
Abstract
Epigenetics is closely related to cardiovascular diseases. Genome-wide linkage and association analyses and candidate gene approaches illustrate the multigenic complexity of cardiovascular disease. Several epigenetic mechanisms, such as DNA methylation, histone modification, and noncoding RNA, which are of importance for cardiovascular disease development and regression. Targeting epigenetic key enzymes, especially the DNA methyltransferases, histone methyltransferases, histone acetylases, histone deacetylases and their regulated target genes, could represent an attractive new route for the diagnosis and treatment of cardiovascular diseases. Herein, we summarize the knowledge on epigenetic history and essential regulatory mechanisms in cardiovascular diseases. Furthermore, we discuss the preclinical studies and drugs that are targeted these epigenetic key enzymes for cardiovascular diseases therapy. Finally, we conclude the clinical trials that are going to target some of these processes.
Similar content being viewed by others

Introduction
Cardiovascular diseases remain a major cause of destruction of human health worldwide.1 In the worldwide, especially China and India have the highest burden of cardiovascular disease.2 It includes coronary heart disease, hypertension, heart failure, vascular calcification and so on. In the Framingham Heart Study,3 which was launched since 1948 covering three generations during the past 70 years, cardiovascular disease is well studied and highly related to multiple factors such as biochemical, environmental, behavioral and genetics factors. Early detection and diagnosis of cardiovascular diseases are essential to improve the treatment and prognosis of cardiovascular disease.
With the rapid development of modern society, the incidence of cardiovascular diseases shows an increasing trend year by year, and the onset age is gradually younger. The causes of cardiovascular diseases such as coronary heart disease, heart failure, and hypertension are closely related to environmental factors and genetic factors. Recent studies have found that epigenetic modification plays an important role in the occurrence and development of cardiovascular diseases. Epigenetics is a regulation mechanism that can perpetuate alternative gene function/expression/activity without changing the content of DNA sequence. It is considered as the major response regulation mechanism for the cell response to the environmental changes.4 Epigenetics mainly regulates cardiovascular disease-related genes function and expression level through DNA methylation, histone modification, and noncoding RNA regulation, thus affecting cardiovascular disease progression. Epigenetic markers are important molecular markers of cardiovascular disease because they occur early in the disease and involve key cardiovascular pathologically related pathways. Most importantly, it can be used as cardiovascular disease biomarkers for cardiovascular disease diagnosis, treatment response prediction and evaluation. As we all know, the pathogenesis of cardiovascular disease remains intricate and complex. Clinically, some cases are still difficult to cure, and the prevalence rate increases with age. Interestingly, because of the reversibility of epigenetic modifications, genes and proteins that control these changes have become new targets for cardiovascular disease treatment. Therefore, new therapeutic strategies based on epigenetic modification have aroused great interest. The development of effective therapies for cardiovascular diseases is an urgent clinical need.
In this review, we illustrate (1) the epigenetics history and general mechanisms, (2) epigenetic regulatory mechanisms in cardiovascular disease, (3) epigenetics as a potential strategy for treating cardiovascular disease. The epigenetics in cardiovascular disease is yet to be cleared. There will be more comprehensive and large-scale epigenetics studies published in future. The novel epigenetics regulation mechanisms will provide additional opportunities to more promising cardiovascular disease diagnostic markers and potential therapeutic solutions. It is hoped that continued research in this area will further enhance the understanding of the cardiovascular disease process, which in order to get a better discovery of the new therapeutic strategy and improve the life quality of cardiovascular disease patients.
Overview of epigenetics
Epigenetics history brief review
The English biologist C.H. Waddington first proposed the term “epigenetics” in 1942, which ostensibly means “changes in non-genetic sequences.5 It was not until the 1980s that the British molecular biologist R Holliday systematically reformulated “Epigenetic” systematically in an academic paper according to the consensus that DNA methylation can change gene activity.6 In 1990, Holliday defined epigenetics as the study of the temporal and spatial mechanisms subjects by which genes activity during the development of complex organisms.7 In 1996, American geneticist Athur D. Riggs and others defined epigenetics as the genetic changes caused by mitosis or meiosis in the function of genes without changing the genetic sequence.8,9 In 2007, the British geneticist S A Bird defined epigenetics as the structural adjustment of the chromosomal region that causes it to express, emit signal, or maintain altered activity state.10 In 2008, at a cold Spring Harbor conference, the nature of epigenetics was recognized as chromosomal changes that caused stable inherited phenotype in the absence of DNA order changes. In addition, according to the extension of epigenetics research, the United States NATIONAL Institutes of Health (NIH) in 2013 believed that epigenetics included both cells or individuals gene activity and expression inherited changes, and stable, long-term, and uninherited changes at the potential level of cell transcription. At present, the widely accepted concept of epigenetics is the study that not DNA sequence changes caused heritable gene expression changes.11 The epigenetic mechanisms are discovered and widely accepted because it regulates gene expression without changing DNA sequence by covalent modifications made to histone proteins and nucleic acids that cooperatively regulate chromatin structure. The epigenetic regulations are reversible and dynamically regulated gene expression. It indicates that epigenetic mechanisms might play more important roles in biology. It also opens the possibility to develop epigenetic drugs for certain diseases. Taken together, the above section is a brief review of epigenetics history. Epigenetics mechanisms will be described below.
Epigenetic regulatory mechanisms
DNA methylation
DNA methylation is a normal and universal modification in eukaryotic cells. It is also the main epigenetic form of gene expression regulation in mammals. There are several ways of methylated modification, but most of them occur on cytosine phosphate guanine (CpG) islands in the gene promoter region. DNA methylation is an important epigenetic mechanism. It can transfer genetic information to offspring DNA through the regulation of DNA methyltransferases (DNMTs) (Fig. 1).

DNA methylation regulation. DNA methylation occurs mainly in the islands of cytosinephosphateguanine (CpG) gene promoter region. It promotes gene transcription in the promoter region by activating DNA methyltransferases. DNA methylases can be divided into three categories according to their roles in DNA methylation: writing enzymes, erasing enzymes, and reading enzymes. Writing enzymes catalyze the addition of methyl groups to cytosine residues. The function of erasing enzymes is to modify and remove methyl groups. Reading enzymes can recognize and bind methyl groups to affect gene expression. This figure was created with the aid of Biorender (https://biorender.com/). DNMTs DNA methyltransferases, MBD methyl-CPG-binding domain, ZBTB zinc finger and broad complex, tramtrack, and bric a brac, TET Ten Eleven Translocation, MECP2 methyl-CpG-binding protein 2, UNG uracil–DNA glycosylase
DNA methylases can be divided into three categories according to their roles in DNA methylation: writing enzymes, erasing enzymes, and reading enzymes. Writing enzymes catalyze the addition of methyl groups to cytosine residues, including DNMT1, DNMT3a, and DNBT3b. Although these enzymes have large N-terminal regulatory domain and c-terminal catalytic domain, there are differences in function and expression patterns.12 DNMT1 can not only accurately mimic the original methylation pattern before DNA replication, but also repair DNA methylation.13 DNMT3a and DNMT3b are also known as de novo DNMT, which can introduce methylation into naked DNA and establish a new methylation pattern for unmodified DNA. DNMT3a and DNMT3b targeting specific DNA sequences may be mediated by transcription factors. They regulate de novo synthetic DNA methylation. DNMTs can bind components of transcription factors or repressor complexes to target DNA methylation.14 In addition, transcription factor binding can help protect CpG sites from de novo methylation. The function of eraser enzyme is to modify and remove methyl groups. DNA demethylation can occur either actively or passively. Passive demethylation means that maintenance of DNMTs during mitosis fails to methylate the newly synthesized DNA strand. However, the molecular mechanism that catalyzes active DNA demethylation has not been elucidated. Reading enzymes can recognize and bind methyl groups to affect gene expression. Proteins in the reading enzymes structural domain (adaptors) are mainly involved in gene expression. The main function of these structural domains is to recruit factors related to DNA metabolism progress, including DNA replication, transcription, DNA recombination, and DNA damage repair. Three protein families recognize DNA methylation: the methyl-CPG-binding domain (MBD) protein, the UHRF protein, and the zinc finger protein. MBD protein contains a conserved methyl-CPG-binding domain (MBD), which has a high affinity for a single methylated CpG site.15 The MBD family includes MeCP2, the first methyl-binding protein to be identified, as well as MBD1, MBD2, MBD3, and MBD4.16 The UHRF family of proteins maintains DNA methylation by binding to DNMT1 and targeting semi-methylated DNA.17 The zinc finger protein family consists of Kaiso, ZBTB4, and ZBTB38. It inhibits transcription mainly through DNA methylation-dependent mode.18
Histone modification
Histone modification refers to the process of histone modification such as methylation, acetylation, phosphorylation, adenylation, ubiquitination, and adenosine diphosphate ribosylation under the action of related enzymes. Modification of histones can change the loose or agglutinating state of chromatin by affecting the affinity between histones and DNA double strands. Gene regulation can also be performed by influencing the affinity between other transcription factors and structural genes promoters.19 Among them, histone methylation and acetylation are well studied (Fig. 2).

Histone-modification regulation. Histone modification refers to the process of histone modification such as methylation, acetylation, phosphorylation, adenylation, ubiquitination, and adenosine diphosphate ribosylation under the action of related enzymes. Among them, histone methylation and acetylation are well studied. Histone methyltransferases are mainly involved in the regulation of histone methylation, which transfers methyl groups to histones lysine residues, whereas, histone demethylases have the opposite effect. The methylation action site is on the N atom of the lysine side chain. Histone acetyltransferases are mainly involved in the regulation of histone acetylation, which transfers acetyl groups to histones lysine residues. However, histone deacetylases have the opposite effect. This figure was created with the aid of Biorender (https://biorender.com/). H3K9 histone H3 lysine 9, H3K4 histone H3 lysine 4
Histone methylation
Histone methylation is one of the most important post-transcriptional modifications. Histone methyltransferase is mainly involved in the regulation of histone methylation, which transfers methyl groups to histones lysine residues using S-adenosine methionine as a substrate. The methylation site is on the N atom of the side chain of lysine and arginine. Common histone methylation includes H3K4 methylation, H3K9 methylation, and H3K27 methylation. Lysine methylation is relatively stable in gene expression regulation. The methylation of lysine residues at the fourth site of H3 is associated with gene activation, while the methylation of lysine residues at the ninth and 27th sites is associated with gene silencing. However, histone arginine methylation is a relatively dynamic marker. Arginine methylation is associated with gene activation, whereas loss of arginine methylation in H3 and H4 is associated with gene silencing. Histone H3K4 methylation mainly concentrates in regions of active transcription, such as the transcription start site, promoter, and enhancer regions. H3K4me1 was enriched in the enhancer region and correlated with H3K27ac or H3K27me3, marking active or inhibitory enhancers, respectively.20 Histone H3K9 methylation, especially H3K9me2 and trimethylated histone 3 lysine 9 (H3K9me3), generally regulates heterochromatin formation and gene inhibition.21 SUV39H1 and SUV39H2 catalyze the formation of H3K9me2 and H3K9me3.22 Enhancer of zeste homolog-2 (EZH2) can methylate H3K27, leading to silencing of related genes.
Histone acetylation
Histone acetylation mainly occurs at the more conserved lysine sites at the N-terminus of H3 and H4. It is coordinated by histone acetyltransferases (HAT) and histone deacetylases (HDACs). Acetylation may regulate gene transcription through its effects on histone charges and interacting proteins. Therefore, histone acetylation is generally considered as an active histone marker. In 1996, researchers discovered the HAT P300 and cyclic AMP response element-binding protein (CBP).23 CBP binds to P300 to form the CBP/P300 complex, which recruits other HAT, such as PCAF (P300/CBP-related factor). According to the structural similarities and substrates of HDAC molecules, the HDACs family can be divided into four categories. HDAC1, 2, 3, and 8 belong to class I RPD3-like proteins. Class II HDACs can be divided into two subclasses: Class IIa (HDAC4, 5, 7, and 9) and Class IIb (HDAC6 and HDAC10). Class III HDACs are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, mainly including Sirtuin (SIRT)1-SIRT7. Class IV protein is HDAC11. There are many lysine residues in histones that can be acetylated, such as H3K4, H3K9, H3K27, etc. Acetylation of lysine regulates functional changes in proteins by altering its structure or affinity for other binding partners. Therefore, lysine acetylation can regulate cancer,24 cardiovascular disease25 and other diseases. In the lysine acetyltransferase (KATs), P300 is a transcription coactivator that regulates gene expression by activating the intrinsic KAT.26 In addition, HDACs have been found to play an important role in the pathological processes of inflammation,27 cardiac hypertrophy, and heart failure.28
Noncoding RNAs
Noncoding RNAs refer to RNAs that do not encode proteins, including ribosomal RNAs, transport RNAs, small nuclear RNAs, small nucleolar RNAs, microRNAs (miRNAs), mRNA, and other known functions, as well as those with unknown functions. These RNAs have common feature that they can be transcribed from the genome but not translated into proteins, performing their respective biological functions at the RNA level (Fig. 3). Noncoding RNAs can be divided into three categories according to length: <50 nt: miRNA, small interfering RNAs, etc. 50–500 nt: ribosomal RNA, transport RNA, nuclear small RNA, nucleolar small RNA, etc. >500 nt: long mRNA-like ncRNAs, long noncoding RNAs (lncRNAs) without polyadenylate tails, etc.

Noncoding RNA regulation. Different mechanisms of action of noncoding RNAs in epigenetic regulations. (1) <50 nt: MicroRNAs (miRNAs): miRNAs complement mRNAs and promote mRNA silencing or degradation. Small interfering RNAs(siRNA): silences gene expression. (2) 50–500 nt: nucleolar small RNA(snoRNA): snoRNA biological function was initially found to modify rRNA. Nuclear small RNA(snRNA): snRNA function is to combine with protein factors to form small nuclear ribonucleoprotein particle and perform the function of splicing mRNA. Transport RNA (tRNA): the main function is to carry amino acids into the ribosome and synthetic proteins with the guidance of mRNA. Ribosomal RNA (rRNA): it binds to proteins to form ribosomes.Its function is to act as a scaffold for mRNA, enabling mRNA molecules to unfold on it to achieve protein synthesis. (3) >500 nt: long noncoding RNAs (lncRNAs): LncRNA acts as mRNA and miRNA endogenous sponges regulating gene expression. Circular RNAs (cirRNAs): circRNA molecules are rich in miRNA-binding sites and act as miRNA sponges in cells, thereby lifting the inhibition of miRNA on target genes and increasing the expression level of target genes. This figure was created with the aid of Biorender (https://biorender.com/)
miRNAs
miRNAs are a major class of small ncRNAs, about 22 nucleotides in length, found in animals, plants, and some viruses. Most miRNAs genes are transcribed primarily by RNA polymerase II (Pol II) into large primary miRNAs (PRI‐miRNAs), which contain one or several stem ring structures, each consisting of about 70 nucleotides. MiRNAs play a role in the post-transcriptional regulation of gene expression. Transcription levels are regulated by tissue-specific epigenetic modifications.29 Thus, miRNA genes are not only targets of epigenetic modifications such as DNA methylation, but also regulators of DNMTs and HDACs.29 miRNAs not only occupy a very important position in the progression of cardiovascular disease, such as cardiac hypertrophy and myocardial cell fibrosis,30 miRNAs can also serve as therapeutic targets for disease.31
LncRNAs
LncRNAs are nucleotides over 200 in length that cannot be converted into proteins. LncRNAs based on function mechanism that are divided into activated ncRNAs with enhancer-like properties (ncRNA-a), competitive endogenous RNAs (ceRNAs), primary transcripts for mi- and piRNAs. NcRNAs-a positively regulate nearby genes, which are the main difference from enhancer RNAs (eRNAs). CeRNAs share a sequence with the transcript encoding the protein. CeRNAs can competitively bind its regulated molecules to perform better function.32 LncRNA regulates gene expression patterns by altering chromatin structure and DNA accessibility through molecular mechanisms such as signaling, bait, guidance and scaffolding.33 LncRNAs have high functional specificity by participating in and regulating various cellular processes such as DNA methylation and histone modification.34 Furthermore, LncRNAs not only regulate cardiovascular diseases, but also are increasingly attached importance in the progression of blood-related diseases.
Circular RNAs
Circular RNAs (CirRNAs) are long, noncoding endogenous RNA molecules with single-stranded covalently closed RNA loops without 5'-Cap and 3’-poly(A) ends. With the development of sequencing technology, several types of cirRNAs have been discovered and identified. There are four main subtypes: exon cirRNAs (ecircRNAs), which are mainly derived from single or multiple exons; circular intron-type cirRNAs, which contain only introns; exon–intron-type cirRNAs (EIciRNAs), which consist of exons and introns. At present, most cirRNAs identified are ecircRNAs.35 Accumulated data suggest that cirRNAs exert their regulatory functions through the following mechanisms: (1) acting as miRNA sponges. CirRNAs regulate the expression of target genes and mRNA translation by interacting with miRNA. (2) as a protein scaffold. CirRNAs can bind to RNA binding proteins or functional proteins to regulate their function and transport. (3) as an important molecule of transcriptional regulation. CircEIF3J and circPAIP2 interact with U1snRNA and RNA polymerase II complexes to enhance transcriptional activity. (4) as a template for protein synthesis. It can participate in protein translation.36 CirRNAs are widespread and diverse in eukaryotic cells,37 but relatively stable in the cytoplasm.38 CirRNAs can be co-transcribed and post-transcribed by a process of reverse splicing or head-tail circular splicing, in which the downstream exon splices in reverse order to the upstream exon.39 CirRNAs are transformed into linear RNA by targeting miRNA and regulate gene expression. CirRNA is more stable than linear RNA isoforms because it lacks an accessible terminal that can resist RNA exonuclease. The functional mechanism of cirRNA is thought to be to change the level of free miRNA in sponges by interacting with miRNA, and then regulate the expression of disease-related proteins.40 In addition, cirRNA is involved in regulating the progression of cardiovascular and autoimmune diseases.40,41,42 Therefore, cirRNA can be used as one of the potential strategies for clinical diagnosis and disease treatment. The epigenetic regulatory mechanisms have been summarized above (Table 1). In the third part, we mainly discuss the research progress of epigenetics in cardiovascular disease in recent years.
