
ABSTRACT
RNA interference (RNAi) is an ancient biological mechanism used to defend against external invasion. It theoretically can silence any disease-related genes in a sequence-specific manner, making small interfering RNA (siRNA) a promising therapeutic modality. After a two-decade journey from its discovery, two approvals of siRNA therapeutics, ONPATTRO® (patisiran) and GIVLAARI™ (givosiran), have been achieved by Alnylam Pharmaceuticals. Reviewing the long-term pharmaceutical history of human beings, siRNA therapy currently has set up an extraordinary milestone, as it has already changed and will continue to change the treatment and management of human diseases. It can be administered quarterly, even twice-yearly, to achieve therapeutic effects, which is not the case for small molecules and antibodies. The drug development process was extremely hard, aiming to surmount complex obstacles, such as how to efficiently and safely deliver siRNAs to desired tissues and cells and how to enhance the performance of siRNAs with respect to their activity, stability, specificity and potential off-target effects. In this review, the evolution of siRNA chemical modifications and their biomedical performance are comprehensively reviewed. All clinically explored and commercialized siRNA delivery platforms, including the GalNAc (N-acetylgalactosamine)–siRNA conjugate, and their fundamental design principles are thoroughly discussed. The latest progress in siRNA therapeutic development is also summarized. This review provides a comprehensive view and roadmap for general readers working in the field.
Similar content being viewed by others

Introduction
Gene therapy is a promising therapeutic platform because it targets disease-causing genes in a sequence-specific manner, which enables more precise and personalized treatment of diverse life-threatening diseases.1 By introducing a certain nucleic acid modality to the desired tissue of the patient, gene expression can be downregulated, augmented or corrected. Small interfering RNA (siRNA), microRNA (miRNA) and inhibitory antisense oligonucleotides (ASOs) are representative molecules used to trigger gene inhibition, whereas plasmid DNA, messenger RNA (mRNA), small activating RNA (saRNA), splicing-modulatory ASOs and CRISPR (clustered regularly interspaced short palindromic repeats)/Cas (CRISPR-associated protein) systems are usually employed to increase or correct target gene expression.2,3,4 Currently, many therapeutic programs have been explored to treat certain diseases.
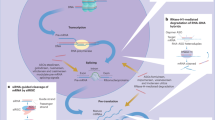
RNA interference (RNAi) is a natural defense mechanism for the invasion of exogenous genes.5,6 RNAi modalities, e.g., siRNA and miRNA, can knockdown the expression of target genes in a sequence-specific way (Fig. 1) by mediating targeted mRNA degradation (for siRNA and miRNA) or mRNA translation repression (for miRNA). As a result of the slight differences between siRNA and miRNA, siRNA can typically trigger more efficient and specific gene silencing than miRNA, whereas one miRNA may compromise the expression of several different target genes simultaneously. Hence, siRNA and miRNA have different roles in pharmaceutical practice.

Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
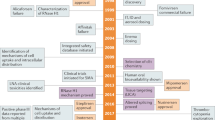
Since the establishment of the RNAi concept in 1998, siRNA therapeutics have experienced many ups and downs. In 2001, Tuschl et al.7 successfully silenced the expression of a specific gene by introducing chemically synthesized siRNA into mammalian cells, leading to the emergence of a developmental upsurge. Although siRNA therapy once suffered due to the obstacles of its stability, specificity and delivery, advances in chemical modification and delivery brought the field to a robust and rapidly developing area of research again in recent years. After a 20-year journey, the United States Food and Drug Administration (FDA) and the European Commission (EC) approved ONPATTRO® (patisiran, ALN-TTR02) as the first commercial RNAi-based therapeutic for the treatment of hereditary amyloidogenic transthyretin (hATTR) amyloidosis with polyneuropathy in adults in 2018.2,8 Recently, the FDA-approved GIVLAARI™ (givosiran, ALN-AS1) for the treatment of adults with acute hepatic porphyria (AHP).9,10,11,12
siRNA has innate advantages over small molecular therapeutics and monoclonal antibody drugs because siRNA executes its function by complete Watson–Crick base pairing with mRNA, whereas small molecule and monoclonal antibody drugs need to recognize the complicated spatial conformation of certain proteins. As a result, there are many diseases that are not treatable by small molecule and monoclonal antibody drugs since a target molecule with high activity, affinity and specificity cannot be identified. In contrast, theoretically, any gene of interest can be targeted by siRNA since only the right nucleotide sequence along the targeting mRNA needs to be selected. This advantage confers the siRNA modality with a shorter research and development span and a wider therapeutic area than small molecule or antibody drugs, especially for those genes that are unfeasible for development with such strategies.
Although siRNA holds promising prospects in drug development, several intracellular and extracellular barriers limit its extensive clinical application. Naked and unmodified siRNA possesses some disadvantages, such as (1) unsatisfactory stability and poor pharmacokinetic behavior and (2) the possible induction of off-target effects. The phosphodiester bond of siRNA is vulnerable to RNases and phosphatases. Once it is systematically administered into circulation, endonucleases or exonucleases throughout the body will quickly degrade siRNA into fragments, thus preventing the accumulation of intact therapeutic siRNA in the intended tissue. In theory, siRNA only functions when its antisense strand is completely base-paired to the target mRNA. However, a few mismatches are tolerated by the RNA-induced silencing complex (RISC), which may lead to undesired silencing of those genes with a few nucleotide mismatches. In addition, the RISC-loaded sense strand of siRNA may also knockdown the expression of other irrelevant genes. Moreover, unformulated and unmodified siRNA may lead to the activation of Toll-like receptor 3 (TLR3) and adversely affect the blood and lymphatic systems.13 These discoveries have raised many concerns about the undesirable effects and pharmaceutical issues of siRNAs.
To maximize the treatment potency and reduce or avoid the side effects of siRNA, researchers have made great efforts to investigate various chemical modification geometries and to develop many different delivery systems. As a result, a series of modification patterns were proposed and evaluated preclinically and clinically with respect to their effects on activity, stability, specificity and biosafety. Delivery materials derived from lipids, lipid-like materials (lipidoids), polymers, peptides, exosomes, inorganic nanoparticles, etc., have been designed and investigated.2,8,14,15,16,17,18,19,20,21,22 As a result, several modification patterns and delivery platforms have been employed in clinical studies. Here, the detailed evolution and advances in the modification and delivery technologies of siRNA are comprehensively summarized and discussed. This review provides an overview and a handbook for reviewing siRNA therapeutic development.
siRNA modification
During the early stages of developing siRNA therapeutics, many agents were designed based on completely unmodified or slightly modified siRNA to arrive at appropriate tissue and then silence the target gene. These molecules can mediate gene silencing in vivo, especially in tissues that receive local drug administration, e.g., eyes. However, limited efficacy and potential off-target effects may be observed with these modalities. For example, Kleinman and colleagues23 observed that bevasiranib and AGN211745 triggered significant activation of toll-like receptor 3 (TLR3) and its adapter molecule TRIF, inducing the secretion of interleukin-12 and interferon-γ. Bevasiranib and AGN211745 are siRNA therapeutics developed for the treatment of age-related macular degeneration.24 The VEGFA-targeting bevasiranib25 and VEGFR1-targeting AGN21174526 are unmodified and slightly modified siRNAs, respectively. Moreover, Kleinman and colleagues further demonstrated that siRNA classes with sequences of 21 nucleotides or longer, regardless of which genes they target, can suppress CNV in mice compared to bevasiranib and AGN211745. These findings eventually led to the termination of the clinical investigation of bevasiranib in 2009.
Chemically modified siRNAs, such as siRNAs with substitution of the 2′-OH with a 2′-O-methyl (2′-OMe)27 or 2′-methoxyethyl (2′-MOE)28 group or the substitution of certain nucleotides with locked nucleic acid (LNA),29 unlocked nucleic acid (UNA)30 or glycol nucleic acid (GNA)31 (Fig. 2), can efficiently suppress immunostimulatory siRNA-driven innate immune activation, enhance activity and specificity, and reduce off-target-induced toxicity. To enhance the potency and reduce the potential toxicity of siRNA, numerous chemical modification geometries have been established and tested.32

Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
According to the natural structure of nucleotides, chemical modifications can be placed at the phosphate backbone, the ribose moiety or the base. Typically, these modifications are simultaneously introduced in siRNA. For example, the combination of 2′-OMe and phosphorothioate (PS) modifications facilitates the systemic administration of cholesterol-conjugated siRNA and achieves efficient gene silencing in vivo.33 In addition, the combination of 2′-OMe and 2′-deoxy-2′-fluoro (2′-F) has been used for ONPATTRO®34,35 (Figs. 2, 3). For QPI-1007, a naked and unformulated siRNA therapeutic,2 2′-OMe was alternatively incorporated into the antisense strand, and an inverted deoxyabasic moiety and an l-DNA cytidine nucleotide were used in the sense strand36 (Figs. 2,3). Moreover, modifications with phosphorothioate (PS), 2′-OMe, 2′-F and 2′-deoxy were employed for inclisiran (ALN-PCSsc), an siRNA therapeutic for the treatment of hypercholesterolemia37,38 (Figs. 2, 3). Overall, the precise modification of siRNA can increase its efficacy, specificity, and stability and reduce its toxicity and immunogenicity. Detailed information will be discussed in the following sections.

Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI™ and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
Phosphonate modification
Phosphorothioate (PS) linkage has previously been used in antisense oligonucleotide (ASO) modification.39 This modification was achieved by leveraging a sulfur atom to replace one nonbridging oxygen of a phosphodiester (Fig. 2). PS linkage endows modified oligonucleotides with resistance to nucleases,40 and these molecules more readily combine with plasma proteins,41 especially albumin42 than their unmodified counterparts, which may result in a longer circulation time. Although some data have shown that PS modification reduces the binding affinity between the oligonucleotide and its target sequence32 to some extent and aggravates chemistry-related toxicities with a high PS content,43,44 PS modification is still very important and necessary for both siRNA and ASO because, compared with unmodified oligonucleotides, PS-modified oligonucleotides are more hydrophobic and stable molecules with higher affinity to certain proteins.44 Appropriate protein binding may prolong the half-life of oligonucleotides in circulation, and the half-life of the elimination phases may be as long as a few days.41 Protein binding is typically beneficial for the cell entry of oligonucleotides; however, too much protein binding showed a positive correlation with in vivo toxicity.45,46 Hence, the position and number of PS linkages are pivotal for their application. For siRNA modification, Alnylam introduced two PS linkages at the first two nucleotides at the 5′-end of the sense strand and two PS linkages at the first two nucleotides at both the 5′-end and the 3′-end of the antisense strand of siRNA (Fig. 3). Moreover, compared to the parent siRNA without modifications, the PS linkage-containing siRNA has a barely changed in vivo biodistribution profile, as it predominantly accumulates in clearance organs, e.g., liver, kidneys and intestines, followed by glandular tissues, bone marrow, adipocytes and lymph nodes.47,48,49,50
Notably, there are two configurations for PS linkages, namely, Rp and Sp isomers (Fig. 2). Verdine and colleagues (team from Wave Life Sciences)51 demonstrated that the stereochemistry of PS significantly influences the pharmacologic performance of ASOs. PS linkages with the Sp configuration are more stable than those with the Rp configuration. More importantly, they unveiled that stereopure ASOs with a triplet stereochemical code, 3′-SpSpRp, make the target RNA more vulnerable to RNase H1 in vitro and achieve a more durable inhibition profile in vivo than stereorandom ASOs. However, Østergaard et al. (team from Ionis Pharmaceuticals)52 disclosed that controlling PS chirality markedly affects the interactions between the ASOs and RNase H1; however, this modulation failed to enhance the overall therapeutic effects. The sequence and design of ASOs are the primary drivers that determine the performances of gapmer ASOs. In addition, recently disclosed patents filed by Alnylam reported that the chirality of PS linkages may markedly influence siRNA performance.53 The terminal chiral PS linkages at the 3′- and 5′-ends of the antisense strand of siRNA prefer Sp and Rp configurations, respectively. The chiral PS number can be 1, 2 or 3 at the 3′-end and 1 or 2 at the 5′-end of the siRNA antisense strand. In addition, the chiral PS linkages at the 5′-end of the siRNA sense strand can adopt either an Sp or an Rp configuration and are positioned at the first internucleotide linkage at the 5′-end of the sense strand. Overall, the chirally modified siRNA can comprise four, five or more terminal, chirally modified PS linkages.
In addition, other residues have been identified and successfully used to replace the phosphodiester group in oligonucleotides and change the properties of intact strands, including phosphorodithioate (PS2),54 methylphosphonate (MP), methoxypropylphosphonate (MOP) and peptide nucleic acid (PNA) (Fig. 2). PS2 modification can increase the affinity between RISC and siRNA. The site-specific incorporation of MP and MOP modifications has been used to reduce ASO protein binding since the PS backbone is negatively charged, whereas alkylphosphonate linkages are charge neutral. As a result, the MOP linkage incorporated at position 2 or 3 from the 5′-end of the DNA gap significantly mitigated the hepatotoxicity of ASOs.46 In addition, both PNA55,56,57 (Fig. 2) and phosphotriesters58 are very meaningful modifications to make siRNA or ASO molecules more druggable, although these modifications are not as predominant as PS. PNA is also typically used to modify detection probes for capturing and detecting certain nucleic acid targets.59
In addition, phosphonate modification with various analogs at the 5′-end of siRNA or single-stranded siRNA (ss-siRNA) is a newly developed but important decoration strategy to enhance siRNA activity. The 5′-phosphate of exogenously introduced siRNA is required for RISC loading. A 5′-phosphate can be modified via either phosphorylation in cells mediated by Clp1 (cleavage and polyadenylation factor I subunit 1) or chemical synthesis in the laboratory.60 However, the natural 5′-phosphate can also be removed by dephosphorylation in cells. Together with the demand to enhance siRNA stability, researchers have identified a series of analogs that have similar conformation and steroidal electronic properties to natural phosphates but are resistant to dephosphorylases. As a result, 5′-(E)-vinyl phosphonate (5′-(E)-VP), 5′-methylphosphonate, (S)-5′-C-methyl with phosphate, 5′-phosphorothioate, etc., were designed and evaluated in vitro and in vivo61 (Fig. 2). Among these strategies, 5′-(E)-VP decoration, which substitutes bridge oxygen and carbon with E-vinyl phosphonate moieties at the 5′-end, is the most potent and metabolically stable mechanism. The replaced hydrophobic substitutions enhance the stability of intact oligonucleotides and are favorable for RISC loading. Ionis Pharmaceuticals first developed and own the intellectual property rights to the 5′-(E)-VP structure. This technology was first applied to ss-siRNA and achieved ideal pharmacodynamic and pharmacokinetic profiles. Modifying ss-siRNA with 5′-(E)-VP and other specific modification structures enables potent repression of targeted gene expression by ss-siRNA in vivo.61,62 Inspired by this finding, the 5′-(E)-VP modification was further applied to double-stranded siRNA. Data have revealed that this modification improves siRNA accumulation and residence time in tissue and enhances siRNA potency in vivo by elevating Argonaute-2 binding.63,64,65
Ribose modification
Ribose modifications at the 2′ position have been widely used to protect siRNA from attacking ribonucleases, which require the 2′-OH group for the hydrolysis of RNA. Notably, 2′-O-methyl (2′-OMe) (Fig. 2) is a naturally occurring ribosugar and the most frequently used modification in drug development so far66 because this molecule can enhance stability67 by blocking the nucleophilic 2′-OH group. In addition, 2′-OMe also shows increased affinity for its target mRNA68 and reduced immunogenicity in the body. Based on 2′-OMe, a series of analogs have been identified, among which 2′-O-methoxyethyl (2′-O-MOE) (Fig. 2) is one of the most useful and popular analogs of 2′-OMe. Moreover, 2′-O-MOE shows higher binding affinity for RNA than 2′-OMe with a 0.9–1.7 °C change in ΔTm per modified nucleotide, which further increases the capability of the modified siRNA to resist nuclease attack.32 Another widely used 2′-OH substitution is 2′-deoxy-2′-fluoro (2′-F)69 (Fig. 2). Highly electronegative fluorine makes the modified siRNA readily adopt a C3′-endo conformation, providing considerable benefits in binding affinity, with a ΔTm of ~2.5 °C per modified nucleotide.32 Interestingly, 2′-arabino-fluoro (2′-Ara-F), an alternative fluoro substitution form, can also be used to modify nucleic acid therapeutics, e.g., antisense oligonucleotides45,70 and siRNA.71 Both 2′-O-benzyl and 2′-O-methyl-4-pyridine (2′-O-CH2Py(4)) were well tolerated when they were incorporated on the guide strand of siRNA in vivo72 (Fig. 2). siRNA with modifications of four 2′-O-benzyl or six 2′-O-CH2Py(4) showed comparable activity with the unmodified siRNA in vivo, and increased activity was even achieved when these modifications were placed at the 8 and 15 positions on the siRNA guide strand.
