
Abstract
Inflammasomes are protein complexes of the innate immune system that initiate inflammation in response to either exogenous pathogens or endogenous danger signals. Inflammasome multiprotein complexes are composed of three parts: a sensor protein, an adaptor, and pro-caspase-1. Activation of the inflammasome leads to the activation of caspase-1, which cleaves pro-inflammatory cytokines such as IL-1β and IL-18, leading to pyroptosis. Effectors of the inflammasome not only provide protection against infectious pathogens, but also mediate control over sterile insults. Aberrant inflammasome signaling has been implicated in the development of cardiovascular and metabolic diseases, cancer, and neurodegenerative disorders. Here, we review the role of the inflammasome as a double-edged sword in various diseases, and the outcomes can be either good or bad depending on the disease, as well as the genetic background. We highlight inflammasome memory and the two-shot activation process. We also propose the M- and N-type inflammation model, and discuss how the inflammasome pathway may be targeted for the development of novel therapy.
Similar content being viewed by others
Introduction
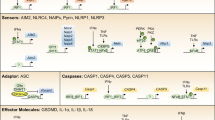
Inflammasomes are intracellular multimeric complex molecules that recognize either pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs).1,2 The nucleotide-binding oligomerization (NOD)-, leucine-rich repeat (LRR)-, and pyrin domain-containing 3 (NLRP3) and the NOD-, lRR-, and CARD-containing 4 (NLRC4) inflammasomes belong to the NOD-like receptor (NLR) family.3,4 Non-NLR proteins, such as absent in melanoma 2 (AIM2), form inflammasomes that can sense cytosolic double-stranded DNA.5,6
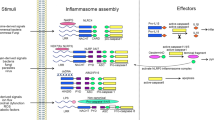
The canonical inflammasomes (NLRP3, NLRC4, and AIM2) serve as a platform to engage pro-caspase-1 (Figs. 1–3 and Box 1), which becomes active caspase-1 through the oligomerization of pro-caspase-1 proteins.7 Activated caspase-1 processes pro-interleukin-1β (IL-1β) and pro-IL-18 to generate their active forms, which induce pyroptosis, a pro-inflammatory form of cell death.2,8,9,10 The non-canonical inflammasomes activate caspase-11 (mouse) or caspase-4/5 (human) in response to Gram-negative bacteria-derived lipopolysaccharide (LPS) without cleaving pro-IL-1β (Fig. 4).1,11,12

Mechanisms of NLRP3 inflammasome assembly and activation. NLRP3 needs to be primed prior to activation. The priming process includes the binding of PAMPs/DAMPs to TLR or CD36, which promotes the transcription of NLRP3 and IL-1β. The priming event also includes de-ubiquitination of NLRP3. The ubiquitination and phosphorylation of the adaptor protein ASC is also necessary for the assembly of inflammasome. Disruption of the lysosome and release of cathepsin enhances the activation of NLRP3 inflammasome. The assembly of activated inflammasome leads to the processing of pro-caspase-1 into mature and active enzyme, which in turn cleaves pro-IL-1β and pro-IL-18 into active cytokines. Inset: activated NLRP3 binds to ASC through PYD–PYD interactions. Pro-caspase-1 binds to ASC through CARD–CARD interactions

Mechanisms of NLRC4 inflammasome assembly and activation. The bacterial ligands such as needle protein, rod, and flagellin bind to NAIP proteins. Ligand-bound NAIP then interacts with NLRC4 to form the inflammasome through oligomerization. NLRC4 recruits pro-caspase-1 to the inflammasome via CARD–CARD interaction. Inset: NLRC4 inflammasome components

The formation and activation of AIM2 inflammasome. The HIN domain of AIM2 binds dsDNA in the cytosol. AIM2 binds to ASC through PYD–PYD interaction. The inflammasome is formed through AIM2-ASC oligomerization. Pro-caspase-1 is recruited by ASC via CARD–CARD interaction. Inset: AIM2 inflammasome components

Mechanisms of non-canonical inflammasome assembly and activation. Intracellular LPS forms a complex with pro-caspase-11 (mouse) or pro-caspases-4 and -5 (human), which become activated through oligomerization. Activated caspase-4/5/11 induces pyroptosis by cleaving intact GSDMD to generate the N-terminal fragment which forms a pore on membrane. The active mCaspase-11/hCaspase-4 is also involved in the assembly and activation of the NLRP3 inflammasome when PAMP/DAMP signals are present
NLRs have three functional domains: the amino-terminal domain includes a pyrin domain (PYD), or a caspase-recruitment domain (CARD); the central nucleotide-binding and oligomerization domain (NACHT) is present in all NLR proteins; and the carboxy-terminal domain is an LRR domain that binds to ligand.13 The PYD interacts with apoptosis-associated speck-like protein containing a CARD (ASC) which in turn binds pro-caspase-1 (Fig. 1).14,15 ASC is an adaptor protein used by many cytoplasmic pattern recognition receptors (PRRs) such as NLRs to recruit pro-caspase-1 to the inflammasomes via its CARD domain. PRRs are sensor proteins that detect signals to activate an inflammatory response.
The membrane-bound PRRs such as Toll-like receptors (TLRs) and cytosolic PRRs such as NLRs recognize PAMPs and host-derived DAMPs, respectively.16 Activation of PRRs by PAMPs leads to the production of type I interferons and chemokines, whereas activation of PRRs by DAMPs activates caspases that eventually lead to the production of pro-inflammatory cytokines. DAMP-induced inflammation is referred to as sterile inflammation because it occurs in the absence of invading microbes.10,13,17 DAMP-triggered sterile inflammation can exaggerate pathology either as a causative or contributing factor in response to host-derived factors such as intracellular molecules released from damaged cells. When the inflammation persists for a long period of time, it becomes a chronic process, leading to sterile inflammatory diseases such as atherosclerosis, myocardial infarction, diabetes, neurodegenerative disease, depression, and cancer.
Discovery and mechanisms of activation of inflammasomes
Different types of inflammasome are briefly discussed in order to understand the role of their activation in disease states.
History of inflammasomes
The term “inflammasome” was first coined in 2002 by Dr. Jurg Tschopp and colleagues.2 They described the inflammasome as a “caspase activating complex” consisting of caspase-1, caspase-5, Pycard (caspase interacting domain), and NALP1 (an old name for NLRP1), which was later referred to as the NLRP1 inflammasome and this complex is responsible for IL-1β maturation. Two years later, Dr. Tschopp’s research team also discovered NLRP3 inflammasome.18 Their study revealed increased activity of the NLRP3 inflammasome in macrophages isolated from patients with Muckle–Wells syndrome. The NLRP3 inflammasome became the best-characterized inflammasome because of its involvement in both innate and adaptive immunity. It is activated by either whole pathogens including fungi,19 bacteria,20 and viruses21,22 or by host-derived molecules such as fibrillar amyloid-β (Aβ) peptide,23 as well as extracellular ATP24 and glucose.25 Unlike NLRP1, NLRP3 does not harbor a CARD domain, but instead recruits pro-caspase-1 through ASC that functions as an adaptor protein connecting NLRP3 and caspase-1. NLRC4 (IPAF) gene was identified by Poyet et al.26 when searching for proteins that can activate pro-caspase-1. They cloned IPAF cDNA using a pair of PCR primers based on sequence similarity to the CARD of pro-caspase-1, and demonstrated that IPAF was able to activate pro-caspase-1 through CARD–CARD interaction. Due to its structural homology to other NLR proteins, IPAF was later renamed as NLRC4.
