFL/NASH, FFAs overload the mitochondria, FAO, and electron flux in the electron transport chain (ETC) increasing and disrupting mitochondrial homeostasis, leading to excessive production of ROS due to the lack of upregulation of ETC complex activity, which generates “electron leakage” and subsequently exacerbates lipid accumulation in hepatocytes.239,240 In addition, the ROS clearance capacity in NAFL/NASH liver is also diminished. For example, glutathione peroxidase (GPx) is one of the most important antioxidant enzymes for maintaining ROS homeostasis; however, in the livers of patients with NASH, GPx activity was greatly reduced.241 Manganese (Mn) is mainly responsible for scavenging ROS in mitochondrial oxidative stress, and deficiency or excess of Mn leads to changes in manganese superoxide dismutase activity, resulting in mitochondrial dysfunction.242,243 Hydrogen peroxide is mostly catabolized by catalase, an enzyme that catalyzes hydrogen peroxide into molecular oxygen and water without the production of free radicals. In fatty liver, the reduced activity of catalase further promotes the accumulation of ROS.244 In addition to ETC, there are other potential sources of ROS in mitochondria, such as mitochondrial flavoenzymes, including pyruvate dehydrogenase, glycerol phosphate dehydrogenase, monoamine oxidase, and α-ketoglutarate dehydrogenase.235 Furthermore, an increase in mitochondrial cytochrome P450 2E1 (CYP2E1) expression also leads to increased lipid peroxidation and ROS production and is associated with the progression of NAFL to NASH.245,246,247 The c2 allele of CYP2E1 gives it higher transcriptional and pro-oxidant activity, which determines the susceptibility to develop NASH at the genetic level.248
Liver mitochondrial DNA (mtDNA) from patients with NAFL/NASH has a higher rate and degree of heterogeneity of mutations, including mutations in the oxidative phosphorylation (OXPHOS) chain genes.249 Mutations in mitochondria encoding cytochrome B, a member of the OXPHOS system, positively correlate with the severity of NAFL/NASH.250 Under lipid overload, mtDNA released from damaged hepatocytes acts as danger-associated molecular patterns (DAMPs), inducing upregulation of IL-33 in macrophages via TLR9 receptor, and enhances lipopolysaccharide (LPS)-induced production of IL-1β and TNFα.251 Moreover, mtDNA also directly activates HSCs, driving liver fibrosis progression.252
ER stress
ER is the primary site of lipid synthesis and protein folding and assembly; however, lipid stress, such as lipid overload and impaired VLDL-TG assembly, activates a specific signaling pathway called the unfolded protein response (UPR).253 UPR consists of three transmembrane proteins: protein kinase RNA-like ER kinase (PERK),254 activating transcription factor 6 (ATF6),255 and inositol-requiring signaling protein-1 (IRE1),256 which all form stable complexes with the regulatory protein glucose regulatory protein 78 (GRP78, also known as Bip) under normal conditions, while upon ER stress, they dissociate from GRP78 and activate downstream signaling pathways.257 Activation of PERK leads to phosphorylation of eukaryotic translation initiation factor-2α (eIF2α) to attenuate global protein translation to reduce the unfolded protein load to ER.254 Meanwhile, transcription of activating transcription factor 4 (ATF4) is upregulated258 and promotes the transcription of the CCAAT/enhancer binding protein homolog (CHOP), which is a transcription factor associated with apoptosis.259 Upon ER stress, ATF6 translocates from the ER to the Golgi where it is cleaved to its active form,260 and activated ATF6 stimulates the expression of ER molecular chaperone-related genes. Phosphorylation of IRE1 activates its endoribonuclease activity to splice XBP-1 mRNA, leading to the upregulation of ER chaperones and ER-associated degradation proteins.261,262,263 The initial activation of UPR is to restore ER homeostasis, whereas unresolved ER stress via long-term lipotoxicity promotes apoptosis through the apoptotic signaling pathway downstream of the UPR.
Each disulfide bond formed during protein folding should generate a single ROS,264 prolonged ER stress increases UPR-mediated ROS production through activation of CHOP.265 In a mouse NASH model, CHOP expression was significantly upregulated.266 CHOP deficiency did not improve steatosis but reduced inflammation and apoptosis in NASH mice induced by the MCD diet, indicating CHOP may play a more predominant role in subsequent liver damage by suppression of apoptosis initiation in addition to affecting steatosis.267 The transcription factor nuclear factor-E2-related factor-2 (Nrf2) is phosphorylated by ER eIF2 and inhibits lipid accumulation and oxidative stress in the liver by interfering with lipogenic pathways and inducing the expression of antioxidative stress genes.268 Deletion of Nrf2 increased oxidative stress, leading to rapid progression of steatosis to NASH in mice fed with an MCD diet.269,270
In addition, the ER is a major intracellular calcium storage site, and prolonged exposure to FFAs causes calcium leakage from the ER.271,272 Alterations in fatty acids and lipid composition decrease sarco/endoplasmic reticulum calcium ATPase (SERCA) activity, which pumps calcium from the cytoplasm into the ER.273 Calcium leaking from the ER may accumulate in the mitochondria, transmitting and amplifying apoptotic signals.271,272
Antioxidative stress agents
Vitamin E was originally found as a dietary factor preventing fetal resorption and had important effects on reproduction in rats.274 Vitamin E has eight natural forms, containing four tocopherols (α-, β-, γ-, and δ-) and tocotrienol (α-, β-, γ-, and δ-). The most abundant of them is -tocopherol, which has strong antioxidant properties.275 In addition, the non-antioxidant effects of α-tocopherol, including specific inhibitory effects such as phosphorylation of protein kinase C, on the growth of certain cells and on the transcription of certain genes (CD36 and collagenase) have been reported.276 Plasma level of vitamin E (α-tocopherol) was decreased in patients with NASH.277 It was also reported that in patients with NAFLD, vitamin E inhibited TGF β expression in the liver, which reduced steatosis, inflammation, and fibrosis.278 In an MCD-induced mouse NASH model, vitamin E supplementation reduced hepatic inflammation and fibrosis by reducing the expression of the proapoptotic BCL2-related X (BAX), TGF-dead, cyclooxygenase-2 (COX-2), and matrix metalloproteinase-2 (MMP-2).279 On the other hand, in HFD-fed mice280 or humans,281 fatty livers produce unrecognized hepatic vitamin E sequestration, which might subsequently drive liver disease, and the sequestered vitamin E might be used to quench oxidants generated within excess fat. These findings indicate that in addition to its antioxidant activity, vitamin E functions in different aspects and mechanisms in NAFL/NASH. Future research should focus more on the detailed molecular mechanism of action of vitamin E to benefit patients with NAFL/NASH.
Currently, vitamin E is recommended to treat NASH patients, associated with reduced serum hepatobiliary enzymes and hepatic steatosis but without improvement of liver fibrosis.62,282,283 In a large-sample, randomized, double-blind, controlled, phase 3 clinical trial (NCT00063622), the efficacy of vitamin E with another antidiabetic agent (Pioglitazone, targeting PPARγ) was confirmed in non-diabetic patients with NASH with histological evaluation as the study endpoint. The results showed that the NASH score improvement in the vitamin E group was significantly higher than that in the placebo group (43% vs. 19%), but not significant between the Pioglitazone group (34% vs. 19%) and the placebo group, while combined Pioglitazone and vitamin E improved histological hepatic steatosis and hepatic lobular inflammation without improvement in fibrosis scores.284
It has been hypothesized that the responsiveness of NASH patients to vitamin E therapy is affected by Haptoglobin (Hp) genotype. Three randomized controlled trials have shown that diabetic individuals with Hp 2-2, of which patients bearing Hp 2-2 mutation are at increased risk of CVD, had higher efficacy from vitamin E intervention. The percentage of NAFL/NASH Chinese patients with Hp 2-2 allele is much higher than that of western patients (65.71% vs. 36%, respectively), suggesting that Chinese patients may better benefit from vitamin E treatment.285 In the long term, vitamin E use is associated with some potential risks such as prostate cancer, stroke, and mortality.283 However, due to the differences in the use form of vitamin E and the analysis methods, the conclusions of the current study are uncertain regarding the assessment of risk. Further investigation is required to fully address the efficacy of vitamin E, especially in long-term studies after appropriate analysis.283,284,285
Currently, there are many antioxidants in addition to vitamin E that are being studied for the treatment of NAFLD. For example, the antioxidant carotenoid beta-cryptoxanthin prevented or reversed the progression of steatosis and fibrosis in NASH mice fed a high-cholesterol and high-fat (CL) diet.286 Melatonin has also exhibited promising results in patients by controlling the progression of NAFL.287 Administration of coenzyme Q10 elevated adiponectin levels and decreased MDA levels, suggesting improved lipid peroxidation in patients with metabolic syndrome.288 Some other natural dietary antioxidants such as curcumin,289,290 green tea, and epigallocatechin gallate291,292 also have positive therapeutic effects on NAFL/NASH. Therefore, we believe that even if there are various mechanisms leading to the development of NAFL/NASH, including oxidative stress, antioxidants have the potential to treat NAFL/NASH. Perhaps antioxidants in combination with other drugs may have an unexpected therapeutic outcome.
Hepatic cell death and pro-survival
Multiple types of cell death, including apoptosis, necroptosis, pyroptosis, and ferroptosis, as well as autophagy, are associated with the development of NAFL/NASH.72,293,294 Among targeting different formats of cell death, inhibition of apoptosis has achieved promising results, here we focus on anti-apoptosis agents (Tables 3 and 4), but other types of cell death will also be discussed below.
Apoptosis
Hepatocyte apoptosis induced by Caspases,295,296 and Bcl-2 family proteins297,298 and c-Jun N-terminal kinase (JNK)299,300,301 plays a driving role in the progression of NAFL/NASH. The cytokeratin-18 sheet (CK18) segment generated by Caspase-3 can be used as a marker to predict the severity of NASH.302,303 Palmitatic acid stimulation activated tumor necrosis factor-related apoptosis-inducing ligand receptor 2 (TRAIL-R2), leading to caspase-dependent cell death in hepatocytes.304 ER stress upregulates proapoptotic proteins, including p53 upregulated modulator of apoptosis (PUMA), Bim, and TRAIL-R2 via CHOP or JNK.305,306,307 PUMA and Bim promote hepatocyte death via the mitochondrial apoptotic pathway. Activated macrophages increase the expression of death receptor ligands, such as Fas ligand (FasL), TNF-related apoptosis-inducing ligand (TRAIL), and proinflammatory cytokines, which further promote hepatocyte apoptosis and inflammatory response.308,309,310,311 Regulation of miR-34a/SIRT1/p53 signaling by UDCA is involved in hepatocyte apoptosis.312 In addition, apoptosis signal-regulated kinase 1 (ASK1), an upstream activator of the JNK and p38 MAPK signaling cascades, can be activated by stress signals, such as ROS, ER stress, and TNFα, and plays a key role in the progression of NASH313 (Fig. 4).

Drugs targeting the apoptosis signaling in NASH. Inflammatory cytokines stimulate hepatocyte apoptosis through different pathways such as TRAIL signaling, Fas signaling and TNFα signaling pathway. TRAIL is a member of the TNF superfamily that can lead to the induction of apoptosis in tumors or infected cells. The Fas receptor induces an apoptotic signal by binding to FasL expressed on the surface of other cells. TNFα is a classical cytokine and its signaling pathway had been well investigated. Antagonists and inhibitors at different trial stages are as indicated. Drugs at different clinical stages are indicated in different colors. Created with BioRender. TRAIL tumor necrosis factor-related apoptosis-inducing ligand, Fas fatty acid synthetase, FADD Fas-associated with death domain protein, FasL Fas ligand, TNFα tumor necrosis factor-alpha, TNFR TNF receptor, TRAF2 TNF receptor-associated factor-2, ASK1 apoptosis signal-regulated kinase 1, JNK c-Jun N-terminal kinase, GSTM2 glutathione s-transferase mu 2, CFLAR caspase 8 and FADD-like apoptosis regulator, TNFAIP3 tumor necrosis factor-alpha-induced protein 3
Anti-apoptotic agents: Emricasan (IDN-6556) is a pan-caspase inhibitor that reduces inflammation and fibrotic apoptosis. In a murine model of NASH, mice that received Emricasan treatment were protected from liver injury and fibrosis, suggesting that inhibition of hepatocytes apoptosis by Emricasan may be an attractive antifibrotic strategy in NASH.314 Furthermore, administration of Emricasan in a phase 2 clinical trials in patients with NASH has shown beneficial results, including the significant reduction of ALT values.315 In addition, biomarkers, such as cleaved cytokeratin-18 (cCK18), full-length cytokeratin-18 (flCK18), and caspase-3/7 were significantly decreased in Emricasan-treated subjects (NCT02077374).315 Consistently, in another multicenter study of 86 patients with cirrhosis (NCT02230670), serum levels of flCK18 (p = 0.02) and caspase-3/7 (p < 0.001) were also decreased with 3 months of Emricasan administration compared to the placebo-treated group. Furthermore, at the 3-month timepoint, Emricasan significantly reduced mean MELD (model for ESLD) (p = 0.003) and Child-Pugh (p = 0.003) scores in subjects whose MELD scores were higher than 15, and significantly reduced international normalized ratio and total bilirubin compared with placebo.316 Despite the positive results that caspase inhibition by Emricasan lowered serum ALT in the short term, it may have directed cells to alternative mechanisms of cell death, resulting in more severe liver fibrosis and hepatocyte ballooning (NCT02686762).317
Selonsertib (GS-4997), an ASK1 inhibitor, prevents hepatocyte apoptosis and can reverse fibrosis and reduce liver inflammation in different preclinical models.318,319 In a 3D in vitro microtissue model, administration of Selonsertib decreased the measurements of specific disease parameters, such as the secretion of the profibrotic factor (procollagen type I), proinflammatory cytokines (TNF-α and IL-6), and chemokines (MCP-1, MIP-1α, IL-8, IP-10), in accordance with clinical observations.320 In a short-term phase 2 trial, the administration of Selonsertib improved NASH and fibrosis (at least a one-stage improvement) in some patients (NCT02466516).247,248
Two randomized, double-blind, placebo-controlled, phase 3 trials of Selonsertib in patients with NASH and bridging fibrosis (F3, STELLAR-3) or compensated cirrhosis (F4, STELLAR-4) were conducted (NCT03053050 and NCT03053063);321 however, neither trial met the primary efficacy endpoint. In STELLAR-3 (NCT03053050), fibrosis improvement without worsening of NASH was observed in 10% (18 mg, p = 0.49 vs. placebo), 12% (6 mg, p = 0.93 vs. placebo), and 13% (placebo) of patients. In STELLAR-4 (NCT03053063), the primary endpoint was achieved in 14% (18 mg, p = 0.56), 13% (6 mg, p = 0.93), and 13% (placebo) of patients.321 Although Selonsertib led to dose-dependent reductions in hepatic levels of phosphorylated p38, an indicator of its pharmacodynamic activity, it does not have a significant effect on liver biochemistry, non-invasive tests of fibrosis, progression to cirrhosis, or adjudicated clinical events, probably due to the advanced fibrosis onset in these patients.321
A small peptide segment in caspase 8 and FADD-like apoptosis regulator (CFLAR) that effectively attenuates the progression of steatohepatitis and metabolic disorders in both mice and monkeys. The dimerization and subsequent autophosphorylation of ASK1 are essential for its activation.322,323 CFLAR directly targets ASK1 and interrupts its N-terminus-mediated dimerization, thereby blocking signaling involving ASK1 and JNK1.324 Tumor necrosis factor-alpha-induced protein 3 (TNFAIP3) is a pivotal endogenous suppressor of ASK1 hyperactivation in the pathogenesis of NASH. Hepatocyte-specific ablation of TNFAIP3 exacerbated NAFLD- and NASH-related phenotypes in mice, including glucose metabolism disorders, lipid accumulation, and enhanced inflammation, in an ASK1-dependent manner.325 In addition, hepatic knockdown of autophagy-related gene 3 (ATG3) by i.v. injection of lentivirus encoding shRNA against ATG3 ameliorates the development of NAFL/NASH by stimulating mitochondrial function.326 Accordingly, ATG3 was identified as a new target of NAFL/NASH downstream of p63, and activation of p63 induced hepatic steatosis in diet-induced obese mice.326,327 In addition, it was reported that the upregulation of the p53 protein in hepatocytes might be a therapeutic strategy in the treatment of NAFL/NASH, for example, pharmacological stimulation of p53 with low-dose Doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis.328,329 Similarly, IGF-I induces senescence of HSCs, inactivates these cells, and limits fibrosis in a p53-dependent manner.
Rapamycin (a thioredoxin interacting protein (TXNIP) agonist) treatment also attenuated MCD-induced steatosis, inflammation, and fibrosis with increased TFEB nuclear translocation and restored FAO in TXNIP-KO mice by inhibiting MTORC1 to promote autophagy.330 Despite the positive preclinical results, more clinical studies should be conducted to verify the efficacy of apoptosis inhibitors in the treatment of patients with NAFL/NASH.