In addition, 2′-C, 4′-C, and even the whole sugar ring can also undergo modifications, resulting in molecules including UNAs, LNAs, GNAs, (S)-cEt-BNAs, tricyclo-DNA (tcDNA) and phosphorodiamidate morpholino oligomers (PMOs) (Fig. 2). UNA, with higher flexibility and thermal destabilization than the unmodified product due to unconnected 2′ and 3′ carbons, can block the entry of passenger strands and promote the RISC loading of the guide strand by introducing chemical asymmetry into duplex siRNAs. Similarly, glycol nucleic acid (GNA),31 another thermally destabilizing nucleotide, can be used to erase off-target effect-induced hepatotoxicity by including it in the seed region of the siRNA guide strand. LNA is a bicyclic structure that contains a bridge between the 2′ oxygen and the 4′ carbon. It “locks” the ribose into its preferred C3′-endo conformation and significantly increases the affinity of base pairing.40 Data have shown that the incorporation of LNAs increases the DNA melting temperature up to 8 °C per LNA.73 Given the successes of LNAs, many more bicyclic and even tricyclic analogs, including ethyl-bridged nucleic acids (ENAs),74 constrained ethyl (cEt) nucleic acids75 and tricyclo-DNA,76 have been engineered and incorporated into RNA or DNA strands in succession. Moreover, PMOs77 do not look like classic nucleotides, as the ribose subunit has been substituted with a morpholine subunit. PMOs are uncharged at physiological pH and are not substrates of RNase H. Therefore, they are mainly used to block RNA splicing or translation.78
Base modification
Base replacement is of great benefit to nucleic acid-based drug development. For instance, the substitution of pseudouridine,79 2-thiouridine,80 N6-methyladenosine,80 5-methylcytidine81 (Fig. 2) or other base analogs of uridine and cytidine residues can reduce innate immune recognition while making ASOs more resistant to nucleases. However, the artificial base substitution of ASOs, similar to siRNA, is basically at the stage of research and development. Pharmaceutical corporations still hold a prudent attitude toward these molecules. Instead, these companies prefer to use naturally occurring base structures, e.g., 5mC and 6 mA, to modify certain base(s), probably because of concerns about the safety of the metabolized unnatural residues that potentially might be incorporated into the genome.
N-ethylpiperidine triazole-modified adenosine analogs with Hoogsteen or Watson–Crick (WC) facial localization have also been used to modify siRNA,82,83,84,85 which can disrupt nucleotide/TLR8 interactions and therefore reduce the immunogenicity of siRNA. N-ethylpiperidine 7-EAA triazole (7-EAA, 7-ethynyl-8-aza-7-deazaadenosine) can pair well with uridine and form an A-form helix structure. This molecule will be recognized as adenosine by Avian myeloblastosis virus reverse transcriptase in RNA strands.83
In addition, 6′-phenylpyrrolocytosine (PhpC) is a cytosine mimic showing excellent base pairing fidelity, thermal stability and high fluorescence.86 PhpC-containing siRNA shows gene-silencing activity comparable to that of the parent molecule, and its fluorescent properties make it useful for fluorescence-based detection or monitoring or for exploring the cellular uptake and trafficking of siRNA in cells.
The internal uridine substitution of 2,4-difluorotolylribonucleoside (rF) is well tolerated by siRNA activity.87 The base pairing between rF and adenosine (A) is relatively unstable compared to the uridine–adenosine pair because the fluorine atom is more hydrophobic than uridine and cannot serve as a hydrogen bond acceptor.
Zhang and colleagues88 reported that a 5-nitroindole modification at position 15 of the siRNA passenger strand greatly reduced the activity of the passenger strand, whereas the effectiveness of the siRNA guide strand was barely affected. This modification provided a practical strategy to reduce the passenger strand-mediated off-target effects.
siRNAs containing 5-fluoro-2′-deoxyuridine (FdU) moieties can suppress targeted gene expression. The effects depend on the locations of the FdU substitution. In addition, these modified siRNAs quickly release FdU after entering the cell, thereby further inducing a variety of DNA-damage repair and apoptosis pathways and ultimately triggering cell death. These findings provide a strategy for siRNA-based cancer therapy.89
Moreover, many other less common base analogs have also been used in siRNA modifications. Their application helps researchers better understand the mechanism of gene silencing and helps develop new methods to mitigate off-target effects caused by harmful protein binding or undesired targeting of mRNA.90
Modification patterns used in clinical studies or for commercial siRNA
Early siRNA therapeutic programs typically employ partial or slight modifications by using 2′-OMe, 2′-F or PS,91,92 e.g., alternative 2′-OMe modifications were placed in the antisense strand of QPI-1007, whereas just one L-DNA was used in the sense strand. After making great efforts to understand the effects of various modifications on siRNA activity, specificity and toxicity, researchers have realized that heavy modification of siRNA, even full-length embellishment, probably do not affect siRNA activity. Furthermore, these judicious modification strategies can significantly enhance siRNA stability or biocompatibility. As a result, several different, interrelated modification patterns have been proposed and validated preclinically and clinically.
Standard template chemistry (STC) (Fig. 3) is a universal modification pattern established by Alnylam Pharmaceuticals. When the lengths of sense and antisense siRNA are 21 and 23 nt, respectively (referred to as “S21nt” and “AS23nt”, respectively, in the following description), 2 PS linkages are placed at the 3′ terminus of the antisense strand, three consecutive 2′-F moieties are incorporated at positions 9, 10 and 11 of the sense strand from the 5′-end, and consecutive 2′-OMe modifications are placed at positions 11, 12 and 13 of the antisense strand from the 5′-end. Moreover, alternative 2′-OMe and 2′-F moieties are employed at other positions in both strands. It is worth mentioning that 2′-OMe and 2′-F are complementarily used for all positions of both strands of siRNA (Fig. 3). It is considered that placing three consecutive 2′-F moieties, instead of larger groups, at the middle of the sense strand aims to facilitate the cleavage and removal of the sense strand in RISC, as release of the sense strand is the prerequisite of RISC activation. PS linkages and OMe positioned at the terminals are used to enhance the capability of siRNA to resist degrading enzyme attack.
The STC design (e.g., as used in revusiran) has been proven to remarkably enhance siRNA stability and affinity without compromising intrinsic RNAi activity.93 ALN-AT3, another STC-modified RNAi therapeutic, also showed potent and durable gene silencing after single subcutaneous (s.c.) administration.94 However, the clinical investigation of revusiran was discontinued due to unbalanced death between revusiran- and placebo-treated groups. Although the detailed investigation report by Alnylam illustrated that the deaths of revusiran-treated patients were not correlated with the test drug, there are still strong concerns regarding STC-modified siRNA therapeutics. Hence, the next generation of modification patterns, called enhanced stabilization chemistry (ESC) (Fig. 3), was proposed. This design includes four more PS linkages at the 5′-end of the antisense strand and the 3′-end of the sense strand. Another important change was the reduction in 2′-F substitutions, probably because heavy use of 2′-F may magnify toxicity.95 These changes markedly enhanced siRNA potency and duration compared with the changes inherent to the STC design.9,96,97 This advancement allowed us to achieve the desired pharmacodynamic effect at a significantly lower dose, with a markedly reduced dosing frequency.98 Cemdisiran (ALN-CC5) achieved potent gene silencing as long as more than 1 year after receipt of a single dose of the testing therapeutic in a phase 1/2 clinical study (Fig. 4f), representing an unprecedented milestone in pharmaceutical history. Moreover, the newly approved GIVLAARI™ is an siRNA therapeutic based on the ESC design (Fig. 3).99

Preclinical and clinical performance of siRNAs with comprehensive modification chemistries. a Sense and antisense strand optimization by using transthyretin (Ttr)-targeting siRNAs and the combinatorial designs explored across 10 siRNAs. The effect relative to the parent is described as the model-adjusted mean difference in the activity of the design variant (DV) compared to the parent. b–d The liver exposure, RISC loading, and silencing activities of the parent, DV18, and fully 2′-OMe conjugates. Data are shown as the mean ± SD. e Gene-silencing duration of antithrombin-targeted siRNAs modified with the designs of patent and DV22 evaluated in Cynomolgus monkeys (n = 3 per group). Asterisks indicate that a significant difference was observed between the parent and DV22. Error is shown as the SD. f C5 knockdown profile after applying a single dose of Cemdisiran that employs ESC modification in a phase 1/2 clinical study. Error is represented as the SEM. a–e Copyright of Elsevier Inc., 2018. f Copyright of Alnylam Pharmaceuticals, 2016
In addition, Alnylam comprehensively optimized the modification design variants (DVs) of the ESC strategy across multiple siRNAs with an iterative screening approach to further enhance stability without compromising intrinsic RNAi activity. As a result, advanced ESC designs, e.g., DV18 and DV22, were proposed (Figs. 3 and 4a–e).96 Compared to the former scheme, the advanced strategy maintains six PS linkages at three strand terminals; however, this modification scheme significantly reduces the proportion of 2′-F moieties. Only 10 and 8 2′-F substitutions are used in two strands for the advanced ESC designs of DV18 and DV22, respectively. For DV18, the 2′-F modifications are positioned at sites 7, 9, 10 and 11 in the sense strand and sites 2, 6, 8, 9, 14 and 16 in the antisense strand (all from the 5′-ends of the strands). Compared with DV18, DV22 has the 2′-F modifications at 8 and 9 in the antisense strand further replaced with 2′-OMe (Figs. 3 and 4a). Both the DV18 and DV22 designs can achieve significantly higher liver exposure and RISC loading and more potent and durable gene silencing than the parent design in preclinical species, including nonhuman primates (Fig. 4b–e). The underlying mechanisms were not fully unveiled in Alnylam’s literature. However, Zheng et al.100 demonstrated that modification of the 14th position of the siRNA guide strand could eliminate its gene-silencing activity by reducing RISC loading and target degradation, and the larger the modification group used was, the higher the reduction efficiency. Song et al.28 further proved that decorations at positions 9 and 10 (for siRNA with a 19 nt/19 nt structure) markedly reduced the activity of the unmodified strand of siRNA without disturbing the potency of the modified strand. In light of this, the specificity and activity of siRNAs could be improved by introducing 2′-MOE at the cleavage site of the siRNA. These observations are in line with the rationales of advanced ESC designs.
Furthermore, investigators from Alnylam have disclosed that the hepatotoxicity of GalNAc (N-acetylgalactosamine)-siRNA conjugates is attributed to off-target gene silencing mediated by miRNA-like recognition between siRNA and a mistargeted RNA.31 Researchers have demonstrated that disorganizing the nucleotides of the seed region without changing the 2′-OMe, 2′-F or PS content or placing a GNA at position 7 of the siRNA antisense strand can markedly alleviate off-target effects and mitigate hepatotoxicity because these modifications affect the binding of siRNA with undesired target mRNA in a seed region-specific manner. The employment of GNA in the siRNA seed region is the main technical characteristic for the ESC+ design compared to advanced ESC designs (Fig. 3). Several investigational siRNA therapeutics of Alnylam, e.g., ALN-AAT02, ALN-HBV02, and ALN-AGT, utilize the ESC+ design as the modification pattern.
In addition to Alnylam, Arrowhead Pharmaceuticals (Arrowhead) proposed a series of new modification designs (Fig. 3) by combining the modification patterns (e.g., inverted bases) of siRNAs with 2′-OMe/2′-F decorations similar to the aforementioned STC and ESC modification patterns. Typically, the modification strategies proposed by Arrowhead are characterized by placing inverted deoxythymine (idT) at the strand terminus and/or termini, including UNA and X (without nucleoside base), flanking the UAU or UAUAU motif(s), conjugating the siRNA with cholesterol or other hydrophobic substrates, etc. These designs are currently being investigated in clinical trials.101 Dicerna Pharmaceuticals (Dicerna)102 employed their proprietary dicer-substrate siRNA (DsiRNA) technology to develop RNAi therapeutics. In addition, these researchers also established a series of DsiRNA modification chemistries. For instance, three consecutive 2′-F moieties may be placed at sites 9, 10, and 11 in the sense strand of siRNA from the 5′-end, and the other sites may use alternative 2′-OMe and 2′-F moieties in the flank sequence. One example of a constant flank sequence is ‘GCAGCCGAAAGGUGC’, which contains inner complementary pairing motifs of ‘GCAGCC’ and ‘GGCUGC’. Consecutive 2′-OMe moieties may be used to modify these motifs, and consecutive RNA or DNA nucleosides without additional 2′ modifications may be employed for the bubble motif of GAAA. Moreover, DNA nucleosides can also be applied at sites 2, 12, 16, 18, 20 and 21 in the antisense strand from the 5′-end. PS linkages may be incorporated into the antisense strand at specific sites, as well as within the constant flank sequence (Fig. 3). The ligands may be placed at the bubble sequence. Silence Therapeutics103 (Fig. 3) and other RNAi-based biotech companies, such as Arbutus Biopharma, OliX Pharmaceuticals and Suzhou Ribo Life Science, have all been devoted to developing state-of-the-art siRNA modification chemistries to establish fruitful drug development pipelines.2
siRNA delivery and drug development
Barriers to siRNA delivery
RNAi modalities (siRNAs and miRNAs) need to employ proteins (such as Argonaute and Tar RNA binding protein) in the cytoplasm. A series of barriers need to be circumvented for systemically administered exogenous siRNA before it can achieve gene silencing,104,105,106 such as nuclease degradation and short-lived circulation, immune recognition in blood circulation, accumulation in desired tissue, effective transmembrane trafficking, and escape from endosomes and lysosomes to the cytoplasm. By incorporating chemical modifications, the challenges of stability against serum nucleases and avoiding immune recognition have been substantially reduced. However, other problems remain to be solved.