AIM2 inflammasome was discovered by several independent groups when searching for new DNA sensors using different approaches. Burckstummer et al.5 identified AIM2 using orthogonal proteomic and genomic screen for proteins that associate with DNA and are transcriptionally regulated by IFN-β. Roberts et al.27 analyzed cyotoplasmic extracts from bone marrow-derived macrophages for binding to double-stranded DNA (dsDNA) by electromobility shift assay and mass spectrometry and identified p202 as a repressor of caspase-1 and caspase-3. p202 is a member of HIN-200 family, which is a cluster of interferon-inducible genes including AIM2. Because AIM2 is the only family member that can form a heterodimer with p202, they investigated the effect of AIM2 silencing on caspase-1 and caspase-3 activation and found that knockdown of AIM2 completely inhibited the activation of the caspases, therefore confirming the role of AIM2 as an activator of caspase-1 and capase-3. Fernandes-Alnemri and colleagues searched the NCBI database for proteins with pyrin and oligonucleotide-binding domains, and identified four human proteins (IFI16, AIM2, IFIX, and MNDA) of the HIN-200 family that meet the criteria. Among these proteins, AIM2 was the only one that can activate caspase-1 when ectopically expressed in 293T-caspase-1-ASC cell line.28 They further showed that cytoplasmic DNA triggers formation of AIM2 inflammasome by inducing AIM2 oligomerization. Hornung et al.29 identified AIM2 by searching for proteins containing a PYD domain and DNA binding domain and demonstrated that AIM2 is a receptor for cytosolic dsDNA and activates caspase-1 by forming a complex with ASC. The NLRP1, NLRP3, NLRC4, and AIM2 inflammasomes were termed as “canonical inflammasomes” because all of these inflammasomes are capable of activating caspase-1. The “non-canonical” inflammasome was recently discovered by several research teams when searching sensors for intracelluar LPS and upstream activator of caspse-11.12,30,31 They showed that, rather than triggering the activation of caspase-1, the non-canonical inflammasome activate caspase-11 in mice and caspase-4 and caspase-5 in humans in response to LPS. Activated caspase-4/5/11 cleaves intact GSDMD to generate the N-terminal fragment which bind to cell membrane, oligomerize, and form pores to induce pyroptosis (Fig. 4).
Canonical inflammasomes
The activation of NLRP3 inflammasome can be triggered by molecules associated with both PAMPs and DAMPs (Fig. 1).9,32 In most cell types, NLRP3 needs to be primed by pattern recognition molecules or other receptors such as scavenger receptor CD36 before it can be activated. Binding of LPS to TLR4 is considered a priming event.13 Nuclear factor-κB (NF-κB) signaling is involved in priming NLRP3 for activation.33 Recent findings have shown that signaling by the PRR primes NLRP3 by inducing its de-ubiquitination in a non-transcriptional fashion.34,35 Once primed, the NLRP3 inflammasome is assembled and activated. In addition, linear ASC ubiquitination is required for NLRP3 inflammasome assembly.36 NLRP3 inflammasome formation can be induced by the products of damaged cells such as ATP, crystalline substances, nucleic acids, or by pathogen-derived molecules from infection.1,3 Of note, molecules formed under pathological conditions such as high glucose, oxidized low-density lipoprotein (oxLDL), and cholesterol can also trigger the activation of inflammasomes (Fig.1).
NLRC4 contains a CARD domain, therefore can directly recruit pro-caspase-1.26 The NLRC4 inflammasome mainly responds to bacterial flagellin and components of the bacterial type III secretory system (T3SS). Certain NLRC4-mediated functions may not require ASC, but do require interactions with the NLR family of apoptosis inhibitory proteins (NAIPs) which are receptors for bacterial protein ligands, and these interactions determine inflammasome specificity (Fig. 2).37,38 There are four NAIPs in mice: NAIP1 binds to the needle protein, NAIP2 detects rod proteins, and NAIP5/6 interact with flagellin.39,40,41,42 There is only one NAIP in humans, and it interact with the needle protein Cprl.42 In light of these findings, NLRC4 may be specific for the needle protein rather than flagellin in human infectious disease.
AIM2 is part of the inflammasome that can induce caspase-1 activation and subsequent cleavage of IL-1β and IL-18, contributing to the immune reaction against bacterial and viral infection. AIM2 inflammasomes directly bind to dsDNA (Fig. 3). Unlike NLRC4 inflammasomes, AIM2 does not contain CARD domains and thus needs to recruit ASC for activation.43 AIM2 not only senses bacteria and viruses derived cytosolic dsDNA, but also from damaged and mislocalized DNA molecules.5,6,29,44,45 Moreover, a minimum DNA length of 80 base pairs is required to activate AIM2.9 AIM2 is also involved in DNA vaccination,46 and may play a role in the development of systemic lupus erythematosus (SLE) due to its ability to recognize host DNA.47 However, more direct evidence is required to establish the role of AIM2 in the pathogenesis of SLE.
Non-canonical inflammasomes
In mice, the non-canonical inflammasome is composed of caspase-11 and activated by cytosolic LPS (Fig. 4). Caspase-11 can activate both caspase-1 and caspase-3.48 Recently, caspase-11 was shown to indirectly activate NLRP3 inflammasome leading to the cleavage of pro-IL-1β or pro-IL-18.30 Importantly, caspase-11 senses intracellular LPS released by Gram-negative bacteria and induces pyroptosis and IL-1β secretion without involvement of the LPS receptor TLR4.1,11,12 Human cells do not possess the gene encoding caspase-11, but express caspase-4 and caspase-5, which have a similar function (Fig. 4).31,49 In response to LPS, caspase-4 can trigger the activation of caspase-1 without the need of second signals to activate the inflammasome.49 Along this line, caspase-11-deficient mice were resistant to a lethal dose of LPS injection.30 Further study of the non-canonical inflammasome will likely yield useful information to understand how human cells react to bacterial infection through PAMP-induced inflammation.
Roles of inflammasomes in sterile inflammatory diseases
The inflammasome is a double-edged sword in various diseases, especially in sterile inflammatory diseases, and the outcomes can be either good or bad depending on the disease, and the genetic background.
Inflammasomes and cardiovascular diseases (CVDs)
The role of the NLRP3 inflammasome in the development of CVDs and the treatment strategies targeting this key regulator of inflammation are discussed.
Inflammasomes and atherosclerosis
IL-1β was one of the first cytokines implicated in the pathogenesis of atherosclerosis. Various NLRP3 inflammasome stimuli that induce the production of mature IL-1β have been shown to contribute to the formation of atherosclerotic plaques.50 The NLRP3–IL-1β pathway has been identified as part of the innate immune memory system because it is a form of “trained immunity”, leading to a robust immune response to secondary infection.