Necroptosis
Necrotizing apoptosis or necroptosis has been identified as a key pathogenic mechanism in NAFL/NASH. For example, upon TNFα stimulation, Receptor-interacting serine/threonine protein kinase-3 (RIP3) RIP3 phosphorylates mixed lineage kinase domain-like (MLKL) and induces cell necroptosis. In general, RIP3, RIP1, and MLKL together form necrosome, and hepatic RIP3 and MLKL phosphorylation and TNFα expression are increased in NAFL/NASH.331 Inhibition of MLKL, RIP3, or RIP1 all improved the NASH characteristics of HFD-fed mouse models.332,333 In addition, MLKL-dependent (but RIP3-independent) noncanonical signaling contributes to western diet-induced liver injury by inhibiting autophagy and inducing necrosis.332,334,335 TN3-19.12, a neutralizing monoclonal antibody against TNFα, could inhibit necroptosis that was activated in NAFLD, with the upregulation of RIP1, RIP3, and MLKL in both CD- and HFD-fed mice. In addition, necroptosis inhibitors necrostatin-1 (Nec-1) and GSK-872 could also inhibit necroptosis in both CD- and HFD-fed mice significantly.336
Pyroptosis
Pyroptosis is characterized by the formation of inflammasome, activation of Caspases and Gasdermin, and the release of proinflammatory cytokines, including IL-1β and IL-18. The canonical pyroptosis pathway recruits and activates caspase-1 upon recognition of PAMPs and DAMPs.337 In noncanonical pyroptosis, caspase-1/11 in mice and caspase-4/5 in human is stimulated directly by LPS in a TLR4-independent manner.338,339 These activated Caspases cleave and activate Gasdermin D (GSDMD) to promote the secretion of proinflammatory cytokines (IL-1β, TNF-α, and MCP-1/CCL2) and activation of NF-ĸB signaling pathway.340 In addition, expression of SREBP1 is reduced in MCD-fed GSDMD-deficient mice, upregulating lipolytic genes and resulting in reduced adipogenesis, subsequently, attenuated MCD-fed induced NASH.341 Increased activity of caspase-1, GSDMD, and inflammasome components was observed in mice with MCD-induced NASH.342 However, there was no activation of inflammasome in mice with simple steatosis, suggesting its role in more severe disease or the progression to NASH. The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome blockade normalized hepatic caspase-1 and IL-1β expression and reduced liver fibrosis in MCD diet-fed mice.343 Cytochalasin B, an endocytic inhibitor, eliminated the increased secretion of inflammatory factor IL-1β and the expression of α-smooth muscle actin (α-SMA) in HSCs that internalized NLRP3 inflammasome particles.344 Ghrelin is a gut hormone with 28-amino acid peptide. Two ghrelin isoforms (acylated ghrelin and desacyl ghrelin) were protective against high-mobility group box 1 (HMGB1) induced pyroptosis in HepG2 cells.345
Ferroptosis
It is reported that patients with NAFL/NASH frequently suffer from hypoferritinemia and are more likely to develop advanced fibrosis, or even have a higher mortality rate compared with NAFL/NASH patients with a normal level of ferritin.346,347 Growing evidence suggest that iron-dependent ferroptosis plays an essential role in the development of multiple liver diseases, including NAFL/NASH.348,349,350 Ferroptosis refers to a type of iron-dependent programmed cell death that is characterized by the accumulation of lipid peroxide and ROS derived from iron metabolism, which is genetically and biochemically distinct from other forms of regulated cell death.351,352 It was reported that scavenging the associated lipid peroxidation in hepatocytes with ferroptosis inhibitors could reduce lipid accumulation and alleviates MCD-induced mouse NASH model and liver fibrosis.350 Glutathione peroxidase 4 (GPX4) is an antioxidant defense enzyme, whose function is to repair oxidative lipids damage and is a leading inhibitor of ferroptosis.353 Activation of GPX4 protected mice from HFD-induced obesity and improved hepatic steatosis in mice.354 Mechanistically, activation of the Keap1/Nrf2 pathway promotes downstream gene expression of heme oxygenase-1 (HO-1), glutathione (GSH), and its peroxidase 4 (GPX4), which eliminate ROS accumulation, inhibit gluconeogenesis and adipogenesis, thereby reducing NAFL/NASH.349,355 Furthermore, activation of GPX4 by Enoyl coenzyme A hydratase 1 (ECH1) suppressed hepatic ferroptosis and significantly alleviated hepatic steatosis, inflammation, fibrogenesis, apoptosis, and oxidative stress in livers of mice fed with MCD diet.356 There are other pathways that alter the susceptibility to ferroptosis, including amino acid and iron metabolism, ferritin autophagy, cell adhesion, p53, and phospholipid biosynthesis.357,358,359,360 In addition, the RNA-binding protein ELAVL1/HuR, ZFP36/TTP, and the bromodomain-containing protein 7 (BRD7) have also been shown to impact liver fibrosis by regulating ferroptosis in HSCs.361,362,363
Hepatic inflammation in NASH/NAFL/NASH and anti-inflammatory therapy
Cell death can be both a consequence and a cause of inflammation.364 While apoptosis induces minimum inflammatory response during the progression of NAFL/NASH, lytic forms of cell death, such as necroptosis and pyroptosis, can trigger the inflammation via DAMPs.293 On the other hand, the extensive release of inflammatory mediators, such as TNFα and TGFβ, will cause more cell death, which collectively promotes the pathogenesis of NAFL/NASH.
Regulation of inflammatory response in NAFL/NASH
Inflammatory response is an essential contributor to the development and progression of NAFL/NASH. Immune cells and proinflammatory cytokines are implicated to play significant roles in NASH pathogenesis (Figs. 2 and 5).

Drugs regulating the inflammatory response in NAFL/NASH. In NASH, extracellular PAMPs or metabolic stress activates proinflammatory signaling pathways through multiple receptors. Drugs regulate inflammation in NAFL/NASH by targeting different inflammatory pathways, such as TNF-α, TLR-IL-1R, IL-17 signaling, and caspase signaling. Drugs at different clinical stages are indicated in different colors. Created with BioRender. PAMP pathogen-associated molecular patterns, TLR Toll-like receptor, IL-R interleukin-receptor, Myd88 myeloid differentiation factor 88, IRAK interleukin-1 receptor-associated kinase 1, TRAF TNF receptor-associated factor, ASK apoptosis signal-regulating kinase, TAK transforming growth factor beta-activated kinase, JNK c-Jun N-terminal kinase, MAPK mitogen-activated protein kinase, ERK extracellular signal-regulated protein kinases, Casp caspase, AP-1 activating protein-1, NFκB nuclear factor kappa B, PTX pentoxifylline, DUSP7 dual specific phosphatase, RGS5 hepatic regulator of G protein signaling 5, TIPE2 tumor necrosis factor-alpha–induced protein 8-like 2. Drugs at different clinical stages are indicated in different colors
KCs, liver-resident macrophages, are the first line of sensors in the liver and are critical in the development of NAFL/NASH.365,366,367 Both the release of gut flora-derived bacterial products (e.g., LPS, bacterial DNA)25 due to increased intestinal permeability and exposure to endogenous substances (e.g., HMGB1, FFAs)368,369 from damaged cells activate hepatic TLRs (e.g., TLR4, TLR2, and TLR9) and NLRs on KCs. The downstream signaling events increase nuclear translocation of NF-κB, further promoting the production of various proinflammatory and profibrogenic cytokines, such as TNFα, IL-1β, CCL5, and TGFβ, which subsequently induce hepatocyte lipid accumulation and apoptosis and HSC activation.365,370 Fatty acids can promote the release of mtDNA or cholesterol crystals leading to activation of NLRP3 inflammasome in KCs that secrete the proinflammatory cytokine IL-1β.343,371 Activated KCs also promote the recruitment of monocyte-derived macrophages (MoMFs) to the liver in a CCR2-dependent manner by secreting CCL2.372,373,374 The recruited inflammatory MoMFs further amplify the inflammatory response. The number of macrophages is positively correlated with NAFL/NASH disease severity, and more macrophages are found even at the early stages of human NASH. Moreover, during NASH, hepatocytes upregulate the main neutrophil recruiting chemokines (CXCL1 and IL-8),375 and neutrophil infiltration around the lipotoxic hepatocytes is considered a hallmark of NASH.376,377 Neutrophil abundance correlates with the degree of steatosis and fibrosis. Inhibition of Neutrophils derived granule proteins and neutrophil extracellular traps could reduce macrophage infiltration and inflammatory cytokine secretion, decreasing the progression of NASH to HCC in NASH mice induced by neonatal streptozotocin and HFD.378 Myeloperoxidase (MPO), an important neutrophil enzyme, contributes to HSC activation and is proapoptotic and profibrotic in NASH.379 Increased expression and activity of neutrophil elastase (NE) in NASH lead to hepatocyte IR and are partially associated with the regulation of hepatic ceramide metabolism.380,381,382 Neutrophil-derived lipocalin 2 (LCN2) exacerbates steatohepatitis by inducing CXCR2 expression to promote crosstalk between neutrophils and hepatic macrophages.383 In addition, adaptive immunity is also involved in the development of NAFL/NASH.384 Interestingly, Th17 development is dependent on ACC1-mediated de novo fatty acid synthesis and potentially glycolytic lipogenic metabolic pathways, demonstrating crosstalk between pathways like lipogenesis and inflammation.385 Increased Th17 cells and expression of IL-17A are markers of progression from NAFL to NASH.386,387 CD4+ T cells may also regulate the inflammatory response of macrophages through IFNγ, affecting liver fibrosis.388 In NASH, pathogenic CD8+ T cells accumulate in the liver.389 These accumulation and activation of T cells and myeloid cells are largely associated with a type I interferon (IFN-I) response in the liver, leading to increased production of the proinflammatory cytokines IFNγ and TNFα.390 At the early stage of NAFL, B-cell responses precede T cells and are accompanied by upregulation of hepatic expression of B-cell-activating factor (BAFF).391 In addition, activated B cells produce pro- or anti-inflammatory cytokines, immunoglobulins, activate T cells, and may also exacerbate tissue damage and liver fibrosis.392,393,394 In short, there are complex immune mechanisms during NAFL/NASH; however, the current understanding of the inflammatory response driving NASH is fragmented. Therefore, more intensive research and conclusive evidence are needed to understand the role of immune cells during disease progression.
Anti-inflammatory therapy
Treatment with C-C chemokine receptors (CCR) antagonists to avoid the abnormal infiltration of inflammatory leukocytes can also be used as a therapy for NASH.395 In a mouse model of diet-induced NASH and a rat model of thioacetamide (TAA)-induced liver fibrosis, CCR2/5 antagonists (Cenicriviroc, CVC) effectively reduced fibrosis as measured by reducing protein expression of collagen I and α-SMA, and collagen deposition in the region around the liver lobules by targeting hepatic-pathogenic monocytes/macrophages and HSCs.396 CVC further underwent a phase 2 clinical evaluation (NCT02217475) and the results indicated that CVC improved fibrosis and had greater efficacy in advanced fibrosis.397,398 CVC at year 2 achieved ≥1-stage fibrosis improvement (24% of patients in the CVC group and 17% in the placebo group) and no worsening of NASH (p = 0.37).396,397 In addition, 60% of patients on CVC who achieved fibrosis response at year 1 maintained benefit at year 2, including 86% on CVC who had stage 3 fibrosis at baseline.397 Moreover, CVC treatment has antifibrotic and anti-inflammatory functions by reducing multiple systemic inflammation biomarkers, including C-reactive protein, IL-6, IL-1β, and fibrinogen.397
Expanding evidence suggested that targeting proinflammatory cytokines exhibited protective effects on NAFL/NASH in preclinical trials, including IL-27, IL-17, IL-11, IL-1, and TNFα, ensemble a significant portion of anti-inflammatory strategy to treat NAFL/NASH. The IL-27 receptor (IL-27Ra, or WSX-1) expresses on adipocytes and plays an important role in promoting metabolic diseases. In addition, IL-27 expressed on T cells also provides new targets for the treatment of obesity-related metabolic syndrome, such as NAFL/NASH.399 Administration of recombinant mouse IL-27 ameliorated metabolic morbidities in obese mice by activating p38 MAPK-PGC-1α signaling axis and stimulating the production of uncoupling protein-1 (UCP1).400 IL-17 stimulates KCs to express the major fibrogenic cytokine TGF-β1, directly induced production of collagen type I in HSCs by activating the signal transducer and activator of transcription 3 (Stat3) signaling pathway, and may serve as an attractive target for antifibrotic therapy.401 Upon anti-IL-17 antibody treatment, the development of hepatic fibrosis was alleviated in NASH mice.401 Anti-IL-11 (X203) and anti-IL-11RA (X209) antibodies reduced fibrosis, steatosis, hepatocyte apoptosis, and inflammation in mice with diet-induced liver steatosis and fibrosis by IL-11-induced ERK-mediated signaling pathway.402 Moreover, it was reported that IL-1 was a key proinflammatory factor, including IL-1α and IL-1β, which promoted IR by impairing adipocyte function and promoting inflammation.403 Hepatic macrophage KCs upregulate IL-1α expression and recruit neutrophils and monocytes to sites of inflammation, exacerbating inflammation and injury. Thus, IL-1 became a key target for the treatment of obesity-related inflammatory diseases, such as NASH. As a major antagonist, IL-1Ra inhibits the IL-1R signaling cascade by preventing the binding of IL-1α/IL-1β, and subsequently attenuates the inflammatory response.403,404 Various natural and synthetic drugs have been found to exert anti-NASH effects by targeting the IL-1 signaling pathway. Natural flavones with anti-inflammatory effects showed the protective properties of NASH in obese mouse models by targeting IL-1 and IL-18.405,406 A recent clinical trial has demonstrated that diacerein (1,8-diacetoxy-9,10-dioxo-dihydroanthracene-3-carboxylic acid), an immune-modulator anti-inflammatory drug, mainly acting on IL-1 pathways, remarkably decreased liver fibrosis in diabetic patients with NAFLD.407 Pentoxyfylline (PTX), an anti-TNFα drug, failed to reduce transaminases and did not positively affect any of the metabolic markers postulated to contribute to NASH, but improved histological features of NASH patients.408,409,410 Although Infliximab, a TNFα inhibitor, did not prevent the development of NASH, it was able to slightly reverse the NASH histopathology score and was effective on necrosis, inflammation, and fibrosis in the NASH rat induced by the MCD diet.411,412 In addition, an immunosuppressant drug (Thalidomide) that targets the expression of TNFα may also contribute to reductions in the inflammatory markers that were associated with obesity in Swiss mice fed with HFD.413
In addition, other proinflammatory pathways served as potent mediators of inflammation and could be potential targets for therapy. ERK antagonists such as FR180204 and Ravoxertinib could significantly reduce ALT, AST, CHO, and TG values in an MCD mice model, and observably improve fatty degeneration, inflammatory state, and liver injury.414 Sparstolonin B (SsnB), a polyphenol, attenuates liver fibrosis in rat primary HSCs and human transformed HSCs (LX-2) by antagonizing TLR4-mediate TGFβ signaling pathway.415 Sulforaphane (SFN) could suppress NLRP3 inflammasome by regulating AMPK-autophagy axis.416,417 SFN reduced ALT and AST levels in serum, hepatic steatosis scores, and other hepatic indicators such as cholesterol, TG, and FFAs levels in mice fed with an HFD.416 Purinergic receptor P2X7 (P2RX7) is a major driver of NLRP3 inflammasome activation and IL-1β processing. SGM-1019 (EVT-401), a P2RX7 inhibitor, improved histological characteristics of NASH and protected from liver inflammation and fibrosis in a CCl4-induced nonhuman primate model of liver fibrosis.418
Fibrosis
Stressed or damaged hepatocytes and activated macrophages (KCs) lead to the activation of HSCs to myofibroblasts that drive hepatic fibrogenesis. The main feature of liver fibrosis is the deposition of type I collagen in the ECM, which disrupts the normal physiology of the liver and leads to liver dysfunction. HSCs are the main source of ECM-producing myofibroblasts in models of fatty liver disease (Figs. 2 and 6).419 TGF-β is one of the most potent profibrotic cytokines in the liver fibrosis process, which mediates the transformation of HSCs to hepatic fibroblasts.420 In the TGF-β signaling pathway, Smads, activated type I receptor phosphoplasmic proteins, are the direct acting substrates of TGF-β.421 TGF-β binds to the type II receptor TGFβR2 (TβR2) and phosphorylates the N-terminal glycine-serine rich GS domain of type I receptor TGFβR1 (TβR1), whose phosphorylation enables activation of TβR1 and the binding of Smad. Activated TβR1 then phosphorylates and acetylates Smad2/3, which is in complex with Smad4, translocates to the nucleus, and promotes profibrotic gene expression.422 In addition to Smad-dependent pathways, TGF-β can also activate liver fibrosis via noncanonical (non-Smad) pathways such as MAPK, mTOR, PI3K/Akt, IKK, Wnt/β-catenin, and Rho GTPase pathways.423,424,425 Inhibition of TGFβ reduces NASH-induced fibrosis and is more effective when combined with IL-13 inhibition.426 The oxidative hepatic environment in obesity inactivates the STAT1/3 phosphatase TCPTP (T-cell protein tyrosine phosphatase) and increases STAT1 and STAT3 signaling. Of interest, STAT1 and STAT3 have segregated roles in driving NASH, fibrosis, and HCC. Activation of STAT1 promotes T-cell recruitment, NASH, and fibrosis, but not HCC, whereas inhibition of STAT3 signaling prevented HCC without affecting NASH and fibrosis,427 suggesting the possibility of separative treatment of NASH and HCC. IL-17 also directly activates HSCs428 and promotes collagen production via the STAT3 pathway.401 In addition, platelet-derived growth factor (PDGF) is also a fibrosis-promoting cytokine. HSCs express high levels of PDGF receptors, and their activation effectively stimulates HSC proliferation and migration.420,429

Drugs targeting the fibrosis process in NASH. Chronic hepatocyte injury induces the activation of hepatic stellate cells (HSCs) and the recruits of immune cells, which result in the deposition and cross-linking of collagens in the extracellular matrix and eventually progress to fibrosis. TGFβ/SMAD signaling, a key pathway in the development of liver fibrosis and inflammation, activates Smad pathway and no-Smad pathway. Activation of TGF-β signaling with OSM exposure drives a cooperative STAT3/SMAD3 gene transcriptional program. OSMR/JAK-mediated STAT3 signaling promotes liver fibrosis and HSCs activation by phosphorylation of SMAD3, resulting in transcriptional activation of select STAT3/SMAD3 targets. Antagonists are indicated with a red inhibitor. Drugs at different clinical stages are as indicated. Drugs at different clinical stages are indicated in different colors. Created with BioRender. Ncst ASO nicastrin antisense oligonucleotide, CCR2/5 C-C chemokine receptor 2/5, MoMF monocyte-derived macrophages, TGF-β transforming growth factor-β, LOXL2 lysyl oxidase-like 2, HSC hepatic stellate cell, OSM oncostatin M, IL-11 interleukin-11, IL-11R IL-11 receptor, GP130 glycoprotein 130, JNK c-Jun N-terminal kinase, STAT3 signal transducer and activator of transcription 3, hsp47 Heat shock protein-47
Antifibrosis agents
The control or reversal of liver fibrosis is important for NASH treatment. Drugs designed to directly antifibrosis (e.g., galectin-3 inhibitor GR-MD-02) or to increase ECM turnover rates (e.g., Simtuzumab) are currently in clinical trials. Lysine oxidase-like 2 (LOXL2) is a matrix formation enzyme that is highly expressed in fibrotic regions of the liver that promotes liver collagen and elastin cross-linking.430 However, Simtuzumab (a LOXL2 inhibitor) did not significantly decrease fibrosis stage or the progression to cirrhosis in patients. Two phase 2b clinical trials of Simtuzumab were all terminated after 96 weeks due to the lack of efficacy (NCT01672866 and NCT01672879).431,432,433 Galectin-3 (Gal-3) is a β-galactoside binding protein expressed in immune cells that recognizes and binds to galactose residues and is associated with chronic inflammation and fibrogenesis. GR-MD-02 (Belapectin), an inhibitor of Gal-3, reduced hepatic fibrosis and portal hypertension in rats. In a phase 2 clinical trial in patients of NASH with cirrhosis and portal hypertension (NCT02462967), Belapectin was not associated with significant fibrosis reduction.434 The current data indicated that Belapectin might be beneficial in the early stages of cirrhosis, the ongoing phase II b/III study (NCT04365868, NCT02421094) in NASH with cirrhosis or advanced fibrosis patients will further investigate the potential benefits of Belapectin.435 Heat shock protein-47 (Hsp47) is a collagen-specific molecular chaperone, which plays an essential role in collagen synthesis and deposition to promote fibrosis.436 BMS-986263, a lipid nanoparticle delivering small interfering RNA (siRNA) of HSP47 could promote HSC apoptosis, thereby treating advanced fibrosis.437 Several clinical trials of BMS-986263 for advanced fibrosis with different causes have been carried out (NCT02227459, NCT03420768). In addition, Losartan, an angiotensin-II type I receptor antagonist, improved hepatic fibrosis by inhibiting the activation of HSCs in mice models and is now evaluated in a phase 3 trial (NCT01051219) for NASH patients with steatohepatitis and fibrosis (Kleiner fibrosis classification F1-F3).438
As a significant player in NASH fibrosis, TGF-β has been one of the actively studied drug targets for antifibrosis. Multiple anti-TGF-β signaling compounds showed promising efficacy in the preclinical studies. Galunisertib (LY2157299), a TGF-β receptor type I kinase inhibitor, showed similar antifibrotic potency in human and rat livers by inhibiting the phosphorylation of SMAD2.439,440 SP-1154, a synthetic TGF-β inhibitor, significantly improved insulin sensitivity with glucose homeostasis and reduced hepatic steatosis by inhibiting TGF-β/Smad3 signaling pathway.441 Oxy210, a novel inhibitor of hedgehog and TGF-β signaling, effectively ameliorated hypercholesterolemia, hepatic fibrosis, and inflammation in mice fed with a western diet.442 Tetrodecadazinone (a novel tetrodecamycin-pyridazinone hybrid) reduced the ECM proteins (fibronectin and collagen I) and α-smooth muscle actin (α-SMA) levels by regulating TGF-β1/Smad2/3 signaling pathway.443 Yellow loosestrife (Lysimachia vulgaris var. davurica) extracted strongly prevented liver fibrosis by blocking TGFβ/Smad signaling in mice.444 The noncanonical TGF-β-activated kinase 1 (TAK1) was shown to be a major upstream signaling molecule in TGF-β1-induced type I collagen and fibronectin expression.445 TNFα-induced protein 8-like 2 (TIPE2) suppresses NAFL/NASH advancement by blocking TAK1-JNK/p38 pathway and is a promising target molecule for NAFL/NASH therapy.446 Dual specific phosphatase 7 (DUSP7) deletion considerably promoted the activation of TAK1 and might function as a protective factor against NAFL/NASH development through alleviating dyslipidemia, inflammation, and oxidative stress by directly interacting with TAK1 in hepatocytes, which was involved in the suppression of fibrosis.447 Breviscapine, a crude extract of several flavonoids of Erigeron breviscapus (Vant.) Hand.-Mazz., prevented metabolic stress-induced NASH progression and significantly reduced lipid accumulation, inflammatory cell infiltration, liver injury, and fibrosis in mice through direct inhibition of TAK1 signaling.448
Alternatively, reducing the number of activated HSCs by promoting their apoptosis or increasing their clearance is another approach to reduce hepatic fibrosis. The apoptosis inducers, such as fraxetin and 4-hydroxy-2(3H)-benzoxazolone have been shown to induce HSC apoptosis in many animal experiments.363,364 Activated HSCs could be killed by the immune surveillance ability of natural killer (NK) cells, suggesting therapeutic activation of NK cells could be an approach to scavenge activated HSCs.449,450 However, as hyperactivated NK cells could also enhance inflammation and lead to the progression of fibrosis, specifically targeting the key pathogenic HSCs within the liver is important for this strategy. IFNγ, a potent anti-fibrogenic cytokine produced by NK cells, was conjugated to a cyclic peptide recognizing the platelet-derived growth factor beta receptor (PDGFβR) that was found strongly upregulated on activated HSCs. The IFNγ conjugates attenuated local HSC activation in an acute liver injury model and inhibited fibrogenesis in the liver fibrosis model, with no observed IFNγ-related side effects.451 Cyclin-dependent kinase 4 (CDK4) is an important regulator to phosphorylate C/EBPα at Ser193 and increase the formation of C/EBPα-p300 complexes, which regulate the development of hepatic steatosis.452,453 Inhibition of CDK4 by Flavopiridol or PD-0332991 significantly attenuated HFD-induced hepatic steatosis and fibrosis in mice, indicating that targeting CDK4 might be an appropriate strategy for resolving liver fibrosis.453 In addition, Emodin was reported to suppress the activation of HSCs and ECM-related gene expression in HSCs (HSC-T6) via blocking SMAD4 and p38 MAPK signaling pathways.454,455
Novel signaling pathways and pharmacological targets
While a vast body of studies explained the genetic, biochemical, immunological, and molecular mechanisms responsible for the onset and progression of NAFL/NASH, emerging new insights have been described regarding the signaling pathways that participate in NAFL/NASH pathogenesis, which may shed light on individualized approaches for future management of NAFL/NASH.