In blood circulation, unspecific binding and glomerular filtration both hamper the accumulation of siRNA in desired tissues. The neutral surface charge of siRNA-loaded nanoformulations is beneficial for avoiding unfavorable protein binding in circulation.107 However, protein decoration or protein corona formation is required for nanoformulation and attachment to specific tissues or cells for some delivery systems.108,109 For instance, ionizable lipid nanoparticles (iLNPs) primarily accumulate in the liver by interacting with serum lipoproteins, e.g., apolipoprotein E3 (ApoE3), which leads to specific recognition by the receptor, e.g., low-density lipoprotein receptor (LDLR).108,109 In contrast, ligand–siRNA conjugates can transport siRNA to desired tissues and cells by specific recognition and interactions between the ligand, e.g., a carbohydrate, peptide, antibody, aptamer, small molecule, etc., and the surface receptor. This active targeting strategy not only reduces siRNA accumulation in unintended tissues, thus decreasing or avoiding undesired side effects and toxicity, but also achieves potent gene silencing at a low dosage.104
siRNA is ~7–8 nm in length and 2–3 nm in diameter. This molecule is too large to cross cell membranes but small enough to be freely cleared by glomeruli,110 as molecules with a size smaller than 8 nm111 are easily filtered into the urine. Hence, once siRNAs leave the bloodstream, they will accumulate in the bladder and be excreted out from the body quickly, within a few minutes to half an hour,48,49,50 which prevents them from accumulating in targeted tissues or cells. Encapsulating siRNA into vesicles or conjugating it to certain ligands can effectively avoid renal clearance and, more importantly, can deliver siRNA to the desired tissues or cells. Lipid nanoparticles (LNPs),106 dynamic polyconjugates (DPC™), and GalNAc–siRNA conjugates have all achieved efficient siRNA delivery to hepatocytes (Figs. 5, 6).112

Schemes of representative clinically studied siRNA formulations and their pharmacodynamic performance. a–g Schemes of a lipid nanoparticles, b DPC™ or EX-1™, c TRiM™, d GalNAc–siRNA conjugates, e LODER™, f iExososme and g GalXC™. h Efficacy of ONPATTRO® (a liposome formulation) in healthy volunteers, in which siRNA targeting PCSK9 is formulated in lipid nanoparticles.34 Copyright Massachusetts Medical Society, 2013. i Levels of urinary aminolevulinic acid (ALA) and porphobilinogen (PBG) after once-monthly and once quarterly doses of GIVLAARI™ in acute hepatic porphyria (AHP) patients. Copyright Alnylam Pharmaceuticals, 2019. j Reduction of plasma PCSK9 after a single increasing dose of inclisiran.38 Copyright Massachusetts Medical Society, 2017. k Serum HBsAg reduction in hepatitis B patients who received the treatment of a single dose of ARC-520 (1–4 mg/kg) (formulated with DPC2.0) on a background of daily oral NUCs.264 PBO, patients on NUC therapy receiving placebo injection. NUCs nucleos(t)ide viral replication inhibitors. The error is shown as the SEM. Copyright American Association for the Advancement of Science, 2017. l Antitumor effect of siG12D-LODER™ combined with chemotherapy in locally advanced inoperable pancreatic cancer in a patient. A computed tomography (CT) scan was performed before and after (nine months later) the administration of siG12D-LODER™. The tumor measured 35.42 and 26.16 mm in the longest diameter, respectively. m PANC-1 tumor growth in animals receiving iExosomes or other control formulations.238N = 3 mice per group. Copyright Macmillan Publishers Limited, part of Springer Nature, 2017. n The 24-h Uox (urinary oxalate) values of healthy volunteers and patients treated with a single dose of Nedosiran (DCR-PHXC, a GalXC™ therapeutic) (3 and 6 mg/kg).265 Copyright Dicerna Pharmaceuticals, 2020. o Reduction of serum AAT in volunteers receiving a single dose of ARO-AAT (a TRiM™ therapeutic, 35–300 mg).266 The error bars show the SEM. Copyright Arrowhead Pharmaceuticals, 2019

siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL™ and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL™ RNAi/oligonucleotide nanoparticle delivery
The relatively high molecular weight (~13–16 kD) and net negative charge prevent artificial siRNA from crossing the cell membrane. Hence, we attempted to determine whether any cell can internalize siRNA without a carrier, leading to the conclusion that naked siRNA can only be taken up by a few cell types, e.g., retinal ganglion cells (RGCs) and neurons. In addition, researchers have also been devoted to identifying various carriers to achieve efficient transmembrane delivery. As a result, cationic cell-penetrating peptides (CPPs) become a choice at the early stage. CPPs, typically tailed with arginine-rich sequences, can form bidentate bonds by the interaction between the guanidinium groups and the negative phosphates, sulfates and carboxylates on the cell surface.113 This interaction causes membrane pore formation, leading to the cellular uptake of siRNA. As another strategy, the negative charge of siRNA can be neutralized by positively charged lipids or polymers, conferring siRNA the ability to more readily bind to the membrane and become easily internalized via adsorptive pinocytosis.
Finally, siRNAs must effectively escape from endosomes and lysosomes to the cytoplasm, where antisense strands of siRNAs need to be loaded into RISCs. Many delivery systems employ a pH-sensitive unit to respond to pH changes in the endosome and lysosome,114,115 where they will absorb H+, presenting a positive charge on the surface. Then, the osmotic pressure will increase in the endosome or lysosome, resulting in the internal flow of Cl− and H2O. Finally, these changes may cause membrane disruption and siRNA release to the cytoplasm. This so-called ‘proton sponge effect’ or ‘colloid osmotic pressure effect’ results in membrane destabilization116,117,118,119 or membrane swelling,120,121 respectively. However, the underlying mechanism of endosomal release remains to be further illuminated. Only 1–2% of internalized LNP-loaded siRNAs were released into the cytoplasm, and this only occurred within a limited time frame after internalization.122,123 Hence, further understanding the escape mechanism and how to enhance the escape efficiency is of great importance for siRNA drug development. Recently, Wang and colleagues124 developed novel endoplasmic reticulum (ER) membrane-modified hybrid nanoplexes (EhCv/siRNA NPs). Compared with unmodified nanoplexes, they showed much higher RNAi activity in vitro and in vivo. The functional proteins on the ER membrane have an important role in intracellular trafficking of siRNA, helping siRNA reach the cytoplasm through the endosome–Golgi–ER pathway instead of the endosome–lysosome pathway, thereby avoiding the lysosomal degradation of siRNA. In addition, electroporation enables siRNA to directly cross the cell membrane, which also constitutes an effective approach to circumvent the endosomal escape issue.125,126,127,128,129,130,131
To date, two RNAi therapeutics, ONPATTRO® and GIVLAARI™, have been approved for commercial application, and two siRNAs, lumasiran (ALN-GO1) and inclisiran, have been submitted for new drug application (NDA) to the FDA. Seven siRNAs are undergoing phase 3 clinical studies, and more candidates are in the early developmental stage. Various delivery systems, e.g., LNPs, DPC™, TRiM™, GalNAc–siRNA conjugates, LODER™ polymers, exosomes and polypeptide nanoparticles (PNPs), have been explored (Figs. 5, 6). Based on these systems, plentiful drug pipelines have been established. We discuss this information in the following sections.
Naked siRNA-based therapeutics
Naked siRNA can be defined as a system that contains no delivery system that is associated with siRNA either covalently or noncovalently.40 Because no protective effect is offered by the delivery vehicle, siRNA should be modified carefully to be resistant to enzyme degradation and extend its circulation time in the bloodstream. As siRNA is naturally filtered to the kidney, it can be used to silence kidney-expressed genes and treat renal diseases.132 Alternatively, a feasible strategy for naked siRNA drug development is the local injection of this molecule into specific organs that are relatively closed off and contain few nucleases, e.g., the eye.
QPI-1002 (I5NP)132,133 and QPI-100736,134 were developed by Quark Pharmaceuticals (Table 1). QPI-1002 is a 19-base pair, 2′-O-methylated, blunt and naked siRNA135 targeting p53 for treating acute kidney injury (phase 2) and delayed graft function (phase 3). The sequence of the sense strand is 5′-GAAGAAAATTTCCGCAAAA-3′.135 Because of their small sizes, most siRNAs accumulate in the kidney after being intravenously administered, achieving concentrations 40 times greater than those in other organs, followed by rapid entry into proximal tubule cells. An in vitro study determined that QPI-1002 elicited near-complete p53 mRNA elimination at a concentration of ~1 nM, with an IC50 of ~0.23 nM. This molecule also effectively inhibited p53 protein expression, even at a transfection concentration of 0.5 nM. Furthermore, bilateral renal-clamp studies were conducted to identify the effect of siRNA on the preservation of kidney function. Compared with PBS-treated animals, siRNA-treated animals showed ischemia with decreased serum creatinine levels from 3.7 mg/dL to 1.9 mg/dL. Adverse effects (AEs) were found only when the dose was higher than 1000 mg/kg in nonhuman primates and higher than 800 mg/kg in rats.133 A phase 1 clinical study (NCT00554359) of QPI-1002 revealed an ideal safety profile.36,132
QPI-1007 is a caspase-2-targeted siRNA without formulation that was developed to treat acute primary angle closure glaucoma (phase 2) and nonarteritic anterior ischemic optic neuropathy (NAION, phase 3). This molecule is administered via intravitreal injection. It is modified by 2′-OMe in the antisense strand, an l-DNA cytidine nucleotide is located before the last nucleotide at the 3′-end, and the sense strand has an inverted deoxyabasic residue at the 5′-end36 (Fig. 3).
Preclinical investigations have validated the potent gene silencing of QPI-1007 in cells and have demonstrated its curative effects on optic nerve-damaged animal models. Data136 have shown that QPI-1007 triggered over 80% gene suppression in HeLa cells and exhibited an IC50 of ~0.8 nM against human caspase-2 mRNA. In animal models, eyes treated with siRNA showed a dose-dependent increase in RGC survival from 5 to 20 μg. In particular, in animals dosed with 20 and 35 μg QPI-1007, RGC densities in the injured eye recovered to close to healthy levels (~98%). Furthermore, a phase 1/2a clinical trial (NCT01064505) reported that most common AEs, such as conjunctival hemorrhage, conjunctival edema, eye irritation, and eye pain, were typical of intravitreal injection, and no serious adverse effects (SAEs) were observed. By comparing the best-corrected visual acuity (BCVA) following a single dose of QPI-1007 with natural history historical controls from the Ischemic Optic Neuropathy Decompression Trial (IONDT, 1998), which is the most comprehensive survey of the disease and serves as the gold standard in the field, it was concluded that QPI-1007 significantly protected the optic nerve as the proportions of subjects who lost 3 lines of visual acuity were markedly decreased. The proportions were 0%, 0% and 3.6% at months 3, 6 and 12, respectively, following a single dose of QPI-1007. In contrast, the proportions for natural history controls were 9%, 15% and 16% at months 3, 6 and 12, respectively. Currently, an international multi-centered phase 2b/3 clinical trial is being conducted in the United States, China, Israel and other countries and regions.
ALN-RSV01 (asvasiran sodium), a naked siRNA that targets the respiratory syncytial virus (RSV) nucleocapsid (N) gene and inhibits viral replication, was explored for the potential treatment or prevention of RSV infection. The sequences of ALN-RSV01 are as follows: sense strand (5′–3′): GGCUCUUAGCAAAGUCAAGdTdT; and antisense strand (5′–3′): CUUGACUUUGCUAAGAGCCdTdT.137 ALN-RSV01 was administered via inhalation with an investigational electronic nebulizer. Although the clinical study of ALN-RSV01 has been terminated, clinical data indicate that it is well tolerated in vivo138 and may have beneficial effects on long-term allograft function in lung transplant patients infected with RSV (NCT00658086).139,140 Another clinical result reported in 2016 showed that ALN-RSV01 may help prevent bronchiolitis obliterans syndrome (BOS) after lung syncytial virus infection in lung transplant recipients.141
Lipid and lipidoid-based nanoparticles
Representative lipid materials
Lipid or lipidoid nanoparticles (LNPs) (Figs. 5a and 6) were originally developed as vehicles for DNA-based drugs. Subsequently, LNPs have been increasingly used for siRNA delivery because these vesicles can protect entrapped siRNA from nuclease attack and renal clearance and transport siRNA to targeted tissues and cells.142,143,144 Canonical siRNA-LNPs comprise similar components, e.g., cationic or ionizable lipids, DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), and cholesterol and polyethylene glycol (PEG) lipids.145 siRNA-LNPs predominantly accumulate in the liver, spleen and kidney after being intravenously injected.
According to the different charge properties of siRNA-binding lipids under neutral conditions, LNPs may be divided into several classes: ionizable LNPs, cationic LNPs and neutral LNPs. Ionizable LNPs are nearly uncharged during circulation but become protonated in a low pH environment, e.g., in the endosomes and lysosomes. These molecules may interact with apolipoprotein E3 (ApoE3), which transports lipids to hepatocytes. Cationic LNPs exhibit a constitutive positive charge in blood circulation and in endosomes or lysosomes. Cationic LNPs also primarily accumulate in hepatocytes; however, this is independent of the ApoE3 interaction. Electrostatic-induced nonspecific binding with plasma protein and relatively higher immunogenicity may cause cationic LNPs to be less efficacious146 and more toxic in vivo than ionizable LNPs;147 as a consequence, most pharmaceutical firms and institutes have made efforts to develop novel ionizable lipids to achieve efficient hepatocyte-targeted delivery of siRNA with minimal side effects. For example, research teams from Arbutus Biopharma, Alnylam Pharmaceuticals and the Massachusetts Institute of Technology continue to establish lipid-based delivery systems. Three generations of lipid delivery systems have been developed by Arbutus and Alnylam (Fig. 6). They employed DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane),148 DLin-MC3-DMA ((6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate)107 and L319 (di((Z)-non-2-en-1-yl) 9-((4-(dimethylamino) butanoyl) oxy) heptadecanedioate)149 as the key lipids and exhibited median effective doses (ED50s) of 1, 0.005 and <0.01 mg/kg, respectively, when loaded with anti-factor VII siRNA and injected via the tail vein. DLin-DMA was used to develop TKM-080301,150 ALN-VSP151 and ALN-TTR01,34 whereas DLin-MC3-DMA was employed in ALN-TTR0234 and ALN-PCS152 development. L319 was derived from DLin-MC3-DMA by incorporating a biocleavable ester linkage within hydrophobic alkyl chains.149 It is readily degraded in vivo, and the metabolite of L319 is a potential substrate for the β-oxidation pathway of fatty acids. L319 is rapidly eliminated from intracellular compartments (plasma, liver and spleen) and is excreted in vivo, which suggests high tolerance of L319-LNPs throughout the body.
