
Abstract
The current treatment strategies in advanced malignancies remain limited. Notably, immunotherapies have raised hope for a successful control of these advanced diseases, but their therapeutic responses are suboptimal and vary considerably among individuals. Tumor-associated macrophages (TAMs) are a major component of the tumor microenvironment (TME) and are often correlated with poor prognosis and therapy resistance, including immunotherapies. Thus, a deeper understanding of the complex roles of TAMs in immunotherapy regulation could provide new insight into the TME. Furthermore, targeting of TAMs is an emerging field of interest due to the hope that these strategies will synergize with current immunotherapies. In this review, we summarize recent studies investigating the involvement of TAMs in immune checkpoint inhibition, tumor vaccines and adoptive cell transfer therapies, and discuss the therapeutic potential of targeting TAMs as an adjuvant therapy in tumor immunotherapies.
Similar content being viewed by others

Introduction
Given the association of malignancies with subverted immune surveilliance,1 immunotherapies provide options for advanced cancer patients, and multiple clinical trials are underway.2 Despite the impressive results achieved in several clinical trials,3,4,5,6 obstacles have been encountered with the current immune checkpoint inhibitor (ICI)-based immunotherapies.7,8,9 Suboptimal efficacy is among the major concerns because previous trials suggest that the response rate to ICI monotherapy is limited, and the responses vary significantly across multiple tumors and among individuals.10,11,12,13
Accumulating evidence has suggested that immune suppression in the tumor microenvironment (TME) represents a major barrier to maximizing the clinical potential of immunotherapies.14 The TME is complex with diverse populations of nontumor stromal cells that impact tumor immune evasion, response to immunotherapy, and patient survival.15 In addition to cytotoxic lymphocytes (CTLs) and natural killer cells (NKs) that are generally considered effective antitumor immune cells, the TME contains a range of other cell types that are involved in the crosstalk with anti-tumor immune cells, including cancer-associated fibroblasts (CAFs),16 endothelial cells (ECs),17 and tumor-associated macrophages (TAMs).18 CAFs can induce a robust stromal reaction characterized by fibrotic extracellular matrix (ECM) and make the TME convert to an immune-excluded type via the transforming growth factor-β (TGF-β) signaling pathway.19,20,21 The tumor-associated vasculature is another hallmark of advanced solid tumors.22,23 ECs of tumor vasculature can not only inhibit antitumor immunity by establishing a selective immune barrier via the vascular endothelial growth factor (VEGF)/prostaglandin E2 (PGE2)-FASL pathway,24 but can also exacerbate the hypoxia condition with low pH and cause high interstitial fluid pressure, which is unfavorable for the infiltration and activation of CTLs and NKs.19,20,22
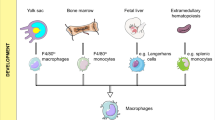
Macrophages are involved in various processes in both homeostasis and disease.25,26 With effector functions such as phagocytosis, antigen presentation, and the plasticity to secrete a wide variety of signaling molecules, they serve as an efficient “firewall” in regulating homeostasis.26,27,28,29,30 They are also dynamic populations, and the resident macrophage pool can be rapidly expanded by infiltrating monocytes under pathological states such as tissue damage, inflammation and malignancy.20,31,32,33,34 Macrophages in the TME can be roughly induced into two contrasting groups: classically activated “M1” macrophages and alternatively activated “M2” macrophages.32 M2 and small populations of M1 cells, also known as tumor-associated macrophages (TAMs), have been generally thought to be involved in tumor initiation, progression, angiogenesis and metastasis.35 Most relevant for patients, a high TAM infiltration is often correlated with poor clinical outcomes in a wide variety of tumors and is believed to decrease responses to standard-of-care therapeutics, including radiotherapy, chemotherapy and targeted therapy.27,36,37,38,39,40,41,42,43,44 However, the “M1-M2” macrophage dichotomy is too simple to describe their complicated roles in the TME.32 Recent data acquired using unbiased large-scale techniques might help discriminate among macrophage subpopulations and have unraveled a previously unrecognized complexity in macrophage polarization, far beyond the old dogma of the binary “M1-M2” binary system.45 Furthermore, significant dynamic changes in macrophage subpopulations were observed during tumor development and were correlated with the efficacy of immunotherapy.37,46,47,48,49 These findings suggest a better understanding of heterogeneous TAMs and their roles in immunotherapy will be critical for developing effective immunotherapies.50,51
Here, we attempt to illustrate the regulatory functions of TAMs in the TME and different immunotherapies. We further discuss the therapeutic potential of targeting TAMs to improve current immunotherapies (immunotherapy classifications are summarized in Fig. 1).

Classification of current tumor immunotherapies. The current tumor immunotherapies can be roughly divided into three types: a checkpoint inhibitors, including anti-PD-1/L1 and anti-CTLA-4 monoclonal antibodies; (b) tumor vaccines, including various dendritic cell-based vaccines and oncolytic virus-based vaccines; and (c) adoptive cell transfer, including CAR-T or TCR-engineered T cells. The antigens released from necrotic tumor cells during surgery, locoregional therapy, chemotherapy or targeted therapy enhance the immune recognition of tumor cells
TAMs in tumor initiation and progression
The strong relationship between inflammation and tumorigenesis has long been recognized.52 Approximately 90–95% of all types of tumors are connected to environmental exposures including tobacco, obesity, smoke, radiation, chemicals, and chronic infections, all of which could induce a smoldering inflammatory state.53 TAMs help establish a pro-inflammatory microenvironment and the link between TAMs and tumor initiation has been extensively studied in various clinical samples and preclinical models.54,55,56 For instance, liver macrophages were found to be the key source of steatosis-induced Wnt expression and the active Wnt/β-catenin signaling in macrophages can promote the growth of tumor progenitor cells, underlying the increased risk of hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) in obese individuals.57,58
TAMs can promote tumor progression by producing mediators that remodel the tumor-supportive TME. Such mediators include growth factors and cytokines that support tumor cell proliferation; NF-κB-mediated factors that protect against apoptosis (for example, interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, C-C motif chemokine (CCL)2, C-X-C motif chemokine (CXCL)8, and CXCL10);35,51 pro-angiogenic growth factors, such as VEGF, platelet derived growth factor (PDGF), TGF-β and fibroblast growth factor (FGF);59,60,61 and other factors that modulate tissue architecture and favor tumor cell migration, invasion and metastasis.39,62,63,64,65
TAMs also subvert local immune surveillance because they can directly reduce the activities of T cells and NKs by expressing cell surface proteins or by releasing soluble factors that display immunosuppressive functions (for example, arginase 1 (ARG1), indoleamine 2,3-dioxygenase (IDO), IL‑10, programmed death ligand 1 (PD-L1), and TGF-β)65,66 or indirectly suppress T cell activities through recruitment of other immune suppressive cells such as regulatory T cells (Tregs).66,67
Overall, TAMs play a dual role as “tumor promoters” and “immune suppressors” because they can promote tumor initiation and act as central drivers of the immunosuppressive TME through their expression of cell surface receptors, secreted cytokines, chemokines, and enzymes that regulate the recruitment and function of multiple immune cell subtypes.