Epigenetic regulatory mechanisms in cardiovascular disease
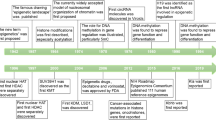
In recent years, epigenetics has been occupied an indispensable position in the cardiovascular diseases historical development progress. The correlation of epigenetics with cardiovascular diseases has primarily been identified in the function and expression of epigenetic-related enzymes found in cardiovascular diseases. To better understand the discovery and research history of epigenetics in cardiovascular diseases, it is helpful to review the timeline of epigenetics43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63 (Fig. 4).

Important historical advances of epigenetics in cardiovascular diseases. This figure was created with the aid of Biorender (https://biorender.com/). MCT3 monocarboxylate transporter 3, HDAC histone deacetylase, SAHA suberoylanilide hydroxamic acid, Lp(A) lipoprotein(A), TET2 TET-methylcytidine dioxygenase-2, DOT1L disruptor of telomeric silencing 1-like, NF-kB nuclear transcription factor-kappa B
The role of DNA methylation in cardiovascular disease
Several studies have shown that DNA methylation plays important roles in cardiovascular diseases (Table 2). In recent years, it has been found that the expression of candidate genes related to coronary heart disease, heart failure, hypertension, and other cardiovascular diseases is associated with DNA methylation. The abnormal methylation status of candidate genes is involved in the mechanism and development of cardiovascular disease and can be used as a marker to assess cardiovascular disease progression.
Coronary heart disease and myocardial infarction
In genome-wide Bonferroni multiple assay correction, Westerman et al.64 found that DNA methylation in three regions (associated with SLC9A1, SLC1A5, and TNRC6C genes) was associated with cardiovascular disease risk. Mendelian randomization analysis showed that one CpG (CG22304262) in SLC1A5 had a causal relationship with incident coronary heart disease (iCHD). The DNA methylation level of CG22304262 may affect SLC1A5 expression.65 Myocardial glutamine storage disorder and SLC1A5 expression were decreased in patients with heart failure. Inhibition of SLC1A5 expression in the myocardium reduced glutamine uptake and impaired glutamine homeostasis in failing myocardium.66 In conclusion, this study explores new blood DNA methylation regions associated with iCHD and has important implications for improving clinical risk prediction.64 In iCHD’s latest Epigenome-wide association studies, Navas-Acien et al.67 identified a biological association between differentially methylated position and atherosclerosis. In this study, blood DNA methylation was associated with coronary heart disease over and above the traditional factors associated with cardiovascular disease. Meanwhile, there were large differences and complex epigenomic characteristics in different populations.67
A study performed a comprehensive analysis of DNA methylation and mRNA expression datasets at a series of time points in a mouse model of acute myocardial infarction (AMI). It was found that the most critical stage of AMI was 6 h. A large number of methylation modification sites were changed during this stage. Five genes (Ptpn6, Csf1r, Col6a1, Cyba, and Map3k14) were identified to participate in AMI process by regulating DNA methylation. These candidate genes are expected to be methylated biomarkers for early clinical diagnosis of acute myocardial infarction in future.68 Framingham offspring’s cohort study of DNA methylation found that four independent underlying factors (9, 19, 21, 27) driven by DNA methylation were associated with cardiovascular disease. In addition, three genes contained in factor 27 were also associated with myocardial infarction factors (CDC42BPB, MAN2A2, and RPTOR). Recent multifactorial approaches integrating DNA methylation and gene expression data provide new insights into the pathogenesis of the cardiovascular disease.69 Thirty-four new DNA methylation sites associated with AMI were identified in two-stage Epigenome-wide association studies. Four of them were associated with coronary heart disease. Cg21566642 was located in the intergenic region, cg05575921, cg04988978, and cg25769469 were labeled AHRR, MPO, and PTCD2, respectively. MPO encodes myeloperoxidase, which promotes atherosclerotic lesions by enhancing APOB oxidation in low-density lipoprotein (LDL)70 and is causally associated with cardiovascular events.71 A CpG located in PTCD2 has previously been associated with hypertension in patients with obstructive sleep apnea.72 Genetic variants of this gene have been associated with blood pressure.73 These differential methylated genes are enriched in various molecular and physiological pathways such as lipid metabolism and inflammatory diseases. They are closely related to the pathogenesis of coronary heart disease and AMI.74
Heart failure
Recently, the regulatory role of DNA methylation in cardiac hypertrophy and heart failure has attracted much attention. However, its exact role in cardiomyocytes remains controversial. Madsen et al.75 showed that DNA methylation of DNA methyltransferase DNMT3a was involved in the homeostasis of human cardiomyocytes. Knockout of DNMT3a not only changed the expression of contractile protein gene in cardiomyocytes, but also resulted in cardiomyocytes mitochondrial damage and impaired glucose metabolism. Therefore, regulating the abnormal DNA methylation process is of great significance for further understanding heart failure pathogenesis.75 Glezeva et al.76 detected 195 distinct regions of differential methylation in a cohort studying heart failure, primarily distributed in hypertrophic obstructive cardiomyopathy, dilated cardiomyopathy, and ischemic cardiomyopathy. In addition, five genes (HEY2, MSR1, MYOM3, COX17, and miRNA-24-1) were found to be hypermethylated in the ventricular septal tissues of heart failure patients that included hypertrophic obstructive cardiomyopathy, ischemic cardiomyopathy, and dilated cardiomyopathy. Three genes (CTGF, MMP2, and miRNA-155) showed hypomethylated state. This study supports the role of DNA methylation in the regulation of heart failure-related genes for different clinical causes. Therefore, these differentially expressed methylated genes in heart failure may be a new markers for the detection and diagnosis of heart failure.76 A study on the effect of genome-wide cardiac DNA methylation on overall gene expression in myocardial samples from patients with end-stage chronic Chagas disease cardiomyopathy (CCC) showed that two differentially expressed methylated genes, KCNA4 and KCNIP4, were involved in the regulation of potassium channels. They were upregulated in CCC and encoded potassium voltage-gated channels Kv1.4 and Kv4.3 to participate in electrical conduction and arrhythmias. The protein encoded by the differentially expressed methylated gene SMOC2 was upregulated in CCC and was involved in CCC matrix remodeling and fibrosis. Therefore, DNA methylation can reveal the pathogenesis and process of CCC by regulating CCC-related cardiac and immune system genes expression.77 In addition, Zhu et al.78 demonstrated that selenium supplementation could inhibit DNMT2-induced DNA methylation of glutathione peroxidase 1 gene promoter in cardiomyocytes, increased glutathione peroxidase 1 expression, further reduced intracellular reactive oxygen species production, and cardiomyocyte apoptosis, and thus played a protective role in heart failure.78
Vascular calcification
DNA methylations related molecules are considered to have the potential to be used as biomarkers for the diagnosis of vascular calcification. Dai et al.79 demonstrated that the expression and activity of S-adenosylhomocysteine hydrolase (SAHH) were reduced in calcified human coronary arteries. SAHH deficiency increased intracellular S-adenosylhomocysteine (SAH) levels, leading to hypomethylation and upregulated expression of H19 gene promoter through competitive inhibition of DNMT3b, Thus promoting H19-mediated runt-related transcription factor-2 (Runx2)-dependent vascular smooth muscle cell (VSMC) osteogenic differentiation and aggravating atherosclerotic calcification. In contrast, SAHH lacks reduced intracellular adenosine levels and AMPK (AmP-Activated protein kinase) activation. Adenosine supplementation activated AMPK. AMPK eliminated H19-mediated Runx2-dependent VSMC osteogenic differentiation by inducing sirt1-mediated low acetylation of histone H3 and DNMT3b-mediated hypermethylation of H19 promoter.79 Ramachandran et al.80 demonstrated that GTPase-activating protein-binding protein (G3BP1) n-terminal nuclear transporter-2 and c-terminal arginine methylation domains were important in activating osteogenic-related transcriptional reactions. G3BP1 methylation was enhanced by the knockout of the typical Wnt receptor LRP6 in mouse aortic vascular smooth muscle (VSM). It was accompanied by activation of osteogenic transcription programs mediated in part by Runx2. Furthermore, G3BP1 activated the transcription of activated T cells nuclear factor (namely NFATc4), and then promoted VSM NFATc4 association with osteopontin (OPN) and alkaline phosphatase (ALP) promoters. Thus G3BP1 could accelerate calcification process.80 High phosphate increases DNMTs activity in the smooth muscle 22a (SM22a) promoter region. SM22a promoter methylation reduces the SM22a gene expression, promotes the osteoblast transcription factor core-binding factor 1 expression and the ALP activity upregulation. Finally, SM22a promoter methylation leads to the VSMCs to osteoblast phenotype transformation and calcification.81 In addition, a study showed that miRNA-34b expression was significantly inhibited in VSMCs treated with high inorganic phosphate. Inhibition of miRNA-34b expression can enhance VSMCs calcification. Elevated DNMT3a induced miRNA-34b hypermethylation in VSMCs and reduced miRNA-34b expression, thereby promoting the occurrence of calcification. After DNMT3a siRNA knockout, the effect of high phosphate on VSMCs calcification disappeared. This is because DNMT3a knockout reduces the miRNA-34b methylation, and the expression level of miRNA-34b increases, acting on its downstream target Notch 1 and reducing VSMCs calcification.82 Lin et al.83 reported that in high phosphate-induced VSMCs, the increase of DNMT3a leaded to miRNA-204 hypermethylation and expression downregulation, thereby promoting VSMCs osteogenic differentiation. After DNMT3a small interfering RNA knockout DNMT3a, miRNA-204 expression was upregulated and VSMCs osteogenic differentiation was alleviated. Thus it eliminates the effect of high phosphate treatment on VSMC calcification.83
Hypertension
DNA methylation was shown to have an important function in hypertension development. A study exploring the association of whole blood DNA methylation with 24-h blood pressure phenotype and clinical blood pressure reported that 72 DNA methylation regions (MRs) were identified as significantly associated with 24-h blood pressure phenotypes (24-h mean, day and night) out of 1,549,368 CpG sites.84 Dwi Putra et al.85 found that mothers with a family history of hypertension had higher mean arterial pressure and lower overall placental DNA methylation in placental samples compared with mothers without a family history of hypertension. However, only in mothers with no family history of hypertension, overall placental DNA methylation was independently negatively associated with maternal mean arterial pressure.85 Jin et al.86 found that mitochondrial fusion 2 can inhibit VSMCs proliferation and is closely related to inflammation, oxidative stress and renin angiotensin system (RAS). Correlation analysis showed that mitochondrial fusion 2 gene methylation level was significantly lower in hypertensive patients than in the control group. Mitochondrial fusion 2 hypomethylation may downregulate the expression of this gene. Consequently, it led to VSMCs proliferation and endothelial cell damage, and then contributed to the development of hypertension.86 Bao et al.87 study result showed that hypomethylation of the interferon γ gene can induce vascular endothelial chemotaxis in a long-term inflammatory state hypertensive patients. At the same time, in vascular tissue, hypomethylation of the interferon γ gene increased VSMCs proliferation and lipid deposition. Therefore, it gave rise to the transformation of blood vessels from normal phenotype to vascular fibrosis, resulting in increased blood pressure.87 In conclusion, these studies indicate that DNA methylation is closely related to the occurrence of cardiovascular diseases.
The role of histone modification in cardiovascular disease
Histone modification is one of the important regulatory mechanisms in epigenetics. Abnormal histone modification results in an imbalance in the expression of genes associated with cardiovascular disease, resulting in changes in cellular phenotypes and cardiac function. Key molecules of histone modification (histone methylation and histone acetylation) may lead to the occurrence and progression of cardiovascular disease through their influence on cardiovascular pathophysiological pathways (Table 3).
The role of histone methylation in cardiovascular disease
Cardiovascular disease can also be regulated by histone methylation (Table 3). Papait et al.88 found that histone methyltransferase G9a had a synergistic effect with the catalytic subunit EZH2 of PRC2 on gene silencing. G9a inhibited cardiomyocytes' different types gene function through dimethylation of lysine 9 on histone H3 and interaction with EZH2. Therefore, G9a was essential to maintain correct gene expression in normal cardiomyocytes and to drive changes in the expression of genes associated with cardiac hypertrophy. The study results suggest that impaired G9a function can lead to cardiac dysfunction. G9a may be a potential target for the treatment of early myocardial hypertrophy in the future.88 Kurozumi et al.89 showed that interleukin (IL)-6/SIL-6R stimulation induces p-STAT3 activation and jumonji domain-containing protein (JMJD)2B recruitment. Runx2 gene expression is increased in human VSMCs (hVSMCs) by bivalent histone modification of the transcription enhancer trimethylation of lysine 4 of histone H3 (H3K4me3) and the transcription suppressor trimethylated histone 3 lysine 9 (H3K9me3). JMJD2B protein was highly expressed in hVSMCs. IL-6/SIL-6R stimulation may result in the recruitment of JMJD2B to stat-targeted sites in the Runx2 promoter region, which demethylated H3K9me3. Therefore it increased the osteoblast differentiation markers (ALP and OPN mRNA) expression, and then enhanced osteoblast differentiation and vascular calcification.89 SM22a encoded by Transgelin (TAGLN) is expressed in mesenchymal cells such as smooth muscle cells. Maleszewska et al.90 reported that TAGLN/SM22a expression was regulated at the epigenetic level by polycomb methyltransferase EZH2. Downregulated IL-1β and transforming growth factor-β (TGFβ)-2 increased the EZH2 expression, inhibited the TAGLN expression, and increased the H3K27me3 level at the proximal promoter of TAGLN. EZH2 regulated the chromatin structure of the TAGLN promoter through trimethylation of H3K27. In addition, activation of EZH2 decreased TGFβ2-induced SM22a and TAGLN expression. SM22a is essential for the maintenance of smooth muscle cell phenotype and function, which may lead to calcification.90 In conclusion, these studies suggest that histone methylation is strongly associated with cardiovascular disease physiopathologic mechanism.
The role of histone acetylation in cardiovascular disease
HATs and HDACs play a crucial effect in regulating histone acetylation. The role of HAT and HDACs-mediated epigenetic processes in vascular homeostasis and cardiovascular disease has received extensive attention (Table 3).
Atherosclerosis and myocardial infarction
Research has found that SIRT1 inhibited the formation of atherosclerotic plaques in ApoE−/− mice by regulating endothelial nitric oxide synthase (eNOS) activation. Thus SIRT1 slowed down the formation of atherosclerosis. However, HDAC3-mediated deacetylation of eNOS K610 promoted atherosclerosis.91,92,93 The level of inducible nitric oxide synthase (iNOS) is also increased in atherosclerotic lesions.94 In endothelial cells, nuclear factor-κB (NF-κB) promotes atherosclerosis by driving the expression of Nos2(encoding iNOS) and proinflammatory genes.95 SIRT1 inhibits NF-κB activity by deacetylating P65 and eliminating the interaction between P300 and NF-κB.96 It has been reported that lysine acetylation can regulate myocardium ischemia–reperfusion injury and myocardial infarction. Overexpression of SIRT1 can alleviate ischemia–reperfusion injury in rat myocardium.97,98 SIRT2 promotes cell apoptosis in renal ischemia–reperfusion injury by deacetylating Forkhead box O3A (FOXO3A).99 SIRT3 can inhibit reperfusion injury by deacetylation of cyclophilin D and prevent the opening of mitochondrial permeability transition pores, resulting in cell death.100 In addition, in rat models of myocardial ischemia–reperfusion injury, HDAC6 reduces the activity of peroxyredoxin 1 by deacetylating its K197 site. In the end, it leads to increased reactive oxygen species production and exacerbate oxidative damage of cardiomyocytes.101
Heart failure
Heart failure is characterized by dysapoptosis of myocardial cells, increased fibrotic scar tissue, and pathological myocardial hypertrophy. SIRT2 deficiency exacerbates angiotensin II-induced myocardial fibrosis.102 SIRT3 can activate GSK3β through K15 deacetylation, which in turn phosphorylates SMAD3 and leads to its degradation, thereby preventing TGF β-induced fibrosis.103 In contrast, SIRT4 depletion attenuated angiotensin II-induced myocardial fibrosis.104 Shen et al.105 found that SIRT6 levels were significantly reduced in phenylephrine-induced neonatal rat cardiomyocytes, which was associated with increased acetyltransferase P300 levels and cardiomyocyte hypertrophy. Overexpressed SIRT6 attenuated phenylephrine-induced cardiac hypertrophy by degrading P300.105 In vitro, knockdown of HDAC4 in human lung fibroblasts inhibits myofibroblast differentiation. Nevertheless, the knockdown of HDAC6 in rat heart fibroblasts blocks cell proliferation.106 These findings suggest that histone acetylation regulates myocardial cell fibrosis in the course of heart failure.
Pathological cardiac remodeling in heart failure is associated with dysregulated myocardial apoptosis.107 SIRT1 overexpression increased apoptosis of neonatal rat cardiomyocytes in vitro by reversing the acetylation of the isodimer receptor NOTCH1. Thus, the stability of NOTCH1 was decreased and the proliferation of cardiomyocytes was inhibited.108 However, overexpression of SIRT2 significantly increased cardiomyocyte hypertrophy, but protected cardiomyocytes from apoptosis under serum starvation in vitro.109 SIRT7 inhibited myocardial apoptosis in vitro by deacetylating p53 and increasing stress resistance.110 These results suggest that histone acetylation may be involved in the proliferation and apoptosis of cardiomyocytes.