A recent study showed that the trained immunity can be induced not only by microbes but also by non-infectious agents such as a Western diet (WD). This study showed that WD-induced systemic inflammation returned to normal levels when mice were shifted back to a chow diet (CD).51 However, the inflammatory response of myeloid cells isolated from WD-fed mice to subsequent innate immune stimuli such as LPS remained elevated even after switching back to CD, suggesting that the myeloid cells retain memories of the WD stimuli. This trained immunity resembles the immune response to infectious agents or a vaccine. The investigators further showed that the trained immunity was due to reprogramming of myeloid progenitor cells at both transcriptional and epigenetic levels induced by the WD. The trained immunity did not occur in mice lacking NLRP3. Quantitative trait locus analysis in human monocytes confirmed that the NLRP3–IL-1β pathway is the mediator of trained immunity. These findings suggest that the harmful effects of a high-cholesterol diet are long-lasting and are mediated by the NLRP3–IL-1β pathway (Fig. 5).

Inflammasome activation contributes to the development of diseases. The NLRP3 inflammasome is activated by DAMPs such as ATP, excess glucose, cholesterol, and high fat, leading to the activation of caspase-1 and production of active IL-1β and IL-18 in cardiomyocytes, endothelial cells, and smooth muscle cells. These risk factors are also involved in the development of depression and T2D. CM cardiomyocyte, EC endothelial cell, SMC smooth muscle cell, T2D type 2 diabetes
The release of IL-1 induces the secretion of IL-6, which in turn promotes the synthesis of fibrinogen and plasminogen activator inhibitor to augment thrombosis and inhibit fibrinolysis, leading to the accumulation of thrombi in arteries.52 The critical role of IL-1 in the development of atherosclerosis highlights this cytokine as a potential target for intervention.53,54 Based on these findings, several clinical trials targeting the IL-1 signaling pathway have shown promising results (Table 1). A double-blind trial of canakinumab, designed to assess the therapeutic effect of a monoclonal antibody against IL-1β, showed beneficial effect in preventing the recurrence of myocardial infarction. Anakinra (an antagonist of the IL-1 receptor that inhibits the action of both IL-1α and IL-1β) and an antibody against IL-1α also showed beneficial effects.55,56 Animal studies demonstrated that IL-1α and IL-1β are involved at different stages during the development of atherosclerosis: neutralization of IL-1α limits early atherogenesis in the aortic root,57 whereas IL-1β blockade decreased the area of established atheromata. Their data indicate that IL-1α is involved in the remodeling of arteries during early atherogenesis, while IL-1β contributes to the formation of advanced atheroma.57
A clinical study showed that IL-18 expression in human atherosclerotic plaques is much higher than that of the normal arteries.58 Importantly, higher IL-18 mRNA levels are found in unstable plaques than stable plaques. IL-18 and its receptor have been identified in endothelial cells, smooth muscle cell, and mononuclear phagocytes within human atheroma, and it is able to stimulate the expression of IFN-gamma in cultured smooth muscle cells and mononuclear phagocytes.58,59,60 MCC950, an inhibitor of the NLRP3 inflammasome, was shown to reduce atherosclerogenic lesions in WD-fed apoE−/− mice.61 MCC950 also improved cardiac function in a pig myocardial infarction model.62 Thus, the inhibition of inflammasome NLRP3 signaling represents a novel interventional therapy to prevent the development of atherosclerosis.61,62,63
Inflammasomes and other cardiovascular dysfunctions
IL-1β is a crucial cytokine promoting the inflammatory reaction in heart failure. Tet2 mutation in blood cells can increase atherosclerotic plaque size due to elevated IL-1β signaling in mice, and heart failure can be prevented by treating the mice with a specific NLRP3 inflammasome inhibitor.64,65 Activation of the inflammasome is also associated with aging. Furman et al. showed that individuals over 85 years of age, and those with increased levels of IL-1β, had a higher mortality rate due to nucleotide metabolism dysfunction. Interestingly, caffeine was shown to inhibit the metabolite-induced IL-1β production.66,67
NLRP3 inflammasome also plays an important role in the development of atrial fibrillation (AF). One recent study showed that the activity of NLRP3 inflammasome is increased in atrial cardiomyocytes (CMs) from patients with AF.68 CM-specific knock-in mice expressing constitutively active NLRP3 (CM-KI) developed spontaneous premature atrial contractions, which were blunted by the NLRP3 inhibitor MCC950. The CM-KI mice exhibited larger atria, abnormal calcium release from the sarcoplasmic reticulum, and electrical remodeling, which were prevented by CM-specific NLRP3 knockdown. Finally, genetic inhibition of NLRP3 prevented spontaneous AF in CREM (cAMP responsive element modulator) transgenic mice.68 These data demonstrate that both the electrical and structural remodeling associated with AF can be inhibited by targeting the NLRP3 inflammasome. Thus, NLRP3 inflammasome is a new therapeutic target for AF.
The NLRP3 inflammasome has been implicated in the development of pressure overload-induced cardiomyopathy. Suetomi et al. demonstrated that the activity of NLRP3 and caspase-1 is increased in the CM but not in the non-CM fraction containing fibroblasts, endothelial cells, and immune cells in response to transverse aortic constriction (TAC). These results indicate that the CM is the original site of NLRP3 inflammasome activation. Using CM-specific Ca2+-calmodulin-dependent protein kinase II (CaMKIIδ)-knockout (CKO) mice, they further showed that the increased levels of IL-1β, IL-18, and caspase-1 activity induced by TAC were attenuated in the CKO mice, suggesting that NLRP3 inflammasome activation in CMs is mediated through the CaMKIIδ signaling pathway. Mice treated with a CaMKII inhibitor in the first one or 2 weeks after TAC showed decreased fibrosis and ventricular dilation, but inhibition of CaMKIIδ after two weeks did not, suggesting that early intervention is required to prevent the development of cardiac remodeling and subsequent heart failure.69
In summary, the NLRP3 inflammasome plays a critical role in the development of CVDs, and NLRP3, caspase-1, and IL-1β can be exploited as new therapeutic targets. Some existing CVD medications have been shown to act via the NLRP3 inflammasome (Table 1), and further clinical assessment of these therapies is needed.
Inflammasomes and depression
Depression is a common and serious mood disorder that affects 264 million people worldwide. Unlike short-lived mood-swings in response to stressful situations in everyday life, the symptoms of depression persist for a long time. Depression not only affects daily life, it can also lead to suicide. Stress is an important risk factor for depression. It is known that stress can induce inflammation, and depressive symptoms can be curbed by anti-inflammatory agents. The NLRP3 inflammasome functions as a bridge that links stress to immune activation.70
The role of the NLRP3 inflammasome in the pathogenesis of depression and mood disorders has been demonstrated in both animal models and patients.70,71,72,73,74,75 Peripheral diseases can activate NLRP3 inflammasome in the brain, leading to depression in rats.71 The expression of both NLRP3 and caspase-1 was shown to be increased in peripheral mononuclear cells from patients with major depressive disorder.73 Of note, depression is also associated with CVDs. High fat, cholesterol crystals, extracellular ATP, and excess glucose are risk factors that contribute to the development of CVDs by activating NLRP3 inflammasome, and these factors are also involved in the development of depression (Fig. 5).76 Dysregulated autophagy and mitophagy have been implicated in inflammasome activation and the development of both depression and CVDs.77,78,79,80 Several specific NLRP3 inflammasome inhibitors are being investigated for the treatment of both depression and CVDs. We anticipate that some of the inhibitors will prove useful in treating both diseases.