Anderson and colleagues153 synthesized and screened thousands of lipidoids through a combinatorial chemistry strategy. Consequently, three generations of lipidoid materials were selected from these libraries: 98N12-5(I)-based,154 C12-200-based155 and cKK-E12-based153 LNPs (Fig. 6). In particular, the ED50 of LNPs containing cKK-E12 is as low as 0.002 mg/kg, which is the lowest ED50 in the liver for all reported LNPs currently.153 In addition, many firms, including Dicerna Pharmaceuticals (Dicerna),156 Silence Therapeutics,157 Sirna Therapeutics,158 Nitto Denko Corporation,159 Life Technologies160 and Suzhou Ribo Life Science,49,142 have explored proprietary delivery technologies for preclinical and clinical investigations.
Tumor treatment is another application of LNPs. The discontinuous and fenestrated endothelium in rapidly growing malignant tumors and the poor lymphatic system allow LNPs and internalized siRNAs greater accumulation and slower clearance in solid tumors, referred to as the enhanced permeation and retention (EPR) effect.161 The EPR effect has become a mainstay of antitumor nanodrug delivery. However, increasing evidence has shown that LNPs are still primarily enriched in hepatocytes; hence, it is difficult to achieve complete remission merely based on the EPR effect for cancer treatment. An advanced design adds active tumor-targeting agents on the surfaces of LNPs that can target (i) the tumor cell surface, (ii) the tumor extracellular matrix and (iii) the endothelial cell surface receptors of tumor vessels by biological or chemical means.162 Tumor-targeting moieties can be selected from antibodies or proteins (e.g., transferrin163,164), peptides (e.g., RGD165 or octreotide166), aptamers or small molecules (e.g., hyaluronan167,168,169 or folate170). These ligands, to some extent, facilitate siRNA-loaded LNP accumulation in tumor tissues.
After more than 10 years of technological evolution, some theories have been established to guide the rational design of novel artificial lipids. Manoharan et al.107 showed that the pKa of lipids or lipidoids is of great importance for delivering siRNA to hepatocytes in vivo. In addition, these researchers elucidated the influences of the PEG-lipid content of siRNA-LNPs on delivery, concluding that PEG-C14 showed the highest delivery efficiency compared with PEG-C16 and PEG-C18, and the incorporation of 1.5 mol% PEG-lipid exhibited ideal potency, as higher proportions of PEG-lipid may decrease the potency.171 Moreover, Anderson and colleagues172 identified four essential structural and pKa criteria for the design of degradable lipidoids: (i) tertiary amine, (ii) O13 tail, (iii) more than 2 hydrophobic tails and (iv) pKa ≥ 5.5. This finding is of importance for designing and developing liver-targeted delivery systems based on lipid-like materials.
In addition, Arcturus Therapeutics,173 Dicerna Pharmaceuticals,174 Silence Therapeutics,175,176 Sirna Therapeutics (acquired by Alnylam from Merck & Co., Inc. in 2014)177,178 and Suzhou Ribo Life Science142 also made great efforts to develop lipid-based siRNA delivery systems, some of which have been advanced to the clinical stage.
Lipid-employed siRNA therapeutics
ONPATTRO®78 is a chemically modified anti-transthyretin (TTR) siRNA formulated in liposomes (Figs. 5a, h and 7). The lipid components of ONPATTRO® include DLin-MC3-DMA (Fig. 6), DSPC, cholesterol and PEG-DMG ((R)-2,3-bis(octadecyloxy)propyl-1-(methoxy polyethylene glycol 2000) carbamate) at a molar ratio of 50/10/38.5/1.5.107 The active pharmaceutical ingredient (API, siRNA) of ONPATTRO® is modified with eleven 2′-OMe (nine in the sense strand and two in the antisense strand) and four 2′-deoxy thymidine (two in the sense strand and two in the antisense strand, all at the 3′-end) moieties. Before ONPATTRO®, Alnylam advanced ALN-TTR01 to a phase 1 clinical study (NCT01148953). The API of ALN-TTR01 is the same as that of ONPATTRO®, whereas the delivery system for ALN-TTR01 comprises Dlin-DMA,148,179 cholesterol, DSPC and PEG-DMG. A phase 1 study of ONPATTRO® (NCT01617967) showed a good dose-dependent pattern for TTR protein reduction.180 Overall, this drug was well tolerated by subjects, although pretreatment with antihistamines, nonsteroidal antihistamines or glucocorticoids was needed to reduce the incidence of infusion-related reactions. Phase 2 and 3 studies reached both the primary and second endpoints of the studies, according to the outcomes regarding the modified Neuropathy Impairment Score +7 (mNIS+7) and the Norfolk Quality of Life Diabetic Neuropathy (QoL-DN) score. Based on these achievements, the FDA and the EC approved the commercial use of ONPATTRO® in August 2018.

Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
In addition, TKM-080301 (TKM-PLK1) is another DLin-DMA-based LNP formulation.181 TKM-080301 was developed by Tekmira Pharmaceuticals and contains anti-PLK1 (polo-like kinase 1) siRNA for the treatment of solid cancer. Preclinical studies have shown ideal antineoplastic activity in xenograft tumor models, and the inhibition of PLK1 expression was maintained at 7–10 days after receipt of a single dose of TKM-080301.182 No measurable immune activation or myelosuppressive effects were manifested, and limited toxicity was observed in the liver and spleen. In the phase 1/2 clinical test (NCT02191878), the patients received 28 days of treatment at escalating doses of TKM-080301 from 0.15 to 0.9 mg/kg through a 30-min i.v. infusion once a week. Dose-limiting toxicities (DLTs) and adverse grade 3 reactions were observed in two patients at a dose of 0.9 mg/kg/week, and the maximum tolerated dose (MTD) was 0.75 mg/kg/week.36,150 In another clinical trial (NCT01262235), TKM-080301 was dosed at 0.6 or 0.75 mg/kg/week, and the treatment lasted for 18 cycles (4 weeks per cycle). As a result, tumor reduction was achieved, but some serious AEs (SAEs) were also observed.150
Moreover, several other RNAi therapeutics also employ liposomes as delivery materials, e.g., ALN-VSP,151 ALN-PCS,152 MRX34,183 Atu027,175,184 ARB-1467,185 ARB-1740,186,187 etc. The clinical studies of all these therapeutics are currently discontinued. ARB-1467 and ARB-1740 are generation 1 and generation 2 anti-HBV modalities, respectively, developed by Arbutus Pharma. ARB-1740 is a clinically investigated and liposome-formulated RNAi agent that contains three siRNAs targeting different regions of the HBV genome.186,187 However, the study of ARB-1740 has been suspended because Arbutus transferred their liver-targeted delivery system from liposomes to GalNAc–siRNA conjugates. Accordingly, another anti-HBV siRNA therapeutic, AB-729, is undergoing a phase 1a/1b study,188 and positive preliminary clinical results have been disclosed recently.
DPC™ and TRiM™ delivery platforms
Characteristics of DPC™ and TRiM™ delivery platforms
DPC™ or EX-1™112,189,190,191,192 is a delivery system established by Arrowhead (Fig. 5b). The first-generation DPC (DPC 1.0) technology is characterized by a polymer backbone (polymer composed of butyl and amino vinyl ether, PBAVE) reversibly attached to siRNA, a shielding agent (PEG), and the targeting ligand by a bifunctional maleamate linkage.112 In detail, PEG and the targeting ligand are alternatively linked to polymers with carboxylated dimethyl maleic acid (CDM) bonds (Fig. 6). These bonds will be cleaved within the acidic environment in endosomes and lysosomes, which will expose the positively charged amino groups of the polymer backbone and trigger the influx of H+ and Cl−, resulting in the elevation of endosomal osmotic pressure and the inflow of H2O. The import of H+, Cl− and H2O causes the destabilization and swelling of the endolysosomal membrane, which are benefits of the ‘proton sponge effect’193 and ‘increased colloidal osmotic pressure’.120 In addition, siRNAs are covalently conjugated to the polymer backbone by disulfide linkage, which will also be degraded in the environment with glutathione, releasing siRNA into the cytoplasm. Increased endolysosomal escape was accomplished by selective activation112 in endosomes and lysosomes, which ensures an effective interaction with other membranes before endocytosis is prevented.
For the second-generation DPC (DPC 2.0) platform, siRNA is modified with cholesterol instead of being conjugated with the polypeptide (melittin-like peptide, a kind of membrane-lytic peptide, MLP) (Figs. 5b and 6). It is worth mentioning that efficacious siRNA delivery could be achieved following the intravenous coinjection of GalNAc-modified MLP and cholesterol-conjugated siRNA (chol-siRNA). In addition, chol-siRNA can also be used in combination with GalNAc and PEG-masked PBAVE,189 a formulation that constitutes generation 1.5 of DPC technology. Regrettably, although biomarkers were reduced by more than 99% in HBV patients receiving a single dose of ARC-520, an siRNA therapeutic employing DPC2.0 technology, Arrowhead stopped the clinical development of this drug because one monkey died during the long-term toxicity evaluation. In addition, clinical studies of two other DPC2.0-based siRNA therapies were discontinued, and Arrowhead transferred their delivery platform from DPC to TRiM™ (Targeted RNAi Molecule) (Fig. 5c). The TRiM™ platform comprises a highly potent RNA trigger (siRNA), targeting ligands, linkers, and structures that enhance pharmacokinetic performance if needed. The targeting moiety is covalently conjugated to the siRNA directly. The targeting ligands can be selected from GalNAc, RGD motifs (ligands of integrin αvβ3 and αvβ5) and the αvβ6 ligand, which are designed to transport siRNA to hepatocytes, cancer cells and lung epithelial cells, respectively. These molecules can trigger efficacious gene silencing in the targeted tissue and potentially may reduce the risk of intracellular metabolite accumulation, thus reducing the in vivo toxicity.
DPC or TRiM employed therapeutics
ARC-520194,195 is a DPC2.0-based siRNA therapeutic containing two siRNAs. siHBV-74 and siHBV-77 elicited the greatest level of gene knockdown among ~140 candidates. Coadministration of these two siRNAs showed further gene knockdown and could be used to target 99.64% of all genotypes of HBV transcripts. Thus, ARC-520 containing these two siRNAs was proposed to combat HBV infection. In preclinical studies,190 ARC-520 significantly inhibited the expression of the 2.4/2.1 kb preS1/S transcripts and the 3.5 kb transcripts and reduced HBsAg levels in a dose-dependent manner, which was well maintained for ~1 month in HBV-transgenic mice.
A phase 1 study (NCT01872065) of ARC-520 showed that it was well tolerated at a dose of up to 2 mg/kg after a single dosing of ARC-520 intravenously. There was no difference between the placebo and ARC-520 groups with respect to adverse event frequency and severity. Only mild or moderate adverse events were observed. In another phase 1 study (NCT02535416), healthy adult volunteers were administered ARC-520 from 4 to 6 mg/kg at varying infusion rates, and no treatment-emergent adverse events were observed. A phase II study of ARC-520194 (Heparc-2001) demonstrated that HBsAg was markedly decreased in treatment-naïve and HBeAg-positive patients (Fig. 5k); however, if the patients were HBeAg-negative or previously had received treatment with nucleos(t)ide viral replication inhibitors (NUCs), the therapeutic effect was significantly reduced. More importantly, this study observed that HBsAg could be expressed from integrated HBV DNA in the host genome, in addition to expression from cccDNA. These findings uncovered an underrecognized source of HBsAg, leading to changes in the trial design and expectations for the end point of new treatments for chronic HBV.
ARC-521, another siRNA therapeutic based on the DPC2.0 delivery system, was designed as a complement to ARC-520, targeting HBV mRNA transcripts from both cccDNA and integrated DNA. This drug is expected to be most suitable for patients who have lower levels of viral cccDNA. In a clinical trial (NCT02797522) of ARC-521, healthy volunteers received single escalating doses of 0.6, 1.0, 2.0, 4.0, 5.0 and 6.0 mg/kg ARC-521,196 and chronic HBV patients received up to three doses (Q28 days) of ARC-521 at multiple dose levels of 2.0, 4.0 and 6.0 mg/kg. All participants were pretreated with oral antihistamine and acetaminophen 2 h prior to ARC-521 dosing. No deaths or dropouts due to AEs or SAEs were reported in healthy volunteers. No infusion reactions or laboratory abnormalities were reported as adverse effects, and no clinically significant alanine transaminase (ALT) elevations were reported in healthy volunteers. One SAE of elevated transaminases occurred in a patient at one month after a single ARC-521 dose, which may have been due to viral flare secondary to fluctuating HBV DNA and NUC nonadherence. In addition, the expression of HBsAg and HBV DNA was efficiently reduced in response to ARC-521. This effect can be elevated by combination with NUCs.
ARC-AAT was developed to treat alpha-1 antitrypsin deficiency that severely damages the liver and lungs. Two clinical trials (NCT02363946 and NCT02900183) were performed to evaluate the safety and treatment effect on intrahepatic and circulating AAT levels after ARC-AAT injection. Unfortunately, deaths were reported at the highest dose in a nonhuman primate toxicology study of ARC-520. As a result, Arrowhead stopped all three programs based on the DPC2.0 or EX-1 delivery system. Instead, Arrowhead launched programs based on the TRiM™ platform, including ARO-AAT and ARO-HBV (Table 1, Fig. 7). According to the data disclosed by Arrowhead, with the exception of being well tolerated at all doses tested, three monthly doses of 300 mg ARO-AAT197 led to the suppression of AAT in serum to a level below quantitation in all subjects. In addition, this suppression was constant for more than 14 weeks, suggesting that it is feasible for patients to receive dosing quarterly or even less frequently. ARO-HBV employs two siRNAs that reduce all measurable viral products and interfere with the upstream reverse transcription process in HBV patients. Data from a phase 1/2 study (NCT03365947)198 showed that a single dose or multiple doses of up to 400 mg of ARO-HBV was well tolerated in the patients and volunteers. Furthermore, patients receiving 3-month doses of ARO-HBV and entecavir or tenofovir showed up to −3.8 log10 HBsAg reduction. More comprehensive studies are ongoing.
GalNAc–siRNA conjugates
Characteristics of GalNAc–siRNA conjugate platforms
Although patisiran, a liposome-based RNAi formulation, reached commercial application, Alnylam still stopped all other liposome-based programs, as the GalNAc–siRNA conjugate delivery platform is superior to that of liposomes regarding both potency and safety for the liver-targeted delivery of siRNA. GalNAc is a ligand of the asialoglycoprotein receptor (ASGPR), which is an endocytic receptor that is highly and specifically expressed on the membrane surface of hepatocytes (~500,000 copies/cell) but barely expressed by other cells.199 ASGPR- and clathrin-mediated endocytosis can efficiently transport galactose-derived ligands from the cell surface to the cytoplasm. In this process, ASGPR is presented on the cell surface within 15 min. Early structure–activity studies revealed that tetravalent and trivalent ligands exhibit orders of magnitude higher affinity than monovalent and bivalent ASGPR when the spacing is ~20 Å. Hence, trivalent or tetravalent GalNAc moieties are covalently conjugated to siRNA with a proprietary linker structure (Figs. 5d and 6). Targeting ligands are placed at the 3′ and 5′ ends of the siRNA sense strand for Alnylam and Arrowhead formulations, respectively.31,200 In contrast, based on the specific DsiRNA structure, four GalNAc residues are positioned at the four unpaired tetraloop-hairpin nucleotides of Dicer on the sense strand (Figs. 3 and 5g).201
Evidence200 showed that subcutaneously administered GalNAc–siRNAs achieved better performances than intravenously administered GalNAc–siRNAs. It is hypothesized that subcutaneously administered siRNAs need to cross connective tissue and capillary and lymphatic endothelial cells to accumulate in circulation. This process changes the pharmacokinetic profile of GalNAc–siRNA. Higher siRNA explosion in the liver was achieved with subcutaneously injected than intravenously injected GalNAc–siRNA because intravenously injected siRNA was excreted quickly from circulation to the kidneys.49 Meanwhile, the cellular entry of GalNAc–siRNA is mediated by ASGPR via a naturally existing mechanism, which constitutes a safer way for siRNA delivery than liposomes, which may trigger detrimental lipid interactions between the cell membrane and the liposome. As a result, GalNAc–siRNA conjugates show excellent pharmaceutical properties, such as allowing subcutaneous administration, with high tissue-targeting specificity, a higher therapeutic index and minimal or limited AEs. More importantly, these conjugates show an extremely long duration of action when combined with enhanced stabilization modification chemistry, e.g., ESC or ESC-plus (Fig. 3), supporting quarterly and even twice-yearly dosing of these RNAi therapeutics.