TAMs as regulators in immunotherapies
Numerous studies have shown the contribution of TAMs to immunotherapy resistance,68 while the precise mechanisms are still unclear. How these heterogeneous populations exert their distinct regulatory capability in response to different immunotherapies remains poorly defined.
TAMs in checkpoint inhibitor therapy
Inhibition of immune checkpoints, such as PD-1/L1 and CTLA-4, removes inhibitory signals of T cell activation, which enabling tumor-reactive T cells to overcome regulatory mechanisms and mount an effective antitumor response.69,70 However, the underlying cellular mechanisms remain unclear, namely, the expression patterns of checkpoint molecules and the interplay among ICIs and different components within the TME. In the setting of ICI therapy, the impact of TAMs should be carefully considered (Fig. 2, left).

TAMs as regulators of tumor immunotherapies. TAMs exert their distinct regulatory functions in response to different immunotherapies: a In checkpoint inhibitor therapy, TAMs can suppress effective T cells directly via the expression of various checkpoint molecules and immunosuppressive cytokines and indirectly through crosstalk with Tregs and hijacking of anti-PD-1 antibodies. b In tumor vaccine therapy, TAMs can inhibit the antigen-presenting efficiency of dendritic cells; c In adoptive cell transfer therapy, TAMs prevent immune infiltration via build up the highly fibrotic and angiogenic TME
Upregulation of checkpoint molecules
Early in the 2000s, the overexpression of checkpoint ligand B7-H1 (PD-L1) on tumor cells was considered a key mechanism of immune evasion.71,72 However, it was not until 2009 that two independent studies first demonstrated that macrophages are the predominant immune cells that express PD-L1 in HCC.73,74 These PD-L1+ TAMs, activated by tumor-derived IL-10, could mediate CD8+T cell dysfunction via the PD-1/PD-L1 interaction.73,74 Similar results were also observed in other tumor types including head and neck squamous cell carcinoma (HNSC), ovarian cancer, soft tissue sarcoma, bladder cancer and CCA.75,76,77,78,79,80 In the last decade, other B7 family checkpoints ligands were found to be expressed on TAMs, including B7-DC (PD-L2) and B7-H4 (B7-S1), as well as alternative checkpoints ligands such as galectin-9 and V-domain Ig-containing suppressor of T cell activation (VISTA).81,82,83,84,85,86 Thus, TAMs have been regarded as carriers of checkpoint ligands that are upregulated in response to TME-derived factors, resulting in immune exhaustion via the checkpoint ligand/receptor interaction in a cell-to-cell contact manner.
However, these findings do not provide a comprehensive picture. The expression of checkpoint molecules on macrophages might reflect the immune status within the TME. PD-L1 expression on macrophages, rather than on tumor cells, was positively correlated with patients’ overall survival (OS) and might be used as an independent prognostic factor based on a cohort study of 453 HCC patients.87 Surprisingly, PD-L1+ TAM-enriched tumors exhibited an activated immune status, with high levels of CD8+ T cell infiltration and immune-related gene expression, which indicates that a substantial proportion of tumor might be a amenable to ICI therapy.87,88,89
Moreover, the blocking effect of ICIs on the checkpoint molecules expressed on TAMs is increasingly attracting attention. Gordon et al. found that the phagocytic potency of PD-1+ macrophages is rescued by PD-1/PD-L1 inhibition, which lengthens the survival of colon cancer preclinical models in a macrophage-dependent manner.90 Another recent study based on a myeloid cell-specific PD-1 silencing model revealed the vital role of elevated intracellular cholesterol during anti-PD-1 treatment, which is required for differentiation of inflammatory macrophages and the promotion of antigen-presenting function.91 These findings show the complexity behind the checkpoint molecules expressed on TAMs. A further understanding of their intracellular regulatory mechanisms will be helpful for precise classification of TAMs and providing guidance for ICI treatment.
Crosstalk with regulatory T cells
Tregs are critical components of the TME and contribute to different aspects of tumor progression.92 Recent studies have revealed the compensation between TAMs and Tregs that derived immune evasion and ICI resistance.93
TAMs favor chemokine/cytokine-mediated recruitment of Tregs to the TME.94,95,96,97,98 TAM-derived CCL20 was found to promote the infiltration of CCR6+Tregs in colorectal cancer (CRC) and HCC, which may be an essential mechanism of anti-PD-L1 therapy resistance.67,99 Specifically, TREM-1+ TAMs elevate the expression of the chemokine CCL20 via the extracellular signal-regulated kinase (ERK)/NF-κβ pathway in response to hypoxia and tumor metabolites promoting infiltration of CCR6+ FOXP3+ Tregs.67 Thus, blocking the TAM-specific TREM-1 pathway could significantly reduce immunosuppressive Tregs recruitment, as well as restore the efficacy of anti-PD-L1 therapy.67 TAM-derived factors also play a central role in the induction of induced Tregs (iTregs) in the TME. It was shown that iTregs could be induced from CD4+CD25− T cells co-cultured with M2-TAMs.100 A recent study by Zhou et al. highlighted the critical roles of TAM-derived exosomes in the induction of iTregs. They identified miRNAs enriched in exosomes, including miR-29a-3p and miR-21–5p which directly suppressed T cell-intrinsic STAT3 and regulated Treg/Th17 in ovarian cancer.101
Moreover, Tregs can further enhance the immunosuppressive properties of TAMs. In laryngeal squamous cell carcinoma (LSCC), malignant pleural effusion (MPE), and CRC, Tregs were found to promote the differentiation of monocytes into immunosuppressive TAMs directly.100,102,103 Tregs can also modulation of lipid metabolism in M2-like TAMs. Liu et al. found that Tregs could suppressed CD8+ T cell secretion of IFN-γ, which would otherwise block the activation of sterol regulatory element binding protein-1 (SREBP1)-mediated fatty acid synthesis in M2 TAMs. Thus, Tregs indirectly but selectively sustained M2-like TAM metabolic fitness, mitochondrial integrity and survival.104
Therefore, a positive feedback loop exists between TAMs and Tregs that further enhances their immunosuppressive effects in the TME.