The NLRP3 inflammasome is involved in the development of diabetes, which is characterized by chronic elevation of blood glucose. Diabetes is due to either the failure of pancreatic β cells to make enough insulin (type 1) or the cells that express insulin receptors fail to respond to insulin (type 2; type 2 diabetes (T2D)). Depression and T2D may be mutually causal. People with depression tend to eat unhealthy food and have sedentary lifestyles that are risk factors for diabetes and CVDs. Conversely, diabetes can cause mood changes, anxiety, and frustration, which lead to depression.81
Two studies shed new light on the mechanisms underlying NLRP3 inflammasome activation. Extracellular ATP and neutrophils are abundant in the early stages of inflammation. ATP—induced activation of NLRP3 inflammasome and secretion of IL-1β is mediated by ionotrophic P2X7 receptors (P2X7R).82 IL-1β interfere with insulin signaling in depressed patients, leading to insulin resistance and T2D. Moreover, increased levels of IL-1β can trigger inflammation of the central nervous system, leading to reduced concentrations of serotonin (Fig. 5).83 Because P2X7R -mediated NLRP3 inflammasome activation is implicated in the development of both diabetes and depression, we expect that novel interventions targeting the P2X7–NLRP3–IL-1β pathway will reduce insulin resistance, leading to potential treatments for both depression and diabetes.
Inflammasomes and neurodegenerative diseases
Alzheimer’s disease (AD) is a neurodegenerative disorder caused by the accumulation of misfolded Aβ peptide in the brain. Recent studies suggest that the deposition of Aβ peptide causes chronic inflammation.84 It is now clear that Aβ-induced activation of NLRP3 inflammasome leads to the synthesis of neurotoxic factors in microglia, which ultimately cause the development of AD.23 This study further showed that NLRP3 inflammasome activation is caused by the action of cathepsin B released from the lysosome rather than the direct actions of Aβ.23 These findings were confirmed by a separate study showing that cathepsin B inhibitors improve the memory deficit in transgenic AD mice.85 Subsequently, it was shown that the disruption of lysosome and release of cathepsin B are caused by CD36 which converts soluble ligands into insoluble fibrils.86 The connection between the NLRP3 inflammasome and the pathogenesis of AD was further confirmed using the gene-knockout approach in mice. NLRP3−/− or caspase-1−/− mice carrying AD mutations did not show loss of spatial memory.87 Furthermore, in APP/PS1/NLRP3−/− mice (transgenic mice carrying an AD mutation crossed with NLRP3−/− mice), caspase-1 cleavage was not seen and the brain IL-1β level was similar to wild-type mice. Importantly, qPCR analysis revealed increased mRNA levels of FIZZ1, arginase-1, and IL-4, suggesting that the microglial cells from APP/PS1/NLRP3−/− mice are skewed to an M2 phenotype leading to increased Aβ clearance and enhanced tissue remodeling.87 Clinical studies have demonstrated increased active caspase-1 level in the brains of AD patients. Therefore, both animal and clinical studies (Table 1) revealed an important role for NLRP3 inflammasome in the pathogenesis of AD and strategies targeting the NLRP3–caspase-1 pathway are being investigated to find a cure for AD.88,89,90,91,92,93,94,95,96
Parkinson’s disease (PD) is a neurodegenerative disorder affecting dopaminergic motor neurons in the midbrain. The cause of the death of these neurons remains poorly understood, but is thought to be due to the abnormal uptake of aggregated α-synuclein (αSyn),97 which forms insoluble fibrils.98 Intracellular αSyn can also be transferred into extracellular fluid through exocytosis,99 which is a mechanism for spreading the misfolded αSyn to surrounding tissues, and the extracellular αSyn aggregates induce microglia activation and the secretion of IL-1β from astrocytes.99,100 Injection of an adenoviral vector expressing IL-1β into substantia nigra has been shown to trigger dopaminergic neuron death accompanied by microglia activation, infiltration of inflammatory cells, and akinesia.101 The fibrillary form of αSyn is able to activate caspase-1 and induce the production of mature IL-1β.102 The production of IL-1β is dependent on inflammasomes, which are activated by the phagocytosis of fibrillar αSyn, which in turn induces the production of reactive oxygen species (ROS) and excessive release of cathepsin B into the cytosol.102 NLRP3−/− mice are resistant to neurotoxin MPTP-induced death of dopaminergic neurons and display reduced serum levels of IL-1β and IL-18 compared to the control mice.103 Dopamine prevents NLRP3 inflammasome activation by releasing the secondary messenger cAMP, which promotes the ubiquitination and degradation of NLRP3 through the E3 ubiquitin ligase MARCH7.103 Furthermore, mice lacking dopamine D1 receptors are more vulnerable to MPTP-induced neuroinflammation.103 These studies suggest that the dopamine signaling pathway and the NLRP3 inflammasome are mutually regulated, and PD patients may benefit from inhibition of the inflammatory process in the neurons.104,105,106,107,108
Inflammasomes and insulin resistance
There are >500 million cases of T2D globally and this number is projected to increase significantly in the coming years. T2D is a chronic disease characterized by hyperglycemia due to insulin resistance.
The role of chronic inflammation in the development of T2D has been well documented,109,110,111 and the NLRP3 inflammasome was shown to mediate obesity-induced inflammation and insulin resistance.112,113 A number of molecules such as high glucose, islet amyloid polypeptide, saturated fatty acids, and mitochondrial ROS are involved in activating the NLRP3 inflammasome and contribute to the pathogenesis of T2D.114,115 The inflammasome and caspase-1 regulate adipocyte differentiation and insulin signaling, and a caspase-1 inhibitor improves insulin sensitivity in obese mice.25,116,117,118 Importantly, elevated inflammasome activation and caspase-1 have been found in both adipose tissue and myeloid cells from patients with T2D119,120 and high concentrations of IL-1β and IL-18 in the circulation are associated with an increased risk of T2D.121,122
One of the complications associated with T2D is the development of CVDs (Fig. 5). It has been shown that inhibitors of sodium-glucose cotransporter 2 (SGLT2) reduce the risk of developing CVDs in patients with T2D, and the beneficial effect is mediated by inhibiting NLRP3 inflammasome activity.123 The investigators showed that treatment with an SGLT2 inhibitor attenuates the activation of NLRP3 inflammasome and IL-1β production in patients with T2D and high cardiovascular risk by increasing serum β-hydroxybutyrate.123 These results opened a new door for the treatment of T2D and its complications by targeting the SGLT2-NLRP3-IL-1β pathway.