In addition to Alnylam, other biotechnological companies have also established GalNAc–oligonucleotide conjugate platforms, e.g., Arrowhead,202 Dicerna,203 Arbutus,188,204 Silence,205 Ionis,206 Wave Life Sciences,207 Regulus208 and Suzhou Ribo Life Science209 (Fig. 6). Moreover, Mascitti and colleagues210 from Pfizer developed a variety of GalNAc analogs as alternative ligands for ASGPR. Compared to the GalNAc-based conjugate, conjugating a compact and stable bicyclic bridged ketal to small-molecule cargo with an oxanorbornadiene linkage could achieve increased ASGPR-mediated cellular uptake and prolonged retention. A proprietary stereopure anti-ApoC3 ASO developed by Wave Life Sciences also achieved efficient liver-targeted delivery of ASO and showed robust gene repression.211
GalXC™94 (Fig. 5g) is a proprietary RNAi delivery platform engineered by Dicerna Pharmaceuticals that facilitates the establishment of plentiful therapeutic pipelines. Dicer-substrate siRNA (DsiRNA) is composed of a sense strand with tetraloop hairpin, a constant sequence, and a 21–23 nt antisense strand complementing the target mRNA. Multivalent GalNAc moieties are linked to DsiRNA at the region of the tetraloop hairpin. The prominent feature of this platform is the introduction of a constant complementary structure in the siRNA sense strand that makes it difficult to be loaded by RISC. Thus, sense strand-mediated off-target effects might be efficiently erased. In addition, a region of DsiRNA was engineered into the GalXC™ molecules to provide additional pharmaceutical functionality. For example, this modification could enhance silencing activity, inducing higher affinity for serum albumin than no modification and improving the circulating half-life of GalXC™ molecules. There are no fixed modification strategies for GalXC™. Several representative modification patterns are shown in Fig. 3, in which a few DNA nucleotides are introduced into the DsiRNA. Another typical characteristic is to use naturally occurring or FDA-approved modification motifs to reduce toxic side effects. As a result, high efficacy (ED50 < 0.3 mg/kg) and good tolerability (no clinical adverse events recorded when administered at 100 mg/kg weekly for 6 weeks) can be achieved.
GalNAc–siRNA conjugate-based therapeutics
Many GalNAc-conjugated oligonucleotide therapeutics are undergoing clinical studies (Table 1, Fig. 7). Recently, only four months after receiving new drug application (NDA) from Alnylam, the FDA-approved GIVLAARI™ for the treatment of AHP,99 a genetic disorder resulting in the buildup of toxic porphyrin molecules that are formed during the production of heme (which helps bind oxygen in the blood). GIVLAARI™, an aminolevulinic acid synthase 1 (ALAS1)-targeting RNAi therapeutic, is the first GalNAc-conjugate RNAi therapeutic worldwide. As mentioned above, ESC modification confers siRNA more stability, which allows quarterly dosing at significantly lower dosages. According to the phase 1 clinical data disclosed by Alnylam,9 givosiran dosed at 2.5 mg/kg once-monthly significantly reduced the levels of ALAS1 mRNA, delta aminolevulinic acid (ALA) and porphobilinogen (PBG), with higher than 80% inhibition efficiency, which was accomplished with a 79% decrease in the mean annualized attack rate compared to that of the placebo. In the ENVISION phase 3 study, patients receiving givosiran showed a 74% mean reduction in the annualized rate of composite porphyria attacks compared to those receiving placebo, with an acceptable overall safety and tolerability profile (Fig. 5i).10
In addition, inclisiran and lumasiran have been submitted to the FDA for new drug commercialization evaluation, and fitusiran (ALN-AT3) and vutrisiran (ALN-TTRsc02) are undergoing phase 3 clinical trials (Table 1, Fig. 7). Fitusiran was developed for the treatment of hemophilia and rare bleeding disorders (RBDs) and functions by inhibiting the expression of antithrombin (AT). This drug employs the ESC-GalNAc delivery platform developed by Alnylam. A phase 2 study (NCT02554773)94,212,213 showed that 50 or 80 mg of fitusiran (lower than 2.5 mg/kg) per month resulted in an antithrombin reduction of 78–88% from baseline. The phase 2 trial of inclisiran (NCT02963311),38,214,215,216 an siRNA targeting proprotein convertase subtilisin kexin type 9 (PCSK9), also exhibited potent and durable gene silencing and a reduction in low-density lipoprotein cholesterol (LDL-C) (Fig. 5j). Lumasiran is an RNAi modality for treating primary hyperoxaluria type 1 (PH1) that functions by inhibiting the expression of glycolate oxidase. Phase 1/2 studies of lumasiran revealed a mean maximal reduction in urinary oxalate (Uox) of 75% relative to baseline across all cohorts (1 mg/kg monthly, 3 mg/kg monthly, and 3 mg/kg quarterly; N = 20). All lumasiran-treated patients experienced a mean maximal decrease of 77% in the ratio of urinary oxalate to creatinine, an additional measure of oxalate reduction that addresses the variability inherent in 24-h urine collections. An acceptable safety and tolerability profile was also observed in phase 1/2 studies. Moreover, a phase 1 clinical investigation of vutrisiran (NCT02797847)217 showed that the reductions in serum TTR were well maintained for as long as 1 year after a single dose of vutrisiran for all six dose groups from 5 to 300 mg.
Cemdisiran, ALN-AAT02, ARO-HBV and DCR-PHXC are undergoing phase 2 trials (Table 1, Fig. 7). Cemdisiran (Fig. 4f) and ALN-AAT02 are two programs developed by Alnylam for the treatment of complement-mediated diseases and AAT deficiency-associated liver disease (alpha-1 liver disease) and function by targeting the C5 component of the complement pathway and alpha-1 antitrypsin (AAT), respectively. Cemdisiran and ALN-AAT02 utilize the ESC-GalNAc delivery platform and the ESC-Plus (ESC+)-GalNAc delivery platform, respectively, developed by Alnylam (Figs. 3, 5d and 6). ARO-HBV and nedosiran (DCR-PHXC) were developed by Arrowhead and Dicerna for curing hepatitis B and primary hyperoxaluria (PH), respectively. DCR-PHXC can inhibit the expression of lactate dehydrogenase A (LDHA), thus blocking the excess production of oxalate, leading to a cure for all three genetic forms of PH (type 1, 2 and 3), a rare, inherited, liver metabolic disorder. The current treatment approaches for PH are either poorly effective or highly invasive.218 In an undergoing study of DCR-PHXC-101 in healthy volunteers and patients219 (phase 1, NCT03392896), the patients were divided into three cohorts that received different doses of DCR-PHXC via subcutaneous administration. The 24-h Uox values for three of the five patients who received 1.5 mg/kg of DCR-PHXC reached near-normalization levels at one or more postdose time points, and their mean maximal 24-h Uox reduction was 51% (range: 28–72%). When the dose was doubled, the 24-h Uox values for 4 out of 5 subjects reached normalization at one or more postdose time points, and their mean maximal 24-h Uox reduction was 71% (range: 62–80%). For the third cohort receiving 6 mg/kg DCR-PHXC, the mean maximal 24-h Uox reduction reached 76% (range: 58–100%). The 24-h Uox values were not disclosed because the values for 2 patients did not return to within 80% of the lowest baseline 24-h Uox measurement (Fig. 5n).
AMG 890 (formerly ARO-LPA), ARO-ANG3, ARO-APOC3, ARO-AAT (Fig. 5o), DCR-HBVS, ALN-AGT01 and AB-729 were evaluated in phase 1 clinical trials (Table 1, Fig. 7). These therapeutics were developed to treat cardiovascular disease, dyslipidemia, hypertriglyceridemia, alpha-1 liver disease, hepatitis B, hypertension and hepatitis B and function by targeting apolipoprotein A (Apo(a)), angiopoietin-like 3 (ANGPTL3), apolipoprotein C3 (ApoC3), AAT, the HBV gene, angiotensinogen (AGT) and the HBV gene, respectively. Moreover, many other candidates have been explored preclinically for liver-related diseases, e.g., ALN-HBV02 (VIR-2218), SLN360, SLN226, RBD1016, RB-HLP002 and DCR-A1AT.2
LODER™ and siG12D-LODER
LOcal Drug EluteR (LODER™)220,221 is a biodegradable polymeric matrix derived from poly(lactic-co-glycolic) acid (PLGA) with a small cylindrical rod ~1 nm in diameter and ~5 nm in length (Figs. 5e and 6). According to Silenseed, LODER™ is the first siRNA delivery system that allows RNAi drugs to be directly delivered into solid tumors. LODER™ enables the slow and stable release of locally delivered drugs. The siRNA in LODER™ was protected from degradation when incubated in phosphate buffer solution or in mouse liver tissue homogenate for over 2 months.
siG12D-LODER is a G12D-mutated KRAS-targeting siRNA therapeutic entrapped in LODER™. Preclinical studies performed in rats and mice showed robust tumor treatment effects at a dose of 0.32 mg/kg. According to preclinical evaluation data collected from 192 rats, siG12D-LODER revealed ideal safety profiles in all animals receiving repeated subcutaneous doses of the studied formulation at days 1, 14 and 28.222 In phase 1/2a studies (NCT01188785 and NCT01676259),223 siG12D-LODER in combination with chemotherapy (gemcitabine or FOLFIRINOX) was used as a first-line treatment for inoperable locally advanced pancreatic cancer (LAPC) patients. Three escalating dose cohorts (0.025, 0.75 and 3.0 mg) were employed in this study. The main adverse events were grade 1 or 2 in severity (89%), and serious adverse events (SAEs) occurred in five patients. Tumor progression was not observed for 12 patients according to the CT scan data (Fig. 5l). Stable disease was achieved in 10 of 12 patients, and partial response was observed in 2 patients. The median overall survival of siG12D-LODER-treated patients was 15.12 months, and the 18-month survival reached 38.5% of patients.
Based on LODER™ delivery technology, Silenseed developed si-PT-LODER224 aiming to cure prostate cancer and si-GBMT-LODER as a treatment for brain cancer, which are in the preclinical and research stages, respectively.
Other platforms
Polymers17,114,115,225,226,227,228,229,230,231,232,233 (e.g., PEI,231 PTMS,115 GDDC4114 and PAsp(DET)232) constitute additional important siRNA delivery platforms. Similar to LNPs, polymers can achieve high cellular uptake and endosomal escape because they are positively charged. Besides LODER™, the cyclodextrin-based RONDEL™ is another polymer platform that was developed by Calando Pharmaceuticals and was once investigated in a phase I clinical study234,235 (Fig. 6). Repeatability, manufacture-controllability, safety and biodegradability are the key issues that researchers need to pay attention to when studying polymers. Therefore, designing simple, biodegradable, targeted or naturally occurring polymers is a potential development direction for polymer-based platforms.
In addition, some other systems are also important for siRNA therapeutic development, which include (i) the EDV™ nanocell platform developed by EnGeneIC;236 (ii) the HKP delivery system employed by Sirnaomics, Inc.237 (Fig. 6); (iii) extracellular vesicles or exosomes derived from various cell types (Fig. 5f, m);16,238,239 (iv) peptides;240,241,242,243 (v) dendrimers;244,245,246 (vi) RNA nanoparticles (e.g., three way-junction, 3WJ);247,248,249,250,251,252,253,254 and (vii) inorganic nanoparticles229,255 (Fig. 6). These platforms also exhibited satisfactory delivery efficiency in the preclinical and/or clinical stages.
To date, hepatocyte-targeted delivery of siRNA has been well resolved by leveraging GalNAc–siRNA conjugates. In the following days, researchers will develop extrahepatic siRNA delivery strategies. Recently, central nervous system (CNS) and eye-targeted delivery platforms have been established by conjugating a lipophilic moiety to the siRNA, according to the descriptions in Alnylam’s patent (WO2019217459A1).2,256 Arrowhead successfully delivered siRNA to renal cancer cells and lung epithelial cells probably by employing RGD motifs or small molecules as the targeting moieties.2 In addition, various lipids or fatty acids have also been used to modify oligonucleotides, including siRNA257,258,259,260 and ASOs,261,262,263 to achieve robust gene silencing in extrahepatic tissues, e.g., heart, lung, kidney, skeletal muscle, brain, etc. This progress remarkably extends the diseases that can be treated by RNAi therapeutics.
Conclusions
Due to the mechanism of action of siRNA, this molecule can be used to target nearly all disease-related genes of interest. The duration of drug research and development is also much shorter than that for small molecules, monoclonal antibodies and proteins. After the successful commercialization of ONPATTRO® and GIVLAARI™ for the treatment of rare diseases, siRNA therapy will achieve another milestone soon for treating common diseases, e.g., dyslipidemia, as excellent phase 3 data have been collected from several clinical studies of inclisiran.
Benefiting from long-term and constant innovations in exploring modification geometries and delivery systems, the effective dose and half-life of the siRNA modalities were reduced from milligrams to 10−3 mg and prolonged from minutes to months, respectively. GalNAc–siRNA conjugates with sophisticated chemical modifications enable quarterly or twice-yearly subcutaneous administration of siRNA therapeutics, which is a great achievement that had never been realized in the history of pharmaceuticals. These improvements will exert widespread and far-reaching impacts on the whole medical industry, e.g., on drug development, government management, patient treatment, payment patterns of medical insurance, financial investments from the capital market, etc.
After studying GalNAc–siRNA conjugation for hepatic delivery of siRNA, researchers will devote themselves to developing extrahepatic delivery platforms. Several important advances have been achieved for renal, CNS and ocular targeted delivery of siRNA. Therefore, state-of-the-art siRNA technologies will definitely change our life in the future.
References
Anguela, X. M. & High, K. A. Entering the modern era of gene therapy. Annu Rev. Med.70, 273–288 (2019).
Weng, Y. et al. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol. Adv.37, 801–825 (2019).
Weng, Y. et al. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv.40, 107534 (2020).
Li, H. et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target Ther.5, 1 (2020).
Setten, R. L., Rossi, J. J. & Han, S. P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov.18, 421–446 (2019).
Carolyn Napoli, C. L. & Richard, J. lntroduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell2, 279–289 (1990).