Extracellular vesicles
EVs have shown an increasingly important role as a mode of intercellular communication in regulating tissue and intercellular metabolic signaling.456 Circulating EVs have increased in human NASH samples as well as mouse models of NASH.457,458 The critical role of EVs in the progression of NAFL/NASH has also been described in more detail in other reviews.459,460 Under both normal and pathological conditions, EVs act as communication mediators between the liver and other organs, carrying various bioactive molecules, including lipids, proteins, DNA, coding, and non-coding RNAs.461 The increased level of hepatocyte-derived EVs correlates with the severity of NASH;462 therefore, the number of circulating EVs and their protein and miRNA composition might potentially be a powerful tool for NAFL/NASH diagnosis and treatment.462,463 During lipid overload, miRNA-containing EVs derived from hepatocytes, including let-7e-5p and miR-210-3p, target adipocytes to promote lipid accumulation.464 miRNAs in EVs released from lipotoxic hepatocytes (miR-128-3p is most effective) suppress PPARγ expression in HSC, leading to significantly upregulated expression of fibrotic genes, including Col1a1 and Atca1.465 Interestingly, neutrophil-derived miR-223-rich EVs target hepatocytes to limit the progression of NASH.466 Bone marrow-specific IL-6 signaling also inhibits liver fibrosis through exosomal miR-223 transfer to hepatocytes.467 Notably, human liver stem cell (HLSC)-derived EVs have therapeutic actions to NASH,468 and serum EVs from normal healthy individuals can decrease the fibrosis-associated molecule expression in activated HSC.469 In the liver of patients with NASH, hepatocytes release ceramide-rich inflammatory EVs to recruit MoMFs after activation of IRE1α in ER leading to inflammation and injury.470 miR-192-5p-rich exosomes released from hepatocytes induce activation of proinflammatory macrophages by regulating Rictor/Akt/FoxO1 signaling.471 In addition, hepatocyte-derived EVs activate NLRP3, inhibit KLF4 and activate NF-κB to increase vascular permeability and promote vascular endothelial inflammation.472,473 This suggests a novel mechanism of the association of NAFL/NASH with CVD. Overall, EVs in NAFL/NASH act as a signaling mediator, leading to lipid accumulation, macrophage, and HSC activation, promoting inflammation and liver fibrosis progression, EVs may serve as new targets for the treatment of NAFL/NASH.
Gut–liver axis
The gut–liver axis is bidirectional communication between the gut and the liver.474 Liver influences the composition and function of the gut microbiota and regulates the intestinal barrier through bile duct excretion of BAs and inflammatory mediators. On the other hand, however, the gut microbiota and its metabolites act on the liver through the portal vein to regulate BAs synthesis and hepatic glucose and lipid metabolism.475 Disturbances in the gut–liver axis have a key role in the pathogenesis of NAFL/NASH, including alteration in the gut microbiota,476,477 disruption of the gut barrier,478 and the liver inflammatory response479 (Fig. 7).

Gut–liver axis in NAFL/NASH. Unhealthy diet, such as high fat and high sugar, induces changes in the intestinal microbiome. This in turn affects alterations in metabolites, such as a decrease in beneficial SFAs and an increase in LPS, ethanol, TMA, etc. The impaired gut barrier allows increased and easier translocation of these dangerous substances to the liver, accelerating the progression of NAFL/NASH. BAs bile acids, SCFAs short-chain fatty acids, LPS lipopolysaccharide, TMA trimethylamine, IAA indole-3-acetic acid
Changes in the levels and composition of BAs modulate the composition of the gut microbiota.480 External administration of BAs simplified the composition of the microbiota and resulted in a significant decrease in serum levels of lipocalin in rats.187 Genetic and pharmacological interference with BA synthesis and excretion can alter the bacterial composition of the intestine. For example, oral administration of glycine-β-muricholic acid (Gly-MCA) alters the gut microbial community structure, decreases the ratio of Firmicutes to Bacteroidetes, reduces the substrate for hepatic DNL, and improves obesity-related metabolic dysfunction.481 In addition, inhibition of intestinal bacterial bile salt hydrolases activity also improved glucose and adipogenesis in the organism.482
Gut microbiota regulates the synthesis of BAs through FXR.483 It was reported that BA diversity is lower in sterile and antibiotic-treated rats, whereas the proportion of BAs bound to taurine, a conjugated form that can increase the solubility and lipid-emulsification properties of BAs, is elevated in peripheral tissues.484 During the use of acarbose in the treatment of diabetes, the microbiome in the gut of patients changed, typically the ratio of bound BA to unbound BA in plasma increased.485 The use of taurine deoxycholic acid, a BA derivative, improved ER stress and increased insulin sensitivity in the liver and muscle.486 Germ-free mice fed with an HFD of cholesterol-rich lard have increased energy expenditure and fecal fat excretion, prioritized carbohydrate oxidation, and reduced fat accumulation.487
Although Firmicutes and Bacteroidetes remain the dominant phylum, the proportions of Bacteroidetes to Firmicutes were higher in patients with NAFL/NASH compared to healthy individuals.488 Development of hepatic steatosis in germ-free mice receiving transplants of gut microbiota from high-fat-fed mice demonstrates that the gut microbiome affects the progression of NAFLD.489 Patients with steatosis have a low microbial diversity, which is shown to associate with the decreased presence of “beneficial” Coprococcus and increased “harmful” Ruminococcus Gnavus.477 Gram-negative Proteobacteria are increased in advanced liver fibrosis, whereas Gram-positive Firmicutes significantly decreased, suggesting microbial profile might serve as a non-invasive marker for advanced fibrosis.488,490 It was reported that oral administration of Faecalibacterium prausnitzii indeed reduced liver fat content as well as ALT and AST levels, while FAO levels were elevated in mice fed with HFD.491 Oral administration of Bifidobacterium longum was able to significantly reduce blood glucose levels and TC levels and improve liver fat accumulation in mice fed with HFD.492 This positive effect may have been achieved by modulating the mRNA expression of components of the renin-angiotensin system.492 Lactobacillus, such as L. acidophilus, L. fermentum, and L. plantarum, reduced hepatic steatosis by lowering cholesterol in mice fed with a western diet.493 Lactobacillus lactis and Pediococcus pentosaceus also restored metabolite dysregulation in the gut of western diet‐induced NAFLD mice.494
Metabolites of the gut flora such as ethanol, short-chain fatty acids (SCFAs), and amino acids have also been demonstrated to be involved in regulating the disease process of NAFLD. In a Chinese cohort, high-ethanol-producing Klebsiella pneumoniae has been linked to up to 60% of patients with NAFLD.495 Excess alcohol produced by Klebsiella pneumoniae in the gut could enter the liver via the portal vein, causing hepatic lesions that was similar to the anatomy and histopathology of alcoholic fatty liver. Increased SCFAs were detected in the feces of patients with NAFL/NASH including acetate, propionate, and butyrate.496 Other studies, on the other hand, imply that SCFAs help to alleviate intestinal injury and reduce hepatic steatosis and inflammatory damage through various mechanisms. This apparent contradiction highlights the need of distinguishing between circulation and fecal SCFAs, as circulating SCFA levels are more closely linked to lipid metabolism and insulin sensitivity.497 Among them, butyrate has been most extensively studied and can improve NAFLD through various mechanisms. Sodium butyrate (SoB) prevented the progression of NAFL to NASH induced by HFD in mice by promoting hepatic GLP-1R expression.498 SoB can also alleviate hepatic steatosis in mice fed HFD through correcting the imbalance of gut microbiota and improving the intestinal barrier.499,500 Oral supplementation of SoB also showed efficacy in a diet rich in fat, fructose, and cholesterol or a western-style diet-induced murine NAFLD models.501,502 However, one study found that the gut microbiome of patients with NAFLD-associated liver cancer promoted butyrate production, and excessive butyrate could be detrimental to patients with liver cancer by blocking the normal immune system function.503 Amino acids that are fermented by intestinal microorganisms produce a number of metabolites, some of which exhibit protective effects on liver function. Glycine analogs (DT-109) with dual hypolipidemic/hypoglycemic properties attenuated experimental NAFLD by stimulating hepatic FAO and glutathione synthesis, inhibiting NF-κB target genes and TGFβ/SMAD signaling to diminish inflammatory infiltration and liver fibrosis, and may serve as potential therapies for NAFLD.504 The metabolism of tryptophan, an essential aromatic amino acid, produces indoles that enhance the barrier function of the intestine.505 Another tryptophan metabolite, indole-3-acetic acid, reduced the inflammatory response in the liver, downregulated adipogenesis-related genes, and alleviated steatosis in mice fed HFD. Other amino acids such as arginine and citrulline can also protect mice from the development of NAFL/NASH.506,507,508
Several amino acid metabolites, on the other hand, accelerate the development of NAFLD. Imidazole propionate is a microbially produced histidine-derived metabolite that is present in higher concentrations in patients with T2DM. Imidazole Propionate activates p38γ MAPK, which promotes p62 phosphorylation and subsequent activation of mTORC1.509 In patients with hepatic steatosis, the levels of N,N,N-trimethyl-5-aminopentanoic acid (TMAVA, a metabolite from intestinal bacterial metabolism of trimethyl lysine) in plasma were increased.510 TMAVA reduces carnitine synthesis and FAO and promotes HFD-induced steatosis in mice. Long-term treatment with phenylacetic acid, a metabolite of phenylalanine, successfully induced steatosis.511 In addition, ingested choline can only be converted to trimethylamine (TMA) by intestinal bacteria, and subsequently, TMA is converted to trimethylamine–N-oxide (TMAO) in the liver.512,513 The HFD enhanced choline catabolism of Escherichia coli and thus increased circulating TMAO levels.514 The level of circulating TMAO was positively correlated with the presence and severity of NAFLD,515 increased circulating TMAO inhibited hepatic FXR to exacerbate steatosis.516 These observations suggested that microbiota regulation targeting metabolic homeostasis and intestinal barrier is expected to provide new strategies for NASH treatment. The precise mechanisms of hepatic immunity mediated by flora metabolites and the elaborate regulatory network among metabolites need to be further investigated.
The intestinal barrier effectively prevents harmful intestinal bacteria and their metabolites from entering the circulation, which causes bacteremia or other tissue damage.517 Altered intestinal barrier triggers inflammatory response and remarkably affects the gut–liver axis. Increased intestinal permeability is found in patients with NAFL/NASH, accompanied by elevated ethanol-producing bacteria, which further promote the disruption of gut tight junction by stimulating the production of proinflammatory cytokines and correlate with the degree of hepatic steatosis.518,519 NAFL/NASH patients have elevated levels of endotoxin in their blood.520 High-fat feeding increases intestinal permeability521 and the proportion of LPS-containing microbiota in the gut.522 Alterations of gut microbiota and disruption of intestinal barrier increase the circulating levels of LPS.523,524 The levels of serum LPS are higher in patients with NAFL/NASH,525 and higher LPS hepatocyte localization is observed in patients with NASH.526 Subsequently, high levels of LPS recruited more TLR4-positive macrophages and activated TLR4 platelets.526 LPS activates TLR signaling in the liver, resulting in persistent and chronic inflammation and hepatocellular injury.479,527 Probiotic intervention reduced lowered plasma LPS levels and liver inflammation in turn delaying the progression of NAFL/NASH.528 Taken together, the gut–liver axis offers a new direction for the treatment of NAFL/NASH.
Gut microbiota targets
Gut microbiota manipulation yielded encouraging results for the treatment of different metabolic disorders in experimental models (Tables 3 and 4).529 Relevant clinical trials have been conducted (NCT02158351 and NCT04130321) with no results posted yet. Probiotics treatment may reduce intrahepatic triglyceride content from 22.6% to 14.9% (p = 0.034) and AST level (p = 0.008) in patients with histology-proven NASH (NCT00870012).530 Steatohepatitis was alleviated after fecal microbiota transplantation (FMT), as indicated by a significant decrease in intrahepatic lipid accumulation, intrahepatic proinflammatory cytokines, and the NAS score in mice.531 However, in a randomized, double-blind, placebo-controlled trial (NCT04074889), ultrasound-diagnosed NAFL/NASH patients supplemented with a probiotics sachet (MCP® BCMC® strains) containing six different Lactobacillus and Bifidobacterium species have no significant changes at the end of the study in terms of hepatic steatosis and fibrosis levels as measured by transient elastography.532 In further studies, more attention should be paid to the sample sizes, the duration of treatment, and different probiotic strains to evaluate the real benefits of probiotics in NAFL/NASH.
Cell therapy
Cell treatment for NASH fibrosis has recently gained extensive attention. Mesenchymal stem cells (MSCs) have the potential for self-renewal and differentiation into multiple cell lineages, including hepatocytes. The migration and engraftation of MSCs into targeted lesions may exert clinical efficacy, such as the improvement of liver fibrosis in patients with NASH.533 It was reported that MSCs reduce hepatocyte damage through immune suppressive pathways, which in turn prevent HSC activation.534,535 Furthermore, MSCs increase the phagocytosis of hepatocyte debris by regulating macrophage polarity, while increasing matrix metalloproteinase synthesis to reduce ECM534 (Fig. 8).

Stem cell therapy in liver fibrosis. MSCs isolated from different sources, including umbilical cord, bone marrow, placental, adipose tissue, placental, hair follicle, function to improve liver fibrosis. MSCs reduced hepatocyte damage through immune suppressive pathways, which in turn prevents HSC activation. Furthermore, MSCs increased the phagocytosis of hepatocyte debris by changing macrophage polarity, while increasing matrix metalloproteinase synthesis to remodel extracellular matrix. Created with BioRender
It has been proposed that MSCs isolated from different tissues, including umbilical cord, bone marrow, placental, adipose tissue, placental, hair follicle, function to improve liver fibrosis. Infused human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) are able to differentiate into hepatocytes in vivo, and transplantation of hUC-MSCs to CCl4-treated rats improved liver transaminase, reduced liver histopathology, and reversed hepatobiliary fibrosis.536 Exosomes of MSCs may also be a promising strategy for the treatment of NAFL/NASH. The hUC-MSC‑exosomal miR‑627‑5p improved glucose and lipid metabolism in a human normal liver cell line (LO2) and alleviated liver damage in high-fat high-fructose–fed induced NAFL rat model by repressing fat mass and obesity-associated gene expression.537 HLSC-Evs were shown to attenuate the activated phenotype of HSCs by delivering antifibrotic miRNAs, such as miR-146a-5p.538 Human adipose-derived MSCs or their EVs significantly increased anti-inflammatory macrophages in the liver and improved liver fibrosis.539
The therapeutic efficacy of MSCs mainly relies on the paracrine actions on different cell type, differentiation of MSCs, and inflammatory actions in the liver.540 In the preclinical studies, hUC-MSCs transplantation improves liver function, the degree of fibrosis, and promotes liver repair in acute-on-chronic liver failure/acute-on-chronic liver injury rats by inhibiting Notch signaling and reversing the imbalance of the Stat1/Stat3 pathway.541 Contrarily, adipose-derived stem cells (ADSCs) treatment reduces apoptosis of hepatocytes through Notch signaling activation and contributes to the repair and regeneration of the liver in NASH mouse model.542 The role of the Notch signaling pathway in the treatment of stem cell needs to be underlined.