Inflammasomes and cancer
Chronic inflammation mediated by inflammasomes plays a central role in tumorigenesis by altering the microenvironment and leading to neoangiogenesis, the proliferation of tumor cells, and metastasis. However, under certain conditions, inflammasome signaling also inhibits tumor growth by maintaining intestinal barrier integrity.124,125
Inflammasomes and the tumor microenvironment
The tumor microenvironment includes blood vessels, immune cells, extracellular matrix, and fibroblasts. Cancer-associated fibroblasts (CAFs) play a critical role in tumor growth and metastasis by activating inflammatory pathways.126
A recent study showed that NLRP3 inflammasomes are upregulated in human breast CAFs. They demonstrated that CAF-derived NLRP3/IL-1β facilitates tumor growth by recruiting myeloid-derived suppressor cells (MDSCs), thus altering the tumor microenvironment toward an immune-suppressive milieu.126 They further showed that tumor growth is delayed when NLRP3 or IL-1β is ablated. Activation of the inflammasome also contributes to resistance to chemotherapy. One study showed that although gemcitabine and 5-fluorouracil impede tumor-induced immunosuppression by killing MDSCs, they also activate NLRP3 inflammasomes in these cells, leading to IL-1β release that accelerates tumor growth.127 In contrast, other studies have shown that depletion of CAFs accelerates pancreatic cancer by recruiting the immunosuppressive FoxP3+ Treg cells.128 Others have shown that NLRP3 is required for suppression of metastasis of colorectal cancer (CRC) in the liver by promoting natural killer cell tumoricidal activity.129 In light of these conflicting reports, the NLRP3/IL-1β pathway can be either friend or foe in tumorigenesis, depending on the tumor type and context.128,129,130,131,132,133,134,135,136
Inflammasomes and metastasis
Tumor metastasis is a multi-step process in which both the primary tumor and the local microenvironment of the metastatic site play roles in tumor cell dissemination, metastatic colonization, and the formation of new tumors in specific distant organs.137 The metastatic sites of target organs form an immune-suppressive environment and a pre-metastatic niche to prepare for the arrival of metastatic cells.138 Circulating tumor cells must be able to adhere to capillary endothelia for metastasis to occur. It has been shown that IL-1β promotes the metastasis of melanoma cells to liver by up-regulating vascular cell adhesion molecule.139 Tumor growth and metastasis requires a sufficient blood supply. IL-1β is a necessary cytokine to induce tumor angiogenesis, and IL-1β−/− mice exhibit impaired blood vessel growth and tumor development.140 The role of IL-1β in tumor metastasis is further supported by data showing that an IL-1R antagonist suppresses metastases of melanoma and improves survival of mice.134 The effects of inflammasome activation on tumor growth and metastasis may be mediated by different cytokines, depending on the cancer type and metastatic site. In the CRC model of intrasplenic injection of colon cancer cells, inflammasome-mediated IL-18 production is responsible for the suppression of tumor growth in the liver.129 However, another study showed that NLRP3-mediated IL-1β secretion promotes the dissemination of colon cancer cells.141 In a carcinogen-induced tumor model, NLRP3 promotes metastasis by suppressing natural killer cell activity.142
Inflammasomes and tumor-suppression
The tumor-suppressive role of the NLRP3,143,144 AIM2,145,146 and NLRC4147,148 inflammasomes is well documented in colon cancer. The NLRP3 inflammasome deficient mice are more susceptible to colitis-induced colorectal cancer due to reduced production of IL-18, and subsequent inactivation of tumor suppressor STAT1. The expression of AIM2 is reduced in cancer cells and AIM2 inhibits tumor growth by interacting with DNA-dependent protein kinase, leading to inactivation of AKT145,149 and inhibition of the proliferation of colonic stem cells.146,149 Caspase-1-deficient mice shows enhanced tumor growth in inflammation-induced colorectal cancer model and the tumor-suppressive function of caspase-1 is mediated by NLRC4.147,148 Of note, ASC is a tumor suppressor in keratinocytes, but a tumor-promoter in myeloid cells.150 These findings suggest that the roles of inflammasomes in tumor-suppression are cell type dependent.
Taken together, the activation of inflammasomes can either enhance or inhibit tumor progression and metastasis depending on tumor type and context. Further studies are required to understand how inflammasomes are regulated in each tumor type and to determine their contribution to tumor development.
Gene polymorphisms of inflammasomes and disease
Gene polymorphisms of inflammasomes and related cytokines are associated with the pathogenesis and prognosis of various diseases such as cancer, CVDs, and T2D.
Gene polymorphisms of inflammasomes in tumors
Gene polymorphisms of inflammasomes are associated with an increased risk of developing cancers. Several single-nucleotide polymorphisms (SNPs) in the NLRP3 region is associated with increased production of IL-1β that contribute to susceptibility to Crohn’s disease,151,152 which is a risk factor for the development of CRC. A heterozygous NLRP3 (Q705K) variant is associated with poor survival in patients with invasive CRC.153 The AIM2 gene contains a coding region frame-shift mutation caused by microsatellite instability in CRC.154,155 In addition, regression analysis has shown that AIM2 can promote the development of endometrial cancer.156
The polymorphisms of genes involved in the NLRP3 inflammasome pathway may be associated with the prognosis of chronic myeloid leukemia (CML). One study investigated the association of genetic polymorphisms of NLRP3, IL-1β, IL-18, and CARD8 with CML. They showed that the AT genotype of CARD8 (rs2043211) was more frequent in high- and intermediate-risk CML. CARD8 is an NLRP3 binding partner that inhibits tumor development by suppressing NF-κB. The rs2043211 variant is translated to a truncated CARD8 protein that is unable to inhibit NF-κB. The gene polymorphisms of both IL-18 (rs1946518) and IL-1β (rs16944) are associated with CML. Therefore, polymorphisms of the NLRP3 inflammasome may provide useful information for predicting the clinical outcome in response to treatment.157
Gene polymorphisms of inflammasomes in other diseases
Gene polymorphisms of inflammasome components have also been implicated in the development of CVDs. One study evaluated the impact of NLRP3 (exon 3) gene polymorphisms and serum NLRP3 levels on the risk of developing myocardial infarction (MI) in 69 patients and 53 controls. They identified five SNPs that were associated with a high incidence of MI. Therefore, genetic polymorphisms of NLRP3 may be used as indicators for predicting the risk of developing MI.158
The association between the genotypes of SNPs in the NLRP3 downstream regulatory region and NLRP3 mRNA expression was investigated in the Swedish First-ever myocardial Infarction study consisting of DNA from 555 MI patients and 1016 healthy individuals. Expression of NLRP3 inflammasome components was significantly higher in carotid artery plaques than in normal arteries. The rs6672995 and rs10733113 variants were associated with NLRP3 mRNA levels in blood cells. These results suggest that genetic variants of the NLRP3 inflammasome are involved in the development of atherosclerosis.159
Chronic inflammation driven by excess body fat is a common feature of T2D. A randomized dietary intervention with Mediterranean (Med) and low-fat diets was conducted to determine whether insulin sensitivity is associated with diet and genetic factors. After 3 years of intervention, SNPs located at the NLRP3 gene were determined in both diabetic and non-diabetic patients. The results showed that insulin sensitivity was improved in non-diabetic patients carrying the SNPs rs4612666 and rs10733113, whereas no effects were found in the diabetic patients. The results suggest that gene polymorphisms of NLRP3 are associated with insulin sensitivity in non-diabetic patients.160
Taken together, SNP analysis may provide useful information for understanding how individual components of the inflammasome contribute to the pathogenesis and prognosis of diseases. Future studies investigating how SNPs contribute to disease phenotypes and treatment outcomes will delineate the molecular mechanisms whereby inflammasomes influence the development and progression of disease.