Vascular calcification
In recent years, increasing evidence has accumulated for histone acetylation in vascular calcification progress. Li et al.111 showed that SIRT6 can deacetylate Runx2, further promoted Runx2 to go out of the nucleus in an exportin 1-dependent manner, and then degraded Runx2 through the ubiquitin–proteasome system, thereby reducing osteogenic differentiation of VSMCs and inhibiting vascular calcification. Therefore, this study illustrates the new potential of longevity protein SIRT6 in the treatment of vascular calcification, which provides a new intervention target and strategy for its clinical prevention and treatment.111 Abend et al.112 found that HDAC4 was upregulated in the early stage of VSMCs calcification. HDAC4 binding and its activity-induced osteocalcin upregulation in vitro VSMCs and aortic rings via the adaptor protein ENIGMA (Pdlim7). Overexpression of HDAC4 can upregulate SOX9, Runx2, ALP, proteoglycan, and calcium mineral accumulation. Therefore, these data identify HDAC4 as a positive regulator of the vascular calcification process.112 HDAC9, a member of the HDAC IIa family, also plays a role in vascular calcification. Malhotra et al.59 reported that HDAC9 increased expression in human aortic smooth muscle cells and promoted osteogenic phenotype and calcification of VSMCs by upregulating Runx2 gene expression. These results suggest that HDAC9 may be a potential therapeutic target for vascular calcification in the future. However, the exact mechanism by which HDAC9 regulates vascular calcification needs further study.59 In addition, Bartoli-leonard et al.113 reported that SIRT1 activation inhibited smooth muscle cells osteogenic transdifferentiation and reduced the diabetic vascular calcification progression by downregulating the Runx2 and osteocalcin expression.113
Hypertension
Existing studies have shown that histone acetylation is closely related to the occurrence and development of hypertension. A study to investigate the role of HDAC6 in hypertension found that Ang II upregulated HDAC6 mRNA and protein expression. HDAC6 induced the deacetylation of Cystathionine γ-lyase (CSEγ), leading to CSEγ degradation through the ubiquitin–albumin pathway. CSEγ is the main enzyme in the production of physiological vasodilator hydrogen sulfide. The degradation of CSEγ reduced the production of hydrogen sulfide, which accelerated hypertension and impaired endothelial function.114 Several studies have shown that changes in HDACs, SIRT1, SIRT3, and bromodomain-containing protein 4 (BRD4) protein expression levels are associated with cell proliferation, inflammation, and pathological vascular remodeling, thereby regulating the pathological processes of hypertension and pulmonary hypertension.115 Downregulation of SIRT3 expression and SIRT3 redox inactivation result in superoxide dismutase 2 (SOD2) inactivation, which promotes the occurrence of hypertension.116 In addition, Arise et al.117 found that angiotensin II(Ang II) enhanced the activity of class I HDAC1/2, reduced histone acetylation of H3K9/14ac and H4K8ac, further inhibited Npr1 (encoding natriuretic peptide receptor-A) transcription, and decreased natriuretic peptide receptor-A protein and cGMP levels, thereby diminishing renal and vascular reactivity and attenuating atrial natriuretic peptide-mediated aortic ring relaxation. Therefore, the study suggests that Ang-II-mediated Npr1 transcription and receptor function inhibition may provide new molecular targets and an important theoretical basis for the treatment and prevention of hypertension.117 In conclusion, these studies indicate that histone acetylation is closely related to cardiovascular diseases mechanism.
The role of noncoding RNAs regulation in cardiovascular disease
A large number of studies have shown that noncoding RNAs plays a key regulatory role in cardiovascular disease. Identification of specific noncoding RNAs will provide new ideas and directions for early diagnosis and prevention of diseases. So far, many noncoding RNAs have been found to be associated with the physiological and pathophysiological processes of cardiovascular diseases such as coronary heart disease, myocardial infarction, and vascular calcification. Noncoding RNA expression is cell- and organ-specific. Noncoding RNAs related to cardiovascular diseases exist in human blood, urine, and other body fluids. Moreover, due to its high sensitivity, stability, easy acquisition, and detection, it is expected to be a novel biomarker for assessing the risk stratification, diagnosis, and prognosis of cardiovascular disease in the future.
The role of miRNAs in cardiovascular disease
In recent years, a number of studies have confirmed that miRNAs can regulate the pathophysiological process of cardiovascular diseases (Table 4).
Coronary heart disease and acute coronary syndrome
At present, miRNA has been implicated in the development of coronary heart disease and acute coronary syndrome. One study reported that miRNA-SNP rs41291957 increased the expression of miRNA-143 and miRNA-145 in human coronary smooth muscle cells and modulated the phenotypic conversion of VSMCs. The study suggested that miRNA-SNP rs41291957 can be regarded as a important gene of assessing coronary heart disease risk and prognosis.118 Jiang et al.119 reported that inhibition of mir-1 not only reduced the inflammatory response of endothelial cells in vitro but also alleviated the occurrence of atherosclerosis.119 In addition, miRNA-106a-3p and miRNA-342-5p also have anti-atherosclerotic effects on endothelial cells,120,121 whereas miRNA-92a promotes the occurrence of atherosclerosis.122 Some studies have shown that miRNA-125b, miRNA-21a-5p, miRNA-25-3p, and miRNA-144 have protective effects on cardiac myocytes, and can be used as potential therapeutic targets for AMI in the future.123,124,125,126 Ling et al.127,128 found that serum levels of exosome miRNA-122-5P and miRNA-126 were positively correlated with coronary artery stenosis in patients with unstable angina and AMI. Therefore, studies have demonstrated that serum exosome miRNA-21, miRNA-122-5p, and miRNA-126 were novel biomarkers for the diagnosis of the acute coronary syndrome.127,128 Studies demonstrated that miRNA-590-3p and miRNA-199a-3p could promote the proliferation of myocardial cells in the mice infarct area. It implied that targeted miRNA-590-3p and miRNA-199a-3p treatment could restore the function of myocardial cells after myocardial infarction.129
Heart failure
According to recent studies, exosome miRNAs play an important role in myocardial remodeling and heart failure.130 Wu et al.131 observed that elevated serum exosome miRNA-92b-5p levels in patients with acute heart failure were negatively correlated with left ventricular ejection fraction. Serum exosome miRNA-92b-5p can be used as a biomarker of heart failure with reduced ejection fraction.131 Wang et al.132 reported downregulation of miRNA-425 and miRNA-744 levels in angiotensin-treated cardiac fibroblasts (CFs). miRNA-425 and miRNA-744 inhibited angiotensin-induced collagen and cellulose synthesis, reduced CFs activation, and improved cardiac remodeling by targeting TGF-β. Therefore, miRNA-425 and miRNA-744 were expected to be therapeutic targets and heart failure diagnostic markers for reversing cardiac remodeling. In conclusion, exosome miRNAs are looked forward to being a new tool for the diagnosis and treatment of heart failure.132
Vascular calcification
miRNA is strongly associated with the occurrence and development of vascular calcification. Xu et al.133 established vascular calcification animal models in vitro and in vivo. They found that miRNA-30b was a potential endogenous regulatory factor of vascular calcification, which had a protective effect on calcification. The main mechanism is that miRNA-30b increases MMPs and autophagy in VSMCs by inhibiting mTOR signaling pathway, maintains mitochondrial homeostasis, and attenuates the phenotypic transformation of VSMCs to osteogenic phenotype.133 In addition, in β-glycerophosphorate-treated VSMCs, miRNA-30b reduced the VSMC calcification occurrence by targeting to inhibit SOX9, decreasing the activation of bone morphogenetic protein 2 (BMP2) and preventing osteoblast differentiation.134 A clinical study showed that plasma miRNA-204 levels were significantly lower in patients with coronary artery calcification than in controls. Plasma miRNA-204 level was significantly and independently correlated with coronary artery calcification. Therefore, plasma miRNA-204 level can be used as a predictor to evaluate coronary artery calcification severity.135 However, Wang et al.136 reported that miRNA-128‐3p promoted Wnt‐1, β‐catenin, GSK‐3β, and Bax expression by downregulating Islet1 (ISL1) through activation of the Wnt pathway, thereby exacerbating cardiovascular calcification in type 2 diabetic rats.136 Chen et al.137 showed that miRNA-19A-3p inhibited HDAC4 expression, upregulated Runx2 and osteocalcin levels, and enhanced ALP activity by targeting the 3’UTR of HDAC4. Thus promoting human mesenchymal stem cells (hMSCs) osteogenic differentiation and calcification.137
Hypertension
miRNA can be an important factor for regulating the pathogenesis of pulmonary hypertension and hypertension. Sind et al.138 demonstrated that miRNA-181A-5p and miRNA-324-5p could reduce pulmonary vascular remodeling to resist the occurrence of pulmonary hypertension.138 Upregulation of miRNA-34C-5p, miRNA-449b, miRNA-571, miRNA-765, miRNA-483-3p, miRNA-143/145, miRNA-21, miRNA-126, miRNA-196a, miRNA-132, miRNA-212, and miRNA-451 may result RAS imbalance, which raises blood pressure.139 Study found that cyclic adenosine phosphate and sex hormones could stimulate the expression of renin mRNA and the secretion of renin proprotein in primary trophoblast cells. After transfection of miRNA-181A-5p and miRNA-663 into trophoblast cells, the expression of renin mRNA and the production of renin proprotein were declined. Thus reducing renin activity and preventing it from lysing angiotensinogen to produce Ang I. In the end, it inhibited RAS response and resulted in decreased blood pressure.140
The role of lncRNAs in cardiovascular disease
LncRNAs is a double-edged sword in cardiovascular disease progression, being both a positive regulator and a negative regulator (Table 4). Studies have shown that Linc1405, LncRNA PANCR, LncRNA Handdown (Hdn) reprograms cardiac fibroblasts into cardiomyocytes, activates cardiomyocyte differentiation, and participates in cardiac development.141,142,143,144 LncRNA CAREL inhibits cardiac regeneration and differentiation by targeting miRNA-296.145 LncRNA NR_045363 and SIRT1 antisense LncRNA activate cardiac regeneration by targeting miRNA-216a and SIRT1 mRNA, respectively.146,147 LncRNA CRRL and LncRNA AZIN2-sv regulate the proliferation of cardiomyocytes by sponges miRNA-199a-3p and miRNA-214.148,149 REN et al.150 found that compared with patients without any obvious complications type II diabetic cardiovascular disease and healthy controls, plasma lncRNA-SRA levels in patients with type II diabetic cardiovascular disease were significantly reduced. The mechanism may be that LncRNA-SRA participates in the regulation of VSMCs proliferation, thus regulating the occurrence and development of cardiovascular diseases.150 Chen et al.151 first revealed the expression profile of lncRNA in a chronic intermittent hypoxia rat model through lncRNA chip experiment and bioinformatics analysis, providing a new idea for exploring the pathogenesis of cardiovascular diseases induced by obstructive sleep apnea.151 Furthermore, the potential value of lncRNAs as diagnostic biomarkers has been widely explored. LncRNA SENCR has certain value in the diagnosis of early-onset coronary artery disease (CAD).152 LncRNA DKFZP434I0714 can be used as a biomarker to predict cardiovascular adverse events in uremia patients.153
Atherosclerosis
LncRNAs play a key role in atherosclerosis. LncRNAs mainly regulate atherosclerosis occurrence and progression by affecting inflammatory response, apoptosis and autophagy of vascular endothelial cells, foam cells formation, lipid metabolism and other mechanisms. One study showed that LncRNA ANRIL could be used as a biomarker of atherosclerosis.154,155 LncRNA LEENE can enhance the anti-inflammatory ability of endothelial cells.156 LincRNA-p21 and smooth muscle-induced lncRNA enhances replication (SMILR) affect the development of atherosclerosis by inducing cell apoptosis and regulating cell proliferation, respectively.157,158 In addition, LncRNA Mexis overexpression has been reported to promote cholesterol efflux by increasing ATP-binding cassette transporter A1 gene (ABCA1) expression, thereby reducing the probability of atherosclerosis in mice.159 Hu et al.160 reported that lncRNA, Nexilin F-actin-binding protein antisense RNA 1 (NEXN-AS1), regulated the expression of actin-binding protein NEXN. Expression microarray analysis showed that the expressions of NEXN-AS1 and NEXN were both decreased in human atherosclerotic plaques. Downregulation of NEXN-AS1 and NEXN expression can enhance the activity of TLR-4 oligomer and NF-κB, upregulate the expression of endothelial cell adhesion molecules and inflammatory factors, and increase the number of macrophages in atherosclerotic lesions, thereby promoting the development of atherosclerosis. These findings suggest that NEXN- AS1 and NEXN have a protective effect on atherosclerosis and may serve as potential therapeutic targets for atherosclerotic diseases in future.160
Myocardial infarction
LncRNAs are not only early indicators of myocardial infarction,161,162 but also act as a crucial element in the pathogenesis of myocardial infarction by controlling autophagy, apoptosis and other processes. LncRNA APF, lncRNA CAIF, and lncRNA Mirf affect the injury of myocardial infarction by regulating cardiac autophagy.163,164,165 Meanwhile, lncRNA Gpr19 inhibition and lncRNA UCA1 overexpression blocked apoptosis by miRNA-324-5p/Mtfr1 axis and miRNA-143/MDM2/p53 axis, respectively, to alleviate myocardial injury after myocardial infarction.166,167 In addition, lncRNA CPR, MALAT1, lncRNA AK139128 participate in cardiac repair and cardiac function development after myocardial infarction by regulating cell proliferation.168,169,170,171 These results suggest that lncRNAs may be potential therapeutic targets for myocardial infarction.
Heart failure
In recent years, increasing evidence has accumulated for lncRNA in heart failure progress. Studies have demonstrated that lncRNA LIPCAR, lncRNA COL1A1, and lncRNA H19 can be regarded as biomarkers to predict and evaluate the risk of heart failure.172,173,174 Piccoli et al.175 reported that lncRNA Meg3 was downregulated in late cardiac remodeling. Meg3 inhibition downregulated expression and activity of MMP2, leading to disminish cardiac fibrosis and hypertrophy.175 Another type of myocardial fibroblast enriched lncRNA Whisper (Wisper 2 superenhance-associated RNA) is primarily used to regulate myocardial fibrosis after injury. The results shows that silencing Wisper could alleviate myocardial infarction-induced fibrosis and cardiac dysfunction through upregulating myocardial fibroblasts Col3a1, Fn1, Tgfb2, and aSma expression.176 These results provide new insights into the pathogenesis of heart failure and contribute to enhance diagnostic performance and treatment strategies for heart failure, thereby improving the long-term prognosis of heart failure.