In other novel studies with cell therapy implication, bone marrow-derived MSCs (BMSCs) were shown to regulate mitochondrial quality control, improve mitochondrial dysfunction, lower the AST/ALT ratio and alleviate the steatosis and histological lesions in livers of diabetic NAFL mice via mitochondrial transfer between stem cells and steatosis hepatocytes.540 BMSCs engrafted within the liver tissue prevented histological alterations and collagen accumulation via upregulating hepatic Nrf2/HO-1 signaling pathway in CCl4-intoxicated rats.543 In addition, BM-MSCs also attenuated hepatic fibrosis by secreting IL-4 and IL-10 to promote macrophage phenotypic switch from profibrotic Ly6Chi subset to restorative Ly6Clo subpopulation, which significantly blocked the source of fibrogenic cytokines (TGF-β, PDGF, TNF-α, and IL-1β) from Ly6Chi macrophages.544 MiR-375 encapsulated by Evs secreted from BMSCs inhibited HCC development via modulating the HOXB3/Wnt/β-catenin signaling axis.545 ADSCs alleviated liver fibrosis in NASH mice by suppressing IL-17-mediated inflammation.546,547
In clinical trials, MSCs have also achieved promising results in the treatment of NASH. Umbilical cord-derived MSC (UC-MSC) treatment markedly improved liver functions, as indicated by the levels of serum albumin, prothrombin activity, cholinesterase, and total bilirubin after 48 weeks follow-up (NCT01220492). UC-MSC treatment is safe and does not increase the occurrence of HCC events, and it has a long-term effect on improving liver function and survival in patients with decompensated liver cirrhosis.548 In addition, CCR2 overexpression in MSCs improves the treatment effect of MSCs by enhancing the targeted migration of stem cells to damaged livers,533 and may provide a novel strategy for improving the efficacy of cell therapy for NASH.
Immunotherapy for NASH-HCC
NASH is rapidly becoming one of the important drivers of HCC,549 and immunotherapy is considered to activate T cells or reinvigorate immune surveillance against cancer, with a response rate of 15–30% in patients with HCC.550,551,552,553 Recently, a study was performed to investigate the roles of immune checkpoint blockade (ICB) in NASH-HCC and non-NASH-HCC mice.554 It was observed that the liver tumors in NASH-HCC mice did not regress after anti-PD1 treatment, while tumors in non-NASH-HCC cancer models were shrunk by the same treatment. Unfortunately, liver fibrosis was exacerbated after anti-PD1 treatment in NASH-HCC models.554 Mechanistically, unconventionally activated CD8+PD1+ T cells accumulate progressively in NASH liver, but anti-PD1 treatment promotes the infiltration of CD8+PD1+ T cells population and secretion of TNF-α, leading to the increase of inflammation, fibrosis, and tumorigenesis.554,555 Cytotoxic CD8T+ cells that are suppressed in cancer accumulate in NASH and increase more after anti-PD1 therapy. The failure of PD1 therapy in NASH-HCC may be due to the loss of the original immune surveillance function of CD8T+ cells. Furthermore, a meta-analysis including three large randomized controlled phase 3 clinical trials of anti-PD(L)1 immunotherapy comprising over 1600 patients with advanced HCC (CheckMate-459,556 Imbrave150,550 and KEYNOTE-240551) revealed that although immunotherapy improved survival in the overall population, but not improve survival in patients with non-viral HCC.554 Notably, the authors investigated two other cohorts which recruited small groups of NASH-HCC patients and found that compared with other malignant tumor patients, the overall survival time of patients with NASH-HCC after immunotherapy was shortened. The abundance and complex signature of innate and adaptive immune cells in the liver may directly or indirectly affect the immunotherapy of NASH-HCC, which may cause less responsiveness to anti-PD1 immunotherapy.554,555,557 These observations suggest that ICB therapy may not be as effective as desired, future works might be required to better understand ICB resistance in NASH-HCC.
Genetic approaches
Inherited factors have been proved to be associated with susceptibility to different stages of NAFL/NASH, suggesting that genetic mapping approaches might be a powerful tool to identify genes with diagnostic and therapeutic potential.558 In the DiscovEHR human genetics study, truncated loss-of-function variant in hydroxysteroid 17β-dehydrogenase 13 (HSD17B13 rs72613567: TA), a lipid droplet-associated protein that is enriched in liver and hepatocyte with retinol dehydrogenase activity, was found to associate with decreased levels of ALT and AST and improved liver histology of NASH.559 The carriers of the HSD17B13 variant have a lower risk of developing NASH and cirrhosis but increased phospholipids that is opposite to the characteristics of the choline-deficient model of liver fibrosis.560 Interestingly, Hsd17b13 whole-body deficiency had no effect on protecting the liver from obesity diet damage in mice suggesting that interspecies differences need to be considered in the drug development process.561 However, some clinical trials in patients with NAFL/NASH have positive and hopeful results. ARO-HSD, a ribonucleic acid interference (RNAi) therapy, significantly reduced the mRNA and protein levels of hepatic HSD17B13, leading to a decrease in serum ALT in the phase 1 trial (NCT04202354).558,562
It was reported that PNPLA3-I148M variant was associated with hepatic steatosis, advanced fibrosis, and cirrhosis in patients with NAFL/NASH.563 The Pnpla3-I148M variant promoted steatosis by inhibiting adipose triglyceride lipase in mice fed high sucrose diet.564 The liver-targeted GalNAc3-conjugated antisense oligonucleotide (ASO)-mediated silencing of Pnpla3 reduced hepatic lipogenesis and steatosis in mice carrying the human I148M mutant fed a NASH-inducing diet.565 Recently, ION839 (AZD2693), an ASO targeting PNPLA3, is treated to participants with NASH and homozygous for PNPLA3 I148M risk allele in a phase 1 clinical trial (NCT04483947). In addition, IONIS-DGAT2Rx, an ASO targeting diacylglycerol acyltransferase 2 (DGAT2, an enzyme that catalyzes the final step in TG synthesis in the liver), improved liver steatosis in patients with NASH in phase 2 clinical trial (NCT03334214).566 GalNAc-siTAZ, a stabilized TAZ (the transcriptional coactivator with PDZ-binding motif) siRNA bearing the hepatocyte-specific ligand N-acetylgalactosamine, partially reversed hepatic inflammation, injury, and fibrosis in mice.567 It is reported that the symptoms (fibrosis and hepatic glucose production) of NASH mice are improved by Notch inhibitors such as Nicastrin antisense oligonucleotide (Ncst ASO)568 and γ-secretase inhibitors (GSI).455 These results suggest that ASO or RNAi therapy as a genetic approach may have promising clinical applications for NAFL/NASH treatment.
Combined therapies
Given the complexity of the pathophysiology of NASH, it will necessitate the involvement of multiple targets and pathways to improve outcomes of pharmacological intervention, which justifies combination therapies in the treatment of NASH. European Medicines Agency guidelines published in 2018 recommend that combination therapy should use drugs with different mechanisms of action.569 FXR agonists are most often combined with other agents in the treatment of NASH to minimize side effects and to increase the likelihood of success by targeting different metabolic pathways.204 The combination of Cilofexor (an FXR agonist) and Firsocostat (an ACC inhibitor) has shown more effective in reducing the NA) (p = 0.040) and minimizing dose-dependent complications of FXR activation (e.g., pruritus and LDL elevation) than Cilofexor used alone in patients with F3-F4 fibrosis or compensated cirrhosis in a phase 2b trial (NCT03449446).570
A randomized, placebo-controlled, multicenter phase 2b clinical trial will evaluate the safety and tolerability of Tropifexor (TXR, a non-BA agonist of FXR) and Cenicriviroc (a CCR2/5 antagonist, CVC) combination therapy in patients with NASH and liver fibrosis (NCT03517540). Dual antagonism of CCR2 and CCR5 by CVC may complement antisteatotic, anti-inflammatory, and antifibrotic actions of TXR. Given this, combination therapy is likely to show additional benefits compared with monotherapy.571 In addition, studies to evaluate Tropifexor alone or in combination with Licogliflozin (a dual SGLT1/2 inhibitor) in patients with NASH and liver fibrosis (NCT04065841) are recruiting. A preclinical study revealed that ZLY18 (a quadruple FFA1/PPAR-α/γ/δ agonist) significantly reduced steatosis, hepatocyte balloon, inflammation, and liver fibrosis in the NASH models, and represented a novel and highly promising quad FFA1/PPAR-α/γ/δ agonist warranting further investigation and development.572
Conclusion and future perspectives
The increased prevalence of NAFL/NASH and global health burden have provoked concentrated research and interest in therapeutics for patients with NAFL/NASH. There are still no approved drugs for the treatment of NAFL/NASH, despite numerous pathophysiological mechanisms and genetic variants having been identified. However, several clinical trials have achieved promising improvement in histology-proven NAFL/NASH, which encourages researchers and pharmaceutical industries to further develop safe and durable therapeutic strategies. For example, antidiabetic agents showed some promising results in NAFL/NASH patients with T2DM; for those patients who had defined genetic variants, direct interventions can be made by targeted therapies such as ASO or RNAi. Furthermore, considering the complexity of the pathogenesis of NAFL/NASH, the synergistic use of drugs with different mechanisms and personalized treatment plan may better benefit patients, especially for the combination of lifestyle changes and pharmacological interventions. Novel drugs, such as ASO and RNAi, targeting gut microbiome and stem cell therapy, have also been demonstrated to have the potential for the treatment of NASH and need to be further explored. In addition, standardized clinical trials and tailored animal models should be thoroughly conducted to improve the clinical success of new therapeutic applications. In summary, future studies are needed to better understand the pathogenic mechanisms linking liver metabolism, response to inflammation, and injury in NAFL/NASH development, with the goal of safe and consistent improvements.
References
Asrani, S. K., Devarbhavi, H., Eaton, J. & Kamath, P. S. Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019).
Charlton, M. R. et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 141, 1249–1253 (2011).
Estes, C. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 69, 896–904 (2018).
Mitra, S., De, A. & Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 5, 16 (2020).
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M. & Sanyal, A. J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 (2018).
Estes, C., Razavi, H., Loomba, R., Younossi, Z. & Sanyal, A. J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018).
EASL-EASD-EASO. Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 59, 1121–1140 (2016).
Boyle, M., Masson, S. & Anstee, Q. M. The bidirectional impacts of alcohol consumption and the metabolic syndrome: cofactors for progressive fatty liver disease. J. Hepatol. 68, 251–267 (2018).
Ismaiel, A. & Dumitraşcu, D. L. Cardiovascular risk in fatty liver disease: the liver-heart axis-literature review. Front. Med. (Lausanne) 6, 202 (2019).
Inan-Eroglu, E. et al. Joint associations of adiposity and alcohol consumption with liver disease-related morbidity and mortality risk: findings from the UK Biobank. Eur. J. Clin. Nutr. 76, 74–83 (2022).
Rinella, M. E. Nonalcoholic fatty liver disease: a systematic review. JAMA 313, 2263–2273 (2015).
Kupcova, V., Fedelesova, M., Bulas, J., Kozmonova, P. & Turecky, L. Overview of the pathogenesis, genetic, and non-invasive clinical, biochemical, and scoring methods in the assessment of NAFLD. Int. J. Environ. Res. Public Health 16, 3570 (2019).
Hardy, T., Oakley, F., Anstee, Q. M. & Day, C. P. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev. Pathol. 11, 451–496 (2016).
Geier, A., Tiniakos, D., Denk, H. & Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 70, 1570–1579 (2021).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016).
Sheka, A. C. et al. Nonalcoholic steatohepatitis: a review. JAMA 323, 1175–1183 (2020).
Bertot, L. C. & Adams, L. A. Trends in hepatocellular carcinoma due to non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 13, 179–187 (2019).
Mittal, S. et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 14, 124–131.e121 (2016).
Bowman, W. Observations on the minute anatomy of fatty degeneration of the liver. Lancet 37, 560–561 (1842).
Iturriaga, H., Bunout, D., Hirsch, S. & Ugarte, G. Overweight as a risk factor or a predictive sign of histological liver damage in alcoholics. Am. J. Clin. Nutr. 47, 235–238 (1988).
Connor, C. L. Fatty infiltration of the liver and the development of cirrhosis in diabetes and chronic alcoholism. Am. J. Pathol. 14, 347–364.349 (1938).
Ludwig, J., Viggiano, T. R., McGill, D. B. & Oh, B. J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 55, 434–438 (1980).
Schaffner, F. & Thaler, H. Nonalcoholic fatty liver disease. Prog. Liver Dis. 8, 283–298 (1986).
Younossi, Z. et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69, 2672–2682 (2019).
Stepanova, M. & Younossi, Z. M. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin. Gastroenterol. Hepatol. 10, 646–650 (2012).
Lee, H., Lee, Y. H., Kim, S. U. & Kim, H. C. Metabolic dysfunction-associated fatty liver disease and incident cardiovascular disease risk: a nationwide cohort study. Clin. Gastroenterol. Hepatol. 19, 2138–2147.e2110 (2021).
Musso, G. et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med. 11, e1001680 (2014).
Cao, Y. et al. The association between NAFLD and risk of chronic kidney disease: a cross-sectional study. Ther. Adv. Chronic Dis. 12, 20406223211048649 (2021).
Musso, G. et al. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes. Rev. 14, 417–431 (2013).
Pulixi, E. A. et al. Risk of obstructive sleep apnea with daytime sleepiness is associated with liver damage in non-morbidly obese patients with nonalcoholic fatty liver disease. PLoS One 9, e96349 (2014).
Vancells Lujan, P., Viñas Esmel, E. & Sacanella Meseguer, E. Overview of non-alcoholic fatty liver disease (NAFLD) and the role of sugary food consumption and other dietary components in its development. Nutrients 13, 1442 (2021).
Boutari, C., Lefkos, P., Athyros, V. G., Karagiannis, A. & Tziomalos, K. Nonalcoholic fatty liver disease vs. nonalcoholic steatohepatitis: pathological and clinical implications. Curr. Vasc. Pharm. 16, 214–218 (2018).
Younossi, Z. M. et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J. Hepatol. 71, 793–801 (2019).
Arner, P. et al. Adipose lipid turnover and long-term changes in body weight. Nat. Med. 25, 1385–1389 (2019).
Arner, P. Turnover of human fat cells and their lipid content. Acta Vet. Scand. 57, K1 (2015).
Younossi, Z. M. et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 64, 1577–1586 (2016).
Targher, G., Tilg, H. & Byrne, C. D. Non-alcoholic fatty liver disease: a multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol. Hepatol. 6, 578–588 (2021).
Targher, G., Byrne, C. D., Lonardo, A., Zoppini, G. & Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J. Hepatol. 65, 589–600 (2016).
Mantovani, A. et al. Risk of heart failure in patients with nonalcoholic fatty liver disease: JACC review topic of the week. J. Am. Coll. Cardiol. 79, 180–191 (2022).
Mantovani, A. et al. Association between nonalcoholic fatty liver disease and colorectal tumours in asymptomatic adults undergoing screening colonoscopy: a systematic review and meta-analysis. Metabolism 87, 1–12 (2018).
Lee, J. M. et al. The association between nonalcoholic fatty liver disease and esophageal, stomach, or colorectal cancer: national population-based cohort study. PLoS One 15, e0226351 (2020).
Chang, C. F. et al. Exploring the relationship between nonalcoholic fatty liver disease and pancreatic cancer by computed tomographic survey. Intern Emerg. Med. 13, 191–197 (2018).
Allen, A. M., Hicks, S. B., Mara, K. C., Larson, J. J. & Therneau, T. M. The risk of incident extrahepatic cancers is higher in non-alcoholic fatty liver disease than obesity – a longitudinal cohort study. J. Hepatol. 71, 1229–1236 (2019).
Kwak, M. S. et al. Nonalcoholic fatty liver disease is associated with breast cancer in nonobese women. Dig. Liver Dis. 51, 1030–1035 (2019).
Rinella, M. E., Tacke, F., Sanyal, A. J. & Anstee, Q. M. Report on the AASLD/EASL joint workshop on clinical trial endpoints in NAFLD. J. Hepatol. 71, 823–833 (2019).
Heck, A. M., Yanovski, J. A. & Calis, K. A. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy 20, 270–279 (2000).
Day, C. P. & James, O. F. Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845 (1998).
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Tilg, H. & Moschen, A. R. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846 (2010).
Neuschwander-Tetri, B. A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52, 774–788 (2010).
Hirsova, P., Ibrabim, S. H., Gores, G. J. & Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J. Lipid Res. 57, 1758–1770 (2016).
Mota, M., Banini, B. A., Cazanave, S. C. & Sanyal, A. J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 65, 1049–1061 (2016).
Powell, E. E., Wong, V. W. & Rinella, M. Non-alcoholic fatty liver disease. Lancet 397, 2212–2224 (2021).
Peng, C., Stewart, A. G., Woodman, O. L., Ritchie, R. H. & Qin, C. X. Non-alcoholic steatohepatitis: a review of its mechanism, models and medical treatments. Front. Pharm. 11, 603926 (2020).
Shi, Y. W. & Fan, J. G. Current status and challenges in the drug treatment for fibrotic nonalcoholic steatohepatitis. Acta Pharmacol. Sin. 43, 1191–1199 (2022).
Neuschwander-Tetri, B. A. Therapeutic landscape for NAFLD in 2020. Gastroenterology 158, 1984–1998.e1983 (2020).
Fraile, J. M., Palliyil, S., Barelle, C., Porter, A. J. & Kovaleva, M. Non-alcoholic steatohepatitis (NASH) – a review of a crowded clinical landscape, driven by a complex disease. Drug Des. Devel Ther. 15, 3997–4009 (2021).
Vachliotis, I., Goulas, A., Papaioannidou, P. & Polyzos, S. A. Nonalcoholic fatty liver disease: lifestyle and quality of life. Hormones (Athens) 21, 41–49 (2022).
Hannah, W. N. Jr. & Harrison, S. A. Effect of weight loss, diet, exercise, and bariatric surgery on nonalcoholic fatty liver disease. Clin. Liver Dis. 20, 339–350 (2016).
Vilar-Gomez, E. et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149, 367–378.e365 (2015). quiz e314-365.
Promrat, K. et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 51, 121–129 (2010).
EASL-EASD-EASO. Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 64, 1388–1402 (2016).
George, E. S. et al. Impact of a Mediterranean diet on hepatic and metabolic outcomes in non-alcoholic fatty liver disease: The MEDINA randomised controlled trial. Liver Int. 42, 1308–1322 (2022).
Yurtdaş, G., Akbulut, G., Baran, M. & Yılmaz, C. The effects of Mediterranean diet on hepatic steatosis, oxidative stress, and inflammation in adolescents with non-alcoholic fatty liver disease: a randomized controlled trial. Pediatr. Obes. 17, e12872 (2022).
Baratta, F. et al. Adherence to Mediterranean diet and non-alcoholic fatty liver disease: effect on insulin resistance. Am. J. Gastroenterol. 112, 1832–1839 (2017).
Baratta, F. et al. High compliance to mediterranean diet associates with lower platelet activation and liver collagen deposition in patients with nonalcoholic fatty liver disease. Nutrients 14, 1209 (2022).