Hijacking of anti-PD-1 antibodies
The constitutive expression of Fcγ receptor (FcγR) on monocytes/macrophages plays a crucial role in the antibody-dependent phagocytosis (ADCP) of tumor cells.105 However, it is worth noting that TAMs may have a significant impact on the pharmacokinetics and efficacy of ICI via Fc-FcγR binding. In mouse models and primary human immune cells, anti-PD-1 antibodies were observed to be seized by macrophages depending on the Fc domain of the antibody and the FcγR expressed by macrophages, which led to ICI therapy resistance.106 Moreover, a recent study has shown that Fc-FcγR binding-mediated TAM reprogramming can even induce hyperprogression in an non-small cell lung cancer (NSCLC) cohort and NSCLC patient-derived xenograft (PDX) models, although the mechanism remains unclear.107 Thus, how to interfere with the constitutive expression of FcγR on macrophages should be explored out in the tumor immunotherapy field. The effects mediated by the Fc domain of checkpoint molecular antibodies should be carefully evaluated and mechanistically understood.108
Macrophages in adoptive cell transfusion
Adoptive cell transfer therapies such as chimeric antigen receptor T cell (CAR-T) or TCR-engineered T cell therapies enhanced the anti-tumor response in different advanced malignancies.109,110,111 The transferred cells must be trafficked and infiltrate into tumor sites to exert their cytolytic effects.112 However, this approach is not feasible for solid tumor treatment because of the relatively limited blood distribution and abnormal structure of tumor neo-vessels.22 In addition, tumors that develop from cirrhosis are highly fibrotic and difficult to penetrate physically.16 These features complicate the infiltration of adoptively transferred cells into tumor sites. In addition, TAMs contribute to the angiogenic and fibrotic TME (Fig. 2, lower right).
TAMs support tumor angiogenesis mainly by the production of factors such as VEGFA, PDGF, TGF-β and FGF.59,60,61 The subpopulation of TAMs characterized by the expression of angiopoietin 1 (TIE2+) in the blood or TME were considered to be close associated with intratumor neovessel formation.113,114 The molecular events of TAM-mediated angiogenesis were identified in a study based on a chronic HBV infection cohort, which showed that the individuals who finally developed HCC had higher serum levels of IL-23.115 IL-23, which is produced by inflammatory macrophages, enhanced macrophage-mediated angiogenesis by upregulating IL-23 receptor expression in macrophages. This “chronic inflammation-macrophage-IL-23” positive feedback loop might partially explain the significant role of macrophages in formation of the TME. Furthermore, high TAM infiltration correlated with a small number of IFN-γ-expressing active NKs in HCC,116 which might have a negative impact on the activation or survival of adoptive transferred NKs.
Macrophages in tumor vaccines therapy
The identification of tumor antigens led to the development of tumor vaccination strategies in the 1980s. The host anti-tumor immune response is induced by tumor-antigen-pulsed dendritic cells (DC-based vaccines) or tumor-derived antigens released from lysed tumor cells (oncolytic virus).117,118,119 However, the results from clinical trials, were not as striking as expected.120,121,122,123 The limited efficacy of tumor vaccines in solid tumors may be ascribed to different possible causes, one of these being the strong immunosuppressive TME.124 As in other immunotherapies, studies have shown the accumulation of immune suppressive CD11b+ myeloid cells in response to the tumor vaccine treatment, which may result in therapy resistance125,126,127(Fig. 2, upper right).
Currently, TAM-targeting strategies combined with tumor vaccination are under evaluation. Anti-CD11b antibody-mediated depletion of myeloid cells showed a synergistic effect along with the vaccine by further prolonging the survival of tumor-bearing mice, although no significant reduction in tumor burden was observed.128 Injection of tumor lysate-pulsed DC also prolonged the survival of mouse models, and this therapeutic effect was further enhanced by injection of PLX3397, a CSF1R inhibitor that reprograms macrophages.126
Therapeutic targeting of TAMs in tumor
Given that TAMs have a profound impact on tumor immunotherapies, there is considerable interest in the therapeutic targeting of TAMs to synergize current ICI-based immunotherapy. The different approaches that have been explored for targeting TAMs can be roughly sorted into three major categories: (1) eliminating TAMs already present in the TME; (2) inhibition of monocyte recruitment; and (3) reprogramming of TAMs (Fig. 3).31,35,129 These strategies have been investigated in preclinical models, and some have been translated into the clinical setting as adjuvant to immunotherapy.130 Here, we summarize current preclinical and clinical studies and discuss the potential strengths and weaknesses of these approaches in different solid tumors (Table 1).

TAM-targeted Strategies in Tumors. TAM-targeted strategies can be roughly divided as follows: a elimination of macrophages already present in tumor tissue; (b) inhibition of monocyte/macrophage recruitment; (c) reeducation of TAMs toward an “immune-supportive” phenotype characterized by restored phagocytic and antigen presenting ability
Macrophage elimination
The clearance of TAMs is an option to counter their negative impact directly during immunotherapy. Bisphosphonates, which are traditionally been used to prevent the bone metastases or excessive bone resorption, can be taken up by phagocytes and have cytotoxic effects on myeloid cells.131,132 Based on their structure, bisphosphonates can be divided into two categories: nonnitrogen-containing and nitrogen-containing bisphosphonates.131
Clodronate belongs to the family of non-nitrogen bisphosphonates. In early studies, clodronate-loaded liposomes (clodrolip) were often used to deplete liver macrophages.133,134 Liposomes are artificially prepared vesicles that undergo phagocytosis by macrophages after injection, and then, the intracellular release and accumulation of clodronate can induce apoptosis of macrophages.135 Administration of clodrolip depleted TAMs resulted in reduced tumor growth in preclinical models.134,136 The benefits of macrophage elimination have not only been seen with clodrolip, but also with other bisphosphonates, such as zoledronate.132 Zoledronate belongs to the third-generation nitrogen-containing bisphosphonate that has been shown to exhibit selective cytotoxicity towards matrix metalloproteinase-9 (MMP9)-expressing TAMs and to impair differentiation of monocytes into TAMs.137 Zoledronate acid (ZA) reduced the infiltration of TAMs, decreased tumor angiogenesis and inhibited tumor progression in different preclinical tumor models.134,138,139,140
A possible major barrier to this therapeutic approach might be the fact that general depletion of monocytes/macrophages is not TAM-specific and coincides with loss of tissue-resident macrophages that are crucial for maintaining homeostasis, and bacterial clearance, especially in the liver.26
Macrophage recruitment inhibition
Another TAM-targeting strategy is to cut off their replenishment by circulating monocytes. The recruitment of circulating monocytes is highly dependent on several chemokine signals,31 and thus, interference with chemokine signaling using monoclonal antibodies or small molecule inhibitors might be an effective way to prevent TAM accumulation in the TME.