Vascular calcification
LncRNA is of the utmost importance in the development of vascular calcification. Yu et al.177 found that LncRNATUG1 was significantly expressed in human aortic valve and primary valvular interstitial cells (VICs). LncRNATUG1 activation downregulated the expression of miRNA-204-5p by sponging and reversed the inhibition of Runx2 by LncRNATUG1 short hairpin RNA (shRNA). Thereby, the levels of osteoblastic-specific proteins (such as osteocalcin, OPN, and osterix) were upregulated to promote calcific aortic valve disease osteoblastic differentiation. Therefore, LncRNATUG1 can serve as a positive regulator of osteogenic differentiation in the calcific aortic valve disease pathogenesis.177 Jeong et al.178 reported that after knockdown Lrrc75a-as1, calcium deposition increased, while its overexpression inhibited calcium deposition in A10 cells. Lrrc75a-as1 overexpression reduced the expression levels of osteogenic factors Runx2, msh homeobox 2 and BMP2 in VSMCs by regulating transcription factors SRF, CREB1, and STAT3. Finally, it slowed down vascular calcification process. Taken together, studies have indicated that Lrrc75a-as1 is a negative regulator of vascular calcification.178 Huang et al.179 reported that lncRNA-SNHG29 expression was downregulated and miRNA-200b-3p expression was upregulated in vitro calcification model. LncRNA-SNHG29 could activate the α-Klotho/FGFR1/FGF23 axis in VSMCs by downregulating miRNA-200b-3p, upregulate the α-Klotho target of miRNA-200b-3p, and inhibit the Wnt/β-catenin signaling pathway. Meanwhile, it also promoted FGFR1 and FGF23 expression, which significantly inhibited osteogenic factors (Runx2 and BMP2) and prevented VSMC calcification. These results suggest that LncRNA-SNHG29 can be a novel therapeutic target for vascular calcification-related diseases.179 A study revealed that Bhlhe40 overexpression inhibited the lncRNA-ES3 expression by binding to the promoter region of lncRNA-ES3 gene (LINC00458), and then upregulated the expressions of miRNA-95-5p, miRNA-6776-5p, miRNA-3620-5p, and miRNA-4747-5p, decreased ALP activity and secretion of osteocalcin, which alleviated human aortic VSMCs calcification induced by high glucose.180
Hypertension
LncRNA has been implicated in the development of hypertension. Yang et al.181 reported that HAS2-AS1 (an extracellular matrix-associated lncRNA) and C/EBPβ were highly expressed in hypoxic HFL-1 cells. C/EBPβ bound to the promoter region of HAS2-AS1 to activate its transcription and promoted the inflammatory response of HFL-1 cells. Downregulation of HAS2-AS1 expression inhibited the HFL-1 cells proliferation, migration, and inflammatory response. Thus, the study reveals that HAS2-AS1 may be involved in the pathophysiology of hypoxic pulmonary hypertension.181 Specific siRNA was used to knockdown MRAK048635_P1 from VSMCs isolated from the thoracic aorta of hypertensive rats. It was found that the downregulation of MRAK048635_P1 could stimulate the proliferation and migration of VSMCs and induce the transformation from contractile to the secretory phenotype of VSMCs. It suggests that decreased expression of LncRNA MRAK048635_P1 could be used as an important factor for vascular remodeling in hypertension.182
The role of circRNAs in cardiovascular disease
Coronary heart disease and myocardial infarction
More and more attention has been paid to the regulation of cirRNAs on coronary heart disease and myocardial infarction. Lin and Pan et al.183,184 found that cirRNAs might regard as a potential clinical marker for the diagnosis of coronary heart disease through high-throughput technology and competitive endogenous ceRNA chip analysis.183,184 CirRNA-SATB2, cirRNA-0044073, cirRNA-284, and cirRNA_RUSC2 participate in the development of atherosclerosis by targeting miRNA-939, miRNA-107, miRNA-221, and miRNA-661 to regulate VSMCs proliferation and migration, respectively.185,186,187,188 CirRNA cdr1as, MFACR, and Cir_Nfix play key roles in promoting cardiac regeneration repair and apoptosis by targeting miRNA-7a, miRNA-652-3p, and miRNA-214, respectively, providing new evidence for further research on myocardial infarction. Circ-Ttc3 protects myocardial infarction-induced myocardial apoptosis by inhibiting the activity of miRNA-15b-5p. CircNfix regulates the Gsk3b signaling pathway through miRNA-214. The downregulation of circNfix expression can ameliorate myocardial infarction.189,190,191,192 CircSlc8a1, CircRNA_010567, CircHIPK3, and CircNFIB regulate the proliferation and migration of cardiac fibroblasts through sponges miRNA-133a, miRNA-141, miRNA-29b-3p, and miRNA-433. Thus changing the cardiac structure and the development of cardiac dysfunction.193,194,195,196 CircNCX1 promotes myocardial apoptosis and myocardial ischemia–reperfusion injury through competitively binds miRNA-133A-3p. Circ ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy.197,198
Heart failure
CirRNAs are deemed as novel regulatory genes in cardiomyocyte hypertrophy, fibrosis, autophagy, and apoptosis, which are involved in the development of heart failure.199 Mmu_circ_0005019 regulates the expression of its target gene Kcnn3 by targeting miRNA-499-5p, thereby inhibiting cardiac fibrosis and reversing electrical remodeling of cardiac myocytes.200 In addition, circNfix inhibit the development of heart failure by regulating the proliferation of cardiomyocytes after myocardial infarction. CirRNA CDYL overexpression can promote myocardial cell proliferation in vitro by targeting miRNA-4793-5p.201 Therefore, circNfix and CDYL have the potential to be used as key modulatory factors to ameliorate the prognosis of myocardial infarction and delay the progression of heart failure. Furthermore, reduced cirRNA 010567 expression alleviates myocardial fibrosis by blocking the TGFβ1 signaling pathway, thereby improving cardiac function.202 Circ_LAS1L inhibits cardiac fibroblasts proliferation by increasing the expression of SFRP5 through sponge miRNA-125b.203 Therefore, cirRNA 010567 and circ_LAS1L slow the progression of heart failure by reducing myocardial fibrosis and preventing ventricular remodeling. Nevertheless, cirRNA_000203 aggravate cardiac hypertrophy through sponge miRNA-26b-5p and miRNA-140-3p to aggravate cardiac hypertrophy, which may increase the risk of heart failure.204 CircFndc3b overexpression increased angiogenic activity and reduced cardiomyocytes and endothelial cells apoptosis by interacting with RNA binding protein Fused. Therefore, these findings suggest that CircFndc3b can ameliorate cardiac remodeling and cardiac function after myocardial infarction.205 CirRNA ACAP2 induces myocardial apoptosis after myocardial infarction by binding to miRNA-29.206 Thus, cirRNAs are closely relevant to heart failure development.
Vascular calcification and hypertension
The role of the cirRNA in the development of vascular calcification and hypertension will be discussed. CDR1as is an important cirRNA. Recently, a study showed that CDR1as might act as a molecular sponge for miRNA-7-5p. Under hypoxia induction, the expression of CDR1as and its target genes CAMK2D and CNN3 were upregulated, while the expression of miRNA-7-5p was downregulated. The co-transfection of siCDR1as and miRNA-7-5p antagonist can promote human pulmonary artery smooth muscle cell (HPASMC) mineralization. It suggests that the CDR1as regulatory role in HPASMC calcification may be related to the inhibition of miRNA-7-5p function. Overexpression of CAMK2D and CNN3 enhances HPASMC mineralization under hypoxia induced by CDR1as and miRNA-7-5p agonists. In conclusion, the study demonstrated that in hypoxic HPASMCs, CDR1as upregulated CAMK2D and CNN3 expression through sponge miRNA-7-5p, accelerating HPASMC osteoblasts differentiation and calcification.207 Yu et al.208 reported that TGFBR2 expression was downregulated and miRNA-25-3p expression was upregulated in osteogenic induced aortic VIC. TGFBR2 sponge miRNA-25-3p regulates TWIST1 expression in osteogenic induced VIC. Overexpression of miRNA-25-3p or TWIST1 knockdown increases osteoblast markers Runx2 and OPN expression and ALP activity, leading to calcium nodular formation. Therefore, overexpression TGFBR2 can inhibit VIC osteoblasts differentiation and calcification by interacting with miRNA-25-3p and TWIST1.208 Research data showed that cirRNA-vgll3 originated from the vgll3 site acted as a miRNA-326-5p sponge. CircRNA-vgll3 overexpression attenuates miRNA-326-5p-mediated integrinα5 (Itga5) inhibition by targeting miRNA-326-5p. The mRNA expression levels of osteogenic marker genes in adipose-derived mesenchymal stem cells including Runx2, osterix, OPN, osteocalcin and BMP2 were markedly enhanced. Thus cirRNA-vgll3 overexpression significantly accelerated adipose-derived mesenchymal stem cells osteogenic differentiation.209 In addition, Sun et al.210 demonstrated that circACTA2 interacts with miRNA-548F-5p targeting α-SMA mRNA3'-UTR, which alleviated the inhibition of α-smooth muscle actin(α-SMA) expression by miRNA-548F-5P. Thereby upregulating the expression of α-SMA, promoting the contraction of VSMCs and regulating vascular tension, and participating in the occurrence of hypertension.210 These results suggest that circRNAs may be potential diagnostic biomarkers for vascular calcification and hypertension.
In conclusion, these findings provide a new perspective for the study of cirRNA in cardiovascular disease (Table 4). After understanding the regulation of epigenetics in different cardiovascular diseases. To sum up, the following comments mainly describe the related drugs and potential targets of epigenetic therapy for cardiovascular disease. It will create more possibilities for the future clinical application of epigenetic drugs for cardiovascular disease.
The role of epigenetics regulation in other cardiovascular diseases
In recent years, epigenetics has been gradually studied in other cardiovascular diseases. Bahado-singh et al.211 found 165 significantly differentially methylated CpG loci in the tetralogy of Fallot cases. Among 165 CpG sites with differential methylation, cg05273049, cg02540011, cg08404201, and cg00687252 had the highest predictive accuracy. These methylation sites can be used as biomarkers to predict tetralogy of Fallot with good accuracy. Therefore, this study shows that there is a significant correlation between DNA methylation changes in the placenta and tetralogy of Fallot.211 As an important member of histone lysine methyltransferases, the SET domain (SETD) family plays a key role in histone modification. SETD1B activates Notch signaling by upregulating the level of H3K4me3 in endothelial cells and exacerbates endothelial inflammation and apoptosis. These findings suggest that SETD1B-based epigenetic reprogramming may potentially improve the course and prognosis of endothelial inflammation-related cardiovascular disease.212 In one study, atrial fibrillation was associated with decreased HDAC2 expression and increased neuron-restrictive silencer factor (NRSF) expression. HDAC2 gene knockdown and increased NRSF expression resulted in decreased KCNJ2 K+ ion channel expression and prolonged action potential duration in neonatal rat cardiomyocytes. These new insights into the mechanisms of epigenetic remodeling may provide the theoretical basis for the treatment of atrial fibrillation.213 Yao et al.214 found that the expression of noncoding RNA myocardial infarction-associated transcript (MIAT) was significantly increased and miRNA-133A-3p was significantly decreased in the rat model induced by atrial fibrillation. MIAT knockdown significantly alleviates atrial fibrillation and reduces atrial fibrillation-induced atrial fibrosis by targeting miRNA-133A-3p and inhibiting fibrosis-related gene expression of collagen I, collagen III, connective tissue growth factor, and TGF-β1.214 Costantino et al.215 demonstrated that in diet-induced obese mouse hearts, AP-1 transcription factor directly bound to the PPARγpromoter, which resulted in activation of PPARγand increased transcription of Fas, Cd36, Lpl and Plin5. Thereby it promoted lipid accumulation, cardiac dysfunction, and ultimately led to metabolic cardiomyopathy. JunD is a direct target of miRNA-494-3p. miRNA-494-3p overexpression can reduce lipid accumulation and obesity-related metabolic cardiomyopathy by inhibiting JunD/PPARγ signaling pathway. These findings open new therapeutic strategies for metabolic cardiomyopathy and left ventricular dysfunction in obese patients.215 Although there is still much unknown about the mechanism of epigenetics in other cardiovascular diseases, its important significance in biology has been highlighted. In future, we look forward to exploring more new breakthroughs in epigenetics in other cardiovascular diseases.
Epigenetic therapy for cardiovascular disease
DNA methylation
DNA methyltransferase inhibitors or related drugs play a role in the treatment of cardiovascular diseases such as coronary heart disease and heart failure by regulating target genes methylation status and expression level (Figs. 5–7). Currently, though, research on DNA methylation as a treatment for cardiovascular disease is still in the development stage. However, because DNA methylation changes are reversible, which offers an optimistic prospect for the treatment of the disease (Table 5).

Epigenetics-related targets and drugs in atherosclerosis and hypertension. This figure was created with the aid of Biorender (https://biorender.com/). LXRa, liver X receptor a, PPARγ1 peroxisome proliferator-activated receptorγ1, DNMTs DNA methyltransferases, ER estrogen receptor, COL15A1 collagen, type XV, alpha 1, ABCA1 ATP-binding cassette transporter A1 gene, SIRT1 sirtuin1, HDAC histone deacetylase, ICAM-1 intercellular adhesion molecule-1, TLR-4 toll-like receptor-4, vWF von Wilebrand factor, ANGPTL3 angiopoietin-like 3, H3K27me3 trimethylated histone 3 lysine 27, TNF-a tumor necrosis factor (TNF)-a, ACE1 angiotensin-converting enzyme 1

Epigenetics-related targets and drugs in coronary heart disease and vascular calcification. This figure was created with the aid of Biorender (https://biorender.com/). DNMTs DNA methyltransferases, ER estrogen receptor, COL15A1 collagen, type XV, alpha 1, ABCA1 ATP-binding cassette transporter A1 gene, SIRT1 sirtuin1, HDAC histone deacetylase, ICAM-1 intercellular adhesion molecule-1, TLR-4 toll-like receptor-4, vWF von Wilebrand factor, MMP matrix metalloproteinase

Epigenetics-related targets and drugs in myocardial infarction and heart failure. This figure was created with the aid of Biorender (https://biorender.com/). FOXO3a forkhead box O3a, AKT-1 protein kinase B-1, TNF-a tumor necrosis factor (TNF)-a, HDAC histone deacetylase, NP natriuretic peptide, HSF1 heat-shock transcription factor 1, SIRT1 sirtuin1, COX-2 cyclooxygenase-2, PCSK9 proprotein convertase subtilisin-kexin type 9, IL-6 interleukin 6, VEGF vascular endothelial growth factor, eNOS endothelial nitric oxide synthase, ANG-1 angiopoietin-1, ANP atrial natriuretic peptide, BNP brain natriuretic peptide, STAT3 signal transducer and activator of transcription 3, β-SMA β-smooth muscle actin
Atherosclerosis and coronary heart disease
Studies have shown that 5-Aza-dC (decitabine) treatment of Ldlr−/− mice can inhibit the migration and adhesion of macrophages to epithelial cells, reduce the infiltration of macrophages into atherosclerotic plaques and the expression of inflammatory genes in macrophages,216 thus alleviating atherosclerotic lesions and reducing the development of atherosclerosis.217 DNMT inhibitor RG108 plays an important role in atherosclerosis and coronary heart disease by inhibiting DNMT1 and DNMT3a activity.218,219 5-AZa-2-deoxycytidine (DAC) demethylation has been demonstrated to treat atherosclerosis and coronary heart disease by upregulating the expression of normal smooth muscle cells and endothelial cells ERa, ERb and COL15A1.220,221 A recent study reported that Cocoa extract improved atherosclerosis and coronary heart disease by inhibiting DNMTs and methylenetetrahydrofolate reductase (MTHFR) gene expression levels in vitro.222 In adults with cardiovascular risk factors, Cocoa in combination with statins reduce cholesterol levels and thus exert a protective effect on the cardiovascular system. (ClinicalTrials.-gov identifier: NCT00502047).222 High methylation levels of the ABCA1 have been found to be associated with coronary heart disease and aging. Acetylsalicylic acid (ASA) treatment can decrease ABCA1 DNA methylation level, thereby reducing the occurrence of atherosclerosis and coronary heart disease.223
Heart failure
DNA methylation is closely related to the treatment of heart failure. It is reported that RG108 can reduce the progression of myocardial hypertrophy and myocardial fibrosis by inhibiting DNA methyltransferase.224 Studies have shown that DNA methylation inhibitor 5-azacytidine can decrease the harmful effects of tumor necrosis factor-a on SECRA2a expression.225 5-azacytidine may improve cardiac hypertrophy and reduce myocardial fibrosis by inhibiting DNA methyltransferase and blocking the expression of hypertrophic cardiomyopathy genes.226,227 Xiao et al.228 demonstrated that 5-AZa-2-deoxycytidine reversed the changes of the myocardial proteome in rats by inhibiting DNA methyltransferase, reduced myocardial hypertrophy, improved myocardial contractility, and eliminated the susceptibility to ischemic injury.228
Vascular calcification
DNA methyltransferase inhibitor can be used as a potential drug to prevent or treat vascular calcification. Tanaka et al.229 demonstrated that DNA methyltransferase inhibitor decitabine might enhance Krüppellike factor-2 (KLF2) mRNA and protein expression by demethylation of KLF2. KLF2 overexpression enhanced transcription of IL-10 and TGFβ1 genes, which inhibited osteoclast differentiation and interacts with Runx2 to induce osteogenic differentiation and calcification.229
In summary, DNA methylation inhibitor(decitabine), has a therapeutic effect in basic studies of atherosclerosis and calcification. DNMT inhibitor RG108 and 5-AZa-2-deoxycytidine can slow the progression of atherosclerosis, coronary heart disease and heart failure. Therefore, in future, large-scale multi-center clinical trials of DNA methylation inhibitors decitabine, RG108, and 5-AZa-2-deoxycytidine are expected to carry out to verify their efficacy in patients with cardiovascular disease. These are promising drugs for the treatment of cardiovascular disease. The changes of DNA methylation is reversible, and methylation inhibitors can change the methylation status and expression level of some genes, thus bringing into the corresponding biological effects. In terms of drug effects in epigenetic pharmacology, the influence of DNA methylation is being explored, which provides us with a new perspective to understand and treat cardiovascular diseases. At present, the research of DNA methylation-related drugs in the treatment of various cardiovascular diseases is still in the development stage, which needs to be further excavated in a deeper level.
Histone modifications
According to recent research findings, histone methyltransferase inhibitors or HAT/HDAC inhibitors are still rarely used in clinical treatment of cardiovascular diseases. However, the development of drugs targeting the mechanisms of histone methylation and histone acetylation has achieved some effect in basic experimental research on the treatment of cardiovascular disease (Figs. 5–7). In future, these drugs are looking forward to being more applied in clinical trials, in order to better meet the needs of ameliorating cardiovascular disease patients' symptoms and prognosis (Table 5).