Ma, G. L. & Chen, Y. T. Polyphenol supplementation benefits human health via gut microbiota: a systematic review via meta-analysis. J. Funct. Foods 66, 103829 (2020).
Rosés, C. et al. Gut microbiota bacterial species associated with Mediterranean diet-related food groups in a northern Spanish population. Nutrients 13, 636 (2021).
Yaskolka Meir, A. et al. Effect of green-Mediterranean diet on intrahepatic fat: the DIRECT PLUS randomised controlled trial. Gut 70, 2085–2095 (2021).
Centis, E. et al. Stage of change and motivation to healthier lifestyle in non-alcoholic fatty liver disease. J. Hepatol. 58, 771–777 (2013).
Samuel, V. T. & Shulman, G. I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41 (2018).
Bessone, F., Razori, M. V. & Roma, M. G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol. Life Sci. 76, 99–128 (2019).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Ameer, F., Scandiuzzi, L., Hasnain, S., Kalbacher, H. & Zaidi, N. De novo lipogenesis in health and disease. Metabolism 63, 895–902 (2014).
Sanders, F. W. & Griffin, J. L. De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 91, 452–468 (2016).
Samsoondar, J. P. et al. Prevention of diet-induced metabolic dysregulation, inflammation, and atherosclerosis in Ldlr(−/−) mice by treatment with the ATP-citrate lyase inhibitor bempedoic acid. Arterioscler. Thromb. Vasc. Biol. 37, 647–656 (2017).
Ahrens, M. et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302 (2013).
Wang, Q. et al. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology 49, 1166–1175 (2009).
Sanjay, K. V. et al. ATP citrate lyase inhibitor Bempedoic Acid alleviate long term HFD induced NASH through improvement in glycemic control, reduction of hepatic triglycerides & total cholesterol, modulation of inflammatory & fibrotic genes and improvement in NAS score. Curr. Res. Pharm. Drug Disco. 2, 100051 (2021).
Ray, K. K. et al. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 380, 1022–1032 (2019).
Agha, A. M., Jones, P. H., Ballantyne, C. M., Virani, S. S. & Nambi, V. Greater than expected reduction in low-density lipoprotein-cholesterol (LDL-C) with bempedoic acid in a patient with heterozygous familial hypercholesterolemia (HeFH). J. Clin. Lipido. 15, 649–652 (2021).
Surapaneni, K. M. & Jainu, M. Pioglitazone, quercetin and hydroxy citric acid effect on hepatic biomarkers in non alcoholic steatohepatitis. Pharmacogn. Res. 6, 153–162 (2014).
Shara, M. et al. Physico-chemical properties of a novel (−)-hydroxycitric acid extract and its effect on body weight, selected organ weights, hepatic lipid peroxidation and DNA fragmentation, hematology and clinical chemistry, and histopathological changes over a period of 90 days. Mol. Cell Biochem. 260, 171–186 (2004).
Bates, J. et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 73, 896–905 (2020).
Loomba, R. et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 155, 1463–1473.e1466 (2018).
Lawitz, E. J. et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 16, 1983–1991.e1983 (2018).
Kim, C. W. et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406.e396 (2017).
Calle, R. A. et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 27, 1836–1848 (2021).
Stiede, K. et al. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: a randomized, double-blind, crossover study. Hepatology 66, 324–334 (2017).
Auguet, T. et al. Altered fatty acid metabolism-related gene expression in liver from morbidly obese women with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 15, 22173–22187 (2014).
Loomba, R. et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology 161, 1475–1486 (2021).
Beysen, C. et al. Inhibition of fatty acid synthase with FT-4101 safely reduces hepatic de novo lipogenesis and steatosis in obese subjects with non-alcoholic fatty liver disease: results from two early-phase randomized trials. Diabetes Obes. Metab. 23, 700–710 (2021).
Harrison, S. A., Fecht, W., Brunt, E. M. & Neuschwander-Tetri, B. A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: a randomized, prospective trial. Hepatology 49, 80–86 (2009).
Zhu, X. et al. Berberine attenuates nonalcoholic hepatic steatosis through the AMPK-SREBP-1c-SCD1 pathway. Free Radic. Biol. Med. 141, 192–204 (2019).
Safadi, R. et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 12, 2085–2091.e2081 (2014).
Bhattacharya, D. et al. Aramchol downregulates stearoyl CoA-desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis. JHEP Rep. 3, 100237 (2021).
Ratziu, V. et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 27, 1825–1835 (2021).
Deng, X. et al. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 287, 20132–20143 (2012).
Shimomura, I., Bashmakov, Y. & Horton, J. D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 274, 30028–30032 (1999).
Shimomura, I. et al. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl Acad. Sci. USA 96, 13656–13661 (1999).
Porstmann, T. et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 (2008).
Wada, S. et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564 (2016).
Gosis, B. S. et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 376, eabf8271 (2022).
Parlati, L., Regnier, M., Guillou, H. & Postic, C. New targets for NAFLD. JHEP Rep. 3, 100346 (2021).
Liu, H. J. et al. Rotundic acid ameliorates non-alcoholic steatohepatitis via SREBP-1c/ SCD1 signaling pathway and modulating gut microbiota. Int. Immunopharmacol. 99, 108065 (2021).
Yang, D. et al. Oral administration of Jinan Red Ginseng and licorice extract mixtures ameliorates nonalcoholic steatohepatitis by modulating lipogenesis. J. Ginseng Res. 46, 126–137 (2022).
Hwahng, S. H., Ki, S. H., Bae, E. J., Kim, H. E. & Kim, S. G. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology 49, 1913–1925 (2009).
Shimozono, R. et al. Nrf2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol. Pharm. 84, 62–70 (2013).
Kim, W. et al. Randomised clinical trial: the efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment Pharm. Ther. 45, 1073–1083 (2017).
Kim, S. G. et al. Pharmacokinetics of oltipraz and its major metabolite (RM) in patients with liver fibrosis or cirrhosis: relationship with suppression of circulating TGF-beta1. Clin. Pharm. Ther. 88, 360–368 (2010).
Musso, G., Gambino, R. & Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 52, 175–191 (2013).
Oteng, A. B., Loregger, A., van Weeghel, M., Zelcer, N. & Kersten, S. Industrial trans fatty acids stimulate SREBP2-mediated cholesterogenesis and promote non-alcoholic fatty liver disease. Mol. Nutr. Food Res. 63, e1900385 (2019).
Park, H. S. et al. Statins increase mitochondrial and peroxisomal fatty acid oxidation in the liver and prevent non-alcoholic steatohepatitis in mice. Diabetes Metab. J. 40, 376–385 (2016).
Wang, W. et al. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS One 8, e76538 (2013).
Ioannou, G. N. et al. Cholesterol-lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J. Lipid Res. 56, 277–285 (2015).
Lee, J. I., Lee, H. W., Lee, K. S., Lee, H. S. & Park, J. Y. Effects of statin use on the development and progression of nonalcoholic fatty liver disease: a nationwide nested case-control study. Am. J. Gastroenterol. 116, 116–124 (2021).
German, M. N., Lutz, M. K., Pickhardt, P. J., Bruce, R. J. & Said, A. Statin use is protective against hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: a case-control study. J. Clin. Gastroenterol. 54, 733–740 (2020).
Chalasani, N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology 41, 690–695 (2005).
Bril, F. et al. Liver safety of statins in prediabetes or T2DM and nonalcoholic steatohepatitis: post hoc analysis of a randomized trial. J. Clin. Endocrinol. Metab. 102, 2950–2961 (2017).
Pockros, P. J. et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 39, 2082–2093 (2019).
Clark, J. M. & Diehl, A. M. Hepatic steatosis and type 2 diabetes mellitus. Curr. Diab Rep. 2, 210–215 (2002).
Lonardo, A., Nascimbeni, F., Mantovani, A. & Targher, G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J. Hepatol. 68, 335–352 (2018).
Francque, S. et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 63, 164–173 (2015).
Zarei, M. et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 8, 117–131 (2018).
Montagner, A. et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65, 1202–1214 (2016).
Régnier, M. et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 10, 6489 (2020).
Kersten, S. et al. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498 (1999).
Brocker, C. N. et al. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 59, 2140–2152 (2018).
Matsusue, K. et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 111, 737–747 (2003).
Skat-Rørdam, J., Højland Ipsen, D., Lykkesfeldt, J. & Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharm. Toxicol. 124, 528–537 (2019).
Morán-Salvador, E. et al. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 59, 1045–1053 (2013).
Braissant, O., Foufelle, F., Scotto, C., Dauça, M. & Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137, 354–366 (1996).
Sanderson, L. M., Boekschoten, M. V., Desvergne, B., Müller, M. & Kersten, S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol. Genomics 41, 42–52 (2010).
Wang, Y. X. et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113, 159–170 (2003).
Tanaka, T. et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl Acad. Sci. USA 100, 15924–15929 (2003).
Liu, S. et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 502, 550–554 (2013).
Francque, S. et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 18, 24–39 (2021).
Lefere, S. et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 73, 757–770 (2020).
Fougerat, A., Montagner, A., Loiseau, N., Guillou, H. & Wahli, W. Peroxisome proliferator-activated receptors and their novel ligands as candidates for the treatment of non-alcoholic fatty liver disease. Cells 9, 1638 (2020).
Bril, F. et al. Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin. Gastroenterol. Hepatol. 16, 558–566.e552 (2018).
Cusi, K. et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann. Intern. Med. 165, 305–315 (2016).
Staels, B. et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58, 1941–1952 (2013).
Ratziu, V. et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e1145 (2016).
Feng, Z. et al. Design, synthesis, and biological evaluation of triazolone derivatives as potent PPARalpha/delta dual agonists for the treatment of nonalcoholic steatohepatitis. J. Med. Chem. 65, 2571–2592 (2022).
Gawrieh, S. et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology 74, 1809–1824 (2021).
Francque, S. M. et al. A randomized, controlled trial of the Pan-PPAR agonist lanifibranor in NASH. N. Engl. J. Med. 385, 1547–1558 (2021).
Nakano, S. et al. Bezafibrate prevents hepatic stellate cell activation and fibrogenesis in a murine steatohepatitis model, and suppresses fibrogenic response induced by transforming growth factor-beta1 in a cultured stellate cell line. Hepatol. Res. 38, 1026–1039 (2008).
Okishio, S. et al. PPARalpha agonist and metformin co-treatment ameliorates NASH in mice induced by a choline-deficient, amino acid-defined diet with 45% fat. Sci. Rep. 10, 19578 (2020).
Boeckmans, J. et al. Human hepatic in vitro models reveal distinct anti-NASH potencies of PPAR agonists. Cell Biol. Toxicol. 37, 293–311 (2021).
Oniciu, D. C., Hashiguchi, T., Shibazaki, Y. & Bisgaier, C. L. Gemcabene downregulates inflammatory, lipid-altering and cell-signaling genes in the STAM model of NASH. PLoS One 13, e0194568 (2018).
Haczeyni, F. et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 1, 663–674 (2017).
Holst, J. J. The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439 (2007).
Liu, Y., Wei, R. & Hong, T. P. Potential roles of glucagon-like peptide-1-based therapies in treating non-alcoholic fatty liver disease. World J. Gastroenterol. 20, 9090–9097 (2014).
Armstrong, M. J. et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690 (2016).
Armstrong, M. J. et al. Liraglutide efficacy and action in non-alcoholic steatohepatitis (LEAN): study protocol for a phase II multicentre, double-blinded, randomised, controlled trial. BMJ Open 3, e003995 (2013).
Armstrong, M. J. et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 64, 399–408 (2016).
Tang, A. et al. Effects of insulin glargine and liraglutide therapy on liver fat as measured by magnetic resonance in patients with type 2 diabetes: a randomized trial. Diabetes Care 38, 1339–1346 (2015).
O’Neil, P. M. et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 392, 637–649 (2018).
Kolotkin, R. L. et al. Validation of a new measure of quality of life in obesity trials: impact of weight on quality of life-lite clinical trials version. Clin. Obes. 9, e12310 (2019).
Harrison, S. A. et al. Semaglutide for the treatment of non-alcoholic steatohepatitis: trial design and comparison of non-invasive biomarkers. Contemp. Clin. Trials 97, 106174 (2020).
Dickson, I. Semaglutide is safe and efficacious for NASH resolution. Nat. Rev. Gastroenterol. Hepatol. 18, 6 (2021).
Newsome, P. N. et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med. 384, 1113–1124 (2021).
Pirro, V. et al. Effects of tirzepatide, a dual GIP and GLP-1 RA, on lipid and metabolite profiles in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 107, 363–378 (2022).
Hartman, M. L. et al. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care 43, 1352–1355 (2020).
Frias, J. P. et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 26, 343–352.e342 (2017).
Nahra, R. et al. Effects of cotadutide on metabolic and hepatic parameters in adults with overweight or obesity and type 2 diabetes: a 54-week randomized phase 2b study. Diabetes Care 44, 1433–1442 (2021).
Patel, C. C., Cusi, K. & Kadiyala, S. The emerging role of glucagon-like peptide-1 receptor agonists for the management of NAFLD. J. Clin. Endocrinol. Metab. 107, 29–38 (2022).
Long, J. et al. Effect of GLP-1R rs2254336 and rs3765467 polymorphisms on gastrointestinal adverse reactions in type 2 diabetes patients treated with liraglutide. Eur. J. Clin. Pharmacol. 78, 589–596 (2022).
AlSaadoun, A. R., AlSaadoun, T. R. & Al, G. A. K. Liraglutide overdose-induced acute pancreatitis. Cureus 14, e21616 (2022).
Ala, M. SGLT2 inhibition for cardiovascular diseases, chronic kidney disease, and NAFLD. Endocrinology 162, bqab157 (2021).
Raj, H. et al. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a systematic review. World J. Diabetes 10, 114–132 (2019).
Xu, L. & Ota, T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: focus on fat browning and macrophage polarization. Adipocyte 7, 121–128 (2018).
Sumida, Y. et al. Antidiabetic therapy in the treatment of nonalcoholic steatohepatitis. Int. J. Mol. Sci. 21, 1907 (2020).
Meng, Z. et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int. Immunopharmacol. 94, 107492 (2021).
Prikhodko, V. A., Bezborodkina, N. N. & Okovityi, S. V. Pharmacotherapy for non-alcoholic fatty liver disease: emerging targets and drug candidates. Biomedicines 10, 274 (2022).
Takahashi, H. et al. Ipragliflozin improves the hepatic outcomes of patients with diabetes with NAFLD. Hepatol. Commun. 6, 120–132 (2022).
Miyake, T. et al. Ipragliflozin ameliorates liver damage in non-alcoholic fatty liver disease. Open Med. (Wars.) 13, 402–409 (2018).
Pokharel, A. et al. The effect of empagliflozin on liver fat in type 2 diabetes mellitus patients with non-alcoholic fatty liver disease. Cureus 13, e16687 (2021).
Chehrehgosha, H. et al. Empagliflozin improves liver steatosis and fibrosis in patients with non-alcoholic fatty liver disease and type 2 diabetes: a randomized, double-blind, placebo-controlled clinical trial. Diabetes Ther. 12, 843–861 (2021).
Lai, L. L., Vethakkan, S. R., Nik, M. N. R., Mahadeva, S. & Chan, W. K. Empagliflozin for the treatment of nonalcoholic steatohepatitis in patients with type 2 diabetes mellitus. Dig. Dis. Sci. 65, 623–631 (2020).
Seko, Y. et al. Efficacy and safety of canagliflozin in type 2 diabetes mellitus patients with biopsy-proven nonalcoholic steatohepatitis classified as stage 1-3 fibrosis. Diabetes Metab. Syndr. Obes. 11, 835–843 (2018).
Aso, Y. et al. Impact of dapagliflozin, an SGLT2 inhibitor, on serum levels of soluble dipeptidyl peptidase-4 in patients with type 2 diabetes and non-alcoholic fatty liver disease. Int. J. Clin. Pract. 73, e13335 (2019).
Tobita, H., Sato, S., Miyake, T., Ishihara, S. & Kinoshita, Y. Effects of dapagliflozin on body composition and liver tests in patients with nonalcoholic steatohepatitis associated with type 2 diabetes mellitus: a prospective, open-label, uncontrolled study. Curr. Ther. Res. Clin. Exp. 87, 13–19 (2017).
Marjot, T. et al. Sodium-glucose cotransporter 2 inhibition does not reduce hepatic steatosis in overweight, insulin-resistant patients without type 2 diabetes. JGH Open 4, 433–440 (2020).
Chávez-Talavera, O., Tailleux, A., Lefebvre, P. & Staels, B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152, 1679–1694.e1673 (2017).
Mouzaki, M. et al. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS One 11, e0151829 (2016).
Islam, K. B. et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141, 1773–1781 (2011).
Chen, J., Thomsen, M. & Vitetta, L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J. Cell Biochem. 120, 2713–2720 (2019).
Nimer, N. et al. Bile acids profile, histopathological indices and genetic variants for non-alcoholic fatty liver disease progression. Metabolism 116, 154457 (2021).
Chow, M. D., Lee, Y. H. & Guo, G. L. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Asp. Med. 56, 34–44 (2017).
Wang, H., Chen, J., Hollister, K., Sowers, L. C. & Forman, B. M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 (1999).
Panzitt, K. & Wagner, M. FXR in liver physiology: multiple faces to regulate liver metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166133 (2021).
Jiao, N. et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 67, 1881–1891 (2018).
Li, T. & Chiang, J. Y. Bile acid signaling in metabolic disease and drug therapy. Pharm. Rev. 66, 948–983 (2014).
Ferrell, J. M., Pathak, P., Boehme, S., Gilliland, T. & Chiang, J. Y. L. Deficiency of both farnesoid X receptor and Takeda G protein-coupled receptor 5 exacerbated liver fibrosis in mice. Hepatology 70, 955–970 (2019).
Shen, H. et al. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell Physiol. Biochem. 22, 1–14 (2008).
Molinaro, A., Wahlström, A. & Marschall, H. U. Role of bile acids in metabolic control. Trends Endocrinol. Metab. 29, 31–41 (2018).
Chiang, J. Y. Recent advances in understanding bile acid homeostasis. F1000Res 6, 2029 (2017).
Sinal, C. J. et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000).
Degirolamo, C. et al. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology 61, 161–170 (2015).
Yang, F. et al. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 67, 863–867 (2007).
Younossi, Z. M. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019).
Neuschwander-Tetri, B. A. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965 (2015).
Fiorucci, S. et al. The identification of farnesoid X receptor modulators as treatment options for nonalcoholic fatty liver disease. Expert Opin. Drug Disco. 16, 1193–1208 (2021).
Patel, K. et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology 72, 58–71 (2020).
Ratziu, V. et al. EDP-305 in patients with NASH: a phase II double-blind placebo-controlled dose-ranging study. J. Hepatol. 76, 506–517 (2022).
Kremoser, C. FXR agonists for NASH: how are they different and what difference do they make? J. Hepatol. 75, 12–15 (2021).
Song, K. H., Li, T., Owsley, E., Strom, S. & Chiang, J. Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 49, 297–305 (2009).
Dongiovanni, P. et al. β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 72, 411–419 (2020).
Jahn, D., Rau, M., Hermanns, H. M. & Geier, A. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 26, 625–635 (2015).