CCL2/CCR2 signaling plays a central regulatory role in circulatory monocytes and their infiltration into the TME, making it a promising TAM-targeted therapy.141,142 Inhibition of CCL2/CCR2 signaling has shown antitumor efficiency in different experimental animal models.29 Genetic silencing and administration of a CCL2 neutralizing antibody or CCR2 antagonist reduced the recruitment of circulatory monocytes, subsequently lowered the number of TAMs, and downregulated the secretion function of M2-like TAMs.41,143,144,145 More importantly, an enhancement of the function of tumor-infiltrating CD8+ T cells and NKs was observed during the treatment,41,143,144 which may suggest a good immunotherapy response. Thus, several phase I/II clinical trials are in progress to assess the therapeutic effect of BMS-813160, a small molecule inhibitor of CCR2/5, in combination with Nivolumab and/or the tumor vaccine GVAX in several solid tumors including HCC, NSCLC, renal cell carcinoma (RCC) and pancreatic ductal adenocarcinoma (PDAC).
Hypoxia-induced upregulation of stromal cell-derived factor 1 alpha (SDF-1α/CXCL12) also contributes to the recruitment of the suppressive M2 macrophages.146 Inhibition of the SDF-1α receptor (CXCR4) using the CXCR4 antagonist AMD3100 relieved regional immunosuppression and facilitated anti-PD-1 antibody treatment in a sorafenib-resistance HCC model.147 This study is of great translation value because hypoxia and HIF-1α activation are the most common and significant features of solid tumors and are usually aggravated during conventional treatments including chemotherapy, transcatheter arterial chemoembolization (TACE) and sorafenib treatment.148 Moreover, inhibition of CXCR4 might have synergistic effects with anti-angiogenesis drugs because TAMs can regulate the expression of CXCR4 via the ERK pathway, which is a novel vascular marker for angiogenesis.149 Therefore, CXCR4 antagonists, such as AMD3100 and BL-8040 should be judiciously considered in the future design of clinical trials for immunotherapies.
Although the efficacy of immunotherapies could be enhanced by myeloid cell recruitment inhibition, preclinical evidence from PDAC suggests that the resistance mechanism against this therapeutic approach may lie in the rapid compensation by tumor-associated neutrophils (TANs) and a lack of effect on tissue-resident TAM populations.150,151 Moreover, withdrawal of CCL2/CCR2 inhibitors may lead to a dramatically release of monocytes previously trapped within the bone marrow, which was shown to accelerate metastasis in a preclinical model of breast cancer.152 Although these limitations have not been reported in completed or ongoing clinical trials, considering them in the design of future clinical trials is critical, and alternative targets that overcome these limitations may be required for optimal and stable therapeutic responses.
Macrophage reprogramming
An inevitable drawback to macrophage clearance and recruitment inhibition is the loss of their potential immune-stimulatory role as the major phagocytes and professional antigen-presenting cells (APCs) within the TME.153 Despite generally being tumor-supportive, TAMs may be phagocytic and suppress tumor growth by activating antitumor immune responses. This suggests that macrophage plasticity can be therapeutically exploited to restore the antitumor properties to TAMs.25 Thus, switching TAMs toward an “immune-supportive” phenotype provides an opportunity to reshape the immune-suppressive or exclusive TME and therefore presents a more effective approach to optimizing current ICI-based immunotherapies. This can be achieved by using therapeutics that promote macrophage polarization and/or using nanoparticles that can selectively reprogram macrophages to a restorative phenotype.130
Restoring phagocytic capacity
In homeostasis, normal cells can avoid self-elimination by phagocytes through the expression of anti-phagocytosis molecules,154,155 which are therefore called “phagocytosis checkpoints.” However, many studies have shown that tumor cells depend even more on phagocytosis checkpoints to evade immune surveilliance.156 Therefore, identification and intervention with phagocytosis checkpoints might provide a new approach for restoring the phagocytic capacity of TAMs to eliminate tumor cells.157
Signal regulatory protein alpha (SIRPα) is an ITIM-bearing inhibitory receptor expressed on myeloid cells, including macrophages.157 SIRPα recognizes CD47, which acts as a “don’t eat me” signal and is found to be overexpressed tumor cells and correlate with patients’ poor survival.158,159 Macrophage phagocytosis of tumor cells was restored after treatment with CD47 antibodies,160 and this macrophage-mediated phagocytosis was further enhanced in the presence of chemotherapeutic drugs, suggesting that patients with lower CD47 expression were more likely to benefit from adjuvant TACE treatment.158 It is worth noticing that CD47 is highly expressed in CCA.161 Interfering with the CD47-SIRPα interaction promotes phagocytosis in TAMs and consequently suppresses the progress of CCA.161 The unique overexpression of CD47 in CCA offers an exceptional opportunity for CD47-targeted therapy.