Elbashir, S. M. et al. Duplexes of 21±nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature411, 494–498 (2001).
Huang, Y. Y. Approval of the first-ever RNAi therapeutics and its technological development history. Prog. Biochem. Biophys.46, 313–322 (2019).
Sardh, E. et al. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N. Engl. J. Med.380, 549–558 (2019).
Bissell, D. M. et al. ENVISION, a phase 3 study of safety and efficacy of givosiran, an investigational RNAi therapeutic, in acute hepatic porphyria patients. Hepatology70, 100A–101A (2019).
de Paula Brandao, P. R., Titze-de-Almeida, S. S. & Titze-de-Almeida, R. Leading R. N. A. Interference therapeutics part 2: silencing delta-aminolevulinic acid synthase 1, with a focus on givosiran. Mol. Diagn. Ther.24, 61–68 (2019).
Agarwal, S. et al. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid (siRNA), givosiran, in patients with acute hepatic porphyria. Clin. Pharmacol. Ther. https://doi.org/10.1002/cpt.1802, (2020).
Cho, W. G. et al. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. PNAS106, 7137–7142 (2009).
Hu, B. et al. Clinical advances of siRNA therapeutics. J. Gene Med.21, e3097 (2019).
Guo, D. X. et al. Photostable and biocompatible fluorescent silicon nanoparticles for imaging-guided co-delivery of siRNA and doxorubicin to drug-resistant. Cancer Cells Nano-Micro Lett.11, 13 (2019).
Zheng, Z. et al. Folate-displaying exosome mediated cytosolic delivery of siRNA avoiding endosome trapping. J. Control. Release311-312, 43–49 (2019).
Zheng, M. et al. ROS-responsive polymeric siRNA nanomedicine stabilized by triple interactions for the robust glioblastoma combinational RNAi therapy. Adv. Mater.31, e1903277 (2019).
Kim, M., Kim, G., Hwang, D. W. & Lee, M. Delivery of high mobility group box-1 siRNA using brain-targeting exosomes for ischemic stroke therapy. J. Biomed. Nanotechnol.15, 2401–2412 (2019).
Liu, J. et al. Effective gene silencing mediated by polypeptide nanoparticles LAH4-L1-siMDR1 in multi-drug resistant human breast cancer. J. Biomed. Nanotechnol.15, 531–543 (2019).
Wang, Y., Li, C., Du, L. & Liu, Y. A reactive oxygen species-responsive dendrimer with low cytotoxicity for efficient and targeted gene delivery. Chin. Chem. Lett.31, 275–280 (2020).
Chen, Y. et al. A supramolecular co-delivery strategy for combined breast cancer treatment and metastasis prevention. Chin. Chem. Lett.31, 1153–1158 (2020).
Ma, J. et al. Preparation of poly(glutamic acid) shielding micelles self-assembled from polylysine-b-polyphenylalanine for gene and drug codelivery. Chinese Chem. Lett. https://doi.org/10.1016/j.cclet.2020.02.034 (2020).
Kleinman, M. E. et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature452, 591–597 (2008).
Barakat, M. R. & Kaiser, P. K. VEGF inhibitors for the treatment of neovascular age-related macular degeneration. Expert Opin. Investig. Drugs18, 637–646 (2009).
Reich, S. J. et al. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol. Vis.9, 210–216 (2003).
Shen, J. et al. Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther.13, 225–234 (2006).
Sioud, M., Furset, G. & Cekaite, L. Suppression of immunostimulatory siRNA-driven innate immune activation by 2’-modified RNAs. Biochem Biophys. Res. Commun.361, 122–126 (2007).
Song, X. et al. Site-specific modification using the 2’-methoxyethyl group improves the specificity and activity of siRNAs. Mol. Ther. Nucleic Acids9, 242–250 (2017).
Fluiter, K., Mook, O. R. & Baas, F. The therapeutic potential of LNA-modified siRNAs: reduction of off-target effects by chemical modification of the siRNA sequence. Methods Mol. Biol.487, 189–203 (2009).
Bramsen, J. B. et al. A screen of chemical modifications identifies position-specific modification by UNA to most potently reduce siRNA off-target effects. Nucleic Acids Res.38, 5761–5773 (2010).
Janas, M. M. et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun.9, 723 (2018).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol.35, 238–248 (2017).
Ju¨rgen Soutschek, A. A. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. nature432, 173–178 (2004).
Coelho, T. et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med.369, 819–829 (2013).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med.379, 11–21 (2018).
Titze-de-Almeida, R., David, C. & Titze-de-Almeida, S. S. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm. Res.34, 1339–1363 (2017).
Khvorova, A. Oligonucleotide therapeutics—a new class of cholesterol-lowering drugs. N. Engl. J. Med. 376, 4–7 (2017).
Fitzgerald, K. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med.376, 41–51 (2017).
Bennett, C. F. Therapeutic antisense oligonucleotides are coming of age. Annu Rev. Med.70, 307–321 (2019).
Rozema, D. B. in Annual Reports in Medicinal Chemistry, Vol 50: Platform Technologies in Drug Discovery and Validation Vol. 50 Annual Reports in Medicinal Chemistry (ed. Goodnow, R. A.) 17–59 (Elsevier Academic Press Inc, 2017).
Yu, R. Z. et al. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab. Dispos.35, 460–468 (2007).
Geselowitz, D. A. & Neckers, L. M. Bovine serum albumin is a major oligonucleotide-binding protein found on the surface of cultured cells. Antisense Res. Dev.5, 213–217 (1995).
Liang, X. H., Sun, H., Shen, W. & Crooke, S. T. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res.43, 2927–2945 (2015).
Crooke, S. T. et al. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol.35, 230–237 (2017).
Shen, W. et al. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol.37, 640–650 (2019).
Migawa, M. T. et al. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res.47, 5465–5479 (2019).
Uyechi, L. S., Gagné, L., Thurston, G. & Szoka, F. C. Jr Mechanism of lipoplex gene delivery in mouse lung: binding and internalization of fluorescent lipid and DNA components. Gene Ther.8, 828–836 (2001).
Huang, Y. et al. Elimination pathways of systemically delivered siRNA. Mol. Ther.19, 381–385 (2011).
Huang, Y. et al. Pharmacokinetic behaviors of intravenously administered siRNA in glandular tissues. Theranostics6, 1528–1541 (2016).
Huang, Y. & Liang, Z. Pharmacokinetic profiles of naked and nano-formulated siRNAs in glandular tissues. Nanomed. Nanotechnol. Biol. Med.14, 1773 (2018).
Iwamoto, N. et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol.35, 845–851 (2017).
Ostergaard, M. E. et al. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res.48, 1691–1700 (2020).
Alnylam. Chirally-enriched double-stranded RNA agents. World Intellect. Prop. Organ.WO2019126651, 1–293 (2019).
Marshall, W. S. & Caruthers, M. H. Phosphorodithioate DNA as a potential therapeutic drug. Science259, 1564–1570 (1993).
Nielsen, P. E., Egholm, M., Berg, R. H. & Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science254, 1497–1500 (1991).
Ndeboko, B. et al. Role of cell-penetrating peptides in intracellular delivery of peptide nucleic acids targeting hepadnaviral replication. Mol. Ther. Nucleic Acids9, 162–169 (2017).
Zeng, Z. et al. A Tat-conjugated peptide nucleic acid Tat-PNA-DR inhibits hepatitis B virus replication in vitro and in vivo by targeting LTR direct repeats of HBV RNA. Mol. Ther. Nucleic Acids5, e295 (2016).
Meade, B. R. et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol.32, 1256–1261 (2014).
Singh, R. P., Oh, B. K. & Choi, J. W. Application of peptide nucleic acid towards development of nanobiosensor arrays. Bioelectrochemistry79, 153–161 (2010).
Weitzer, S. & Martinez, J. The human RNA kinase hClp1 is active on 3’ transfer RNA exons and short interfering RNAs. Nature447, 222–226 (2007).
Prakash, T. P. et al. Identification of metabolically stable 5’-phosphate analogs that support single-stranded siRNA activity. Nucleic Acids Res.43, 2993–3011 (2015).
Lima, W. F. et al. Single-stranded siRNAs activate RNAi in animals. Cell150, 883–894 (2012).
Haraszti, R. A. et al. 5-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res.45, 7581–7592 (2017).
Parmar, R. et al. 5’-(E)-vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. Chembiochem.17, 985–989 (2016).
Elkayam, E. et al. siRNA carrying an (E)-vinylphosphonate moiety at the 5 end of the guide strand augments gene silencing by enhanced binding to human Argonaute-2. Nucleic Acids Res.45, 3528–3536 (2017).
Shen, X. & Corey, D. R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res.46, 1584–1600 (2018).
Monia, B. P. et al. Evaluation of 2” modified oligonucleotides containing 2’-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem.268, 14514–14522 (1993).
Hideo Inoue, Y. H., Hnura, A., Iwai, S., Miura, K. & Ohtsuka, E. Synthesis and hybridiztion studies on two complementary nona(2’-O-methyl)ribonucleotides. Nucleic Acids Res.15, 6131–6148 (1987).
Fucini, R. V. et al. Adenosine modification may be preferred for reducing siRNA immune stimulation. Nucleic Acid Ther.22, 205–210 (2012).
Ostergaard, M. E. et al. Fluorinated nucleotide modifications modulate allele selectivity of SNP-targeting antisense oligonucleotides. Mol. Ther. Nucleic Acids7, 20–30 (2017).
Dowler, T. et al. Improvements in siRNA properties mediated by 2’-deoxy-2’-fluoro-beta-d-arabinonucleic acid (FANA). Nucleic Acids Res.34, 1669–1675 (2006).
Kenski, D. M. et al. siRNA-optimized modifications for enhanced in vivo activity. Mol. Ther. Nucleic Acids1, e5 (2012).
Christensen, U., Jacobsen, N., Rajwanshi, V. K., Wengel, J. & Koch, T. Stopped-flow kinetics of locked nucleic acid (LNA)–oligonucleotide duplex formation: studies of LNA–DNA and DNA–DNA interactions. Biochem. J.354, 481–484 (2001).
Koji Morita, C. H. et al. 2’-O,4’-C-ethylene-bridged nucleic acids (ENA) with nucleaseresistance and high affnity for RNA. Nucleic Acids Res. Suppl.1, 241–242 (2001).
Seth, P. P. et al. Synthesis and biophysical evaluation of 2′,4′-constrained 2′O-methoxyethyl and 2′,4′-constrained 2′O-ethyl nucleic acid analogues. J. Org. Chem.75, 1569–1581 (2010).
Leumann, R. S. A. C. J. Synthesis and thermodynamic and biophysical properties of tricyclo-DNA. 121, 3249–3255 (1999).
WELLER, J. Sa. D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev.7, 187–195 (1997).
Crooke, S. T., Witztum, J. L., Bennett, C. F. & Baker, B. F. RNA-targeted therapeutics. Cell Metab.27, 714–739 (2018).
Kariko, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther.16, 1833–1840 (2008).
Anderson, B. R. et al. Nucleoside modifications in RNA limit activation of 2’-5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res.39, 9329–9338 (2011).
Kormann, M. S. et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol.29, 154–157 (2011).
Valenzuela, R. A. et al. Base modification strategies to modulate immune stimulation by an siRNA. Chembiochem.16, 262–267 (2015).
Phelps, K. J. et al. Click modification of RNA at adenosine: structure and reactivity of 7-ethynyl- and 7-triazolyl-8-aza-7-deazaadenosine in RNA. ACS Chem. Biol.9, 1780–1787 (2014).
Ibarra-Soza, J. M. et al. 7-Substituted 8-aza-7-deazaadenosines for modification of the siRNA major groove. Org. Biomol. Chem.10, 6491–6497 (2012).
Peacock, H., Fostvedt, E. & Beal, P. A. Minor-groove-modulating adenosine replacements control protein binding and RNAi activity in siRNAs. ACS Chem. Biol.5, 1115–1124 (2010).
Wahba, A. S. et al. Phenylpyrrolocytosine as an unobtrusive base modification for monitoring activity and cellular trafficking of siRNA. ACS Chem. Biol.6, 912–919 (2011).
Xia, J. et al. Gene silencing activity of siRNAs with a ribo-difluorotoluyl nucleotide. ACS Chem. Biol.1, 176–183 (2006).
Zhang, J. et al. Modification of the siRNA passenger strand by 5-nitroindole dramatically reduces its off-target effects. Chembiochem.13, 1940–1945 (2012).
Wu, S. Y. et al. Development of modified siRNA molecules incorporating 5-fluoro-2’-deoxyuridine residues to enhance cytotoxicity. Nucleic Acids Res.41, 4650–4659 (2013).
Peacock, H., Kannan, A., Beal, P. A. & Burrows, C. J. Chemical modification of siRNA bases to probe and enhance RNA interference. J. Org. Chem.76, 7295–7300 (2011).
Watts, J. K., Deleavey, G. F. & Damha, M. J. Chemically modified siRNA: tools and applications. Drug Discov. Today13, 842–855 (2008).
Dar, S. A., Thakur, A., Qureshi, A. & Kumar, M. siRNAmod: a database of experimentally validated chemically modified siRNAs. Sci. Rep.6, 20031 (2016).
Gillmore, J. D. et al. Phase 2, open-label extension (OLE) study of revusiran, an investigational RNAi therapeutic for the treatment of patients with transthyretin cardiac amyloidosis. Orphanet. J. Rare Dis.10, O21 (2015).
Sehgal, A. et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat. Med.21, 492–497 (2015).
Janas, M. M. et al. Impact of oligonucleotide structure, chemistry, and delivery method on in vitro cytotoxicity. Nucleic Acid Ther.27, 11–22 (2016).
Foster, D. J. et al. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol. Ther.26, 708–717 (2018).
Nair, J. K. et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res.45, 10969–10977 (2017).
Janas, M. M. et al. Safety evaluation of 2’-deoxy-2’-fluoro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res.47, 3306–3320 (2019).
Alnylam. FDA Approves First Treatment for Inherited Rare Disease https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/0212194s000lbl.pdf 1–11 (The U.S. Food and Drug Administration, 2019).
Zheng, J. et al. Single modification at position 14 of siRNA strand abolishes its gene-silencing activity by decreasing both RISC loading and target degradation. FASEB J.27, 4017–4026 (2013).
Turner, A. M. et al. Hepatic-targeted RNA interference provides robust and persistent knockdown of alpha-1 antitrypsin levels in ZZ patients. J. Hepatol.69, 378–384 (2018).
Dicerna. Methods and compositions for the specific inhibition of transthyretin (TTR) by double-stranded RNA. US Pat. Trademark Off.US20190144859, 1–240 (2019).
US. Silence nucleic acid linked to a trivalent glycoconjugate. US Pat.Trademark Off. 1–52 (2019).
Wittrup, A. & Lieberman, J. Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet.16, 543–552 (2015).
Whitehead, K. A., Langer, R. & Anderson, D. G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov.8, 129–138 (2009).
Kanasty, R., Dorkin, J. R., Vegas, A. & Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater.12, 967–977 (2013).
Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl.51, 8529–8533 (2012).
Wolfrum, C. et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol.25, 1149–1157 (2007).