Histone methylation
Xiao et al.230 found that SMYD4 belonged to the lysine methyltransferase family. As a histone methyltransferase, SMYD4 also regulated histone acetylation by interacting with HDAC1. Transcriptome and bioinformatics analysis of smyd4L544Efs*1 and wild-type developing hearts showed that SMYD4 was a critical epigenetic regulator of heart development, involved in the regulation of endoplasmic reticulum mediated protein processing and primary signaling and metabolic pathways during heart development in zebrafish. Therefore, SMYD4 had the potential to be used as a therapeutic target in embryonic development and cardiogenesis.230 SUV39H1 was a histone methyltransferase that catalyzes increased methylation of histone 3 lysine 9. Upregulation of SUV39H1 significantly reduced infarct size and myocardial injury after ischemia–reperfusion injury by decreasing the activity of the mitogen-activated protein kinase family and its downstream transcription factor NF-κB. Therefore, SUV39H1 can be regarded as a treatment strategy for ischemia–reperfusion injury in diabetes mellitus.231 Weng et al.232 reported that IFN-γ treatment enhanced the expression of histone H3K9 trimethyltransferase SUV39H1 in endothelial cells and promoted the recruitment of SUV39H1 to eNOS promoter. SUV39H1 silencing removes IFN-γ inhibition of eNOS by eliminating H3K9Me3 on the eNOS promoter. Therefore, SUV39H1 might be used as a drug target to intervene in endothelial dysfunction.232 Ono et al.233 demonstrated that cardiac chronic stress could gradually promote intron repeat regions excessive heterochromatinization of genes critical to cardiac pumping function, such as those related to mitochondrial function. Excessive heterochromatinization of mitochondrial gene repeat elements in failing hearts may lead to gene silencing and impair cardiac function. The histone H3K9 methyltransferase inhibitor chaetocin can maintain appropriate chromatin structure and reverse excessive heterochromatinization. Chaetocin improves myocardial hypertrophy by inhibiting H3K9 methyltransferase.233 Therefore, chaetocin will be a potential drug for treating chronic heart failure in the future.234 In addition, a study reports that resveratrol may exert a therapeutic effect in DOCA salt hypertension through vascular H3K27me3 methylation.235
Histone acetylation
Atherosclerosis
Histone acetylation is crucial to the treatment of atherosclerosis. Arunachalam et al.236 demonstrated that resveratrol could ameliorate metabolic disorders, atherosclerosis and coronary heart disease by upregulating SIRT1 in endothelial cells.236 Furthermore, it has been reported that statins might prevent the loss of HDAC1 and HDAC2 binding to IL-8 (CXCL8) and MCP1 (CCL2) gene promoters, partially restoring the overall activity of HDAC.236 Statins decreased oxLDL-associated histone modifications (H3S10 phosphorylation; acetylation of H3K14 and H4K8) and recruitment of CREB-binding proteins 300, NF-KB, and RNA polymerase II, which in turn control atherosclerotic inflammation. In the end, it plays a significant impact on the pathogenesis of atherosclerosis.237 In addition, trichostatin A, an HDAC inhibitor, blocks upregulation of uremia environment-induced markers of endothelial dysfunction (intercellular adhesion molecule-1, surface Toll-like receptor-4, von Wilebrand factor) and reactive oxygen species. Thereby trichostatin A could treat atherosclerosis and coronary heart disease.238
Myocardial infarction and ischemia–reperfusion injury
Histone deacetylase inhibitors are deemed as underlying drugs and strategies for the treatment of myocardial infarction and ischemia–reperfusion injury. Study showed that entinostat (MS-275), a class I-specific HDAC inhibitor, increased the expression of SOD2 and catalase in myocardial mitochondria after ischemia–reperfusion via the nuclear FOXO3A transcription factor. Thus, MS-275 significantly reduced the size of myocardial infarction and improved left ventricular function and tissue vitality. Finally, it protected cardiac systolic function after ischemia–reperfusion.239,240 It was found that trichostatin A treatment significantly inhibited the activity of HDAC, increased the phosphorylation of AKT-1, and reduced myocardium and serum TNF-a. Consequently, trichostatin A increased the formation of myocardial cells and cardiac microvessels. Meanwhile, it significantly reduced myocardial infarction size, and generated a protective effect in the heart of patients with myocardial infarction. These results suggest that inhibition of HDAC can maintain cardiac function and attenuate cardiac remodeling by stimulating endogenous cardiac regeneration.241,242,243,244,245,246 In the context of ischemia/reperfusion injury and myocardial infarction, HDAC inhibitors valproic acid, tributyl butyrate, and suberoylanilide hydroxamic acid (SAHA) reduce myocardial infarction size and ventricular remodeling by inhibiting HDAC, and induce increased angiogenic response.47,247,248 Studies have demonstrated that long-term use of low doses of SAHA ameliorated cardiac remodeling after infarction. Finally, it resulted in a lasting protective impact on the heart and no toxic effects on the heart.249,250,251 Therefore, HDAC inhibitor SAHA is expected to be tested in clinical trials in order to better serve as a potential treatment drug for myocardial infarction in future. In addition, vorinostat was shown to delay ischemia–reperfusion injury in mice and rabbits, which might be a significant drug for future clinical treatment of ischemia–reperfusion injury.252 It is reported that long-term use of resveratrol can activate SIRT1, attenuate FOXO1-related pro-apoptotic signaling pathway, increase peroxisome proliferator-activated receptor γ coactivator-1 (PCC-1) α and mitochondrial biogenesis, improve myocardial function and Ang-II-induced cardiac remodeling in aging mice, thereby protecting myocardial cells from ischemia–reperfusion injury.253,254,255,256 Studies have shown that SRT1720 up-regulates SIRT1, activates the COX-2 signaling pathway, and decreases oxidative stress and inflammation, thereby alleviating mice vascular endothelial dysfunction and reducing myocardial infarction in SIRT1(+/−) hearts.257 Phosphodiesterase 5 inhibitors sildenafil and adiponectin play a protective role on the heart by increasing SIRT1 activity in the myocardium.258,259 In a diabetic mouse model, tadalafil was administered to activate SIRT1-PGC-1α activity and eNOS, Akt, AMPK phosphorylation. In the end, it ameliorated myocardial function in diabetic mice.260 Therefore, targeting SIRT may be a promising approach for treating cardiovascular disease strategies.
Heart failure
Histone deacetylase inhibitors are vital for the prevention and treatment of heart failure. Wang et al.261 showed that a high-fat diet enhanced SUV39H, decreased SIRT1, inhibited AMPK and CaM kinase II autophagy and phosphorylation, thereby contributing to ventricular hypertrophy and interstitial fibrosis, and resulting in cardiac systolic dysfunction. Mitochondrial aldehyde dehydrogenase (ALDH2) fights high-fat diet-induced cardiac structural and functional abnormalities through mechanisms related to autophagy regulation and promotion of SUV39H-SIRT1-dependent PGC-1α deacetylation. Thus it generates a protective effect on the heart.261 Morales et al262 demonstrated that inhibition of class I HDACs with apicidin induced the expression of the mTOR inhibitor tuberous sclerosis complex 2 (TSC2), which in turn attenuated cardiac hypertrophy by inhibiting mTOR.262 In addition, one study data suggest that resveratrol disminishes IL-6 activation induced by SIRT1 to protect H9c2 cells from Ang-II-induced hypertrophy.263
Studies have shown that the P300 HAT inhibitor curcumin significantly reduces LDL levels and increases high-density lipoprotein (HDL) levels in healthy volunteers and patients with atherosclerosis.264,265 Curcumin can prevent ventricular hypertrophy and maintain systolic function in heart failure rat models by inhibiting histone acetylation and hypertrophy response transcription factor (GATA4) and decreasing P300 HAT activity by disrupting the P300/GATA4 complex.266 In addition, class I and II HDACs inhibitors (trichostatin A) and apicidin derivative, API-D (selective inhibitors of class I HDAC) ameliorated cardiac function in thoracic aortic constriction mice by preventing cardiomyocyte hypertrophy and myocardial fibrosis.267,268,269 HDAC4 inhibitors have been reported to block cardiomyocyte hypertrophy, improve cardiac function and inhibit cardiac remodeling in mice.270 Trichostatin A and emodin have good clinical value in the treatment of cardiac hypertrophy and heart failure, through deacetylation of histone.271,272 It was found that MPT0E014 generated beneficial impact in heart failure by inhibiting HDAC, increasing cardiac function, and alleviating the effects of heart failure on cardiometabolism and inflammation.273 HDAC class I inhibitor Mocetinostat reverses myocardial fibrosis in heart failure by reducing myocardial fibroblast activation and inducing cell cycle arrest/apoptosis.274,275 Furthermore, valproic acid treatment can reduce myocardial hypertrophy and fibrosis in rats.276 The class II-specific HDAC inhibitor MC1568 inhibits the activity of HDAC4 and HDAC5 in skeletal muscle and heart. Therefore it may have potential clinical significance in the treatment of muscle and heart disease.277
The study reveals that the histone acetylation reader BRD4 undergoes stimulus-dependent, genome-wide redistribution in cardiac fibroblasts. It is enriched on a set of enhancers and super-enhancers, and leads to RNA polymerase II activation and downstream target gene expression. Therefore, BRD4 is a central regulator of the phenotype of cardiac fibroblasts promoting fibrosis. The BRD4 inhibitor JQ1 decreases the expression of activation markers and extracellular matrix proteins in cardiac fibroblasts. In addition, JQ1 also inhibites the contractile activity and β-SMA expression of myocardial fibroblasts. Administration of JQ1 improves cardiac function in preestablished heart failure or myocardial infarction mice.46,278 Thus, BRD4 inhibitor JQ1 will be used as a drug for the clinical treatment of heart failure and myocardial infarction in future.279
Vascular calcification
Histone acetylation-related drugs and strategies are of great importance in vascular calcification treatment progress. HAT P300 is a transcription coactivator involved in gene expression regulation and protein acetylation. Gu et al.280 reported elevated levels of acetylated histone 3 and 4 in human aortic valve calcification. Inhibitors of HAT P300 inhibited acetylated histone 3 and 4, alleviating aortic valve calcification induced by high calcium/high phosphate treatment in vitro.280 In addition, Li et al.281 found that C646 (P300 inhibitor) could downregulate the osteocalcin gene and protein expression. P300 inhibition might regulate Klotho expression and weaken osteogenic transdifferentiation and calcification in VICs by downregulating HAT activity. Therefore, P300 inhibition could be a potential therapeutic target for vascular calcification.281 HDAC6 is a member of the HDAC IIb subfamily. Fu et al.282 demonstrated that the HDAC6 expression in Aortic valve (AoV) tissues of patients with aortic stenosis was significantly decreased. Downregulation of HDAC6 might promote AoV calcification through endoplasmic reticulum stress/activating transcription factor 4 (ATF4) mediated osteogenic pathway. Thus, HDAC6 will be a new target for the prevention and treatment of vascular calcification in the future.282 IL-1β-induced inflammatory response is associated with osteoarthritis (OA) and vascular calcification development. The HDAC inhibitor vorinostat inhibits IL-1β-induced MMPs expression by P38 and ERK1/2 phosphorylation, thereby attenuating VSMC osteoblastic differentiation and calcification. Therefore, HDAC inhibitor vorinostat will be expected to be an important drug in the treatment of vascular calcification in the future.283
Hypertension
Histone deacetylase inhibitors can be used as therapeutic targets for pulmonary hypertension. Boucherat et al.284 reported that HDAC6 maintained Ku70 in a low acetylation state, blocked the translocation of Bax to mitochondria, and prevented cell apoptosis. Inhibition of HDAC6 could reduce the proliferation and anti-apoptotic ability of pulmonary smooth muscle cells with pulmonary hypertension. Therefore, HDAC6 deficient mice had some protective effect against pulmonary hypertension caused by chronic hypoxia. Study results suggest that pharmacological inhibition of HDAC6 ameliorates established pulmonary hypertension and will be a potential therapeutic target for pulmonary hypertension in the future.284 In addition, resveratrol treatment can also alleviate vascular remodeling and prevent the development of pulmonary hypertension.285
Endothelial dysfunction is an important determinant of hypertension and its complications. Therefore, it is of great clinical significance to identify to prevent endothelial dysfunction potential therapeutic targets. New evidence suggests that histone acetylation is closely related to the regulation of endothelial function. SIRT6 is a member of the highly conserved NAD + dependent deacetylase (class III HDAC). SIRT6 has some effects such as increasing the bioavailability of vascular nitric oxide, promoting endothelium-dependent vascular dilation, and reducing endothelial cell permeability. In the experiment of SIRT6 gene knockout hypertensive mice, it was found that the loss of endothel-specific SIRT6 significantly increased blood pressure, aggravated endothelial dysfunction and cardiac renal injury. SIRT6 inhibited transcription and expression of Nkx3.2 by deacetylating histone H3K9. Meanwhile, SIRT6 induced the expression of GATA5, a novel blood pressure regulator, to regulate the GATA5-mediated signaling pathway to prevent endothelial injury, improve endothelial cell permeability, and promote nitric oxide production and endothelium-dependent vasodilation. Thereby ultimately preventing hypertension and its complications. In a word, pharmacological targeting of SIRT6 will be an innovative therapeutic strategy for hypertension patients in future.286 Tubastatin A (TubA), a highly selective HDAC6 inhibitor, can significantly improve Ang-II-induced vasoconstriction and elevate blood pressure by inhibiting the expression and activity of Ang-II-induced HDAC6 and reducing the degradation of CSEγ. Therefore, TubA can be used as an effective strategy to prevent the progression of hypertension.114 Wang et al.287 found that prenatal exposure to lipopolysaccharide could lead to hypertension in young rats. Nevertheless, ascorbic acid could prevent hypertension occurrence and development in offspring of rats prenatal exposed to lipopolysaccharide. Lipopolysaccharide might induce histone H3 acetylation by inhibiting the enrichment of HDAC1 on ACE1 promoter, resulting in increased ACE1 gene expression in rat offspring and promoting hypertension. However, prenatal treatment with ascorbic acid not only disminished oxidative stress but also downregulated ACE1 gene expression through deacetylation of histone H3 in promoter region ACE1, thereby reducing hypertension risk.287 In conclusion, these findings provide potential targets for new antihypertensive therapies that could act as early prevention or treatment for hypertension.
Taken together, statins widely used in clinical practice are histone acetylation inhibitors, which have good efficacy in the treatment of atherosclerosis and coronary heart disease in both basic and clinical trials. Resveratrol has a protective effect on atherosclerosis, coronary heart disease, ischemia–reperfusion injury, heart failure, hypertension, and pulmonary hypertension by regulating histone modification-related factors. Trichostatin A and P300 HAT inhibitor curcumin can improve the progression of atherosclerosis, coronary heart disease and heart failure. But it has not been applied in clinic to treat cardiovascular diseases. Histone modification inhibitors resveratrol, Trichostatin A and Curcumin are expected to be widely used in the treatment of various cardiovascular diseases through further clinical studies. At present, although some inhibitors of HDACs have been applied in the clinical treatment of tumors, their clinical application in cardiovascular diseases is relatively little. Because of the broad spectrum of HDACs substrates, these drugs may lead to nonspecific gene activation or suppression, and they can act on either disease target cells or normal cells with side effects. Therefore, it is necessary to further explore the epigenetic regulation mechanism during the occurrence and development of cardiovascular diseases. The development of histone modification-related drugs with specificity and low side effects on cardiovascular diseases will be the direction of our focus and efforts in the future.
Noncoding RNA
In recent years, increasing evidence has accumulated for noncoding RNAs function in gene regulation and cardiovascular disease pathogenesis (Figs. 5–7). Noncoding RNAs are attractive targets for potential clinical interventions. Currently, the field of nucleotide gene therapy, including antisense oligonucleotide (ASO) and siRNA, is developing rapidly. Analogs or inhibitors of noncoding RNA are easy to synthesize and have low cytotoxicity when transfected in vivo. Therefore, it will be looking forward to being potential treatment drugs for human heart disease. With the exploration of noncoding RNA mechanism in cardiovascular diseases, we believe that the treatment of noncoding RNA in cardiovascular diseases will usher in a new breakthrough (Table 5).