Therapeutic strategies targeting the inflammasome signaling pathway
Several recent studies targeting NLRP3 inflammasome, including IL-1, have shown promising beneficial results.56,124,161
The Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) demonstrated that inflammation has a central role in the pathogenesis of atherothrombosis.162 This randomized clinical trial recruited 10,061 patients who were treated with either Canakinumab, a monoclonal antibody blocking IL-1β, or a placebo. All patients had a previous bout of MI and increased levels of high-sensitivity C-reactive protein, the latter an indicator of ongoing inflammation. The primary end-point was a combination of non-fatal MI, non-fatal stroke, or cardiovascular death. The results showed that Canakinumab reduced the rate of recurrent cardiovascular events. Therapeutic approaches targeting IL-1α and IL-1 receptor also showed beneficial effects.55,56 The beneficial effects were achieved by inhibiting the release of cytokines from leukocytes and abrogating proliferation of smooth muscle cells. Likewise, apoptosis and necrosis of CMs were reduced and leukocyte adhesion to endothelial cells was diminished (Fig. 6).

Mechanisms underlying the beneficial effect of IL-1 blockade in treating cardiovascular diseases and cancer. The CANTOS trial showed a reduced incidence of CVD in patients treated with canakinumab, a monoclonal antibody blocking IL-1β. The potential mechanisms underlying the beneficial effects of IL-1 targeting strategies include inhibition of cardiomyocyte apoptosis and necrosis, reduction of inflammatory mediator activation in endothelial cells, prevention of recruitment of tumor suppressor cells, and inhibition of tumor angiogenesis and tumor growth. CRP C-reactive protein, CVD cardiovascular disease
Blockade of cytokines has shown promising results in delaying or preventing cancer progression. A phase II clinical trial investigated the effect of anakinra, an IL-1 receptor antagonist (IL-1Ra), in patients with indolent multiple myeloma showed that anakinra decreased the myeloma proliferation rate.163,164 IL-1Ra blocks the signaling pathway activated by both IL-1α and IL-1β. Whereas the production of IL-1β is mostly mediated by inflammasome, the release of IL-1α could be either inflammasome-dependent or -independent based on the activator.165 A phase III clinical trial has demonstrated that MABp1, a monoclonal antibody blocking IL-1α, improves the survival rate in patients with refractory CRC.166
Targeting the pro-inflammatory IL-1β pathway with canakinumab has been shown to inhibit the development and progression of lung cancer.167 The results from a phase II clinical trial showed that co-administration of anakinra with 5-fluorouracil and bevacizumab improves the median survival rate in patients with metastatic CRC.168 These promising results demonstrate that the anti-inflammasome strategy is an effective approach for cancer treatment and its efficacy can be improved with a combination therapy. The therapeutic effect of canakinumab is mediated by blocking the IL-1 signaling in CAFs, leading to reduced infiltration of tumor suppressor cells and inhibition of tumor angiogenesis as well as tumor growth (Fig. 6).
Although the anti-IL-1β strategy is successful in preventing recurrent cardiovascular events, it is associated with a higher incidence of fatal infections as shown in the CANTOS trial. This side-effect is likely due to suppression of the immune system. To overcome this problem, Li and colleagues have fabricated platelet microparticles (PMs) armed with the anti-IL-1β antibodies that specifically block the IL-1β signaling pathway in the injured heart. Administration of platelets carrying the anti-IL-1β PMs prevented adverse cardiac remodeling after MI by inhibiting caspase-3 activity and protecting the CMs from apoptosis. Therefore, strategies designed to target specific cells or organs are expected to make IL-1β antibody safer and more efficient.169 Tissue-specific blocking of inflammasome signaling may facilitate the development of more efficient therapeutic strategies.
Conclusions and perspectives
In this review, we summarized the link between inflammasomes and sterile inflammatory diseases, including CVDs, depression, neurodegenerative diseases, diabetes, and cancer. Stress, smoking, poor diet, and a sedentary lifestyle are common risk factors for diabetes, CVDs, and depression. These risk factors activate inflammasomes, which in turn leads to chronic inflammation that contributes to insulin resistance, atherosclerosis, and mood change. However, in the case of cancer, inflammasomes can be either friend or foe depending on the cell type and context. Activation of inflammasomes in acute diseases helps to remove dead cells and initiate tissue repair. However, persistent activation of inflammasomes in chronic diseases is detrimental because it damage tissues (Fig. 7). In order to explore the potential of inflammasomes as therapeutic targets, a greater understanding of the signaling molecules mediating the beneficial and detrimental effects of their activation is needed.

Beneficial effects of acute inflammation and deleterious effects of chronic inflammation. Acute inflammation (M-type) is a beneficial process to destroy pathogens and initiate tissue repair. However, chronic inflammation (N-type) is a pathological condition that lasts for a long time, leading to tissue damage. Both types of inflammation can be modulated by the inflammasome
As strategies targeting IL-1 enter clinical trials, we face new challenges as well as opportunities. The CANTOS trials confirmed that inflammasome activation is the cause rather than a consequence of atherothrombosis. This is an important step toward the clinical application of therapeutic strategies targeting the inflammasome signaling pathway. However, the trial also revealed an increased incidence of fatal infection due to off-target effects, which must be properly addressed before canakinumab can be routinely used to prevent cardiovascular events. To solve this problem, Li et al.169 introduced PMs as carriers to deliver the IL-1β antibody to the injured heart and demonstrated promising results. This approach not only reduces the risk of side-effects but also improves therapeutic efficiency.
In addition to IL-1 blockade, inhibition of NLRP3 could also be beneficial in certain diseases. Along this line, MCC950, a specific and potent inhibitor of NLRP3, has been shown to reduce the development of atherosclerogenic lesions in WD-fed apoE−/− mice.61 MCC950 reduces infarct size in a pig myocardial infarction model,62 protects against diabetes-associated atherosclerosis in mice,170 and improves spatial memory in SAMP8 mice, an animal model of AD.171 In addition, NLRP3 silencing in apoE−/− mice inhibits the induction of IL-1β and IL-18 and prevents the progression of plaques.172 These studies point to a potential new therapeutic avenue for treating sterile inflammatory diseases.
In summary, NLRP3 acts as a nodal factor which can be beneficial after either activation or inhibition. Aberrant NLRP3 activation is implicated in chronic inflammation that leads to the development of cancer, aging, and degenerative diseases. IL-1 blockers such as canakinumab, anakinra, and rilonacept are only effective in treating certain NLRP3-mediated diseases. The development of more effective treatment relies on a thorough understanding of the signaling pathways triggering the activation of the inflammasome (Box 2). In addition, a better understanding the mechanisms underlying the regulation of non-canonical inflammasome responses and the interaction between the non-canonical inflammasome and the canonical inflammasome may lead to the development of effective therapies for a broader range of pathological conditions.
References
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Vanaja, S. K., Rathinam, V. A. & Fitzgerald, K. A. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 25, 308–315 (2015).
Vance, R. E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 32, 84–89 (2015).
Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Schroder, K., Muruve, D. A. & Tschopp, J. Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 19, R262–R265 (2009).
Yang, X., Chang, H. Y. & Baltimore, D. Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell 1, 319–325 (1998).
Dinarello, C. A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 (2009).
Rathinam, V. A., Vanaja, S. K. & Fitzgerald, K. A. Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 (2012).
Thornberry, N. A. et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774 (1992).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Srinivasula, S. M. et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 277, 21119–21122 (2002).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Chen, G., Shaw, M. H., Kim, Y. G. & Nunez, G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–398 (2009).
Chen, G. Y. & Nunez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Gross, O. et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436 (2009).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Kanneganti, T. D. et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 (2006).
Muruve, D. A. et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865 (2008).