Akinc, A. et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther.18, 1357–1364 (2010).
Zuckerman, J. E. & Davis, M. E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov.14, 843–856 (2015).
Wartiovaara, J. et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J. Clin. Invest.114, 1475–1483 (2004).
Rozema, D. B. et al. Dynamic polyconjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl Acad. Sci. USA104, 12982–12987 (2007).
Stanzl, E. G., Trantow, B. M., Vargas, J. R. & Wender, P. A. Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc. Chem. Res.46, 2944–2954 (2013).
Zhou, J. et al. pH-sensitive nanomicelles for high-efficiency siRNA delivery in vitro and in vivo: an insight into the design of polycations with robust cytosolic release. Nano Lett.16, 6916–6923 (2016).
Du, L. et al. The pH-triggered triblock nanocarrier enabled highly efficient siRNA delivery for cancer therapy. Theranostics7, 3432–3445 (2017).
Hafez, I. M., Maurer, N. & Cullis, P. R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther.8, 1188–1196 (2001).
Torchilin, V. P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng.8, 343–375 (2006).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol.28, 172–176 (2010).
Zelphati, O. & Szoka, F. C. Jr. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl Acad. Sci. USA93, 11493–11498 (1996).
Li, J. et al. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release142, 416–421 (2010).
Dominska, M. & Dykxhoorn, D. M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci.123, 1183–1189 (2010).
Wittrup, A. et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol.33, 870–876 (2015).
Sahay, G. et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol.31, 653–658 (2013).
Qiu, C. et al. Regulating intracellular fate of siRNA by endoplasmic reticulum membrane-decorated hybrid nanoplexes. Nat. Commun.10, 2702 (2019).
Huang, D. et al. Continuous vector-free gene transfer with a novel microfluidic chip and nanoneedle array. Curr. Drug Deliv.16, 164–170 (2019).
Huang, H. et al. An efficient and high-throughput electroporation microchip applicable for siRNA delivery. Lab Chip.11, 163–172 (2011).
Huang, D., Huang, Y. & Li, Z. Transdermal delivery of nucleic acid mediated by punching and electroporation. Methods Mol. Biol.2050, 101–112 (2020).
Huang, D. et al. Efficient delivery of nucleic acid molecules into skin by combined use of microneedle roller and flexible interdigitated electroporation array. Theranostics8, 2361–2376 (2018).
Wei, Z. et al. A pliable electroporation patch (ep-Patch) for efficient delivery of nucleic acid molecules into animal tissues with irregular surface shapes. Sci. Rep.5, 7618–7618 (2015).
Zhao, D. et al. A flow-through cell electroporation device for rapidly and efficiently transfecting massive amounts of cells in vitro and ex vivo. Sci. Rep.6, 18469–18469 (2016).
Wei, Z. et al. A laminar flow electroporation system for efficient DNA and siRNA delivery. Anal. Chem.83, 5881–5887 (2011).
Demirjian, S. et al. Safety and tolerability study of an intravenously administered small interfering ribonucleic acid (siRNA) post on-pump cardiothoracic surgery in patients at risk of acute kidney injury. Kidney Int. Rep.2, 836–843 (2017).
Thompson, J. D. et al. Toxicological and pharmacokinetic properties of chemically modified siRNAs targeting p53 RNA following intravenous administration. Nucleic Acid Ther.22, 255–264 (2012).
Solano, E. C. et al. Toxicological and pharmacokinetic properties of QPI-1007, a chemically modified synthetic siRNA targeting caspase 2 mRNA, following intravitreal injection. Nucleic Acid Ther.24, 258–266 (2014).
Molitoris, B. A. et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol.20, 1754–1764 (2009).
Ahmed, Z. et al. Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis.2, e173 (2011).
Alvarez, R. et al. RNA interference-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent antiviral strategy. Antimicrob. Agents Chemother.53, 3952–3962 (2009).
DeVincenzo, J. et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antivir. Res.77, 225–231 (2008).
Zamora, M. R. et al. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am. J. Respir. Crit. Care Med.183, 531–538 (2011).
DeVincenzo, J. et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl Acad. Sci. USA107, 8800–8805 (2010).
Gottlieb, J. et al. ALN-RSV01 for prevention of bronchiolitis obliterans syndrome after respiratory syncytial virus infection in lung transplant recipients. J. Heart Lung Transpl.35, 213–221 (2016).
Zheng, S. et al. siRNA knockdown of RRM2 effectively suppressed pancreatic tumor growth alone or synergistically with doxorubicin. Mol. Ther. Nucleic Acids12, 805–816 (2018).
Kim, B., Park, J.-H. & Sailor, M. J. Rekindling RNAi therapy: materials design requirements for in vivo siRNA delivery. Adv. Mater.31, e1903637–e1903637 (2019).
Weng, Y. et al. Improved nucleic acid therapy with advanced nanoscale biotechnology. Mol. Ther. Nucleic Acids19, 581–601 (2019).
Wan, C., Allen, T. M. & Cullis, P. R. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res.4, 74–83 (2014).
Sato, Y. et al. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. J. Control. Release163, 267–276 (2012).
Bottega, R. E. & R, M. Inhibition of protein kinase C by cationic amphiphiles. Biochemistry31, 9025–9030 (1992).
Morrissey, D. V. et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol.23, 1002–1007 (2005).
Maier, M. A. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther.21, 1570–1578 (2013).
El Dika, I. et al. An open-label, multicenter, phase I, dose escalation study with phase II expansion cohort to determine the safety, pharmacokinetics, and preliminary antitumor activity of intravenous TKM-080301 in subjects with advanced hepatocellular carcinoma. Oncologist24, 747–e218 (2019).
Tabernero, J. et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov.3, 406–417 (2013).
Fitzgerald, K. et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet383, 60–68 (2014).
Dong, Y. et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl Acad. Sci. USA111, 3955–3960 (2014).
Akinc, A. et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol.26, 561–569 (2008).
Love, K. T. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl Acad. Sci. USA107, 1864–1869 (2010).
Ganesh, S. et al. Direct pharmacological inhibition of beta-catenin by RNA interference in tumors of diverse origin. Mol. Cancer Ther.15, 2143–2154 (2016).
Aleku, M. et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res.68, 9788–9798 (2008).
Mihaila, R. et al. Lipid nanoparticle purification by spin centrifugation-dialysis (SCD): a facile and high-throughput approach for small scale preparation of siRNA-lipid complexes. Int. J. Pharm.420, 118–121 (2011).
Sato, Y. et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol.26, 431–442 (2008).
Eguchi, A. et al. Liver Bid suppression for treatment of fibrosis associated with non-alcoholic steatohepatitis. J. Hepatol.64, 699–707 (2016).
Maeda, H., Wu, J., Sawa, T., Matsumura, Y. & Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release65, 271–284 (2000).
Liu, R., Li, X., Xiao, W. & Lam, K. S. Tumor-targeting peptides from combinatorial libraries. Adv. Drug Deliv. Rev.110-111, 13–37 (2017).
Ryschich, E. et al. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer40, 1418–1422 (2004).
Prutki, M. et al. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett.238, 188–196 (2006).
Kimura, R. H., Levin, A. M., Cochran, F. V. & Cochran, J. R. Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity. Proteins Struct. Funct. Bioinformatics77, 359–369 (2009).
Gill, M. R., Falzone, N., Du, Y. & Vallis, K. A. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol.18, e414–e423 (2017).
Cohen, Z. R. et al. Localized RNAi therapeutics of chemoresistant grade IV glioma using hyaluronan-grafted lipid-based nanoparticles. ACS Nano.9, 1581–1591 (2015).
Mizrahy, S. et al. Tumor targeting profiling of hyaluronan-coated lipid basednanoparticles. Nanoscale6, 3742–3752 (2014).
Eliaz, R. E. & Szoka, F. C. Jr Liposome-encapsulated doxorubicin targeted to CD44: a strategy to kill CD44-overexpressing tumor cells. Cancer Res.15, 2592–2601 (2001).
Parker, N. et al. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem.338, 284–293 (2005).
Mui, B. L. et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic Acids2, e139 (2013).
Whitehead, K. A. et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun.5, 4277 (2014).
Yanagi, T. et al. Lipid nanoparticle-mediated siRNA transfer against PCTAIRE1/PCTK1/Cdk16 inhibits in vivo cancer growth. Mol. Ther. Nucleic Acids5, e327 (2016).
Ganesh, S. et al. RNAi-mediated beta-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol. Ther.26, 2567–2579 (2018).
Schultheis, B. et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol.32, 4141–4148 (2014).
Fehring, V. et al. Delivery of therapeutic siRNA to the lung endothelium via novel Lipoplex formulation DACC. Mol. Ther.22, 811–820 (2014).
Mihaila, R. et al. Modeling the kinetics of lipid-nanoparticle-mediated delivery of multiple siRNAs to evaluate the effect on competition for Ago2. Mol. Ther. Nucleic Acids16, 367–377 (2019).
Mihaila, R. et al. Mathematical modeling: a tool for optimization of lipid nanoparticle-mediated delivery of siRNA. Mol. Ther. Nucleic Acids7, 246–255 (2017).
Zimmermann, T. S. et al. RNAi-mediated gene silencing in non-human primates. Nature441, 111–114 (2006).
Suhr, O. B. et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet. J. Rare Dis.10, 109 (2015).
Wu, S. Y., Lopez-Berestein, G., Calin, G. A. & Sood, A. K. RNAi therapies: drugging the undruggable. Sci. Transl. Med.6, 240ps247 (2014).
Liu, X. Targeting polo-like kinases: a promising therapeutic approach for cancer treatment. Transl. Oncol.8, 185–195 (2015).
Beg, M. S. et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. N. Drugs35, 180–188 (2017).
Strumberg, D. et al. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int J. Clin. Pharm. Ther.50, 76–78 (2012).
Streinu-Cercel, A. et al. A phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg positive and negative virally suppressed subjects with hepatitis B. J. Hepatol.66, S688–S689 (2017).
Thi, E. P. et al. ARB-1740, a RNA interference therapeutic for chronic hepatitis B infection. ACS Infect. Dis.5, 725–737 (2019).
Ye, X. et al. Hepatitis B virus therapeutic agent ARB-1740 has inhibitory effect on hepatitis delta virus in a new dually-infected humanized mouse model. ACS Infect. Dis.5, 738–749 (2019).
Lee, A. C. H. et al. Function and drug combination studies in cell culture models for AB-729, a subcutaneously administered siRNA investigational agent for chronic hepatitis B infection. J. Hepatol.70, E471–E471 (2019).
Wong, S. C. et al. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther.22, 380–390 (2012).
Wooddell, C. I. et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther.21, 973–985 (2013).
Sebestyen, M. G. et al. Targeted in vivo delivery of siRNA and an endosome-releasing agent to hepatocytes. Methods Mol. Biol.1218, 163–186 (2015).
Rozema, D. B. et al. Protease-triggered siRNA delivery vehicles. J. Control. Release209, 57–66 (2015).
Jin, L. et al. Current progress in gene delivery technology based on chemical methods and nano-carriers. Theranostics4, 240–255 (2014).
Wooddell, C. I. et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 9, eaan0241 (2017).
Gish, R. G. et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antivir. Res.121, 97–108 (2015).
Gane, E. et al. A phase 1 study to evaluate safety and tolerability of escalating single doses of the hepatitis B virus RNA interference drug ARC-521 in a healthy volunteer population. J. Hepatol.66, S265–S265 (2017).
Schwabe, C. et al. A phase 1 single and multiple dose-escalating study to evaluate the safety, tolerability, pharmacokinetics and effect of ARO-AAT on serum alpha-1 antitrypsin levels in normal adult volunteers. Hepatology68, 1451A–1452A (2018).
Gane, E. J. et al. First results with RNA interference (RNAi) in chronic hepatitis B (CHB) using ARO-HBV. Hepatology68, 1463A–1463A (2018).
Khorev, O. et al. Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor. Bioorg. Med. Chem.16, 5216–5231 (2008).
Nair, J. K. et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc.136, 16958–16961 (2014).
Dicerna. Ligand-modified double-stranded nucleic acids. World Intellect. Prop. Organ.WO2016100401A1, 1–426 (2016).
Arrowhead. Targeting ligands. World Intellect. Prop. Organ.WO2018044350, 1–254 (2018).
Craig, K., Abrams, M. & Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv.15, 629–640 (2018).
Lee, A. C. H. et al. Durable inhibition of hepatitis B virus replication and antigenemia using a subcutaneously administered siRNA agent in preclinical models. J. Hepatol.68, S18–S18 (2018).
Silence. Advanced GalNAc-siRNA Platform and Its Therapeutic Applications https://www.silence-therapeutics.com/media/1799/2018-boston-tides.pdf 1–36 (Silence, 2018).
Prakash, T. P. et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res.42, 8796–8807 (2014).
Apponi, L. et al. Stereochemistry enhances pharmacological properties of APOC3 antisense oligonucleotides. J. Hepatol.68, S137–S137 (2018).
van der Ree, M. H. et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet389, 709–717 (2017).
SuzhouRiboLifeScience. Compound, conjugates and use and kit thereof. World Intellect. Prop. Organ.WO2019128611, 1–271 (2019).
Sanhueza, C. A. et al. Efficient Liver targeting by polyvalent display of a compact ligand for the asialoglycoprotein receptor. J. Am. Chem. Soc.139, 3528–3536 (2017).
WaveLifeSciences. Oligonucleotide compositions and methods of use thereof. World Intellect. Prop. Organ.WO2018223073, 1–773 (2018).
Pasi, K. J. et al. A subcutaneously administered investigational RNAi therapeutic, fitusiran (ALN-AT3), targeting antithrombin for treatment of hemophilia: interim results in patients with hemophilia A or B. Haemophilia22, 76–76 (2016).
Pasi, K. J. et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N. Engl. J. Med.377, 819–828 (2017).
Strat, A. L., Ghiciuc, C. M., Lupusoru, C. E. & Mitu, F. New class of drugs: therapeutic RNAi inhibition of PCSK9 as a specific LDL-c lowering therapy. Rev. Med Chir. Soc. Med. Nat. Iasi120, 228–232 (2016).
Fitzgerald, K. et al. ALN-PCSsc, an RNAi investigational agent that inhibits PCSK9 with potential for effective quarterly or possibly bi-annual dosing: results of single-blind, placebo-controlled, phase 1 single-ascending dose (SAD), and multi-dose (MD) trial in adults with elevated LDL-C, on and off statins. Circulation132, 2275–2275 (2015).
Hassan, M. FOURIER & PCSK9 RNAi: towards enhancing durability and efficacy of PCSK9 inhibitors. Glob. Cardiol. Sci. Pr.2017, 13 (2017).
Alnylam. Phase 1 Study of ALN-TTRsc02, a Subcutaneously Administered Investigational RNAi Therapeutic for the Treatment of Transthyretin-Mediated Amyloidosis http://www.alnylam.com/wp-content/uploads/2018/03/10.-TTR-SCO2_FINAL.pdf (2018).
Dindo, M. et al. Molecular basis of primary hyperoxaluria: clues to innovative treatments. Urolithiasis47, 67–78 (2019).
Dicerna. Corporate Overview. Jefferies Global Healthcare Conference 2019. http://investors.dicerna.com/static-files/6ca8fc33-2696-459a-b8ca-a8a39ec68903 1–22 (2019).