The bridging between innate and adaptive immune cells provides the rationale for combining phagocytosis checkpoint inhibitors with current ICI-based immunotherapies that boost the adaptive immune response.157 The potential for such combinations was initially observed when anti-CD47 therapy was shown to have synergistic effect with PD-L1 inhibitor in a mouse model bearing the B16F10 melanoma.162 Similarly, a bispecific antibody targeting PD-L1 on tumor cells and SIRPα on APCs showed a more significant antitumor effect against murine colon cancer compared with either anti-PD-L1 or anti-SIRPα monotherapy.163 Overall, these preclinical results along with earlier observations in ICIs confirm the notion that the conventional boundary between the innate immune checkpoint and adaptive immune checkpoints is becoming unclear, because more of these checkpoints have been found to function at both the innate and adaptive levels.157
Unleashing the immune-stimulatory capacity
The CSF1/CSF1R axis has been heavily investigated for its role in defining the survival, proliferation, differentiation and function of macrophages.164,165,166 Targeting CSF1/CSF1R signaling in protumoral TAMs represents an attractive strategy to eliminate CSF1R-dependent or reprogram M2-like TAMs.167 The altered TAM’s polarization will be key to reshaping the immunosuppressive TME and boosting a preexisting antitumor immune response.167,168 In preclinical models, CSF1/CSF1R blockade has been shown to improve the efficacy of different immunotherapies, including ICIs and adoptive cell transfer therapy.169,170,171 The positive results of these studies have led to clinical trials combining CSF1 and/or CSF1R inhibitors with ICIs or other immunotherapies.172,173
CD40, a receptor that belongs to the TNF receptor superfamily, is primarily expressed on APCs. The CD40-CD40L interaction upregulates the expression of MHC and promotes the secretion of pro-inflammatory cytokines, such as IL-12, which plays a significant role in T cell priming.129 Macrophage treatment with CD40 agonists in combination with anti-CSF1R antibodies resulted in profound TAM reprogramming before their depletion; these reprogrammed TAMs created a pro-inflammatory environment that elicited effective T cell responses, even in tumors that were nonresponsive to ICIs.174,175
Phosphatidylinositol 3-kinase γ (PI3Kγ) acts as a molecular switch that turns on an “immunosuppressive program” while shutting down “immune-stimulatory program”.176 Kaneda et al. showed that PI3Kγ determines the immunosuppressive properties of TAMs.177 It was shown that the lack of PI3Kγ activity in TAMs induced the expression of MHC-II and pro-inflammatory cytokines while reducing the immunosuppressive molecules including IL-10 and arginase.177 This dramatic shift of TAMs also enhanced adaptive immunity in the TME and significantly inhibited tumor progression.177 Another critical study by De Henau et al. also showed the potential of targeting myeloid-intrinsic PI3Kγ in overcoming ICI resistance.178 Further analysis is required to determine whether PI3Kγ inhibition could exert similar immunomodulatory function in other solid tumors.
The Lmdd-MPFG (LM) vaccine activates the NF-κB pathway in TAMs through the Toll-like receptor (TLR)2-MyD88 pathways, and recruits p62 to activate the autophagy pathway.179,180 The overall effect of LM skews the TAMs from the M2-like state into the M1-like state.181 Most importantly, this approach skewed the TME cytokine profile to anti-tumor profile, and this change restored the T cell reactivity to the anti-PD-1 blockade.180
Nanoparticles in the optimization of macrophages reprogramming
Systemic targeting of TAMs using nanomedicines is an attractive approach because TAMs are ideal therapeutic targets due to their considerable propensity to phagocytose nanoparticles.182 Notably, it has been reported that myeloid cells could take up ten-fold more nanoparticles than tumor cells in a preclinical model.183
Several recent studies have used nanoparticles loaded with TLR agonists or tumor peptides to promote reprogramming of the TAMs, exploiting the capacity of nanoparticles to both target TAMs and promote antitumor immunity. For example, the TLR7/TLR8 agonist R848 loaded nanoparticles preferentially accumulated in TAMs in mouse models and promoted macrophage reeducation.184 Another study based on immunotherapy resistance tumors showed that codelivery of a long peptide antigen, which induced antigen-presenting activity of TAMs, and TLR agonists to TAMs using a nanosized hydrogel (nanogel) can transform the resistant tumors into tumors sensitive to adaptive immune cell transfer.185
However, the development of TAM-reeducating therapies based on nanoparticles is still facing great challenges, such as how to preferentially deliver them to the protumoral M2-like TAMs or how to acquire a long-lasting and sufficient antitumor response.
Fortunately, engineering of new nanomedicines provides new opportunities by (1) applying nanoparticles modified with ligands that could recognize M2 TAM’s specific markers to achieve target delivery; and (2) preparing nanoparticles to reduce the number of TAMs in the tumor via specific cytotoxicity, or reeducating TAMs in a long-lasting manner with the carriers possessing drug controlled release properties.186 Effective development of such nanomedicines could lead to a breakthrough in the field of tumor immunotherapy.
Conclusion and perspectives
TAMs are primary immune cells within the TME with high heterogeneity and complex roles as regulators of tumor immunity and immunotherapy. Thus, it is fundamental to reveal their exact regulatory mechanisms and identify macrophage-specific targets to optimize the efficacy of current immunotherapies. Recent studies have partially revealed the regulatory mechanisms and have highlighted three major TAM-targeting strategies: macrophage elimination, recruitment inhibition and reprogramming. Early clinical trials have focused on the first two approaches. Regardless, organ homeostasis disruption induced by resident macrophages and the potential metastasis-promoting withdrawal reaction remain key barriers to practical application in clinical settings. Going forward, a better strategy for macrophage reprogramming that attenuates their immune-suppressive ability while enhancing their potential immune-stimulatory functions is favorable for current ICI-based immunotherapy. However, the actual synergistic effect of macrophage-reprogramming agents, such as PI3Kγ inhibitors and CD40 agonists needs further evaluation. Moreover, macrophage reprogramming using nanoparticles has therapeutic potential in several preclinical models, but nanoparticle efficiency, safety and tolerability should be carefully evaluated in the human body.
Despite the recent progress in clinical and preclinical studies, some questions remain unanswered. For example, studies have highlighted the molecular events and signaling pathways of TAMs. Still, less is known about the intracellular metabolic switch during tumor progression and its potential impact on immunotherapy. Novel checkpoint receptors, such as T Cell Immunoglobulin and ITIM domain (TIGIT), VISTA or Lymphocyte-activation-gene-3 (LAG-3), have attracted broad interest, but what is the significance of macrophage populations expressing these different checkpoint receptors? Finally, because macrophages in the digestive system are direct sentinel cells for changes in the gut microbiota, understanding the exact mechanisms of these interactions and their consequences could potentially aid in tailoring an antitumor microbial cocktail. This concept is based on emerging studies suggesting that the manipulation of the gut microbiome can alter cancer incidence and the responses to immunotherapy.180,187,188,189
References
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013).
Egen, J. G., Ouyang, W. & Wu, L. C. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity 52, 36–54 (2020).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Ribas, A. et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA315, 1600–1609 (2016).