Mariathasan, S. ASC, Ipaf and Cryopyrin/Nalp3: bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. 9, 664–671 (2007).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Poyet, J. L. et al. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313 (2001).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Ratsimandresy, R. A., Dorfleutner, A. & Stehlik, C. An update on PYRIN domain-containing pattern recognition receptors: from immunity to pathology. Front. Immunol. 4, 440 (2013).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Juliana, C. et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 287, 36617–36622 (2012).
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Rodgers, M. A. et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333–1347 (2014).
Van Opdenbosch, N. et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 5, 3209 (2014).
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E. & Monack, D. M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Rayamajhi, M., Zak, D. E., Chavarria-Smith, J., Vance, R. E. & Miao, E. A. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 191, 3986–3989 (2013).
Yang, J., Zhao, Y., Shi, J. & Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl Acad. Sci. USA 110, 14408–14413 (2013).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Schattgen, S. A. & Fitzgerald, K. A. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 243, 109–118 (2011).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Suschak, J. J., Wang, S., Fitzgerald, K. A. & Lu, S. Identification of Aim2 as a sensor for DNA vaccines. J. Immunol. 194, 630–636 (2015).
Panchanathan, R. et al. Cell type and gender-dependent differential regulation of the p202 and Aim2 proteins: implications for the regulation of innate immune responses in SLE. Mol. Immunol. 49, 273–280 (2011).
Kang, S. J. et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol. 149, 613–622 (2000).
Kajiwara, Y. et al. A critical role for human caspase-4 in endotoxin sensitivity. J. Immunol. 193, 335–343 (2014).
Grebe, A., Hoss, F. & Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 122, 1722–1740 (2018).
Christ, A. et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 172, 162–175. e114 (2018).
Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J. Am. Coll. Cardiol. 70, 2278–2289 (2017).
Dinarello, C. A., Simon, A. & van der Meer, J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652 (2012).
Van Tassell, B. W., Toldo, S., Mezzaroma, E. & Abbate, A. Targeting interleukin-1 in heart disease. Circulation 128, 1910–1923 (2013).
Abbate, A. et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 9, e014941 (2020).
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
Vromman, A. et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Heart J. 40, 2482–2491 (2019).
Mallat, Z. et al. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 104, 1598–1603 (2001).
Gerdes, N. et al. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J. Exp. Med. 195, 245–257 (2002).
Wang, J. et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 21, 820–826 (2015).
van der Heijden, T. et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice—brief report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461 (2017).
van Hout, G. P. et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 38, 828–836 (2017).
Toldo, S. et al. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 22, 1146–1161 (2015).
Swirski, F. K. Inflammation and CVD in 2017: from clonal haematopoiesis to the CANTOS trial. Nat. Rev. Cardiol. 15, 79–80 (2018).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Furman, D. et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 23, 174–184 (2017).
Westerterp, M. et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 138, 898–912 (2018).
Yao, C. et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242 (2018).
Suetomi, T. et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/Calmodulin-dependent Protein Kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 138, 2530–2544 (2018).
Wang, D. et al. P2X7 receptor mediates NLRP3 inflammasome activation in depression and diabetes. Cell Biosci. 10, 28 (2020).
Hirshman, N. A. et al. Cyclophosphamide-induced cystitis results in NLRP3-mediated inflammation in the hippocampus and symptoms of depression in rats Am. J. Physiol. Ren. Physiol. 318, F354–F362 (2020).
Li, D. X. et al. NLRP3 inflammasome-dependent pyroptosis and apoptosis in hippocampus neurons mediates depressive-like behavior in diabetic mice. Behav. Brain Res. 391, 112684 (2020).
Alcocer-Gomez, E. et al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 36, 111–117 (2014).
Alcocer-Gomez, E. et al. Stress-induced depressive behaviors Require a functional NLRP3 inflammasome. Mol. Neurobiol. 53, 4874–4882 (2016).
Fleshner, M., Frank, M. & Maier, S. F. Danger signals and inflammasomes: stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacology 42, 36–45 (2017).
Goldberg, E. L. & Dixit, V. D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 265, 63–74 (2015).
Zhong, Z. et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Yamaguchi, O., Murakawa, T., Nishida, K. & Otsu, K. Receptor-mediated mitophagy. J. Mol. Cell. Cardiol. 95, 50–56 (2016).
Gassen, N. C. et al. Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: evidence in cells, mice, and humans. PLoS Med. 11, e1001755 (2014).
Iwata, M., Ota, K. T. & Duman, R. S. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain, Behav. Immun. 31, 105–114 (2013).
Reus, G. Z. et al. Pathophysiological mechanisms involved in the relationship between diabetes and major depressive disorder. Life Sci. 183, 78–82 (2017).
Karmakar, M., Katsnelson, M. A., Dubyak, G. R. & Pearlman, E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat. Commun. 7, 10555 (2016).
Moulton, C. D., Pickup, J. C. & Ismail, K. The link between depression and diabetes: the search for shared mechanisms. Lancet Diabetes Endocrinol. 3, 461–471 (2015).
Heneka, M. T., Golenbock, D. T. & Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236 (2015).
Hook, V. Y., Kindy, M. & Hook, G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753 (2008).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 (2013).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013).
Zhang, X. et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 6, eabb8680 (2020).
Zhang, Y., Zhao, Y., Zhang, J. & Yang, G. Mechanisms of NLRP3 inflammasome activation: its role in the treatment of Alzheimer’s disease. Neurochem. Res. 45, 2560–2572 (2020).
Lonnemann, N. et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 117, 32145–32154 (2020).
Beyer, M. M. S. et al. Enduring changes in neuronal function upon systemic inflammation are NLRP3 inflammasome dependent. J. Neurosci. 40, 5480–5494 (2020).
Zhang, Y., Dong, Z. & Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target. Ther. 5, 37 (2020).
Wu, A. G. et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 65, 101202 (2021).
Feng, Y. S., Tan, Z. X., Wu, L. Y., Dong, F. & Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 64, 101192 (2020).
Pereira, C. F. et al. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 56, 100966 (2019).
Tejera, D. et al. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 38, e101064 (2019).
Shulman, J. M., De Jager, P. L. & Feany, M. B. Parkinson’s disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222 (2011).
Chiti, F. & Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006).
Lee, S. J. Origins and effects of extracellular alpha-synuclein: implications in Parkinson’s disease. J. Mol. Neurosci. 34, 17–22 (2008).
Beraud, D. & Maguire-Zeiss, K. A. Misfolded alpha-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 18, S17–S20 (2012).
Ferrari, C. C. et al. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol. Dis. 24, 183–193 (2006).
Codolo, G. et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
Tan, E. K. et al. Parkinson disease and the immune system—associations, mechanisms and therapeutics. Nat. Rev. Neurol. 16, 303–318 (2020).
Cheng, J. et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 16, 2193–2205 (2020).
Yan, Y. Q., Fang, Y., Zheng, R., Pu, J. L. & Zhang, B. R. NLRP3 Inflammasomes in Parkinson’s disease and their Regulation by Parkin. Neuroscience 446, 323–334 (2020).
de Araujo, F. M. et al. Role of microgliosis and NLRP3 inflammasome in Parkinson’s disease pathogenesis and therapy. Cell. Mol. Neurobiol. https://doi.org/10.1007/s10571-020-01027-6.