Zorde Khvalevsky, E. et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl Acad. Sci. USA110, 20723–20728 (2013).
Amotz Shemi, E. Z. K. et al. Multistep, effective drug distribution within solid tumors. Oncotarget7, 39564–39577 (2015).
Ramot, Y. et al. Preclinical safety evaluation in rats of a polymeric matrix containing an siRNA drug used as a local and prolonged delivery system for pancreatic cancer therapy. Toxicol. Pathol.44, 856–865 (2016).
Golan, T. et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget6, 24560–24570 (2015).
Shemi, A. & Khvalevsky, Z. RNA interference compositions targeting heat shock protein 90 and methods of use thereof. US Pat. Trademark Off.US20170283803A1, 1–20 (2017).
Takemoto, H. & Nishiyama, N. Functional polymer-based siRNA delivery carrier that recognizes site-specific biosignals. J. Control. Release267, 90–99 (2017).
Guo, S. et al. Ternary complexes of amphiphilic polycaprolactone-graft-poly (N,N-dimethylaminoethyl methacrylate), DNA and polyglutamic acid-graft-poly(ethylene glycol) for gene delivery. Biomaterials32, 4283–4292 (2011).
Lin, D. et al. Structural contributions of blocked or grafted poly(2-dimethylaminoethyl methacrylate) on PEGylated polycaprolactone nanoparticles in siRNA delivery. Biomaterials32, 8730–8742 (2011).
Huang, Y. et al. Binary and ternary complexes based on polycaprolactone-graft-poly (N, N-dimethylaminoethyl methacrylate) for targeted siRNA delivery. Biomaterials33, 4653–4664 (2012).
Lin, D. et al. Intracellular cleavable poly(2-dimethylaminoethyl methacrylate) functionalized mesoporous silica nanoparticles for efficient siRNA delivery in vitro and in vivo. Nanoscale5, 4291–4301 (2013).
Han, S. et al. Effects of hydrophobic core components in amphiphilic PDMAEMA nanoparticles on siRNA delivery. Biomaterials48, 45–55 (2015).
Zhang, T. et al. Fluorinated oligoethylenimine nanoassemblies for efficient siRNA-mediated gene silencing in serum-containing media by effective endosomal escape. Nano Lett.18, 6301–6311 (2018).
Suma, T. et al. Enhanced stability and gene silencing ability of siRNA-loaded polyion complexes formulated from polyaspartamide derivatives with a repetitive array of amino groups in the side chain. Biomaterials33, 2770–2779 (2012).
Liu, Y. et al. Charge conversional biomimetic nanocomplexes as a multifunctional platform for boosting orthotopic glioblastoma RNAi therapy. Nano Lett.20, 1637–1646 (2020).
Davis, M. E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol. Pharm.6, 659–668 (2009).
Davis, M. E. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature464, 1067–1070 (2010).
van Zandwijk, N. et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol.18, 1386–1396 (2017).
Zhou, J. et al. Simultaneous silencing of TGF-beta1 and COX-2 reduces human skin hypertrophic scar through activation of fibroblast apoptosis. Oncotarget8, 80651–80665 (2017).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature546, 498–503 (2017).
Lu, M. & Huang, Y. Bioinspired exosome-like therapeutics and delivery nanoplatforms. Biomaterials242, 119925 (2020).
Huang, Y. et al. Systemic and tumor-targeted delivery of siRNA by cyclic NGR and isoDGR motif-containing peptides. Biomater. Sci.4, 494–510 (2016).
Huang, Y. et al. Systemic administration of siRNA via cRGD-containing peptide. Sci. Rep.5, 12458 (2015).
Kim, S. S. et al. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol. Ther.18, 993–1001 (2010).
Kumar, P. et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature448, 39–43 (2007).
Dong, Y. et al. A dual targeting dendrimer-mediated siRNA delivery system for effective gene silencing in cancer therapy. J. Am. Chem. Soc.140, 16264–16274 (2018).
Zhou, J. et al. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol. Ther.19, 2228–2238 (2011).
Liu, X. et al. Adaptive amphiphilic dendrimer-based nanoassemblies as robust and versatile siRNA delivery systems. Angew. Chem. Int. Ed. Engl.53, 11822–11827 (2014).
Cui, D. et al. Regression of gastric cancer by systemic injection of RNA nanoparticles carrying both ligand and siRNA. Sci. Rep.5, 10726 (2015).
Lee, T. J. et al. RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget6, 14766–14776 (2015).
Stewart, J. M. et al. Programmable RNA microstructures for coordinated delivery of siRNAs. Nanoscale8, 17542–17550 (2016).
Xu, Y. et al. Specific delivery of delta-5-desaturase siRNA via RNA nanoparticles supplemented with dihomo-gamma-linolenic acid for colon cancer suppression. Redox Biol.21, 101085 (2019).
Pi, F. et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol.13, 82–89 (2018).
Smith, J. A. et al. RNA nanotherapeutics for the amelioration of astroglial reactivity. Mol. Ther. Nucleic Acids10, 103–121 (2018).
Xu, C. et al. Favorable biodistribution, specific targeting and conditional endosomal escape of RNA nanoparticles in cancer therapy. Cancer Lett.414, 57–70 (2018).
Jasinski, D., Haque, F., Binzel, D. W. & Guo, P. Advancement of the emerging field of RNA nanotechnology. ACS Nano.11, 1142–1164 (2017).
Guo, S. et al. Enhanced gene delivery and siRNA silencing by gold nanoparticles coated with charge-reversal polyelectrolyte. ACS Nano.4, 5505–5511 (2010).
Alnylam. Extrahepatic delivery. World Intellect. Prop. Organ.WO2019217459A1, 1–271 (2019).
Biscans, A., Coles, A., Echeverria, D. & Khvorova, A. The valency of fatty acid conjugates impacts siRNA pharmacokinetics, distribution, and efficacy in vivo. J. Control. Release302, 116–125 (2019).
Biscans, A. et al. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res.47, 1082–1096 (2019).
Nikan, M. et al. Synthesis and evaluation of parenchymal retention and efficacy of a metabolically stable O-phosphocholine-N-docosahexaenoyl-l-serine siRNA conjugate in mouse brain. Bioconjug. Chem.28, 1758–1766 (2017).
Osborn, M. F. & Khvorova, A. Improving siRNA delivery in vivo through lipid conjugation. Nucleic Acid Ther.28, 128–136 (2018).
Ostergaard, M. E. et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res.47, 6045–6058 (2019).
Prakash, T. P. et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res.47, 6029–6044 (2019).
Wang, S. et al. Lipid conjugates enhance endosomal release of antisense oligonucleotides into cells. Nucleic Acid Ther.29, 245–255 (2019).
Schluep, T. et al. Safety, tolerability, and pharmacokinetics of ARC-520 injection, an RNA interference-based therapeutic for the treatment of chronic hepatitis B Virus Infection, in healthy volunteers. Clin. Pharm. Drug Dev.6, 350–362 (2017).
Dicerna Corporate Presentation February 2020. (2020).
Arrowhead. ARO-AAT for Liver Disease in Alpha-1 Antitrypsin Deficiency: Clinical Development Progress (Arrowhead, 2019).
Liebow, A. et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J. Am. Soc. Nephrol.28, 494–503 (2017).
Hill, A. et al. A subcutaneously administered investigational RNAi therapeutic (ALN-CC5) targeting complement C5 for treatment of PNH and complement-mediated diseases: preliminary phase 1/2 study results in patients with PNH. Blood128, 5 (2016).
Huang, Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol. Ther. Nucleic Acids6, 116–132 (2017).
Huang, Y. Y. & Liang, Z. C. Asialoglycoprotein receptor and its application in liver-targeted drug delivery. Prog. Biochem. Biophys.42, 501–510 (2015).
Haas, M. J. Alnylam interrupts preeclampsia. SciBX: Science-Business eXchange. 7, https://doi.org/10.1038/scibx.2014.1170 (2014).
Wooddell, C. et al. ARO-AAT, a subcutaneous RNAi-based therapeutic for alpha-1 antitrypsin-related liver disease, demonstrates liver exposure-response and efficacy in preclinical studies. J. Hepatol.68, S82–S82 (2018).
Wooddell, C. et al. Development of subcutaneously administered RNAi therapeutic ARO-HBV for chronic hepatitis B virus infection. J. Hepatol.68, S18–S19 (2018).
Butler, A. A. et al. Fructose-induced hypertriglyceridemia in rhesus macaques is attenuated with fish oil or ApoC3 RNA interference. J. Lipid Res.60, 805–818 (2019).
Hamilton, J. Overcoming the challenges of RNAi-based therapy: an interview with James Hamilton. Ther. Deliv.9, 511–513 (2018).
Melquist, S. et al. Targeting apolipoprotein(a) with a novel RNAi delivery platform as a prophylactic treatment to reduce risk of cardiovascular events in individuals with elevated lipoprotein (a). Circulation134, 7 (2016).
Borrelli, M. J., Youssef, A., Boffa, M. B. & Koschinsky, M. L. New Frontiers in Lp(a)-Targeted Therapies. Trends Pharm. Sci.40, 212–225 (2019).
Soule, B. et al. Safety, tolerability, and pharmacokinetics of BMS-986263/ND-L02-s0201, a novel targeted lipid nanoparticle delivering HSP47 siRNA, in healthy participants: A randomised, placebo-controlled, double-blind, phase 1 study. J. Hepatol.68, S112–S112 (2018).
Kimchi-Sarfaty, C. et al. In vitro-packaged SV40 pseudovirions as highly efficient vectors for gene transfer. Hum. Gene Ther.13, 299–310 (2002).
Nishimura, M. et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov.3, 1302–1315 (2013).
Landen, C. N. Jr. et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res.65, 6910–6918 (2005).
Duxbury, M. S. et al. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene23, 1448–1456 (2004).
Jensen, S. A. et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med.5, 209ra152 (2013).
Hwang, J. et al. Development of cell-penetrating asymmetric interfering RNA targeting connective tissue growth factor. J. Invest. Dermatol.136, 2305–2313 (2016).
Leachman, S. A. et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol. Ther.18, 442–446 (2010).
Lee, D. U., Huang, W., Rittenhouse, K. D. & Jessen, B. Retina expression and cross-species validation of gene silencing by PF-655, a small interfering RNA against RTP801 for the treatment of ocular disease. J. Ocul. Pharmacol. Ther.28, 222–230 (2012).
Hobo, W. et al. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood116, 4501–4511 (2010).
van der Waart, A. B. et al. siRNA silencing of PD-1 ligands on dendritic cell vaccines boosts the expansion of minor histocompatibility antigen-specific CD8(+) T cells in NOD/SCID/IL2Rg(null) mice. Cancer Immunol. Immunother.64, 645–654 (2015).
Libertine, L. et al. RXI-109 treatment for proliferative vitreoretinopathy (PVR) and other ocular disorders. Invest Ophth Vis. Sci.55, 3 (2014).
Schultheis, B. et al. Combination therapy with gemcitabine and Atu027 in patients with locally advanced or metastatic pancreatic adenocarcinoma—a Phase Ib/IIa study. Oncol. Res. Treat.41, 64 (2018).
Golan, T. et al. A phase I trial of a local delivery of siRNA against k-ras in combination with chemotherapy for locally advanced pancreatic adenocarcinoma. J. Clin. Oncol.31, 1 (2013).
Moreno-Montañés, J., Bleau, A.-M. & Jimenez, A. I. Tivanisiran, a novel siRNA for the treatment of dry eye disease. Expert Opin. Investig. Drugs27, 421–426 (2018).
Beatriz, J. et al. Safety and efficacy clinical trials for SYL1001, a novel short interfering RNA for the treatment of dry eye disease. Invest. Ophthalmol. Vis. Sci.57, 6447–6454 (2016).
Moreno-Montanes, J. et al. Phase I clinical trial of SYL040012, a small interfering RNA targeting beta-adrenergic receptor 2, for lowering intraocular pressure. Mol. Ther.22, 226–232 (2014).
Martinez, T. et al. In vitro and in vivo efficacy of SYL040012, a novel siRNA compound for treatment of glaucoma. Mol. Ther.22, 81–91 (2014).
Triozzi, P. et al. Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. J. Immunother. Cancer3, P175 (2015).
Loibner, H. et al. Adoptive cellular immunotherapy with APN401, autologous cbl-b silenced peripheral blood mononuclear cells: data from a phase I study in patients with solid tumors. J. Clin. Oncol.36, 1 (2018).
Seto, A. G. et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol.183, 428–444 (2018).
Gallant-Behm, C. L. et al. A microRNA-29 mimic (Remlarsen) represses extracellular matrix expression and fibroplasia in the skin. J. Invest. Dermatol.139, 1073–1081 (2019).
Montgomery, R. L. et al. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med.6, 1347–1356 (2014).
Gallant-Behm, C. L. et al. A synthetic microRNA-92a inhibitor (MRG-110) accelerates angiogenesis and wound healing in diabetic and nondiabetic wounds. Wound Repair Regen.26, 311–323 (2018).
Javanbakht, H. et al. Liver-targeted anti-HBV single-stranded oligonucleotides with locked nucleic acid potently reduce HBV gene expression in vivo. Mol. Ther. Nucleic Acids11, 441–454 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31871003, 31901053), the Hunan Provincial Natural Science Foundation of China (2018JJ1019, 2019JJ50196), the Hu-Xiang Young Talent Program (2018RS3094), the Fundamental Research Funds for the Central Universities (3052018065) and the Beijing Institute of Technology Research Fund Program for Young Scholars. It was also supported, in part, by grants from the National Science and Technology Major Project of China (2019ZX09301-132), Program for Changjiang Scholars and Innovative Research Team in University of China (IRT_15R13).
Author information
Authors and Affiliations
Contributions
Y.H., B.H. and L.Z. wrote the paper. Y.H. organized the figures. Y.W. contributed to summarizing the clinical activities of siRNAs. X.-J.L., Y.Z., and L.P. provided insightful discussions and comments on the manuscript. Y.H., X.-J.L. and Y.Z. revised the manuscript and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, B., Zhong, L., Weng, Y. et al. Therapeutic siRNA: state of the art. Sig Transduct Target Ther 5, 101 (2020). https://doi.org/10.1038/s41392-020-0207-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-0207-x
This article is cited by
-
Microfluidics-enabled fluorinated assembly of EGCG-ligands-siTOX nanoparticles for synergetic tumor cells and exhausted t cells regulation in cancer immunotherapy
Journal of Nanobiotechnology (2024)
-
The siRNA-mediated knockdown of AP-1 restores the function of the pulmonary artery and the right ventricle by reducing perivascular and interstitial fibrosis and key molecular players in cardiopulmonary disease
Journal of Translational Medicine (2024)
-
A versatile engineered extracellular vesicle platform simultaneously targeting and eliminating senescent stromal cells and tumor cells to promote tumor regression
Journal of Nanobiotechnology (2024)
-
Developing hydrogels for gene therapy and tissue engineering
Journal of Nanobiotechnology (2024)
-
Application of nanotechnology in the treatment of glomerulonephritis: current status and future perspectives
Journal of Nanobiotechnology (2024)