Horn, L. et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 379, 2220–2229 (2018).
Lin, Z. et al. Heterogeneous responses in hepatocellular carcinoma: the achilles heel of immune checkpoint inhibitors. Am. J. Cancer Res. 10, 1085–1102 (2020).
Shah, N. N. & Fry, T. J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019).
Baruch, E. N. et al. Adoptive T cell therapy: an overview of obstacles and opportunities. Cancer 123, 2154–2162 (2017).
Okusaka, T. & Ikeda, M. Immunotherapy for hepatocellular carcinoma: current status and future perspectives. ESMO Open 3, e000455 (2018).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017).
Zhu, A. X. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952 (2018).
Qin, S. et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 21, 571–580 (2020).
Haanen, J. B. A. G., Lugowska, I, Garassino, M. C. & Califano, R. ESMO Handbook of Immuno-oncology. OncologyPRO, ESMO (2018).
Rizvi, S., Wang, J. & El‐Khoueiry, A. B. Liver cancer immunity. Hepatology. In Press (2020).
Coulouarn, C. & Clement, B. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J. Hepatol. 60, 1306–1309 (2014).
Morse, M. A. et al. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 25, 912–920 (2019).
Salmon, H., Remark, R., Gnjatic, S. & Merad, M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227 (2019).
Chen, J., Gingold, J. A. & Su, X. Immunomodulatory TGF-beta signaling in hepatocellular carcinoma. Trends Mol. Med. 25, 1010–1023 (2019).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812–830 e814 (2018).
Manzanares, M. et al. Transforming growth factors α and β are essential for modeling cholangiocarcinoma desmoplasia and progression in a three-dimensional organotypic culture model. Am. J. Pathol. 187, 1068–1092 (2017).
Schaaf, M. B., Garg, A. D. & Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 9, 115 (2018).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Motz, G. T. et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 20, 607–615 (2014).
Wynn, T. A., Chawla, A. & Pollard, J. W. Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 (2013).
Krenkel, O. & Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 17, 306–321 (2017).
Ruffell, B. & Coussens, L. M. Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Schmall, A. et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am. J. Respir. Crit. Care Med. 191, 437–447 (2015).
Balmer, M. L. et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 6, 237ra66 (2014).
Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 66, 1300–1312 (2017).
Guillot, A. & Tacke, F. Liver macrophages: old dogmas and new insights. Hepatol. Commun. 3, 730–743 (2019).
Wang, J. & Kubes, P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 165, 668–678 (2016).
Varol, C., Mildner, A. & Jung, S. Macrophages: development and tissue specialization. Annu. Rev. Immunol. 33, 643–675 (2015).
Mantovani, A. et al. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Kitano, Y. et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br. J. Cancer 118, 171–180 (2018).
Zhang, Q. et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845 e820 (2019).
Zhou, S. L. et al. Tumor-associated neutrophils recruit macrophages and t-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150, 1646–1658 e1617 (2016).
Wan, S. et al. Myeloid cells in hepatocellular carcinoma. Hepatology 62, 1304–1312 (2015).
Yeung, O. W. et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 62, 607–616 (2015).
Li, X. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66, 157–167 (2017).
Hasita, H. et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 101, 1913–1919 (2010).
Zhou, Z. et al. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell. Oncol. 42, 81–92 (2019).
Jung, K. Y. et al. Cancers with higher density of tumor-associated macrophages were associated with poor survival rates. J. Pathol. Transl. Med. 49, 318–324 (2015).
Engblom, C., Pfirschke, C. & Pittet, M. J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016).
Gubin, M. M. et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell 175, 1014–1030 e1019 (2018).
Molgora, M. et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell 182, 886–900 (2020).
Xiong, H. et al. Anti-PD-L1 treatment results in functional remodeling of the macrophage compartment. Cancer Res. 79, 1493–1506 (2019).
Spear, P., Barber, A., Rynda-Apple, A. & Sentman, C. L. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J. Immunol. 188, 6389–6398 (2012).
Wang, L. & Wang, F. S. Clinical immunology and immunotherapy for hepatocellular carcinoma: current progress and challenges. Hepatol. Int. 13, 521–533 (2019).
Li, X. et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 18, 177–177 (2019).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41 (2019).
Aggarwal, B. B., Vijayalekshmi, R. V. & Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin. Cancer Res. 15, 425–430 (2009).
Solinas, G., Germano, G., Mantovani, A. & Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 86, 1065–1073 (2009).
Sica, A., Allavena, P. & Mantovani, A. Cancer related inflammation: the macrophage connection. Cancer Lett. 267, 204–215 (2008).
Szebeni, G. J., Vizler, C., Kitajka, K. & Puskas, L. G. Inflammation and cancer: extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediators Inflamm. 2017, 9294018 (2017).
Debebe, A. et al. Wnt/beta-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene 36, 6020–6029 (2017).
Boulter, L. et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest. 125, 1269–1285 (2015).
Lai, Q. et al. Platelets and hepatocellular cancer: bridging the bench to the clinics. Cancers 11, 1568 (2019).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2, 16018 (2016).
Dhanasekaran, R., Venkatesh, S. K., Torbenson, M. S. & Roberts, L. R. Clinical implications of basic research in hepatocellular carcinoma. J. Hepatol. 64, 736–745 (2016).
Ju, C. & Tacke, F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 13, 316–327 (2016).
Jiang, J. et al. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp. Cell Res. 367, 81–88 (2018).
Yin, Z. et al. Macrophages activating chemokine (C-X-C motif) ligand 8/miR-17 cluster modulate hepatocellular carcinoma cell growth and metastasis. Am. J. Transl. Res. 9, 2403–2411 (2017).
Xiao, P. et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology 7, e1440166 (2018).
Yang, J. et al. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via Treg requires TLR4 signaling. World J. Gastroenterol. 18, 2938–2947 (2012).
Wu, Q. et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology 70, 198–214 (2019).
Lin, Z. et al. Heterogeneous responses in hepatocellular carcinoma: the achilles heel of immune checkpoint inhibitors. Am. J. Cancer Res. 10, 1085–1102 (2020).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Dong, H. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8, 793–800 (2002).
Dong, H. & Chen, L. B7-H1 pathway and its role in the evasion of tumor immunity. J. Mol. Med (Berl.) 81, 281–287 (2003).
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009).
Wu, K. et al. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 69, 8067–8075 (2009).
Lyford-Pike, S. et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 73, 1733–1741 (2013).