Dolegowska, K., Marchelek-Mysliwiec, M., Nowosiad-Magda, M., Slawinski, M. & Dolegowska, B. FGF19 subfamily members: FGF19 and FGF21. J. Physiol. Biochem. 75, 229–240 (2019).
Harrison, S. A. et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology 160, 219–231.e211 (2021).
Harrison, S. A. et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391, 1174–1185 (2018).
Sanyal, A. J. et al. Potent suppression of hydrophobic bile acids by aldafermin, an FGF19 analogue, across metabolic and cholestatic liver diseases. JHEP Rep. 3, 100255 (2021).
Sinha, R. A., Bruinstroop, E., Singh, B. K. & Yen, P. M. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 29, 1173–1191 (2019).
Tanase, D. M. et al. Hypothyroidism-induced nonalcoholic fatty liver disease (HIN): mechanisms and emerging therapeutic options. Int. J. Mol. Sci. 21, 5927 (2020).
Kannt, A. et al. Activation of thyroid hormone receptor-β improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br. J. Pharm. 178, 2412–2423 (2021).
Harrison, S. A. et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol. Commun. 5, 573–588 (2021).
Harrison, S. A. et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 394, 2012–2024 (2019).
Luong, X. G. et al. Regulation of gene transcription by thyroid hormone receptor beta agonists in clinical development for the treatment of non-alcoholic steatohepatitis (NASH). PLoS One 15, e0240338 (2020).
Lee, K., Kerner, J. & Hoppel, C. L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286, 25655–25662 (2011).
Perra, A. et al. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 22, 2981–2989 (2008).
Loomba, R. et al. LBP-20-VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: a phase 2 randomized, placebo-controlled trial. J. Hepatol. 70, e150–e151 (2019).
Badman, M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 (2007).
Inagaki, T. et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 (2007).
Klaebel, J. H., Lykkesfeldt, J. & Tveden-Nyborg, P. Efficacy of fibroblast growth factor 21 in non-alcoholic fatty liver disease in guinea pigs. Basic Clin. Pharmacol. Toxicol. 130, 385–393 (2022).
Sanyal, A. et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392, 2705–2717 (2019).
Abdelmalek, M. F. et al. The FALCON program: two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemp. Clin. Trials 104, 106335 (2021).
Harrison, S. A. et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 27, 1262–1271 (2021).
Ibrahim, S. H., Hirsova, P. & Gores, G. J. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67, 963–972 (2018).
Stiuso, P. et al. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: a pilot study. Oxid. Med. Cell Longev. 2014, 169216 (2014).
Loguercio, C. et al. Non-alcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J. Hepatol. 35, 568–574 (2001).
Rui, L. Energy metabolism in the liver. Compr. Physiol. 4, 177–197 (2014).
Sunny, N. E., Bril, F. & Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 28, 250–260 (2017).
Chen, Z., Tian, R., She, Z., Cai, J. & Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 152, 116–141 (2020).
Kakimoto, P. A., Chausse, B., Caldeira da Silva, C. C., Donato Júnior, J. & Kowaltowski, A. J. Resilient hepatic mitochondrial function and lack of iNOS dependence in diet-induced insulin resistance. PLoS One 14, e0211733 (2019).
Patterson, R. E. et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 310, E484–E494 (2016).
Satapati, S. et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 53, 1080–1092 (2012).
Grattagliano, I. et al. Role of mitochondria in nonalcoholic fatty liver disease–from origin to propagation. Clin. Biochem. 45, 610–618 (2012).
Begriche, K., Massart, J., Robin, M. A., Bonnet, F. & Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58, 1497–1507 (2013).
Baskol, G., Baskol, M. & Kocer, D. Oxidative stress and antioxidant defenses in serum of patients with non-alcoholic steatohepatitis. Clin. Biochem. 40, 776–780 (2007).
Rotter, I. et al. Relationship between the concentrations of heavy metals and bioelements in aging men with metabolic syndrome. Int. J. Environ. Res. Public Health 12, 3944–3961 (2015).
Li, L. & Yang, X. The essential element manganese, oxidative stress, and metabolic diseases: links and interactions. Oxid. Med. Cell Longev. 2018, 7580707 (2018).
Hwang, I. et al. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free Radic. Biol. Med. 148, 22–32 (2020).
Weltman, M. D., Farrell, G. C. & Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111, 1645–1653 (1996).
Weltman, M. D., Farrell, G. C., Hall, P., Ingelman-Sundberg, M. & Liddle, C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 27, 128–133 (1998).
Aubert, J., Begriche, K., Knockaert, L., Robin, M. A. & Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 35, 630–637 (2011).
Varela, N. M. et al. Study of cytochrome P450 2E1 and its allele variants in liver injury of nondiabetic, nonalcoholic steatohepatitis obese women. Biol. Res. 41, 81–92 (2008).
Sookoian, S. et al. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 240, 437–449 (2016).
Pirola, C. J., Garaycoechea, M., Flichman, D., Castaño, G. O. & Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b – a key component of the respirasome - drive the severity of fatty liver disease. J. Intern Med. 289, 84–96 (2021).
Gao, Y., Wang, Y., Liu, H., Liu, Z. & Zhao, J. Mitochondrial DNA from hepatocytes induces upregulation of interleukin-33 expression of macrophages in nonalcoholic steatohepatitis. Dig. Liver Dis. 52, 637–643 (2020).
An, P. et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 11, 2362 (2020).
Lebeaupin, C. et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 69, 927–947 (2018).
Harding, H. P., Zhang, Y. & Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 (1999).
Yoshida, H. et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 20, 6755–6767 (2000).
Cox, J. S., Shamu, C. E. & Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 (1993).
Ibrahim, I. M., Abdelmalek, D. H. & Elfiky, A. A. GRP78: a cell’s response to stress. Life Sci. 226, 156–163 (2019).
Vattem, K. M. & Wek, R. C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl Acad. Sci. USA 101, 11269–11274 (2004).
Harding, H. P. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 (2000).
Shen, J., Chen, X., Hendershot, L. & Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111 (2002).
Sidrauski, C. & Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 (1997).
Calfon, M. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002).
Mei, Y., Thompson, M. D., Cohen, R. A. & Tong, X. Endoplasmic reticulum stress and related pathological processes. J. Pharm. Biomed. Anal. 1, 1000107 (2013).
Shimizu, Y. & Hendershot, L. M. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal 11, 2317–2331 (2009).
Song, B., Scheuner, D., Ron, D., Pennathur, S. & Kaufman, R. J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118, 3378–3389 (2008).
Fuchs, C. D., Claudel, T., Scharnagl, H., Stojakovic, T. & Trauner, M. FXR controls CHOP expression in steatohepatitis. FEBS Lett. 591, 3360–3368 (2017).
Toriguchi, K. et al. Attenuation of steatohepatitis, fibrosis, and carcinogenesis in mice fed a methionine-choline deficient diet by CCAAT/enhancer-binding protein homologous protein deficiency. J. Gastroenterol. Hepatol. 29, 1109–1118 (2014).
Okada, K. et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J. Gastroenterol. 48, 620–632 (2013).
Sugimoto, H. et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G283–G294 (2010).
Wang, C. et al. Nrf2 deletion causes “benign” simple steatosis to develop into nonalcoholic steatohepatitis in mice fed a high-fat diet. Lipids Health Dis. 12, 165 (2013).
Deniaud, A. et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299 (2008).
Wei, Y., Wang, D., Gentile, C. L. & Pagliassotti, M. J. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol. Cell Biochem. 331, 31–40 (2009).
Fu, S. et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531 (2011).
Evans, H. M. & Bishop, K. S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 56, 650–651 (1922).
Burton, G. W., Joyce, A. & Ingold, K. U. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes? Arch. Biochem. Biophys. 221, 281–290 (1983).
Azzi, A. & Stocker, A. Vitamin E: non-antioxidant roles. Prog. Lipid Res. 39, 231–255 (2000).
Erhardt, A. et al. Plasma levels of vitamin E and carotenoids are decreased in patients with nonalcoholic steatohepatitis (NASH). Eur. J. Med. Res. 16, 76–78 (2011).
Hasegawa, T., Yoneda, M., Nakamura, K., Makino, I. & Terano, A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharm. Ther. 15, 1667–1672 (2001).
Nan, Y. M. et al. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol. 44, 1121–1131 (2009).
Bartolini, D. et al. Nonalcoholic fatty liver disease impairs the cytochrome P-450-dependent metabolism of α-tocopherol (vitamin E). J. Nutr. Biochem. 47, 120–131 (2017).
Violet, P. C. et al. Vitamin E sequestration by liver fat in humans. JCI Insight 5, e133309 (2020).
Chalasani, N. et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018).
Sumida, Y. et al. Role of vitamin E in the treatment of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 177, 391–403 (2021).
Sanyal, A. J. et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 362, 1675–1685 (2010).
Zang, S. et al. Haptoglobin genotype and vitamin E versus placebo for the treatment of nondiabetic patients with nonalcoholic steatohepatitis in China: a multicenter, randomized, placebo-controlled trial design. Adv. Ther. 35, 218–231 (2018).
Ni, Y. et al. Prevention and reversal of lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice by an antioxidant carotenoid, β-cryptoxanthin. Endocrinology 156, 987–999 (2015).
Akhavan Rezayat, A. et al. The effects of melatonin therapy on the treatment of patients with Non-alcoholic steatohepatitis: a systematic review and meta-analysis on clinical trial studies. Eur. J. Pharm. 905, 174154 (2021).
Dludla, P. V. et al. Coenzyme Q(10) supplementation improves adipokine levels and alleviates inflammation and lipid peroxidation in conditions of metabolic syndrome: a meta-analysis of randomized controlled trials. Int. J. Mol. Sci. 21, 3247 (2020).
Inzaugarat, M. E. et al. New evidence for the therapeutic potential of curcumin to treat nonalcoholic fatty liver disease in humans. PLoS One 12, e0172900 (2017).
Lee, D. E., Lee, S. J., Kim, S. J., Lee, H. S. & Kwon, O. S. Curcumin ameliorates nonalcoholic fatty liver disease through inhibition of O-GlcNAcylation. Nutrients 11, 2702 (2019).
Kuzu, N. et al. Epigallocatechin gallate attenuates experimental non-alcoholic steatohepatitis induced by high fat diet. J. Gastroenterol. Hepatol. 23, e465–e470 (2008).
Xiao, J. et al. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 53, 187–199 (2014).
Gautheron, J., Gores, G. J. & Rodrigues, C. M. P. Lytic cell death in metabolic liver disease. J. Hepatol. 73, 394–408 (2020).
Shojaie, L., Iorga, A. & Dara, L. Cell death in liver diseases: a review. Int. J. Mol. Sci. 21, 9682 (2020).
Feldstein, A. E. et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125, 437–443 (2003).
Johnson, E. S. et al. Metabolomic profiling reveals a role for caspase-2 in lipoapoptosis. J. Biol. Chem. 288, 14463–14475 (2013).
Li, C. P., Li, J. H., He, S. Y., Li, P. & Zhong, X. L. Roles of Fas/Fasl, Bcl-2/Bax, and Caspase-8 in rat nonalcoholic fatty liver disease pathogenesis. Genet Mol. Res. 13, 3991–3999 (2014).
Ramalho, R. M. et al. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol. 18, 21–29 (2006).
Malhi, H., Bronk, S. F., Werneburg, N. W. & Gores, G. J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 281, 12093–12101 (2006).
Kodama, Y. et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 137, 1467–1477.e1465 (2009).
Win, S. et al. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 62, 1367–1374 (2015).
Feldstein, A. E. et al. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 50, 1072–1078 (2009).
Wieckowska, A. et al. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 44, 27–33 (2006).
Cazanave, S. C. et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 286, 39336–39348 (2011).
Cazanave, S. C. et al. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G236–G243 (2010).
Barreyro, F. J. et al. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 282, 27141–27154 (2007).
Akazawa, Y. et al. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 52, 586–593 (2010).
Hirsova, P. & Gores, G. J. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 1, 17–27 (2015).
Item, F. et al. Fas cell surface death receptor controls hepatic lipid metabolism by regulating mitochondrial function. Nat. Commun. 8, 480 (2017).
Idrissova, L. et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J. Hepatol. 62, 1156–1163 (2015).
Cullen, S. P. et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 49, 1034–1048 (2013).
Castro, R. E. et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 58, 119–125 (2013).
Challa, T. D. et al. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol. Med. 11, e10124 (2019).
Barreyro, F. J. et al. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 35, 953–966 (2015).
Shiffman, M. et al. Randomised clinical trial: emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment Pharm. Ther. 49, 64–73 (2019).
Frenette, C. T. et al. Emricasan improves liver function in patients with cirrhosis and high model for end-stage liver disease scores compared with placebo. Clin. Gastroenterol. Hepatol. 17, 774–783.e774 (2019).
Harrison, S. A. et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 72, 816–827 (2020).
Yoon, Y. C. et al. Selonsertib inhibits liver fibrosis via downregulation of ASK1/MAPK pathway of hepatic stellate cells. Biomol. Ther. (Seoul.) 28, 527–536 (2020).
Mukherjee, S. et al. Development and validation of an in vitro 3D model of NASH with severe fibrotic phenotype. Am. J. Transl. Res. 11, 1531–1540 (2019).
Strobel, S. et al. A 3D primary human cell-based in vitro model of non-alcoholic steatohepatitis for efficacy testing of clinical drug candidates. Sci. Rep. 11, 22765 (2021).
Harrison, S. A. et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J. Hepatol. 73, 26–39 (2020).
Hong, S. W. et al. p34 (SEI-1) inhibits ROS-induced cell death through suppression of ASK1. Cancer Biol. Ther. 12, 421–426 (2011).
Bunkoczi, G. et al. Structural and functional characterization of the human protein kinase ASK1. Structure 15, 1215–1226 (2007).
Wang, P. X. et al. Corrigendum: targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 23, 1241 (2017).
Zhang, P. et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 24, 84–94 (2018).
Da, S. L. N. et al. Inhibition of ATG3 ameliorates liver steatosis by increasing mitochondrial function. J. Hepatol. 76, 11–24 (2022).
Porteiro, B. et al. Hepatic p63 regulates steatosis via IKKβ/ER stress. Nat. Commun. 8, 15111 (2017).
Farrell, G. C. et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 24, 443–452 (2009).
Porteiro, B. et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 8, 132–143 (2018).
Park, H. S. et al. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 17, 2549–2564 (2021).
Afonso, M. B. et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. (Lond.) 129, 721–739 (2015).
Majdi, A. et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 72, 627–635 (2020).
Roychowdhury, S. et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 64, 1518–1533 (2016).
Xu, H. et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 23, 14–23 (2019).
Wu, X. et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 73, 616–627 (2020).
Yang, F. et al. TNFα-mediated necroptosis aggravates ischemia-reperfusion injury in the fatty liver by regulating the inflammatory response. Oxid. Med. Cell Longev. 2019, 2301903 (2019).
Fink, S. L. & Cookson, B. T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 8, 1812–1825 (2006).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Beier, J. I. & Banales, J. M. Pyroptosis: an inflammatory link between NAFLD and NASH with potential therapeutic implications. J. Hepatol. 68, 643–645 (2018).
Xu, B. et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 68, 773–782 (2018).
Csak, T. et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54, 133–144 (2011).
Mridha, A. R. et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 66, 1037–1046 (2017).
Gaul, S. et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 74, 156–167 (2021).
Ezquerro, S. et al. Ghrelin reduces TNF-α-induced human hepatocyte apoptosis, autophagy, and pyroptosis: role in obesity-associated NAFLD. J. Clin. Endocrinol. Metab. 104, 21–37 (2019).
Hagström, H. et al. Elevated serum ferritin is associated with increased mortality in non-alcoholic fatty liver disease after 16 years of follow-up. Liver Int. 36, 1688–1695 (2016).
Buzzetti, E. et al. Evaluating the association of serum ferritin and hepatic iron with disease severity in non-alcoholic fatty liver disease. Liver Int. 39, 1325–1334 (2019).
Kowdley, K. V. et al. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 55, 77–85 (2012).
Gao, G. et al. Dehydroabietic acid improves nonalcoholic fatty liver disease through activating the Keap1/Nrf2-ARE signaling pathway to reduce ferroptosis. J. Nat. Med. 75, 540–552 (2021).
Li, X. et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 40, 1378–1394 (2020).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Qi, J., Kim, J. W., Zhou, Z., Lim, C. W. & Kim, B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 190, 68–81 (2020).
Slocum, S. L. et al. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch. Biochem. Biophys. 591, 57–65 (2016).
Liu, B., Yi, W., Mao, X., Yang, L. & Rao, C. Enoyl coenzyme A hydratase 1 alleviates nonalcoholic steatohepatitis in mice by suppressing hepatic ferroptosis. Am. J. Physiol. Endocrinol. Metab. 320, E925–E937 (2021).
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C. & Stockwell, B. R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278 (2016).
Kwon, M. Y., Park, E., Lee, S. J. & Chung, S. W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403 (2015).
Wang, S. J. et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 17, 366–373 (2016).
Capelletti, M. M., Manceau, H., Puy, H. & Peoc’h, K. Ferroptosis in liver diseases: an overview. Int. J. Mol. Sci. 21, 4908 (2020).
Zhang, Z. et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14, 2083–2103 (2018).
Zhang, Z. et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16, 1482–1505 (2020).
Zhang, Z. et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 36, 101619 (2020).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol. Immunol. 18, 1106–1121 (2021).
Wen, Y., Lambrecht, J., Ju, C. & Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 18, 45–56 (2021).
Dixon, L. J., Barnes, M., Tang, H., Pritchard, M. T. & Nagy, L. E. Kupffer cells in the liver. Compr. Physiol. 3, 785–797 (2013).
Remmerie, A., Martens, L. & Scott, C. L. Macrophage subsets in obesity, aligning the liver and adipose tissue. Front Endocrinol. (Lausanne) 11, 259 (2020).
Shi, H. et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 (2006).
Senn, J. J. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J. Biol. Chem. 281, 26865–26875 (2006).
Roh, Y. S. & Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 28(Suppl 1), 38–42 (2013).
Pan, J. et al. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 332, 111–120 (2018).
Baeck, C. et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426 (2012).
Morinaga, H. et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64, 1120–1130 (2015).
Parker, R. et al. CC chemokine receptor 2 promotes recruitment of myeloid cells associated with insulin resistance in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G483–g493 (2018).
Bertola, A. et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 5, e13577 (2010).
Ou, R. et al. Neutrophil depletion improves diet-induced non-alcoholic fatty liver disease in mice. Endocrine 57, 72–82 (2017).
Khoury, T. et al. Neutrophil-to-lymphocyte ratio is independently associated with inflammatory activity and fibrosis grade in nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 31, 1110–1115 (2019).
van der Windt, D. J. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68, 1347–1360 (2018).
Pulli, B. et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid. Redox Signal 23, 1255–1269 (2015).
Chen, J. et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem. Biophys. Res. Commun. 518, 691–697 (2019).
Mirea, A. M. et al. Increased proteinase 3 and neutrophil elastase plasma concentrations are associated with non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. Mol. Med. 25, 16 (2019).
Talukdar, S. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 18, 1407–1412 (2012).
Ye, D. et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 65, 988–997 (2016).