Atherosclerosis
Noncoding RNAs may serve as privotal therapeutic targets in atherosclerosis. Inclisiran (ALN-PCSSC) is a long-acting RNA interference (RNAi) therapeutic agent that inhibits the synthesis of proprotein convertase subtilisin-kexin type 9 (PCSK9). PCSK9 is a target for lowering LDL cholesterol. Inclisiran was observed in phase 1–3 clinical trials with a low incidence of adverse events and significantly reduced LDL cholesterol levels. Inclisiran may provide a novel method for low-density lipoprotein cholesterol (LDL-C) reduction and is also a more successful RNA drug for cardiovascular disease treatment.51,52,53,54,55 AKCEA-APOCIII-LRx is a liver-targeted N-acetylgalactosamine-coupled antisense oligonucleotide that selectively inhibits the synthesis of apolipoprotein C-III protein, thus treating hypertriglyceridemia.60 One study showed that IONIS-ANGPTL3-LRx was an ASO targeting angiopoietin-like 3 (ANGPTL3) mRNA that reduced atherosclerotic lipoprotein levels in mice and humans and delayed the progression of atherosclerosis.56 Haemmig et al.288 found that lncRNA small nucleolar host gene-12 (SNHG12) was highly expressed in vascular endothelium, but gradually decreased with the progression of the disease. SNHG12 expression was reduced in pig and human atherosclerotic specimens and negatively correlated with DNA damage and markers of aging. DNA-dependent protein kinase (DNA-PK) is an important regulator of DNA damage response. Deletion of SNHG12 decreased the interaction of DNA-PK with their binding partners Ku70 and Ku80 and increased DNA damage. Intravenous administration of SNHG12 alleviated atherosclerosis by protecting the intima from DNA damage and slowing endothelial aging.288 Furthermore, volanesorsen was found to treat hypertriglyceridemia by targeting apolipoprotein C3(APOC3).289,290 IONIS-APO(a)-LRx was a novel, tolerable and effective therapy for reducing lipoprotein(a) (Lp[a]) concentration. IONIS-APO(a)-LRx effectively decreased plasma Lp(a) and its associated OxPL, LDL-C, and APOB-100 by targeting apolipoprotein(a). At the same time, IONIS-APO(a)-LRx attenuated adhesion promoting of plasma monocytes, thereby reducing the risk of cardiovascular disease in individuals elevated by Lp(a). Clinical trials had been conducted in patients with cardiovascular disease or calcified aortic stenosis with elevated Lp(a) concentration.49 In terms of lipid regulation, inhibition of miRNA-33a and miRNA-33b can reduce plasma in LDL-C level and increase plasma HDL cholesterol level without significant adverse reactions. It suggests that targeted inhibition of miRNA-33 plays an important role in the treatment of hyperlipidemia.45 In addition, it has been reported that lncRNA RP5-833A20.1 and lncRNA DYN-LRB2-2 can also be used as atherosclerosis therapeutic targets.291,292 Studies have shown that circWDR77 can regulate the proliferation and migration of VSMCs through the sponge-mediated miRNA-124/FGF-2. Therefore, circWDR77 can be used as a therapeutic target for atherosclerosis.293
Myocardial infarction
The role of noncoding RNA and related drugs in the treatment of myocardial infarction have gradually attracted attention. Among patients with acute coronary syndrome treated with high-intensity statins, patients treated with alirocumab had a lower risk of recurrent ischemic cardiovascular events than those treated with placebo.57 The absolute reduction in cardiovascular events in patients (LDL-C concentration 0.65–1.30 mmol/L) with diabetes treated with alirocumab was twice as large as in those without diabetes. Treatment with alirocumab did not increase the risk of new-onset diabetes.294 Liao et al.295 have reported that in patients with myocardial infarction, lncRNA MIAT participates in the shearing of Wnt7b through targeting miRNA-150-5p and VEGF signaling pathways, and is differentially expressed in patients’ peripheral blood. Therefore, lncRNA MIAT can also be applied as a potential strategy and drug to treat myocardial infarction patients.295 Wang et al.296 found that cirRNA MFACR was upregulated in myocardial infarction. CirRNA MFACR promoted hypoxia-induced apoptosis of cardiomyocytes by downregulating miRNA-125b. Therefore, targeted inhibition of cirRNA MFACR might serve as a essential target for the treatment of myocardial infarction and the protection of myocardial cells.296
Heart failure
Some drugs have a crucial function in the treatment of heart failure by regulating noncoding RNA. MRG-110 and CDR132L have been shown to play an important role in the treatment of heart failure by targeting miRNA-92a and miRNA-132, and are currently in phase 1 clinical trials.297 In view of cirRNA as a targeted therapeutic target for cardiovascular disease, cirRNA (HRCR) can block the development of cardiac hypertrophy and heart failure, and will be looked forward to becoming a controlling gene for the treatment of heart failure and cardiac hypertrophy in future.298 In addition, study demonstrated that ectopic expression of circ-FOXO3 inhibited cell cycle progression by binding to CDK2 and cyclin-dependent kinase inhibitor 1 (or P21), and at the same time, disminished the expression of these proteins in the nucleus and promoted cell aging phenotype. Finally, it provided a new therapeutic strategy for delaying cardiac aging and myocardial protection.299
Vascular calcification
Noncoding RNAs will be novel therapeutic genes for the treatment of vascular calcification in the future. Zhang et al.300 demonstrated that lncRNA-ANCR was an important factor regulating osteoblast differentiation. LncRNA-ANCR might significantly reduce the Runx2 and BMP2 expression and mineralized nodules formation by activating β-GP-induced VSMC autophagy, and weaken the VSMCs osteogenic differentiation, thereby protecting vascular calcification. LncRNA-ANCR could be a pivotal strategy for the treatment of vascular calcification.300 Zhu et al.301 found that lncRNAs H19 significantly enhanced the Runx2, osteocalcin, ALP, and β‐catenin expression levels in human renal interstitial fibroblasts (hRIFs) by activating the Wnt-β-catenin pathway. It leaded to the formation of mineralized nodules. Finally, lncRNAs H19 accelerated hRIFs osteogenic differentiation and calcification process. Downregulation of XAV939 (Wnt-β-catenin signaling pathway inhibitor) inhibited H19-induced hRIFs osteogenic differentiation, which will serve as a potential drug for vascular calcification future treatment.301 In addition, a clinical study showed that exosomes hsa_circRNA_0006859 were upregulated in patients with osteoporosis compared with healthy controls. According to bioinformatics analysis, hsa_circRNA_0006859 might be the sponge of miRNA-431-5p. Hsa_circRNA_0006859 overexpression upregulated the target gene ROCK1 of miRNA-431-5p by inhibiting miRNA-431-5p, significantly decreased the osteocalcin and ALP protein levels in human bone marrow mesenchymal stem cells, and reduced the mineralized nodules formation, thus inhibiting osteoblast differentiation. The results showed that hsa_circRNA_0006859 might be an effective therapeutic gene for preventing vascular calcification.302 These studies suggest that RNA therapy is a promising strategy for treating cardiovascular disease (Figs. 5–7). We look forward to applying these findings to clinical disease treatment.
Noncoding RNA plays an important regulatory role in complex life processes and can be considered as a potential therapeutic target for many cardiovascular diseases. Anti-miRNA and antisense oligonucleotides inhibit some specific miRNAs expression in order to regulate the occurrence and development of cardiovascular diseases, which has been used in the cardiovascular diseases clinical treatment. Using the same strategy to regard noncoding RNAs as therapeutic targets in order to improve cardiomyocytes and vessals status and function, which could be a novel approach to treat cardiovascular disease. We hope that in future clinical work, the detection of cardiovascular-related noncoding RNA plasma levels will contribute to determine the disease and severity of patients. It is even hoped to reverse pathological changes in cardiovascular disease and improve the prognosis of patients with cardiovascular disease through gene-targeted therapy.
In Table 5, we report on potential epigenetic therapy drugs for cardiovascular disease. Meanwhile, in Table 6 are included the few most current clinical trials with observational and/or interventional study type for primary and secondary prevention, registered and extracted from the website https://clinicaltrials.gov. These epigenetics-based clinical trials and related epigenetic drug studies are mainly carried out in patients with atherosclerosis, coronary heart disease, heart failure, hypertension, myocardial infarction, and other cardiovascular diseases. However, most researches on the application of epigenetic regulation to cardiovascular disease are still in preclinical trials or early clinical trials stage. Although some epigenetic drugs have not been widely used in clinical practice in cardiovascular diseases. It is believed that through continuous exploration and large-scale clinical research in the future, more emerging epigenetic drugs for the treatment of cardiovascular diseases will be created, in order to better ameliorate the symptoms and prognosis of patients with cardiovascular diseases.
Conclusions
We briefly review the history and mechanisms of epigenetics. At the same time, the mechanisms of epigenetics such as DNA methylation, histone modification, and noncoding RNA in various cardiovascular diseases are comprehensively described. In view of the recent great attention paid to epigenetics in the treatment of cardiovascular diseases, we also summarize the epigenetics drugs related to cardiovascular diseases and all relevant clinical trials. Throughout the current research and application of epigenetics in cardiovascular diseases, it is found that DNA methylation and histone acetylation have been widely and deeply studied in the fields of coronary heart disease, heart failure, and vascular calcification. However, the role of histone methylation in various cardiovascular disease mechanisms remains poorly explored. This can be a new direction worth further excavating in future. In addition, in recent years, more and more studies have confirmed the role of various potential epigenetic drugs in the treatment of cardiovascular disease. At present, in the field of cardiovascular diseases, HDAC inhibitors application in the treatment of atherosclerosis, myocardial infarction, and heart failure has been relatively more studied. The development of HDAC inhibitors has opened up new approaches for the treatment of cardiovascular diseases. However, previous researchers only explored at the animal level. Therefore, by summarizing clinical trials on epigenetic therapy for cardiovascular diseases, we found that DNA methylation and histone acetylation inhibitors have been widely studied in coronary heart disease, myocardial infarction and hypertension, which are currently in phase 1–3 clinical trials. Due to the lack of reliable evidence from large-scale basic and clinical studies, there are still many problems (such as off-target effects) that need to be addressed in the development of noncoding RNA-based therapies. Effective and safe implementation of noncoding RNA therapy into clinical practice remains a major challenge. Large-scale clinical studies are still needed to develop small molecule drugs based on histone methylation and noncoding RNA expression as therapeutic targets.
In summary, epigenetics is a promising field in the diagnosis and intervention of cardiovascular diseases. The rapid development of epigenetics and genomics may serve as a new direction for the precise treatment of cardiovascular diseases. The development of more epigenetics-related drugs with higher specificity, fewer side effects and lower drug resistance for different types of cardiovascular diseases will be the future development goal. The combination of epigenetic regulatory drugs and targeted drugs that drive the regulatory genes of cardiovascular diseases may be an effective target for overcoming drug resistance, which will bring new hope to overcome the difficulties of drug resistance. It is believed that in the near future, the continuous exploration and research, and development of epigenetic regulatory drugs for cardiovascular diseases will have broader application prospects and benefit more patients with cardiovascular diseases. In future, we hope to further explore the molecular mechanism of epigenetics regulating cardiovascular disease, and find more strategies for the prevention and treatment of cardiovascular disease, so as to better guide clinical treatment.
References
Roth, G. et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25 (2017).
Zhao, D. et al. Epidemiology of cardiovascular disease in China: current features and implications. Nat. Rev. Cardiol. 16, 203–212 (2019).
Andersson, C. et al. 70-year legacy of the Framingham Heart Study. Nat. Rev. Cardiol. 16, 687–698 (2019).
Feinberg, A. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med. 378, 1323–1334 (2018).
Waddington, C. The epigenotype. 1942. Int J. Epidemiol. 41, 10–13 (2012).
Holliday, R. Epigenetics: a historical overview. Epigenetics 1, 76–80 (2006).
Holliday, R. Epigenetics: an overview. Dev. Genet. 15, 453–457 (1994).
Riggs, A. D. & TN, P. In: Russo VEA, Martienssen, R. & Riggs, A. D. (eds) Epigenetic Mechanism of Gene Regulation. Ch. Overview of epigenetic mechanisms 29–45 (Cold Spring Harbor Laboratory Press, 1996).
Riggs, A. D. & TN, P. In: Russo VEA, Martienssen, R. & Riggs, A. D. (eds) Epigenetic Mechanism of Gene Regulation Ch. Introduction, 1–4 (Cold Spring Harbor Laboratory Press, 1996).
Bird, A. Perceptions of epigenetics. Nature 447, 396–398 (2007).
Cavalli, G. & Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499 (2019).
Xie, S. et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene 236, 87–95 (1999).
Mortusewicz, O. et al. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl Acad. Sci. USA 102, 8905–8909 (2005).
Brenner, C. et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 24, 336–346 (2005).
Nan, X., Meehan, R. & Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892 (1993).
Hendrich, B. & Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547 (1998).
Achour, M. et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene 27, 2187–2197 (2008).
Filion, G. et al. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 26, 169–181 (2006).
Millán-Zambrano, G., Burton, A., Bannister, A. & Schneider, R. Histone post-translational modifications—cause and consequence of genome function. Nat. Rev. Genet. https://doi.org/10.1038/s41576-022-00468-7 (2022).
Creyghton, M. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Bannister, A. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 (2001).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Ogryzko, V. et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959 (1996).
Saba, N. et al. Acetylated tubulin (AT) as a prognostic marker in squamous cell carcinoma of the head and neck. Head Neck Pathol. 8, 66–72 (2014).
McLendon, P. et al. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc. Natl Acad. Sci. USA 111, E5178–E5186 (2014).
Chan, H. & La Thangue, N. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114, 2363–2373 (2001).
Kee, H. et al. Trichostatin A prevents neointimal hyperplasia via activation of Krüppel like factor 4. Vasc. Pharm. 55, 127–134 (2011).
Yoon, S. et al. PP2A negatively regulates the hypertrophic response by dephosphorylating HDAC2 S394 in the heart. Exp. Mol. Med. 50, 1–14 (2018).
Poddar, S., Kesharwani, D. & Datta, M. Interplay between the miRNome and the epigenetic machinery: implications in health and disease. J. Cell Physiol. 232, 2938–2945 (2017).
Reddy, S. et al. miR-21 is associated with fibrosis and right ventricular failure. JCI Insight 2, e91625 (2017).
Petrovic, N. & Ergun, S. miRNAs as potential treatment targets and treatment options in cancer. Mol. Diagn. Ther. 22, 157–168 (2018).
St Laurent, G., Wahlestedt, C. & Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 31, 239–251 (2015).
Quinn, J. & Chang, H. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 17, 47–62 (2016).
Wei, J., Huang, K., Yang, C. & Kang, C. Non-coding RNAs as regulators in epigenetics (review). Oncol. Rep. 37, 3–9 (2017).
Lou, Z. et al. Minor and major circRNAs in virus and host genomes. J. Microbiol. 59, 324–331 (2021).
Yang, T. et al. Circle the cardiac remodeling with circRNAs. Front. Cardiovasc Med. 8, 702586 (2021).
Bahn, J. et al. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin. Chem. 61, 221–230 (2015).
Hansen, T., Kjems, J. & Damgaard, C. Circular RNA and miR-7 in cancer. Cancer Res. 73, 5609–5612 (2013).
Li, J. et al. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am. J. Cancer Res. 5, 472–480 (2015).
Zhou, Z., Sun, B., Huang, S. & Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 10, 503 (2019).
Bayoumi, A. et al. Circular noncoding RNAs as potential therapies and circulating biomarkers for cardiovascular diseases. Acta Pharm. Sin. 39, 1100–1109 (2018).
Li, J. et al. A novel strategy of identifying circRNA biomarkers in cardiovascular disease by meta-analysis. J. Cell Physiol. 234, 21601–21612 (2019).
Zhu, S., Goldschmidt-Clermont, P. & Dong, C. Inactivation of monocarboxylate transporter MCT3 by DNA methylation in atherosclerosis. Circulation 112, 1353–1361 (2005).
Kong, Y. et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113, 2579–2588 (2006).
Rayner, K. et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478, 404–407 (2011).
Anand, P. et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582 (2013).
Xie, M. et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129, 1139–1151 (2014).
Sniderman, A., Tsimikas, S. & Fazio, S. The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J. Am. Coll. Cardiol. 63, 1935–1947 (2014).
Viney, N. et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 388, 2239–2253 (2016).
Fuster, J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Fitzgerald, K., Kallend, D. & Simon, A. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 376, e38 (2017).
Ray, K. et al. Effect of 1 or 2 doses of inclisiran on low-density lipoprotein cholesterol levels: one-year follow-up of the ORION-1 randomized clinical trial. JAMA Cardiol. 4, 1067–1075 (2019).
Ray, K. et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Engl. J. Med. 376, 1430–1440 (2017).
Ray, K. et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 382, 1507–1519 (2020).
Smith, K. & White, C. Inclisiran: a novel small interfering RNA drug for LDL reduction. J. Clin. Pharmacol. https://doi.org/10.1002/jcph.2045 (2022).
Graham, M. et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N. Engl. J. Med. 377, 222–232 (2017).
Schwartz, G. et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 379, 2097–2107 (2018).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Malhotra, R. et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 51, 1580–1587 (2019).
Alexander, V. et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 40, 2785–2796 (2019).
Potus, F. et al. TET2 novel mutations and decreased expression of the epigenetic regulator in pulmonary arterial hypertension. Circulation 141, 1986–2000 (2020).
Kim, S. et al. Epigenetic reader BRD4 (bromodomain-containing protein 4) governs nucleus-encoded mitochondrial transcriptome to regulate cardiac function. Circulation 142, 2356–2370 (2020).
Farina, F. et al. The epigenetic enzyme DOT1L orchestrates vascular smooth muscle cell-monocyte crosstalk and protects against atherosclerosis via the NF-κB pathway. Eur. Heart J. https://doi.org/10.1093/eurheartj/ehac097 (2022).
Westerman, K. et al. DNA methylation modules associate with incident cardiovascular disease and cumulative risk factor exposure. Clin. Epigenet. 11, 142 (2019).
Battle, A., Brown, C., Engelhardt, B. & Montgomery, S. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Kennel, P. et al. Impairment of myocardial glutamine homeostasis induced by suppression of the amino acid carrier SLC1A5 in failing myocardium. Circ. Heart Fail 12, e006336 (2019).
Navas-Acien, A. et al. Blood DNA Methylation and incident coronary heart disease: evidence from the strong heart study. JAMA Cardiol. 6, 1237–1246 (2021).
Luo, X. et al. Integrative analysis of DNA methylation and gene expression reveals key molecular signatures in acute myocardial infarction. Clin. Epigenet. 14, 46 (2022).
Palou-Márquez, G. et al. DNA methylation and gene expression integration in cardiovascular disease. Clin. Epigenet. 13, 75 (2021).
Teng, N. et al. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 22, 51–73 (2017).
Yao, C. et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 9, 3268 (2018).
Chen, Y. et al. Whole genome DNA methylation analysis of obstructive sleep apnea: IL1R2, NPR2, AR, SP140 methylation and clinical phenotype. Sleep 39, 743–755 (2016).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Fernández-Sanlés, A. et al. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin. Epigenet. 13, 86 (2021).
Madsen, A. et al. An important role for DNMT3A-mediated DNA methylation in cardiomyocyte metabolism and contractility. Circulation 142, 1562–1578 (2020).
Glezeva, N. et al. Targeted DNA methylation profiling of human cardiac tissue reveals novel epigenetic traits and gene deregulation across different heart failure patient subtypes. Circ. Heart Fail 12, e005765 (2019).
Laugier, L. et al. Whole-genome cardiac DNA methylation fingerprint and gene expression analysis provide new insights in the pathogenesis of chronic Chagas disease cardiomyopathy. Clin. Infect. Dis. 65, 1103–1111 (2017).
Zhu, H. et al. Selenium supplementation improved cardiac functions by suppressing DNMT2-mediated GPX1 promoter DNA methylation in AGE-induced heart failure. Oxid. Med. Cell Longev. 2022, 5402997 (2022).
Dai, X. et al. Epigenetic upregulation of H19 and AMPK inhibition concurrently contribute to S-adenosylhomocysteine hydrolase deficiency-promoted atherosclerotic calcification. Circ. Res. 130, 1565–1582 (2022).
Ramachandran, B. et al. A GTPase-activating protein-binding protein (G3BP1)/antiviral protein relay conveys arteriosclerotic Wnt signals in aortic smooth muscle cells. J. Biol. Chem. 293, 7942–7968 (2018).
Montes de Oca, A. et al. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J. Bone Min. Res. 25, 1996–2005 (2010).
Lin, X. et al. Aberration methylation of miR-34b was involved in regulating vascular calcification by targeting Notch1. Aging 11, 3182–3197 (2019).