Duraiswamy, J., Freeman, G. J. & Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 73, 6900–6912 (2013).
D’Angelo, S. P. et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 46, 357–365 (2015).
Gottlieb, C. E., Mills, A. M., Cross, J. V. & Ring, K. L. Tumor-associated macrophage expression of PD-L1 in implants of high grade serous ovarian carcinoma: a comparison of matched primary and metastatic tumors. Gynecol. Oncol. 144, 607–612 (2017).
Kitano, Y. et al. Clinical significance of PD-L1 expression in both cancer and stroma cells of cholangiocarcinoma patients. Ann. Surg. Oncol. 27, 599–607 (2020).
Wang, X. et al. Bladder cancer cells induce immunosuppression of T cells by supporting PD-L1 expression in tumour macrophages partially through interleukin 10. Cell Biol. Int. 41, 177–186 (2017).
Liao, H. et al. Expression of programmed cell death-ligands in hepatocellular carcinoma: correlation with immune microenvironment and survival outcomes. Front. Oncol. 9, 883 (2019).
Li, J. et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity 48, 773–786 e775 (2018).
Li, H. et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 56, 1342–1351 (2012).
Flecken, T. & Sarobe, P. Tim-3 expression in tumour-associated macrophages: a new player in HCC progression. Gut 64, 1502–1503 (2015).
Gao, J. et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 23, 551–555 (2017).
Ni, L. & Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 276, 52–65 (2017).
Liu, C. Q. et al. Expression patterns of programmed death ligand 1 correlate with different microenvironments and patient prognosis in hepatocellular carcinoma. Br. J. Cancer 119, 80–88 (2018).
Galluzzi, L. et al. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 10, eaat7807 (2018).
Fontugne, J. et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 8, 24644–24651 (2017).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Strauss, L. et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 5, eaay1863 (2020).
Paluskievicz, C. M. et al. T regulatory cells and priming the suppressive tumor microenvironment. Front. Immunol. 10, 2453 (2019).
Gyori, D. et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight. 3, e120631 (2018).
Wang, J. et al. Tumor cells induced-M2 macrophage favors accumulation of Treg in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 10, 8389–8401 (2017).
Ringelhan, M. et al. The immunology of hepatocellular carcinoma. Nat. Immunol. 19, 222–232 (2018).
Ren, L. et al. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 7, 75763–75773 (2016).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004).
Zhou, J. et al. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 125, 1640–1648 (2009).
Liu, J. et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 6, e19495 (2011).
Sun, W. et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 117, 1631–1643 (2017).
Zhou, J. et al. Exosomes released from tumor-associated macrophages transfer mirnas that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol. Res. 6, 1578–1592 (2018).
Ma, Q. et al. Co-expression of LAG3 and TIM3 identifies a potent Treg population that suppresses macrophage functions in colorectal cancer patients. Clin. Exp. Pharm. Physiol. 45, 1002–1009 (2018).
Wang, D. et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 452, 244–253 (2019).
Liu, C. et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived interferon-γ. Immunity 51, 381–397.e386 (2019).
Gül, N. & van Egmond, M. Antibody-dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 75, 5008–5013 (2015).
Arlauckas, S. P. et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 9, eaal3604 (2017).
Lo Russo, G. et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 25, 989–999 (2019).
DiLillo, D. J. & Ravetch, J. V. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol. Res. 3, 704–713 (2015).
Desrichard, A., Snyder, A. & Chan, T. A. Cancer neoantigens and applications for immunotherapy. Clin. Cancer Res. 22, 807–812 (2016).
Feng, K.-c et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 10, 4 (2017).
Anwar, M. Y., Williams, G. R. & Paluri, R. K. CAR T cell therapy in pancreaticobiliary cancers: a focused review of clinical data. J. Gastrointest. Cancer. In Press (2020).
Zhang, R. et al. Adoptive cell transfer therapy for hepatocellular carcinoma. Front. Med. 13, 3–11 (2019).
De Palma, M. et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8, 211–226 (2005).
Mazzieri, R. et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19, 512–526 (2011).
Zang, M. et al. IL-23 production of liver inflammatory macrophages to damaged hepatocytes promotes hepatocellular carcinoma development after chronic hepatitis B virus infection. Biochimica et. Biophysica Acta Mol. Basis Dis. 1864, 3759–3770 (2018).
Wu, Y. et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 57, 1107–1116 (2013).
Rosenberg, S. A., Yang, J. C. & Restifo, N. P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915 (2004).
Heo, J. et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 19, 329–336 (2013).
Lawler, S. E., Speranza, M.-C., Cho, C.-F. & Chiocca, E. A. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 3, 841–849 (2017).
Palmer, D. H. et al. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 49, 124–132 (2009).
Chen, Y. et al. Potential therapeutic value of dendritic cells loaded with NY‑ESO‑1 protein for the immunotherapy of advanced hepatocellular carcinoma. Int. J. Mol. Med. 32, 1366–1372 (2013).
El Ansary, M. et al. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. J. Cancer Res. Clin. Oncol. 139, 39–48 (2013).
Yarchoan, M. et al. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med. 9, 1485–1494 (2020).
Buonaguro, L., Consortium, H. Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol. Immunother. 65, 93–99 (2016).
Khan, A. N. et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 6, 11310–11326 (2015).
Dammeijer, F. et al. Depletion of tumor-associated macrophages with a CSF-1R kinase inhibitor enhances antitumor immunity and survival induced by DC immunotherapy. Cancer Immunol. Res. 5, 535–546 (2017).
Fulci, G. et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 67, 9398–9406 (2007).
Yuan, X. et al. Specific cellular immune response elicited by the necrotic tumor cell-stimulated macrophages. Int. Immunopharmacol. 27, 171–176 (2015).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
van der Heide, D., Weiskirchen, R. & Bansal, R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front. Immunol. 10, 2852 (2019).
Roelofs, A. J., Thompson, K., Gordon, S. & Rogers, M. J. Molecular mechanisms of action of bisphosphonates: current status. Clin. Cancer Res. 12, 6222s–6230s (2006).
Russell, R. G. Bisphosphonates: the first 40 years. Bone 49, 2–19 (2011).
Van Rooijen, N., Kors, N., vd Ende, M. & Dijkstra, C. D. Depletion and repopulation of macrophages in spleen and liver of rat after intravenous treatment with liposome-encapsulated dichloromethylene diphosphonate. Cell Tissue Res. 260, 215–222 (1990).