Sutti, S. & Albano, E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 17, 81–92 (2020).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Rau, M. et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J. Immunol. 196, 97–105 (2016).
Gomes, A. L. et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30, 161–175 (2016).
Luo, X. Y. et al. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G891–G899 (2013).
Haas, J. T. et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 1, 604–614 (2019).
Ghazarian, M. et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2, eaai7616 (2017).
Bruzzì, S. et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 124, 249–259 (2018).
Béland, K., Marceau, G., Labardy, A., Bourbonnais, S. & Alvarez, F. Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology 62, 1511–1523 (2015).
Thapa, M. et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology 61, 2067–2079 (2015).
McPherson, S., Henderson, E., Burt, A. D., Day, C. P. & Anstee, Q. M. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 60, 1055–1062 (2014).
Parthasarathy, G. & Malhi, H. Macrophage heterogeneity in NASH: more than just nomenclature. Hepatology 74, 515–518 (2021).
Lefebvre, E. et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 11, e0158156 (2016).
Ratziu, V. et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology 72, 892–905 (2020).
Friedman, S. et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp. Clin. Trials 47, 356–365 (2016).
Dibra, D. et al. Mutant p53 in concert with an interleukin-27 receptor alpha deficiency causes spontaneous liver inflammation, fibrosis, and steatosis in mice. Hepatology 63, 1000–1012 (2016).
Wang, Q. et al. IL-27 signalling promotes adipocyte thermogenesis and energy expenditure. Nature 600, 314–318 (2021).
Meng, F. et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 143, 765–776.e763 (2012).
Widjaja, A. A. et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology 157, 777–792.e714 (2019).
Ghanbari, M. et al. Interleukin-1 in obesity-related low-grade inflammation: From molecular mechanisms to therapeutic strategies. Int. Immunopharmacol. 96, 107765 (2021).
Schreuder, H. et al. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 386, 194–200 (1997).
Abu-Elsaad, N. & El-Karef, A. Protection against nonalcoholic steatohepatitis through targeting IL-18 and IL-1alpha by luteolin. Pharm. Rep. 71, 688–694 (2019).
Schumacher, M., Juncker, T., Schnekenburger, M., Gaascht, F. & Diederich, M. Natural compounds as inflammation inhibitors. Genes Nutr. 6, 89–92 (2011).
Leite, N. C. et al. Efficacy of diacerein in reducing liver steatosis and fibrosis in patients with type 2 diabetes and non-alcoholic fatty liver disease: a randomized, placebo-controlled trial. Diabetes Obes. Metab. 21, 1266–1270 (2019).
Van Wagner, L. B. et al. Pentoxifylline for the treatment of non-alcoholic steatohepatitis: a randomized controlled trial. Ann. Hepatol. 10, 277–286 (2011).
Zein, C. O. et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology 54, 1610–1619 (2011).
Ipsen, D. H. et al. The effect of acetylsalicylic acid and pentoxifylline in guinea pigs with non-alcoholic steatohepatitis. Basic Clin. Pharm. Toxicol. 128, 583–593 (2021).
Yalcin, M. et al. A comparison of the effects of infliximab, adalimumab, and pentoxifylline on rats with non-alcoholic steatohepatitis. Turk. J. Gastroenterol. 25(Suppl 1), 167–175 (2014).
Koca, S. S. et al. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation 31, 91–98 (2008).
Pinto, L. F. et al. The immunosuppressant drug, thalidomide, improves hepatic alterations induced by a high-fat diet in mice. Liver Int. 30, 603–610 (2010).
Boning, L. I. U., Xiang, W. U., Ke, X. U., Huiqin, Y. A. N. & Qinghua, Z. Application of Isothiocyanate to Preparation of Medicine for Preventing and Treating Medicine-resistance Tumor (Tianjin Medical University General Hospital, 2010).
Dattaroy, D. et al. Sparstolonin B (SsnB) attenuates liver fibrosis via a parallel conjugate pathway involving P53-P21 axis, TGF-beta signaling and focal adhesion that is TLR4 dependent. Eur. J. Pharm. 841, 33–48 (2018).
Yang, G., Lee, H. E. & Lee, J. Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 6, 24399 (2016).
Calcagno, D. et al. Nlrp3 activation causes spontaneous inflammation and fibrosis that mimics human NASH. Hepatology (2022). Online ahead of print.
Baeza-Raja, B. et al. Pharmacological inhibition of P2RX7 ameliorates liver injury by reducing inflammation and fibrosis. PLoS One 15, e0234038 (2020).
Mederacke, I. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823 (2013).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Di Gregorio, J. et al. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front Cell Dev. Biol. 8, 607483 (2020).
David, C. J. & Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 19, 419–435 (2018).
Finnson, K. W., Almadani, Y. & Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Semin Cell Dev. Biol. 101, 115–122 (2020).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 (2003).
Mi, X. J. et al. Maltol mitigates thioacetamide-induced liver fibrosis through TGF-β1-mediated activation of PI3K/Akt signaling pathway. J. Agric. Food Chem. 67, 1392–1401 (2019).
Hart, K. M. et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci. Transl. Med. 9, eaal3694 (2017).
Grohmann, M. et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 175, 1289–1306.e1220 (2018).
Tan, Z. et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 191, 1835–1844 (2013).
Kocabayoglu, P. et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 63, 141–147 (2015).
Chen, W. et al. Lysyl oxidase (LOX) family members: rationale and their potential as therapeutic targets for liver fibrosis. Hepatology 72, 729–741 (2020).
Sanyal, A. J. et al. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: data from the simtuzumab trials. Hepatology 70, 1913–1927 (2019).
Younossi, Z. M. et al. The association of histologic and noninvasive tests with adverse clinical and patient-reported outcomes in patients with advanced fibrosis due to nonalcoholic steatohepatitis. Gastroenterology 160, 1608–1619.e1613 (2021).
Harrison, S. A. et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 155, 1140–1153 (2018).
Chalasani, N. et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 158, 1334–1345.e1335 (2020).
Al, A. A., Antaramian, A. & Noureddin, M. Review of galectin-3 inhibitors in the treatment of nonalcoholic steatohepatitis. Expert Rev. Clin. Pharm. 14, 457–464 (2021).
Ito, S. & Nagata, K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 294, 2133–2141 (2019).
Lawitz, E. J. et al. BMS-986263 in patients with advanced hepatic fibrosis: 36-week results from a randomized, placebo-controlled phase 2 trial. Hepatology 75, 912–923 (2022).
Yoshiji, H. et al. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Res. Notes 2, 70 (2009).
Luangmonkong, T. et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharm. 174, 3107–3117 (2017).
Yingling, J. M. et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 9, 6659–6677 (2018).
Pahk, K. et al. SP-1154, a novel synthetic TGF-beta inhibitor, alleviates obesity and hepatic steatosis in high-fat diet-induced mice. Biomed. Pharmacother. 145, 112441 (2022).
Hui, S. T. et al. Oxy210, a novel inhibitor of hedgehog and TGF-beta signalling, ameliorates hepatic fibrosis and hypercholesterolemia in mice. Endocrinol. Diabetes Metab. 4, e00296 (2021).
Liu, R. X. et al. Tetrodecadazinone, a novel tetrodecamycin-pyridazinone hybrid with anti-liver fibrosis activity from Streptomyces sp. HU051. Bioorg. Chem. 119, 105573 (2022).
Son, Y. J. et al. Yellow loosestrife (Lysimachia vulgaris var. davurica) ameliorates liver fibrosis in db/db mice with methionine- and choline-deficient diet-induced nonalcoholic steatohepatitis. BMC Complement Med. Ther. 21, 44 (2021).
Choi, M. E., Ding, Y. & Kim, S. I. TGF-β signaling via TAK1 pathway: role in kidney fibrosis. Semin. Nephrol. 32, 244–252 (2012).
Liu, Y. et al. Tumor necrosis factor alpha-induced protein 8-like 2 alleviates nonalcoholic fatty liver disease through suppressing transforming growth factor beta-activated kinase 1 activation. Hepatology 74, 1300–1318 (2021).
Wu, L., Liu, Y., Zhao, Y., Li, M. & Guo, L. Targeting DUSP7 signaling alleviates hepatic steatosis, inflammation and oxidative stress in high fat diet (HFD)-fed mice via suppression of TAK1. Free Radic. Biol. Med. 153, 140–158 (2020).
Lan, T. et al. Breviscapine alleviates NASH by inhibiting TGF-beta-activated kinase 1-dependent signaling. Hepatology 76, 155–171 (2022).
Melhem, A. et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 45, 60–71 (2006).
Yang, F. et al. Crosstalk between hepatic stellate cells and surrounding cells in hepatic fibrosis. Int. Immunopharmacol. 99, 108051 (2021).
Bansal, R. et al. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology 54, 586–596 (2011).
Jin, J. et al. Increased expression of enzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Rep. 3, 831–843 (2013).
Jin, J. et al. Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Rep. 16, 744–756 (2016).
Wang, X., Niu, C., Zhang, X. & Dong, M. Emodin suppresses activation of hepatic stellate cells through p38 mitogen-activated protein kinase and Smad signaling pathways in vitro. Phytother. Res. 32, 2436–2446 (2018).
Zheng, Q., Li, S., Li, X. & Liu, R. Advances in the study of emodin: an update on pharmacological properties and mechanistic basis. Chin. Med. 16, 102 (2021).
Samuelson, I. & Vidal-Puig, A. J. Fed-EXosome: extracellular vesicles and cell-cell communication in metabolic regulation. Essays Biochem. 62, 165–175 (2018).
Nakao, Y. et al. Circulating extracellular vesicles are a biomarker for NAFLD resolution and response to weight loss surgery. Nanomedicine 36, 102430 (2021).
Povero, D. et al. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One 9, e113651 (2014).
Hernández, A. et al. Extracellular vesicles in NAFLD/ALD: from pathobiology to therapy. Cells 9, 817 (2020).
Ipsen, D. H. & Tveden-Nyborg, P. Extracellular vesicles as drivers of non-alcoholic fatty liver disease: small particles with big impact. Biomedicines 9, 93 (2021).
Morán, L. & Cubero, F. J. Extracellular vesicles in liver disease and beyond. World J. Gastroenterol. 24, 4519–4526 (2018).
Povero, D. et al. Characterization and proteome of circulating extracellular vesicles as potential biomarkers for NASH. Hepatol. Commun. 4, 1263–1278 (2020).
Newman, L. A. et al. Selective isolation of liver-derived extracellular vesicles redefines performance of miRNA biomarkers for non-alcoholic fatty liver disease. Biomedicines 10, 195 (2022).
Zhao, Y. et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat. Commun. 11, 719 (2020).
Povero, D. et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell Mol. Gastroenterol. Hepatol. 1, 646–663.e644 (2015).
He, Y. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J. Clin. Invest. 131, e141513 (2021).
Hou, X. et al. Myeloid-cell-specific IL-6 signaling promotes microRNA-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology 74, 116–132 (2021).
Bruno, S. et al. HLSC-derived extracellular vesicles attenuate liver fibrosis and inflammation in a murine model of non-alcoholic steatohepatitis. Mol. Ther. 28, 479–489 (2020).
Chen, L. et al. Therapeutic effects of serum extracellular vesicles in liver fibrosis. J. Extracell. Vesicles 7, 1461505 (2018).
Dasgupta, D. et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology 159, 1487–1503.e1417 (2020).
Liu, X. L. et al. Lipotoxic hepatocyte-derived exosomal microRNA 192-5p activates macrophages through Rictor/Akt/Forkhead Box Transcription Factor O1 signaling in nonalcoholic fatty liver disease. Hepatology 72, 454–469 (2020).
Zuo, R. et al. Hepatic small extracellular vesicles promote microvascular endothelial hyperpermeability during NAFLD via novel-miRNA-7. J. Nanobiotechnology 19, 396 (2021).
Jiang, F. et al. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 72, 156–166 (2020).
Mandato, C., Delli Bovi, A. P. & Vajro, P. The gut-liver axis as a target of liver disease management. Hepatobiliary Surg. Nutr. 10, 100–102 (2021).
Di Ciaula, A. et al. Liver steatosis, gut-liver axis, microbiome and environmental factors. a never-ending bidirectional cross-talk. J. Clin. Med. 9, 2648 (2020).
Li, Z. et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 37, 343–350 (2003).
Alferink, L. J. M. et al. Microbiomics, metabolomics, predicted metagenomics, and hepatic steatosis in a population-based study of 1,355 adults. Hepatology 73, 968–982 (2021).
Gupta, B. et al. Western diet-induced increase in colonic bile acids compromises epithelial barrier in nonalcoholic steatohepatitis. FASEB J. 34, 7089–7102 (2020).
Fei, N. et al. Endotoxin producers overgrowing in human gut microbiota as the causative agents for nonalcoholic fatty liver disease. mBio 11, e03263–19 (2020).
Wahlström, A., Sayin, S. I., Marschall, H. U. & Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016).
Zhang, L. et al. Farnesoid X receptor signaling shapes the gut microbiota and controls hepatic lipid metabolism. mSystems 1, e00070–16 (2016).
Xie, C. et al. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 66, 613–626 (2017).
Sayin, S. I. et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235 (2013).
Swann, J. R. et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl Acad. Sci. USA 108(Suppl 1), 4523–4530 (2011).
Gu, Y. et al. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat. Commun. 8, 1785 (2017).
Kars, M. et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59, 1899–1905 (2010).
Kübeck, R. et al. Dietary fat and gut microbiota interactions determine diet-induced obesity in mice. Mol. Metab. 5, 1162–1174 (2016).
Loomba, R. et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054–1062.e1055 (2017).
Le Roy, T. et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794 (2013).
Dong, T. S. et al. A microbial signature identifies advanced fibrosis in patients with chronic liver disease mainly due to NAFLD. Sci. Rep. 10, 2771 (2020).
Munukka, E. et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 11, 1667–1679 (2017).
Machado, A. S. et al. Oral probiotic bifidobacterium longum supplementation improves metabolic parameters and alters the expression of the renin-angiotensin system in obese mice liver. Biol. Res. Nurs. 23, 100–108 (2021).
Lee, N. Y. et al. Lactobacillus attenuates progression of nonalcoholic fatty liver disease by lowering cholesterol and steatosis. Clin. Mol. Hepatol. 27, 110–124 (2021).
Yu, J. S. et al. Lactobacillus lactis and Pediococcus pentosaceus-driven reprogramming of gut microbiome and metabolome ameliorates the progression of non-alcoholic fatty liver disease. Clin. Transl. Med. 11, e634 (2021).
Yuan, J. et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 30, 675–688.e677 (2019).
Rau, M. et al. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. U. Eur. Gastroenterol. J. 6, 1496–1507 (2018).
Müller, M. et al. Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci. Rep. 9, 12515 (2019).
Zhou, D. et al. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp. Mol. Med. 50, 1–12 (2018).
Fang, W., Xue, H., Chen, X., Chen, K. & Ling, W. Supplementation with sodium butyrate modulates the composition of the gut microbiota and ameliorates high-fat diet-induced obesity in mice. J. Nutr. 149, 747–754 (2019).
Zhou, D. et al. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 23, 60–75 (2017).
Jin, C. J., Sellmann, C., Engstler, A. J., Ziegenhardt, D. & Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 114, 1745–1755 (2015).
Baumann, A. et al. Oral supplementation of sodium butyrate attenuates the progression of non-alcoholic steatohepatitis. Nutrients 12, 951 (2020).
Behary, J. et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 12, 187 (2021).
Rom, O. et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci. Transl. Med. 12, eaaz2841 (2020).
Shimada, Y. et al. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS One 8, e80604 (2013).
Sellmann, C. et al. Oral arginine supplementation protects female mice from the onset of non-alcoholic steatohepatitis. Amino Acids 49, 1215–1225 (2017).
Jegatheesan, P. et al. Citrulline and nonessential amino acids prevent fructose-induced nonalcoholic fatty liver disease in rats. J. Nutr. 145, 2273–2279 (2015).
Jegatheesan, P. et al. Preventive effects of citrulline on western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 116, 191–203 (2016).
Koh, A. et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175, 947–961.e917 (2018).
Zhao, M. et al. TMAVA, a metabolite of intestinal microbes, is increased in plasma from patients with liver steatosis, inhibits γ-butyrobetaine hydroxylase, and exacerbates fatty liver in mice. Gastroenterology 158, 2266–2281.e2227 (2020).
Hoyles, L. et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 24, 1070–1080 (2018).
Zhu, W. et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165, 111–124 (2016).
Zeisel, S. H., Wishnok, J. S. & Blusztajn, J. K. Formation of methylamines from ingested choline and lecithin. J. Pharm. Exp. Ther. 225, 320–324 (1983).
Yoo, W. et al. High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 373, 813–818 (2021).
Chen, Y. M. et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 6, 19076 (2016).
Tan, X. et al. Trimethylamine N-oxide aggravates liver steatosis through modulation of bile acid metabolism and inhibition of farnesoid X receptor signaling in nonalcoholic fatty liver disease. Mol. Nutr. Food Res. 63, e1900257 (2019).
Portincasa, P. et al. Intestinal barrier and permeability in health, obesity and NAFLD. Biomedicines 10, 83 (2021).
De Munck, T. J. I. et al. Intestinal permeability in human nonalcoholic fatty liver disease: a systematic review and meta-analysis. Liver Int. 40, 2906–2916 (2020).
Kessoku, T. et al. The role of leaky gut in nonalcoholic fatty liver disease: a novel therapeutic target. Int. J. Mol. Sci. 22, 8161 (2021).
Harte, A. L. et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. (Lond.) 7, 15 (2010).
Stenman, L. K., Holma, R. & Korpela, R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J. Gastroenterol. 18, 923–929 (2012).
Cani, P. D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007).
Cani, P. D. et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103 (2009).
Cani, P. D. et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008).
Baratta, F. et al. Poor adherence to mediterranean diet and serum lipopolysaccharide are associated with oxidative stress in patients with non-alcoholic fatty liver disease. Nutrients 12, 1732 (2020).
Carpino, G. et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 72, 470–485 (2020).
Sharifnia, T. et al. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G270–G278 (2015).
Xue, L. et al. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci. Rep. 7, 45176 (2017).
Musso, G., Gambino, R. & Cassader, M. Gut microbiota as a regulator of energy homeostasis and ectopic fat deposition: mechanisms and implications for metabolic disorders. Curr. Opin. Lipido. 21, 76–83 (2010).
Wong, V. W. et al. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann. Hepatol. 12, 256–262 (2013).
Zhou, D. et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 7, 1529 (2017).
Mohamad, N. M. H. et al. The effect of probiotics (MCP((R)) BCMC((R)) Strains) on hepatic steatosis, small intestinal mucosal immune function, and intestinal barrier in patients with non-alcoholic fatty liver disease. Nutrients 13, 3192 (2021).
Xu, R. et al. CCR2-overexpressing mesenchymal stem cells targeting damaged liver enhance recovery of acute liver failure. Stem Cell Res. Ther. 13, 55 (2022).
Lotfy, A., El-Metwaly, S., Soliman, R., Hassan, A. A. & Shiha, G. Umbilical cord mesenchymal stem/stromal cells and low-dose obeticholic acid as a possible combined treatment for liver fibrosis. Stem Cell Rev. Rep. 18, 1875–1877 (2022).