Lin, X. et al. Arterial calcification is regulated via an miR-204/DNMT3a regulatory circuit both in vitro and in female mice. Endocrinology 159, 2905–2916 (2018).
Roberts, M. et al. Unique associations of DNA methylation regions with 24-hour blood pressure phenotypes in black participants. Hypertension 79, 761–772 (2022).
Dwi Putra, S. et al. Association between placental global DNA methylation and blood pressure during human pregnancy. J. Hypertens. 40, 1002–1009 (2022).
Jin, F. et al. Association of mitofusin 2 methylation and essential hypertension: a case-control study in a Chinese population. Hypertens. Res. 41, 605–613 (2018).
Bao, X. et al. Hypomethylation of the interferon γ gene as a potential risk factor for essential hypertension: a case-control study. Tohoku J. Exp. Med. 244, 283–290 (2018).
Papait, R. et al. Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation 136, 1233–1246 (2017).
Kurozumi, A. et al. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 124, 53–61 (2019).
Maleszewska, M., Gjaltema, R., Krenning, G. & Harmsen, M. Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22α expression in endothelial cells in response to interleukin-1β and transforming growth factor-β2. Cell Signal 27, 1589–1596 (2015).
Mattagajasingh, I. et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA 104, 14855–14860 (2007).
Jung, S. et al. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ. Res. 107, 877–887 (2010).
Schermuly, R., Ghofrani, H., Wilkins, M. & Grimminger, F. Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 8, 443–455 (2011).
Miyoshi, T. et al. Deficiency of inducible NO synthase reduces advanced but not early atherosclerosis in apolipoprotein E-deficient mice. Life Sci. 79, 525–531 (2006).
Cortese-Krott, M. et al. Zinc regulates iNOS-derived nitric oxide formation in endothelial cells. Redox Biol. 2, 945–954 (2014).
Shinozaki, S. et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci. Signal 7, ra106 (2014).
Ding, M. et al. SIRT1 protects against myocardial ischemia-reperfusion injury via activating eNOS in diabetic rats. Cardiovasc Diabetol. 14, 143 (2015).
Lu, Y. et al. Thymoquinone attenuates myocardial ischemia/reperfusion injury through activation of SIRT1 signaling. Cell Physiol. Biochem 47, 1193–1206 (2018).
Wang, Y. et al. SIRT2-mediated FOXO3a deacetylation drives its nuclear translocation triggering FasL-induced cell apoptosis during renal ischemia reperfusion. Apoptosis 22, 519–530 (2017).
Bochaton, T. et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell Cardiol. 84, 61–69 (2015).
Leng, Y. et al. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid. Med Cell Longev. 2018, 9494052 (2018).
Tang, X. et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 136, 2051–2067 (2017).
Sundaresan, N. et al. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol. Cell Biol. 36, 678–692 (2015).
Luo, Y. et al. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 38, 1389–1398 (2017).
Shen, P. et al. SIRT6 suppresses phenylephrine-induced cardiomyocyte hypertrophy through inhibiting p300. J. Pharm. Sci. 132, 31–40 (2016).
Tao, H., Yang, J., Shi, K. & Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 65, 415–426 (2016).
Baldi, A. et al. Apoptosis and post-infarction left ventricular remodeling. J. Mol. Cell Cardiol. 34, 165–174 (2002).
Collesi, C. et al. Reversible Notch1 acetylation tunes proliferative signalling in cardiomyocytes. Cardiovasc Res. 114, 103–122 (2018).
Alcendor, R. et al. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 95, 971–980 (2004).
Vakhrusheva, O. et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 102, 703–710 (2008).
Li, W. et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J. Clin. Investig. 132, e150051 (2022).
Abend, A. et al. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 18, 1166–1185 (2017).
Bartoli-Leonard, F. et al. Suppression of SIRT1 in diabetic conditions induces osteogenic differentiation of human vascular smooth muscle cells via RUNX2 signalling. Sci. Rep. 9, 878 (2019).
Chi, Z. et al. Histone deacetylase 6 inhibitor tubastatin A attenuates angiotensin II-induced hypertension by preventing cystathionine γ-lyase protein degradation. Pharmacol. Res. 146, 104281 (2019).
Chelladurai, P. et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 178, 54–71 (2021).
Dikalova, A. et al. Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res. 121, 564–574 (2017).
Arise, K. et al. Angiotensin II represses Npr1 expression and receptor function by recruitment of transcription factors CREB and HSF-4a and activation of HDACs. Sci. Rep. 10, 4337 (2020).
Hall, I. et al. rs41291957 controls miR-143 and miR-145 expression and impacts coronary artery disease risk. EMBO Mol. Med. 13, e14060 (2021).
Jiang, F. et al. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 72, 156–166 (2020).
Liu, Y. et al. Exosome-mediated miR-106a-3p derived from ox-LDL exposed macrophages accelerated cell proliferation and repressed cell apoptosis of human vascular smooth muscle cells. Eur. Rev. Med Pharm. Sci. 24, 7039–7050 (2020).
Xing, X. et al. Adipose-derived mesenchymal stem cells-derived exosome-mediated microRNA-342-5p protects endothelial cells against atherosclerosis. Aging 12, 3880–3898 (2020).
Chang, Y. et al. Extracellular microRNA-92a mediates endothelial cell-macrophage communication. Atertio. Thromb. Vasc. Biol. 39, 2492–2504 (2019).
Zhu, L. et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics 8, 6163–6177 (2018).
Luther, K. et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J. Mol. Cell Cardiol. 119, 125–137 (2018).
Peng, Y. et al. Exosomal miR-25-3p from mesenchymal stem cells alleviates myocardial infarction by targeting pro-apoptotic proteins and EZH2. Cell Death Dis. 11, 317 (2020).
Wen, Z. et al. Mesenchymal stem cell-derived exosomes ameliorate cardiomyocyte apoptosis in hypoxic conditions through microRNA144 by targeting the PTEN/AKT pathway. Stem Cell Res Ther. 11, 36 (2020).
Ling, H. et al. Serum exosomal miR-122-5p is a new biomarker for both acute coronary syndrome and underlying coronary artery stenosis. Biomarkers 25, 539–547 (2020).
Ling, H. et al. Serum exosomal MicroRNA-21, microRNA-126, and PTEN are novel biomarkers for diagnosis of acute coronary syndrome. Front Physiol. 11, 654 (2020).
Eulalio, A. et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381 (2012).
Danielson, K. et al. Plasma circulating extracellular RNAs in left ventricular remodeling post-myocardial infarction. EBioMedicine 32, 172–181 (2018).
Wu, T. et al. Circulating exosomal miR-92b-5p is a promising diagnostic biomarker of heart failure with reduced ejection fraction patients hospitalized for acute heart failure. J. Thorac. Dis. 10, 6211–6220 (2018).
Wang, L. et al. Reduced exosome miR-425 and miR-744 in the plasma represents the progression of fibrosis and heart failure. Kaohsiung J. Med. Sci. 34, 626–633 (2018).
Ma, W. et al. Restoring mitochondrial biogenesis with metformin attenuates β-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol. Cell. Endocrinol. 479, 39–53 (2019).
Xu, T. et al. Restoration of microRNA-30b expression alleviates vascular calcification through the mTOR signaling pathway and autophagy. J. Cell Physiol. 234, 14306–14318 (2019).
Ding, Y. et al. Association of plasma MiRNA-204 and the presence and severity of coronary artery calcification in patients with type 2 diabetes. Angiology 72, 451–458 (2021).
Wang, X., Zhang, X., Li, F. & Ji, Q. MiR-128-3p accelerates cardiovascular calcification and insulin resistance through ISL1-dependent Wnt pathway in type 2 diabetes mellitus rats. J. Cell Physiol. 234, 4997–5010 (2019).
Chen, R. et al. MiRNA-19a-3p alleviates the progression of osteoporosis by targeting HDAC4 to promote the osteogenic differentiation of hMSCs. Biochem Biophys. Res Commun. 516, 666–672 (2019).
Sindi, H. et al. Therapeutic potential of KLF2-induced exosomal microRNAs in pulmonary hypertension. Nat. Commun. 11, 1185 (2020).
Leimena, C. & Qiu, H. Non-coding RNA in the pathogenesis, progression and treatment of hypertension. Int J. Mol. Sci. 19, 927 (2018).
Wang, Y. et al. Regulation of the human placental (pro)renin receptor-prorenin-angiotensin system by microRNAs. Mol. Hum. Reprod. 24, 453–464 (2018).
Guo, X. et al. A Linc1405/Eomes complex promotes cardiac mesoderm specification and cardiogenesis. Cell Stem Cell 22, 893–908.e6 (2018).
Banerjee, P. et al. Long noncoding RNA RP11-380D23.2 drives distal-proximal patterning of the lung by regulating PITX2 expression. Stem Cells 36, 218–229 (2018).
Gore-Panter, S. et al. PANCR, the PITX2 adjacent noncoding RNA, is expressed in human left atria and regulates PITX2c expression. Circ. Arrhythm. Electrophysiol. 9, e003197 (2016).
Ritter, N. et al. The lncRNA locus handsdown regulates cardiac gene programs and is essential for early mouse development. Dev. Cell 50, 644–657.e8 (2019).
Cai, B. et al. The long noncoding RNA CAREL controls cardiac regeneration. J. Am. Coll. Cardiol. 72, 534–550 (2018).
Wang, J. et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell Cardiol. 127, 105–114 (2019).
Li, B. et al. Sirt1 antisense long noncoding RNA promotes cardiomyocyte proliferation by enhancing the stability of Sirt1. J. Am. Heart Assoc. 7, e009700 (2018).
Chen, G. et al. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J. Mol. Cell Cardiol. 122, 152–164 (2018).
Li, X. et al. Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc Res. 114, 1642–1655 (2018).
Ren, S. et al. Downregulation of lncRNA-SRA participates in the development of cardiovascular disease in type II diabetic patients. Exp. Ther. Med. 17, 3367–3372 (2019).
Chen, Q. et al. Expression profile of long non-coding RNAs in rat models of OSA-induced cardiovascular disease: new insight into pathogenesis. Sleep. Breath. 23, 795–804 (2019).
Ziaee, S. et al. Non-invasive diagnosis of early-onset coronary artery disease based on cell type-specific gene expression analyses. Biomed. Pharmacother. 108, 1115–1122 (2018).
Lai, C. et al. Circulating long noncoding RNA DKFZP434I0714 predicts adverse cardiovascular outcomes in patients with end-stage renal disease. Int. J. Cardiol. 277, 212–219 (2019).
Sallam, T., Sandhu, J. & Tontonoz, P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function. Circ. Res. 122, 155–166 (2018).
Holdt, L. et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Atertio. Thromb. Vasc. Biol. 30, 620–627 (2010).
Miao, Y. et al. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 9, 292 (2018).
Wu, G. et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130, 1452–1465 (2014).
Ballantyne, M. et al. Smooth muscle enriched long noncoding RNA (SMILR) regulates cell proliferation. Circulation 133, 2050–2065 (2016).
Sallam, T. et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med. 24, 304–312 (2018).
Hu, Y. et al. Long noncoding RNA NEXN-AS1 mitigates atherosclerosis by regulating the actin-binding protein NEXN. J. Clin. Investig. 129, 1115–1128 (2019).
Vausort, M., Wagner, D. & Devaux, Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ. Res. 115, 668–677 (2014).
Gao, L. et al. Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cell Physiol. Biochem. 44, 1497–1508 (2017).
Wang, K. et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat. Commun. 6, 6779 (2015).
Liu, C. et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 9, 29 (2018).
Liang, H. et al. 2810403D21Rik/MirfLncRNA promotes ischemic myocardial injury by regulating autophagy through targeting. Autophagy 16, 1077–1091 (2020).
Huang, L. et al. Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB Life 72, 373–383 (2020).
Wang, Q., Zhou, J. & Li, X. LncRNA UCA1 protects cardiomyocytes against hypoxia/reoxygenation induced apoptosis through inhibiting miR-143/MDM2/p53 axis. Genomics 112, 574–580 (2020).
Xie, L. et al. The roles of lncRNA in myocardial infarction: molecular mechanisms, diagnosis biomarkers, and therapeutic perspectives. Front. Cell Dev. Biol. 9, 680713 (2021).
Ponnusamy, M. et al. Long noncoding RNA CPR (cardiomyocyte proliferation regulator) regulates cardiomyocyte proliferation and cardiac repair. Circulation 139, 2668–2684 (2019).
Yuan, Z. & Huang, W. New developments in exosomal lncRNAs in cardiovascular diseases. Front. Cardiovasc Med. 8, 709169 (2021).
Wang, L. & Zhang, J. Exosomal lncRNA AK139128 derived from hypoxic cardiomyocytes promotes apoptosis and inhibits cell proliferation in cardiac fibroblasts. Int. J. Nanomed. 15, 3363–3376 (2020).
Omura, J. et al. Identification of long noncoding RNA H19 as a new biomarker and therapeutic target in right ventricular failure in pulmonary arterial hypertension. Circulation 142, 1464–1484 (2020).
Kumarswamy, R. et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 114, 1569–1575 (2014).
Hua, X. et al. Multi-level transcriptome sequencing identifies COL1A1 as a candidate marker in human heart failure progression. BMC Med. 18, 2 (2020).
Piccoli, M. et al. Meg3 inhibition of the cardiac fibroblast-enriched lncRNA prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 121, 575–583 (2017).
Micheletti, R. et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 9, eaai9118 (2017).
Yu, C. et al. LncRNA TUG1 sponges miR-204-5p to promote osteoblast differentiation through upregulating Runx2 in aortic valve calcification. Cardiovasc Res. 114, 168–179 (2018).
Jeong, G. et al. Long noncoding RNAs in vascular smooth muscle cells regulate vascular calcification. Sci. Rep. 9, 5848 (2019).
Huang, C. et al. LncRNA-SNHG29 inhibits vascular smooth muscle cell calcification by downregulating miR-200b-3p to activate the α-Klotho/FGFR1/FGF23 axis. Cytokine 136, 155243 (2020).
Zhong, J. et al. LncRNA-ES3 inhibition by Bhlhe40 is involved in high glucose-induced calcification/senescence of vascular smooth muscle cells. Ann. N. Y. Acad. Sci. 1474, 61–72 (2020).
Yang, X. et al. The transcription factor C/EBPβ promotes HFL-1 cell migration, proliferation, and inflammation by activating lncRNA HAS2-AS1 in hypoxia. Front. Cell Dev. Biol. 9, 651913 (2021).
Fang, G., Qi, J., Huang, L. & Zhao, X. LncRNA MRAK048635_P1 is critical for vascular smooth muscle cell function and phenotypic switching in essential hypertension. Biosci. Rep. 39, BSR20182229 (2019).
Lin, F. et al. circRNA‑miRNA association for coronary heart disease. Mol. Med. Rep. 19, 2527–2536 (2019).
Pan, R. et al. Circular RNAs promote TRPM3 expression by inhibiting hsa-miR-130a-3p in coronary artery disease patients. Oncotarget 8, 60280–60290 (2017).
Mao, Y. et al. Circ-SATB2 upregulates STIM1 expression and regulates vascular smooth muscle cell proliferation and differentiation through miR-939. Biochem. Biophys. Res. Commun. 505, 119–125 (2018).
Shen, L. et al. CircRNA‑0044073 is upregulated in atherosclerosis and increases the proliferation and invasion of cells by targeting miR‑107. Mol. Med Rep. 19, 3923–3932 (2019).
Bazan, H. et al. Carotid plaque rupture is accompanied by an increase in the ratio of serum circR-284 to miR-221 levels. Circ. Cardiovasc. Genet. 10, e001720 (2017).
Sun, J., Zhang, Z. & Yang, S. Circ_RUSC2 upregulates the expression of miR-661 target gene SYK and regulates the function of vascular smooth muscle cells. Biochem. Cell Biol. 97, 709–714 (2019).
Geng, H. et al. The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PLoS ONE 11, e0151753 (2016).
Wang, K. et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 24, 1111–1120 (2017).
Cai, L. et al. Circular RNA Ttc3 regulates cardiac function after myocardial infarction by sponging miR-15b. J. Mol. Cell Cardiol. 130, 10–22 (2019).
Huang, S. et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 139, 2857–2876 (2019).
Lim, T. et al. Targeting the highly abundant circular RNA circSlc8a1 in cardiomyocytes attenuates pressure overload induced hypertrophy. Cardiovasc Res. 115, 1998–2007 (2019).
Zhou, B. & Yu, J. A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-β1. Biochem. Biophys. Res. Commun. 487, 769–775 (2017).
Ni, H. et al. Inhibition of circHIPK3 prevents angiotensin II-induced cardiac fibrosis by sponging miR-29b-3p. Int. J. Cardiol. 292, 188–196 (2019).
Zhu, Y. et al. Upregulation of circular RNA CircNFIB attenuates cardiac fibrosis by sponging miR-433. Front. Genet. 10, 564 (2019).
Li, M. et al. ncx1A circular transcript of gene mediates ischemic myocardial injury by targeting miR-133a-3p. Theranostics 8, 5855–5869 (2018).
Zhou, L. et al. The circular RNA ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy via modulation of the Pink1/ FAM65B pathway. Cell Death Differ. 26, 1299–1315 (2019).
Jiang, L. et al. Advance in circular RNA modulation effects of heart failure. Gene 763, 100036 (2020).
Wu, N. et al. Circular RNA mmu_circ_0005019 inhibits fibrosis of cardiac fibroblasts and reverses electrical remodeling of cardiomyocytes. BMC Cardiovasc Disord. 21, 308 (2021).
Zhang, M. et al. Circular RNA (circRNA) CDYL induces myocardial regeneration by ceRNA after myocardial infarction. Med. Sci. Monit. 26, e923188 (2020).