Zhang, W. et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 16, 3420–3430 (2010).
Van Rooijen, N. & Sanders, A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 174, 83–93 (1994).
Wang, B. et al. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 12, 43 (2011).
Giraudo, E., Inoue, M. & Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 114, 623–633 (2004).
Zhou, D. Y. et al. Zoledronic acid inhibits infiltration of tumor-associated macrophages and angiogenesis following transcatheter arterial chemoembolization in rat hepatocellular carcinoma models. Oncol. Lett. 14, 4078–4084 (2017).
Martin, C. K. et al. Zoledronic acid reduces bone loss and tumor growth in an orthotopic xenograft model of osteolytic oral squamous cell carcinoma. Cancer Res. 70, 8607–8616 (2010).
Lv, J. et al. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced Wnt/β-catenin pathway. Life Sci. 256, 117925 (2020).
Nagarsheth, N., Wicha, M. S. & Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572 (2017).
Li, M. et al. A role for CCL2 in both tumor progression and immunosurveillance. Oncoimmunology 2, e25474 (2013).
Teng, K. Y. et al. Blocking the CCL2-CCR2 axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol. Cancer Ther. 16, 312–322 (2017).
Yao, W. et al. A natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine 22, 58–67 (2017).
Nywening, T. M. et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112–1123 (2018).
Chen, Y. et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology 59, 1435–1447 (2014).
Chen, Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 61, 1591–1602 (2015).
Guo, Y. et al. Hypoxiainducible factors in hepatocellular carcinoma (Review). Oncol. Rep. 8, 46691 (2019).
Meng, Y. M. et al. Monocytes/Macrophages promote vascular CXCR4 expression via the ERK pathway in hepatocellular carcinoma. Oncoimmunology 7, e1408745 (2018).
Zhu, Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338 e326 (2017).
Nywening, T. M. et al. Targeting both tumour-associated CXCR2+neutrophils and CCR2+macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112–1123 (2018).
Bonapace, L. et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133 (2014).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Oldenborg, P. A. et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000).
Medzhitov, R. & Janeway, C. A. Jr Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300 (2002).
MP, C., IL, W. & immunology, M. R. J. C. o. i. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 24, 225–232 (2012).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Chen, J. et al. Macrophages induce CD47 upregulation via IL-6 and correlate with poor survival in hepatocellular carcinoma patients. Oncoimmunology 8, e1652540 (2019).
Willingham, S. B. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 109, 6662–6667 (2012).
Xiao, Z. et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 360, 302–309 (2015).
Vaeteewoottacharn, K. et al. Attenuation of CD47-SIRPalpha signal in cholangiocarcinoma potentiates tumor-associated macrophage-mediated phagocytosis and suppresses intrahepatic metastasis. Transl. Oncol. 12, 217–225 (2019).
Sockolosky, J. T. et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl Acad. Sci. USA 113, E2646–E2654 (2016).
Liu, X. et al. Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep. 24, 2101–2111 (2018).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Otero, K. et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 10, 734–743 (2009).
Lin, H. et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320, 807–811 (2008).
Cannarile, M. A. et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53 (2017).
Ao, J. Y. et al. Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular carcinoma. Mol. Cancer Ther. 16, 1544–1554 (2017).
Neubert, N. J. et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 10, eaan3311 (2018).
Mok, S. et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 74, 153–161 (2014).
Zhu, Y. et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 68, 1653–1666 (2019).
Quail, D. F. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018 (2016).
Kumar, V. et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 32, 654–668 e655 (2017).
Perry, C. J. et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J. Exp. Med. 215, 877–893 (2018).
Hoves, S. et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 215, 859–876 (2018).
Zheng, W. & Pollard, J. W. Inhibiting macrophage PI3Kgamma to enhance immunotherapy. Cell Res. 26, 1267–1268 (2016).
Kaneda, M. M. et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016).
De Henau, O. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447 (2016).
Chen, Y. et al. Development of a Listeria monocytogenes-based vaccine against hepatocellular carcinoma. Oncogene 31, 2140–2152 (2012).
Xu, G. et al. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 39, 1429–1444 (2019).
Hassan, R. et al. Clinical response of live-attenuated, listeria monocytogenes expressing mesothelin (CRS-207) with chemotherapy in patients with malignant pleural mesothelioma. Clin. Cancer Res. 25, 5787–5798 (2019).
Torres Andón, F. & Alonso, M. J. Nanomedicine and cancer immunotherapy-targeting immunosuppressive cells. J. Drug Target. 23, 656–671 (2015).
Turk, M. J., Waters, D. J. & Low, P. S. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 213, 165–172 (2004).
Rodell, C. B. et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2, 578–588 (2018).
Muraoka, D. et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J. Clin. Invest. 129, 1278–1294 (2019).
Andon, F. T. et al. Targeting tumor associated macrophages: the new challenge for nanomedicine. Semin Immunol. 34, 103–113 (2017).
Roy, S. & Trinchieri, G. Microbiota: a key orchestrator of cancer therapy. Nat. Rev. Cancer 17, 271–285 (2017).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (81625003); National Science and Technology Major Project (2017ZX10203205); National Science and Technology Key Program (81930016); Key R&D Project of Zhejiang Province (2019C03050); and the National Natural Science Foundation of China (81801824). X.N.X. collected the related papers and drafted the manuscript; J.G.W. revised the manuscript and drafted the figures; D.L. provided advice to the manuscript; X.X. participated in the design of the review and drafted the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, X., Wang, J., Lu, D. et al. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Sig Transduct Target Ther 6, 75 (2021). https://doi.org/10.1038/s41392-021-00484-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00484-9
This article is cited by
-
In vivo therapy of osteosarcoma using anion transporters-based supramolecular drugs
Journal of Nanobiotechnology (2024)
-
A novel PDPN antagonist peptide CY12-RP2 inhibits melanoma growth via Wnt/β-catenin and modulates the immune cells
Journal of Experimental & Clinical Cancer Research (2024)
-
Translational research on drug development and biomarker discovery for hepatocellular carcinoma
Journal of Biomedical Science (2024)
-
CAR macrophages on a fast track to solid tumor therapy
Nature Immunology (2024)
-
The role of CXCL2-mediated crosstalk between tumor cells and macrophages in Fusobacterium nucleatum-promoted oral squamous cell carcinoma progression
Cell Death & Disease (2024)