Xu, X., Wang, W., Lin, L. & Chen, P. Liraglutide in combination with human umbilical cord mesenchymal stem cell could improve liver lesions by modulating TLR4/NF-kB inflammatory pathway and oxidative stress in T2DM/NAFLD rats. Tissue Cell 66, 101382 (2020).
Zhang, G. Z., Sun, H. C., Zheng, L. B., Guo, J. B. & Zhang, X. L. In vivo hepatic differentiation potential of human umbilical cord-derived mesenchymal stem cells: therapeutic effect on liver fibrosis/cirrhosis. World J. Gastroenterol. 23, 8152–8168 (2017).
Cheng, L. et al. Human umbilical cord-derived mesenchymal stem cell-exosomal miR-627-5p ameliorates non-alcoholic fatty liver disease by repressing FTO expression. Hum. Cell 34, 1697–1708 (2021).
Chiabotto, G., Ceccotti, E., Tapparo, M., Camussi, G. & Bruno, S. Human liver stem cell-derived extracellular vesicles target hepatic stellate cells and attenuate their pro-fibrotic phenotype. Front Cell Dev. Biol. 9, 777462 (2021).
Watanabe, T. et al. Development of a non-alcoholic steatohepatitis model with rapid accumulation of fibrosis, and its treatment using mesenchymal stem cells and their small extracellular vesicles. Regen. Ther. 14, 252–261 (2020).
Bi, Y. et al. Bone marrow derived-mesenchymal stem cell improves diabetes-associated fatty liver via mitochondria transformation in mice. Stem Cell Res. Ther. 12, 602 (2021).
He, Y. et al. Human umbilical cord-derived mesenchymal stem cells improve the function of liver in rats with acute-on-chronic liver failure via downregulating Notch and Stat1/Stat3 signaling. Stem Cell Res. Ther. 12, 396 (2021).
Ishida, K. et al. Restorative effect of adipose tissue-derived stem cells on impaired hepatocytes through Notch signaling in non-alcoholic steatohepatitis mice. Stem Cell Res. 54, 102425 (2021).
Khadrawy, S. M., Mohamed, H. M. & Mahmoud, A. M. Mesenchymal stem cells ameliorate oxidative stress, inflammation, and hepatic fibrosis via Nrf2/HO-1 signaling pathway in rats. Environ. Sci. Pollut. Res. Int. 28, 2019–2030 (2021).
Li, Y. H. et al. Mesenchymal stem cells attenuate liver fibrosis by targeting Ly6C(hi/lo) macrophages through activating the cytokine-paracrine and apoptotic pathways. Cell Death Disco. 7, 239 (2021).
Yu, Z. et al. Extracellular vesicle-encapsulated microRNA-375 from bone marrow-derived mesenchymal stem cells inhibits hepatocellular carcinoma progression through regulating HOXB3-mediated Wnt/beta-catenin pathway. Anal. Cell Pathol. (Amst.) 2022, 9302496 (2022).
Yang, N. et al. Transplantation of adipose-derived stem cells ameliorates Echinococcus multilocularis-induced liver fibrosis in mice. PLoS Negl. Trop. Dis. 16, e0010175 (2022).
Yamato, M. et al. Adipose tissue-derived stem cells prevent fibrosis in murine steatohepatitis by suppressing IL-17-mediated inflammation. J. Gastroenterol. Hepatol. 34, 1432–1440 (2019).
Shi, M. et al. Mesenchymal stem cell therapy in decompensated liver cirrhosis: a long-term follow-up analysis of the randomized controlled clinical trial. Hepatol. Int. 15, 1431–1441 (2021).
Huang, D. Q., El-Serag, H. B. & Loomba, R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18, 223–238 (2021).
Finn, R. S. et al. Atezolizumab plus Bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020).
Finn, R. S. et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 38, 193–202 (2020).
Duffy, A. G. et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 66, 545–551 (2017).
Sangro, B. et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 59, 81–88 (2013).
Pfister, D. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592, 450–456 (2021).
Peng, Y., Wong, C. C. & Yu, J. The paradox of immunotherapy in NASH-HCC. Signal Transduct. Target Ther. 6, 228 (2021).
Yau, T. et al. LBA38_PR - CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 30, v874–v875 (2019).
Hindson, J. T cells in NASH and liver cancer: pathology and immunotherapy. Nat. Rev. Gastroenterol. Hepatol. 18, 367 (2021).
Anstee, Q. M. et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 73, 505–515 (2020).
Abul-Husn, N. S. et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106 (2018).
Luukkonen, P. K. et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight 5, e132158 (2020).
Ma, Y. et al. 17-Beta hydroxysteroid dehydrogenase 13 deficiency does not protect mice from obesogenic diet injury. Hepatology 73, 1701–1716 (2021).
Zhang, H. B., Su, W., Xu, H., Zhang, X. Y. & Guan, Y. F. HSD17B13: a potential therapeutic target for NAFLD. Front Mol. Biosci. 8, 824776 (2021).
Bruschi, F. V., Tardelli, M., Claudel, T. & Trauner, M. PNPLA3 expression and its impact on the liver: current perspectives. Hepat. Med. 9, 55–66 (2017).
Wang, Y., Kory, N., BasuRay, S., Cohen, J. C. & Hobbs, H. H. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 69, 2427–2441 (2019).
Lindén, D. et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 22, 49–61 (2019).
Loomba, R. et al. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 5, 829–838 (2020).
Wang, X. et al. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol. Commun. 3, 1221–1234 (2019).
Zhu, C. et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 10, eaat0344 (2018).
EMA. Draft reflection paper on regulatory requirements for the development of medicinal products for chronic non-infectious liver diseases (PBC, PSC, NASH) Committee for Medicinal Products for Human Use (CHMP) (2018).
Loomba, R. et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 73, 625–643 (2021).
Pedrosa, M. et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 88, 105889 (2020).
Zhou, Z. et al. Discovery of new and highly effective quadruple FFA1 and PPARalpha/gamma/delta agonists as potential anti-fatty liver agents. Eur. J. Med. Chem. 229, 114061 (2022).
Gawrieh, S. et al. Saroglitazar, a PPAR-alpha/gamma agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology 74, 1809–1824 (2021).
Nakajima, A. et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharm. Ther. 54, 1263–1277 (2021).
Lawitz, E. J. et al. Fenofibrate mitigates hypertriglyceridemia in nonalcoholic steatohepatitis patients treated with cilofexor/firsocostat. Clin. Gastroenterol. Hepatol. (2022). Online ahead of print.
Oscarsson, J. et al. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: a double-blind, randomized, placebo-controlled study. J. Clin. Lipido. 12, 1390–1403.e1394 (2018).
Olson, E. J., Pearce, G. L., Jones, N. P. & Sprecher, D. L. Lipid effects of peroxisome proliferator-activated receptor-δ agonist GW501516 in subjects with low high-density lipoprotein cholesterol: characteristics of metabolic syndrome. Arterioscler Thromb. Vasc. Biol. 32, 2289–2294 (2012).
Riserus, U. et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 57, 332–339 (2008).
CymaBay Therapeutics. CymaBay Therapeutics reports topline 12-week data from an ongoing phase 2b study of seladelpar in patients with nonalcoholic steatohepatitis. CymaBay. https://ir.cymabay.com/pressreleases?year=2019&page=2 (2019).
Shao, N. et al. Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 30, 521–529 (2014).
Buse, J. B. et al. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: an interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clin. Ther. 29, 139–153 (2007).
Khoo, J. et al. Randomized trial comparing effects of weight loss by liraglutide with lifestyle modification in non-alcoholic fatty liver disease. Liver Int. 39, 941–949 (2019).
Jepsen, M. M. & Christensen, M. B. Emerging glucagon-like peptide 1 receptor agonists for the treatment of obesity. Expert Opin. Emerg. Drugs 26, 231–243 (2021).
Alba, M., Yee, J., Frustaci, M. E., Samtani, M. N. & Fleck, P. Efficacy and safety of glucagon-like peptide-1/glucagon receptor co-agonist JNJ-64565111 in individuals with obesity without type 2 diabetes mellitus: a randomized dose-ranging study. Clin. Obes. 11, e12432 (2021).
Di Prospero, N. A. et al. Efficacy and safety of glucagon-like peptide-1/glucagon receptor co-agonist JNJ-64565111 in individuals with type 2 diabetes mellitus and obesity: a randomized dose-ranging study. Clin. Obes. 11, e12433 (2021).
Shankar, S. S. et al. Native oxyntomodulin has significant glucoregulatory effects independent of weight loss in obese humans with and without type 2 diabetes. Diabetes 67, 1105–1112 (2018).
Alam, S., Ghosh, J., Mustafa, G., Kamal, M. & Ahmad, N. Effect of sitagliptin on hepatic histological activity and fibrosis of nonalcoholic steatohepatitis patients: a 1-year randomized control trial. Hepat. Med. 10, 23–31 (2018).
Kuchay, M. S. et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial). Diabetes Care 41, 1801–1808 (2018).
Eriksson, J. W. et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia 61, 1923–1934 (2018).
Inoue, M. et al. Effects of canagliflozin on body composition and hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease. J. Diabetes Investig. 10, 1004–1011 (2019).
Ozaki, A. et al. Effect of tofogliflozin and pioglitazone on hepatic steatosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a randomized, open-label pilot study (ToPiND study). Contemp. Clin. Trials Commun. 17, 100516 (2020).
Shibuya, T. et al. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective randomized controlled pilot study. Diabetes Obes. Metab. 20, 438–442 (2018).
Fouqueray, P. et al. Pharmacodynamic effects of direct AMP kinase activation in humans with insulin resistance and non-alcoholic fatty liver disease: a phase 1b study. Cell Rep. Med. 2, 100474 (2021).
Kimura, Y. et al. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 45, 750–757 (2010).
Wang, Y., Li, X. & Ren, S. Cholesterol metabolites 25-hydroxycholesterol and 25-hydroxycholesterol 3-sulfate are potent paired regulators: from discovery to clinical usage. Metabolites 11, 9 (2020).
Stefan, N. et al. Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2, 406–416 (2014).
Fouda, A. et al. A randomized controlled trial comparing the effects of vitamin E, Ursodeoxycholic acid and Pentoxifylline on Egyptian non-alcoholic steatohepatitis patients. Eur. Rev. Med. Pharm. Sci. 25, 7449–7459 (2021).
Nadinskaia, M. et al. Ursodeoxycholic acid as a means of preventing atherosclerosis, steatosis and liver fibrosis in patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 27, 959–975 (2021).
Ratziu, V. et al. REGENERATE: design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 84, 105803 (2019).
Ratziu, V. et al. EDP-305 in patients with NASH: a phase II double-blind placebo-controlled dose-ranging study. J. Hepatol. 76, 506–517 (2021).
Tully, D. C. et al. Discovery of Tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J. Med. Chem. 60, 9960–9973 (2017).
Xiao, Y. et al. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut-liver axis. Liver Int. 41, 2117–2131 (2021).
Fiorucci, S., Biagioli, M., Sepe, V., Zampella, A. & Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 29, 623–632 (2020).
Chianelli, D. et al. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. J. Med. Chem. 63, 3868–3880 (2020).
Charles, E. D. et al. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obes. (Silver Spring) 27, 41–49 (2019).
Hupa-Breier, K. L. et al. Dulaglutide alone and in combination with empagliflozin attenuate inflammatory pathways and microbiome dysbiosis in a non-diabetic mouse model of NASH. Biomedicines 9, 353 (2021).
Lee, J. et al. Dulaglutide ameliorates palmitic acid-induced hepatic steatosis by activating FAM3A signaling pathway. Endocrinol. Metab. (Seoul.) 37, 74–83 (2022).
Kim, E. R. et al. A GLP-1/GLP-2 receptor dual agonist to treat NASH: Targeting the gut-liver axis and microbiome. Hepatology 75, 1523–1538 (2022).
Ma, T. et al. A novel long-acting oxyntomodulin analogue eliminates diabetes and obesity in mice. Eur. J. Med. Chem. 203, 112496 (2020).
Harriman, G. et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl Acad. Sci. USA 113, E1796–E1805 (2016).
Lally, J. S. V. et al. Inhibition of Acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 29, 174–182.e175 (2019).
Gao, Y. S. et al. WZ66, a novel acetyl-CoA carboxylase inhibitor, alleviates nonalcoholic steatohepatitis (NASH) in mice. Acta Pharm. Sin. 41, 336–347 (2020).
Zhang, X. J. et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci. Transl. Med. 13, eabg8116 (2021).
Xiao, W., Ren, M., Zhang, C., Li, S. & An, W. Amelioration of nonalcoholic fatty liver disease by hepatic stimulator substance via preservation of carnitine palmitoyl transferase-1 activity. Am. J. Physiol. Cell Physiol. 309, C215–C227 (2015).
Jiang, S. Y. et al. Discovery of a INSIG binding compound that ameliorates nonalcoholic steatohepatitis by inhibiting SREBP-mediated lipogenesis. Hepatology (2022). Online ahead of print.
Guo, T. et al. LIFR-alpha-dependent adipocyte signaling in obesity limits adipose expansion contributing to fatty liver disease. iScience 24, 102227 (2021).
Sylvester, D. S. et al. Hepatoprotective effect of bisbenzylisoquinoline alkaloid tiliamosine from Tiliacora racemosa in high-fat diet/diethylnitrosamine-induced non-alcoholic steatohepatitis. Biomed. Pharmacother. 108, 963–973 (2018).
Columbano, A., Chiellini, G. & Kowalik, M. A. GC-1: a thyromimetic with multiple therapeutic applications in liver disease. Gene Expr. 17, 265–275 (2017).
Zarei, M. et al. Oral administration of a new HRI activator as a new strategy to improve high-fat-diet-induced glucose intolerance, hepatic steatosis, and hypertriglyceridaemia through FGF21. Br. J. Pharm. 176, 2292–2305 (2019).
Loomba, R. et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology 67, 549–559 (2018).
Garcia-Tsao, G. et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 72, 885–895 (2020).
Vilar-Gomez, E. et al. Vitamin E improves transplant-free survival and hepatic decompensation among patients with nonalcoholic steatohepatitis and advanced fibrosis. Hepatology 71, 495–509 (2020).
Harrison, S. A. et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharm. Ther. 44, 1183–1198 (2016).
Suk, K. T. et al. Transplantation with autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: phase 2 trial. Hepatology 64, 2185–2197 (2016).
Wang, P. X. et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 23, 439–449 (2017).
Lan, T. et al. Hepatocyte glutathione S-transferase mu 2 prevents non-alcoholic steatohepatitis by suppressing ASK1 signaling. J. Hepatol. 76, 407–419 (2022).
Xu, F. et al. Annexin A5 regulates hepatic macrophage polarization via directly targeting PKM2 and ameliorates NASH. Redox Biol. 36, 101634 (2020).
Wang, J., Liu, Z., Gao, M. & Xie, Z. Application of FR180204 in Preparation of Drug for Prevention and/or Treatment of Non-alcoholic Fatty Liver Disease or Hepatitis. 7 (Beijing GigaCeuticals Co., Ltd., P. R. China, 2021).
Wang, J., Liu, Z., Gao, M. & Xie, Z. Application of Ravoxertinib in Preparation of Drugs for Prevention and/or Treatment of Non-alcoholic Fatty Liver Disease or Hepatitis. 7 (Beijing GigaCeuticals Co., Ltd., P. R. China, 2021).
Nishizawa, H. et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 6, 34605 (2016).
Tao, X. et al. Overexpression of PDE4D in mouse liver is sufficient to trigger NAFLD and hypertension in a CD36-TGF-β1 pathway: therapeutic role of roflumilast. Pharm. Res. 175, 106004 (2022).
Xu, Y. et al. Hepatocyte nuclear factor 4alpha prevents the steatosis-to-NASH progression by regulating p53 and bile acid signaling (in mice). Hepatology 73, 2251–2265 (2021).
Gao, X. et al. Matrine attenuates endoplasmic reticulum stress and mitochondrion dysfunction in nonalcoholic fatty liver disease by regulating SERCA pathway. J. Transl. Med. 16, 319 (2018).
Mahzari, A. et al. Matrine protects against MCD-induced development of NASH via upregulating HSP72 and downregulating mTOR in a manner distinctive from metformin. Front Pharm. 10, 405 (2019).
Sugino, H. et al. Polaprezinc attenuates liver fibrosis in a mouse model of non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 23, 1909–1916 (2008).
Murray, J. K. et al. Identification and optimization of a minor allele-specific siRNA to prevent PNPLA3 I148M-driven nonalcoholic fatty liver disease. Nucleic Acid Ther. 31, 324–340 (2021).
Cherubini, A., Casirati, E., Tomasi, M. & Valenti, L. PNPLA3 as a therapeutic target for fatty liver disease: the evidence to date. Expert Opin. Ther. Targets 25, 1033–1043 (2021).
Schwartz, B. E. et al. Discovery and targeting of the signaling controls of PNPLA3 to effectively reduce transcription, expression, and function in pre-clinical NAFLD/NASH settings. Cells 9, 2247 (2020).
Imarisio, C. et al. Adenosine A(2a) receptor stimulation prevents hepatocyte lipotoxicity and non-alcoholic steatohepatitis (NASH) in rats. Clin. Sci. (Lond.) 123, 323–332 (2012).
Chen, L., Sun, B. & Xu, C. Rassf4 as Diabetes Complicated of Alcohol Resistance Fat Alcohol Hepatopathy and Liver Cancer Treatment of Target and Application [Machine Translation]. 19 (Zhuxianyi Memorial Hospital, Tianjin Medical University, P. R. China, 2021).
Koh, E. H. et al. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut 70, 1954–1964 (2021).
Acknowledgements
We thank Dr. Han Wang and Dr. Xiaoqin Wu (Cleveland Clinic, OH, USA) for critical comments and advice on the manuscript. This work was supported by the Major Program of National Natural Science Foundation of China (81991525) and Key R&D Program of Shandong Province (2020CXGC010503). This work was also supported by grants from the National Institutes of Health (R00AA026648 to K.L.P.).
Author information
Authors and Affiliations
Contributions
X.X., L.W., and S.L. wrote the manuscript and created the figures and tables. K.L.P., T.M., Q.S., Q.W., C.Z., and C.L. revised the initial manuscript and figures. J.Y. provided the conceptual idea and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Poulsen, K.L., Wu, L. et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Sig Transduct Target Ther 7, 287 (2022). https://doi.org/10.1038/s41392-022-01119-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01119-3
This article is cited by
-
Exploring the role of genetic variations in NAFLD: implications for disease pathogenesis and precision medicine approaches
European Journal of Medical Research (2024)
-
Integrated traditional Chinese and Western medicine in the prevention and treatment of non-alcoholic fatty liver disease: future directions and strategies
Chinese Medicine (2024)
-
Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases
Signal Transduction and Targeted Therapy (2024)
-
Gut-Brain Axis Modulation of Metabolic Disorders: Exploring the Intertwined Neurohumoral Pathways and Therapeutic Prospects
Neurochemical Research (2024)
-
Mesenchymal stromal/stem cells and their extracellular vesicles in liver diseases: insights on their immunomodulatory roles and clinical applications
Cell & Bioscience (2023)