Bai, M., Pan, C., Jiang, G. & Zhang, Y. CircRNA 010567 improves myocardial infarction rats through inhibiting TGF-β1. Eur. Rev. Med Pharm. Sci. 24, 369–375 (2020).
Sun, L. et al. Circ_LAS1L regulates cardiac fibroblast activation, growth, and migration through miR-125b/SFRP5 pathway. Cell Biochem. Funct. 38, 443–450 (2020).
Li, H. et al. Circular RNA circRNA_000203 aggravates cardiac hypertrophy via suppressing miR-26b-5p and miR-140-3p binding to Gata4. Cardiovasc Res. 116, 1323–1334 (2020).
Garikipati, V. et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 10, 4317 (2019).
Liu, X. et al. CircRNA ACAP2 induces myocardial apoptosis after myocardial infarction by sponging miR-29. Minerva Med. 113, 128–134 (2020).
Ma, C. et al. circRNA CDR1as promotes pulmonary artery smooth muscle cell calcification by upregulating CAMK2D and CNN3 via sponging miR-7-5p. Mol. Ther. Nucleic Acids 22, 530–541 (2020).
Yu, C., Wu, D., Zhao, C. & Wu, C. CircRNA TGFBR2/MiR-25-3p/TWIST1 axis regulates osteoblast differentiation of human aortic valve interstitial cells. J. Bone Min. Metab. 39, 360–371 (2021).
Zhang, D. et al. CircRNA-vgll3 promotes osteogenic differentiation of adipose-derived mesenchymal stem cells via modulating miRNA-dependent integrin α5 expression. Cell Death Differ. 28, 283–302 (2021).
Sun, Y. et al. A novel regulatory mechanism of smooth muscle α-actin expression by NRG-1/circACTA2/miR-548f-5p axis. Circ. Res. 121, 628–635 (2017).
Bahado-Singh, R. et al. Placental DNA methylation changes in detection of tetralogy of Fallot. Ultrasound Obstet. Gynecol. 55, 768–775 (2020).
Katakia, Y. et al. Dynamic alterations of H3K4me3 and H3K27me3 at ADAM17 and Jagged-1 gene promoters cause an inflammatory switch of endothelial cells. J. Cell Physiol. 237, 992–1012 (2022).
Lugenbiel, P. et al. Epigenetic regulation of cardiac electrophysiology in atrial fibrillation: HDAC2 determines action potential duration and suppresses NRSF in cardiomyocytes. Basic Res. Cardiol. 116, 13 (2021).
Yao, L. et al. LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol. Biol. Rep. 47, 2605–2617 (2020).
Costantino, S. et al. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur. Heart J. 40, 997–1008 (2019).
Cao, Q. et al. Inhibiting DNA Methylation by 5-Aza-2’-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology 155, 4925–4938 (2014).
Zhuang, J. et al. The Yin-Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Atertio. Thromb. Vasc. Biol. 37, 84–97 (2017).
Yu, J. et al. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci. Rep. 6, 30053 (2016).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Kim, J. et al. Epigenetic changes in estrogen receptor beta gene in atherosclerotic cardiovascular tissues and in-vitro vascular senescence. Biochim Biophys. Acta 1772, 72–80 (2007).
Connelly, J. et al. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum. Mol. Genet. 22, 5107–5120 (2013).
Crescenti, A. et al. Cocoa consumption alters the global DNA methylation of peripheral leukocytes in humans with cardiovascular disease risk factors: a randomized controlled trial. PLoS ONE 8, e65744 (2013).
Guay, S. et al. Acetylsalicylic acid, aging and coronary artery disease are associated with ABCA1 DNA methylation in men. Clin. Epigenet. 6, 14 (2014).
Stenzig, J. et al. Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J. Mol. Cell Cardiol. 120, 53–63 (2018).
Kao, Y. et al. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 38, 217–222 (2010).
Watson, C. et al. Epigenetic therapy for the treatment of hypertension-induced cardiac hypertrophy and fibrosis. J. Cardiovasc Pharm. Ther. 21, 127–137 (2016).
Fang, X. et al. cAMP induces hypertrophy and alters DNA methylation in HL-1 cardiomyocytes. Am. J. Physiol. Cell Physiol. 309, C425–C436 (2015).
Xiao, D. et al. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res. 101, 373–382 (2014).
Tanaka, U. et al. Decitabine inhibits bone resorption in periodontitis by upregulating anti-inflammatory cytokines and suppressing osteoclastogenesis. Biomedicines 9, 199 (2021).
Xiao, D. et al. The roles of SMYD4 in epigenetic regulation of cardiac development in zebrafish. PLoS Genet. 14, e1007578 (2018).
Yang, B. et al. Suv39h1 protects from myocardial ischemia-reperfusion injury in diabetic rats. Cell Physiol. Biochem. 33, 1176–1185 (2014).
Weng, X. et al. Class II transactivator (CIITA) mediates IFN-γ induced eNOS repression by enlisting SUV39H1. Biochim Biophys. Acta Gene Regul. Mech. 1862, 163–172 (2019).
Papait, R. et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl Acad. Sci. USA 110, 20164–20169 (2013).
Ono, T. et al. The histone 3 lysine 9 methyltransferase inhibitor chaetocin improves prognosis in a rat model of high salt diet-induced heart failure. Sci. Rep. 7, 39752 (2017).
Han, S. et al. Resveratrol affects histone 3 lysine 27 methylation of vessels and blood biomarkers in DOCA salt-induced hypertension. Mol. Biol. Rep. 42, 35–42 (2015).
Arunachalam, G. et al. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: Role of resveratrol. Biochem. Biophys. Res. Commun. 393, 66–72 (2010).
Dje N’Guessan, P. et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Atertio. Thromb. Vasc. Biol. 29, 380–386 (2009).
Palomo, M. et al. Up-regulation of HDACs, a harbinger of uraemic endothelial dysfunction, is prevented by defibrotide. J. Cell. Mol. Med. 24, 1713–1723 (2020).
Aune, S., Herr, D., Mani, S. & Menick, D. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J. Mol. Cell Cardiol. 72, 138–145 (2014).
Herr, D. et al. HDAC1 localizes to the mitochondria of cardiac myocytes and contributes to early cardiac reperfusion injury. J. Mol. Cell Cardiol. 114, 309–319 (2018).
Zhang, L. et al. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J. Pharm. Exp. Ther. 341, 285–293 (2012).
Guo, X. et al. NFκB promotes oxidative stress-induced necrosis and ischemia/reperfusion injury by inhibiting Nrf2-ARE pathway. Free Radic. Biol. Med. 159, 125–135 (2020).
Zhao, T., Zhang, L., Cheng, G. & Liu, J. gp-91 mediates histone deacetylase inhibition-induced cardioprotection. Biochim Biophys. Acta 1803, 872–880 (2010).
Zhang, L. et al. Targeted deletion of NF-kappaB p50 diminishes the cardioprotection of histone deacetylase inhibition. Am. J. Physiol. Heart Circ. Physiol. 298, H2154–H2163 (2010).
Wang, Y. et al. Inhibition of histone deacetylases prevents cardiac remodeling after myocardial infarction by restoring autophagosome processing in cardiac fibroblasts. Cell Physiol. Biochem. 49, 1999–2011 (2018).
Yu, L., Lu, M., Wang, P. & Chen, X. Trichostatin A ameliorates myocardial ischemia/reperfusion injury through inhibition of endoplasmic reticulum stress-induced apoptosis. Arch. Med Res. 43, 190–196 (2012).
Lee, T., Lin, M. & Chang, N. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am. J. Physiol. Heart Circ. Physiol. 293, H968–H977 (2007).
Nagata, S., Marunouchi, T. & Tanonaka, K. Histone deacetylase inhibitor SAHA treatment prevents the development of heart failure after myocardial infarction via an induction of heat-shock proteins in rats. Biol. Pharm. Bull. 42, 453–461 (2019).
Minucci, S. & Pelicci, P. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6, 38–51 (2006).
Kimbrough, D. et al. HDAC inhibition helps post-MI healing by modulating macrophage polarization. J. Mol. Cell Cardiol. 119, 51–63 (2018).
Zhang, L. et al. Inhibition of histone deacetylase-induced myocardial repair is mediated by c-kit in infarcted hearts. J. Biol. Chem. 287, 39338–39348 (2012).
Gillette, T. & Hill, J. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ. Res. 116, 1245–1253 (2015).
Becatti, M. et al. SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes. Cell Mol. Life Sci. 69, 2245–2260 (2012).
Tan, L., Yu, J. & Guan, H. Resveratrol exerts pharmacological preconditioning by activating PGC-1alpha. Med. Hypotheses 71, 664–667 (2008).
Sin, T. et al. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J. Physiol. 592, 2535–2548 (2014).
Biala, A. et al. Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press 19, 196–205 (2010).
Liu, X. et al. SRT1720 promotes survival of aged human mesenchymal stem cells via FAIM: a pharmacological strategy to improve stem cell-based therapy for rat myocardial infarction. Cell Death Dis. 8, e2731 (2017).
Shalwala, M. et al. Sirtuin 1 (SIRT1) activation mediates sildenafil induced delayed cardioprotection against ischemia-reperfusion injury in mice. PLoS ONE 9, e86977 (2014).
Potenza, M. et al. Activation of AMPK/SIRT1 axis is required for adiponectin-mediated preconditioning on myocardial ischemia-reperfusion (I/R) injury in rats. PLoS ONE 14, e0210654 (2019).
Koka, S. et al. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: potential role of NO/SIRT1/PGC-1α signaling. Am. J. Physiol. Heart Circ. Physiol. 306, H1558–H1568 (2014).
Wang, S. et al. ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: role of CaM kinase II, histone H3K9 methyltransferase SUV39H, Sirt1, and PGC-1α deacetylation. Int J. Obes. 42, 1073–1087 (2018).
Morales, C. et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci. Signal 9, ra34 (2016).
Akhondzadeh, F. et al. Resveratrol suppresses interleukin-6 expression through activation of sirtuin 1 in hypertrophied H9c2 cardiomyoblasts. J. Cell Physiol. 235, 6969–6977 (2020).
Soni, K. & Kuttan, R. Effect of oral curcumin administration on serum peroxides and cholesterol levels in human volunteers. Indian J. Physiol. Pharm. 36, 273–275 (1992).
Ramirez Boscá, A. et al. An hydroalcoholic extract of Curcuma longa lowers the abnormally high values of human-plasma fibrinogen. Mech. Ageing Dev. 114, 207–210 (2000).
Pan, M., Lai, C., Wu, J. & Ho, C. Epigenetic and disease targets by polyphenols. Curr. Pharm. Des. 19, 6156–6185 (2013).
Antos, C. et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 278, 28930–28937 (2003).
Gallo, P. et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res. 80, 416–424 (2008).
Kee, H. et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113, 51–59 (2006).
Chen, Y. et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. 14, 99 (2015).
Kashyap, S. et al. HOPX plays a critical role in antiretroviral drugs induced epigenetic modification and cardiac hypertrophy. Cells 10, 3458 (2021).
Evans, L. et al. Emodin and emodin-rich rhubarb inhibits histone deacetylase (HDAC) activity and cardiac myocyte hypertrophy. J. Nutr. Biochem. 79, 108339 (2020).
Lkhagva, B. et al. Novel histone deacetylase inhibitor modulates cardiac peroxisome proliferator-activated receptors and inflammatory cytokines in heart failure. Pharmacology 96, 184–191 (2015).
Nural-Guvener, H. et al. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenes. Tissue Repair 7, 10 (2014).
Nural-Guvener, H. et al. Anti-fibrotic effects of class I HDAC inhibitor, mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int. J. Mol. Sci. 16, 11482–11499 (2015).
Kang, S. et al. Histone deacetylase inhibition attenuates cardiac hypertrophy and fibrosis through acetylation of mineralocorticoid receptor in spontaneously hypertensive rats. Mol. Pharmacol. 87, 782–791 (2015).
Nebbioso, A. et al. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 21, e51028 (2020).
Duan, Q. et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 9, eaah5084 (2017).
Stratton, M. et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ. Res. 125, 662–677 (2019).
Gu, J. et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp. Mol. Med. 51, 1–14 (2019).
Li, S. et al. Activated p300 acetyltransferase activity modulates aortic valvular calcification with osteogenic transdifferentiation and downregulation of Klotho. Int. J. Cardiol. 232, 271–279 (2017).
Fu, Z. et al. Histone deacetylase 6 reduction promotes aortic valve calcification via an endoplasmic reticulum stress-mediated osteogenic pathway. J. Thorac. Cardiovasc Surg. 158, 408–417.e2 (2019).
Zhong, H., Ding, Q., Chen, W. & Luo, R. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-κB nuclear translocation. Int. Immunopharmacol. 17, 329–335 (2013).
Boucherat, O. et al. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci. Rep. 7, 4546 (2017).
Lin, J. et al. Resveratrol downregulates TNF-α-induced monocyte chemoattractant protein-1 in primary rat pulmonary artery endothelial cells by P38 mitogen-activated protein kinase signaling. Drug Des. Devel Ther. 13, 1843–1853 (2019).
Guo, J. et al. Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ. Res. 124, 1448–1461 (2019).
Wang, J. et al. Ascorbic acid protects against hypertension through downregulation of ACE1 gene expression mediated by histone deacetylation in prenatal inflammation-induced offspring. Sci. Rep. 6, 39469 (2016).
Haemmig, S. et al. Long noncoding RNA SNHG12 integrates a DNA-PK-mediated DNA damage response and vascular senescence. Sci. Transl. Med. 12, eaaw1868 (2020).
Kolovou, G., Kolovou, V. & Katsiki, N. Volanesorsen: a new era in the treatment of severe hypertriglyceridemia. J. Clin. Med. 11, 982 (2022).
Parthymos, I., Kostapanos, M., Liamis, G. & Florentin, M. Early investigational and experimental therapeutics for the treatment of hypertriglyceridemia. J. Cardiovasc Dev. Dis. 9, 42 (2022).
Hu, Y. et al. RP5-833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Atertio. Thromb. Vasc. Biol. 35, 87–101 (2015).
Hu, Y. et al. A lincRNA-DYNLRB2-2/GPR119/GLP-1R/ABCA1-dependent signal transduction pathway is essential for the regulation of cholesterol homeostasis. J. Lipid Res. 55, 681–697 (2014).
Chen, J. et al. Circular RNA WDR77 target FGF-2 to regulate vascular smooth muscle cells proliferation and migration by sponging miR-124. Biochem. Biophys. Res. Commun. 494, 126–132 (2017).
Ray, K. et al. Effects of alirocumab on cardiovascular and metabolic outcomes after acute coronary syndrome in patients with or without diabetes: a prespecified analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 7, 618–628 (2019).
Liao, J. et al. LncRNA MIAT: myocardial infarction associated and more. Gene 578, 158–161 (2016).
Wang, S., Li, L., Deng, W. & Jiang, M. CircRNA MFACR is upregulated in myocardial infarction and downregulates miR-125b to promote cardiomyocyte apoptosis induced by hypoxia. J. Cardiovasc Pharm. 78, 802–808 (2021).
Huang, C.-K., Kafert.-Kasting, S. & Thum, T. Preclinical and clinical development of noncoding RNA therapeutics for cardiovascular disease. Circ. Res. 126, 663–678 (2020).
Wang, K. et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 37, 2602–2611 (2016).
Du, W. et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 44, 2846–2858 (2016).
Zhang, X. et al. The protective effects of long non-coding RNA-ANCR on arterial calcification. J. Bone Miner. Metab. 38, 421–431 (2020).
Zhu, Z. et al. Long non-coding RNA H19 promotes osteogenic differentiation of renal interstitial fibroblasts through Wnt-β-catenin pathway. Mol. Cell. Biochem. 470, 145–155 (2020).
Zhi, F. et al. Exosomal hsa_circ_0006859 is a potential biomarker for postmenopausal osteoporosis and enhances adipogenic versus osteogenic differentiation in human bone marrow mesenchymal stem cells by sponging miR-431-5p. Stem Cell Res Ther. 12, 157 (2021).
Acknowledgements
The authors thank to the support by the National Key Research and Development Program [2020YFC2004405], National Natural Science Foundation of China (NSFC) [82073408] to JC, NSFC [82061160372 and 81870506], project of traditional Chinese medicine in Guangdong province [20201062], Basic Research Project of Shenzhen Science and Technology Innovation Committee [JCYJ20180306174648342 and JCYJ20190808102005602], Futian District Public Health Scientific Research Project of Shenzhen [FTWS2019003] and Shenzhen Key Medical Discipline Construction Fund [SZXK002], and Guangdong Basic and Applied Basic Research Foundation 2021B1515120083].
Author information
Authors and Affiliations
Contributions
All the authors participated in the design of the manuscript. Y.S. collected and sorted out materials, wrote and revised manuscripts. H.H. supervised and managed the manuscripts. H.Z. and S.H. modified the manuscript contents. P.L. polished language and correct grammar errors. L.Y. and F.W. conceived the idea and modified tables and figures. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, Y., Zhang, H., Huang, S. et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Sig Transduct Target Ther 7, 200 (2022). https://doi.org/10.1038/s41392-022-01055-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01055-2
This article is cited by
-
Chromatin modifiers in human disease: from functional roles to regulatory mechanisms
Molecular Biomedicine (2024)
-
Structural insights into the binding mechanism of Clr4 methyltransferase to H3K9 methylated nucleosome
Scientific Reports (2024)
-
Cardiovascular Mechano-Epigenetics: Force-Dependent Regulation of Histone Modifications and Gene Regulation
Cardiovascular Drugs and Therapy (2024)
-
Epigenetic analyses in forensic medicine: future and challenges
International Journal of Legal Medicine (2024)
-
Exploring epigenetic strategies for the treatment of osteoporosis
Molecular Biology Reports (2024)