He, X. M. et al. Ellagic acid protects dopamine neurons via inhibition of NLRP3 inflammasome activation in microglia. Oxid. Med. Cell Longev. 2020, 2963540 (2020).
Ambati, J. et al. Repurposing anti-inflammasome NRTIs for improving insulin sensitivity and reducing type 2 diabetes development. Nat. Commun. 11, 4737 (2020).
Pickup, J. C. & Crook, M. A. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41, 1241–1248 (1998).
Donath, M. Y. & Shoelson, S. E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107 (2011).
Masters, S. L., Latz, E. & O’Neill, L. A. The inflammasome in atherosclerosis and type 2 diabetes. Sci. Transl. Med. 3, 81ps17 (2011).
Vandanmagsar, B. et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 (2011).
Masters, S. L. et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11, 897–904 (2010).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Youm, Y. H. et al. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 152, 4039–4045 (2011).
Stienstra, R. et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl Acad. Sci. USA 108, 15324–15329 (2011).
Stienstra, R. et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 12, 593–605 (2010).
Goossens, G. H. et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol. Immunol. 50, 142–149 (2012).
Lee, H. M. et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62, 194–204 (2013).
Spranger, J. et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 52, 812–817 (2003).
Thorand, B. et al. Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA Augsburg Study, 1984–2002. Diabetes 54, 2932–2938 (2005).
Kim, S. R. et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 11, 2127 (2020).
Karki, R. & Kanneganti, T. D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214 (2019).
Terlizzi, M., Casolaro, V., Pinto, A. & Sorrentino, R. Inflammasome: cancer’s friend or foe? Pharmacol. Ther. 143, 24–33 (2014).
Ershaid, N. et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 10, 4375 (2019).
Bruchard, M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64 (2013).
Pallangyo, C. K., Ziegler, P. K. & Greten, F. R. IKKbeta acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 212, 2253–2266 (2015).
Dupaul-Chicoine, J. et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763 (2015).
Kolb, R., Liu, G. H., Janowski, A. M., Sutterwala, F. S. & Zhang, W. Inflammasomes in cancer: a double-edged sword. Protein Cell 5, 12–20 (2014).
Zitvogel, L., Kepp, O., Galluzzi, L. & Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 13, 343–351 (2012).
Voloshin, T. et al. Blocking IL1beta pathway following Paclitaxel chemotherapy slightly inhibits primary tumor growth but promotes spontaneous metastasis. Mol. Cancer Ther. 14, 1385–1394 (2015).
Wu, T. et al. Modulation of IL-1beta reprogrammes the tumor microenvironment to interrupt oral carcinogenesis. Sci. Rep. 6, 20208 (2016).
Vidal-Vanaclocha, F., Amezaga, C., Asumendi, A., Kaplanski, G. & Dinarello, C. A. Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 54, 2667–2672 (1994).
Dinarello, C. A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 29, 317–329 (2010).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging biological principles of metastasis. Cell 168, 670–691 (2017).
Vidal-Vanaclocha, F. et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl Acad. Sci. USA 97, 734–739 (2000).
Voronov, E. et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl Acad. Sci. USA 100, 2645–2650 (2003).
Deng, Q. et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 442, 21–30 (2019).
Chow, M. T. et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 72, 5721–5732 (2012).
Allen, I. C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Zaki, M. H., Vogel, P., Body-Malapel, M., Lamkanfi, M. & Kanneganti, T. D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920 (2010).
Wilson, J. E. et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913 (2015).
Man, S. M. et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 162, 45–58 (2015).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Chen, J., Wang, Z. & Yu, S. AIM2 regulates viability and apoptosis in human colorectal cancer cells via the PI3K/Akt pathway. Onco Targets Ther. 10, 811–817 (2017).
Drexler, S. K. et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl Acad. Sci. USA 109, 18384–18389 (2012).
Villani, A. C. et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Schoultz, I. et al. Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn’s disease in Swedish men. Am. J. Gastroenterol. 104, 1180–1188 (2009).
Ungerback, J. et al. Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 33, 2126–2134 (2012).
Mori, Y. et al. Instabilotyping: comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 61, 6046–6049 (2001).
Schulmann, K. et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology 128, 590–599 (2005).
Woerner, S. M. et al. Pathogenesis of DNA repair-deficient cancers: a statistical meta-analysis of putative Real Common Target genes. Oncogene 22, 2226–2235 (2003).
Zhang, A. et al. The genetic polymorphism and expression profiles of NLRP3 inflammasome in patients with chronic myeloid leukemia. Hum. Immunol. 79, 57–62 (2018).
H, M. J. & H, F. G. NLRP3 inflammasome gene polymorphisms variably associated with its serum levels in acute myocardial infarction. Pak. J. Biol. Sci. 23, 612–618 (2020).
Paramel Varghese, G. et al. NLRP3 inflammasome expression and activation in human atherosclerosis.J. Am. Heart Assoc 5, e003031 (2016).
Roncero-Ramos, I. et al. Mediterranean diet, glucose homeostasis, and inflammasome genetic variants: The CORDIOPREV Study. Mol. Nutr. food Res. 62, e1700960 (2018).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Ridker, P. M. et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Lust, J. A. et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84, 114–122 (2009).
Lust, J. A. et al. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 91, 571–574 (2016).
Gross, O. et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400 (2012).
Hickish, T. et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 18, 192–201 (2017).
Ridker, P. M. et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Isambert, N. et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology 7, e1474319 (2018).
Li, Z. et al. Targeted anti-IL-1beta platelet microparticles for cardiac detoxing and repair. Sci. Adv. 6, eaay0589 (2020).
Sharma, A. et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 70, 772–787 (2021).
Li, J. et al. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 238, 2603–2614 (2020).
Zheng, F., Xing, S., Gong, Z., Mu, W. & Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014, 507208 (2014).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81870194 and 91849122 to Y.L., 81873528 and 81670358 to Y.-H.S., 91839101 to Z.S., and U1601227 to X.-Y.Y.), Jiangsu Province Peak of Talent in Six Industries (BU24600117 to Y.L.), and Introduction Project of Clinical Medicine Expert Team for Suzhou (No. SZYJTD201704). We thank Dr. I.C. Bruce for reading the manuscript. We apologize in advance to colleagues whose work was not directly cited in this Review because of space limitations. Some figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Huang, H., Liu, B. et al. Inflammasomes as therapeutic targets in human diseases. Sig Transduct Target Ther 6, 247 (2021). https://doi.org/10.1038/s41392-021-00650-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00650-z
This article is cited by
-
SARS-CoV-2 vaccination may mitigate dysregulation of IL-1/IL-18 and gastrointestinal symptoms of the post-COVID-19 condition
npj Vaccines (2024)
-
Divergent functions of NLRP3 inflammasomes in cancer: a review
Cell Communication and Signaling (2023)
-
Real-life exposure to Fusarium toxins deoxynivalenol and zearalenone triggers apoptosis and activates NLRP3 inflammasome in bovine primary theca cells
Mycotoxin Research (2023)
-
Role of NLRP3 inflammasome-mediated neuronal pyroptosis and neuroinflammation in neurodegenerative diseases
Inflammation Research (2023)
-
CircSHOC2 Knockdown Alleviates High Glucose-Induced Vascular Endothelial Cell Pyroptosis via Targeting miR-145/FOXO1 Axis In Vitro Condition
Molecular Biotechnology (2023)
