
Abstract
Drug resistance is a major hurdle in cancer treatment and a key cause of poor prognosis. Epitranscriptomics and epiproteomics are crucial in cell proliferation, migration, invasion, and epithelial–mesenchymal transition. In recent years, epitranscriptomic and epiproteomic modification has been investigated on their roles in overcoming drug resistance. In this review article, we summarized the recent progress in overcoming cancer drug resistance in three novel aspects: (i) mRNA modification, which includes alternative splicing, A-to-I modification and mRNA methylation; (ii) noncoding RNAs modification, which involves miRNAs, lncRNAs, and circRNAs; and (iii) posttranslational modification on molecules encompasses drug inactivation/efflux, drug target modifications, DNA damage repair, cell death resistance, EMT, and metastasis. In addition, we discussed the therapeutic implications of targeting some classical chemotherapeutic drugs such as cisplatin, 5-fluorouridine, and gefitinib via these modifications. Taken together, this review highlights the importance of epitranscriptomic and epiproteomic modification in cancer drug resistance and provides new insights on potential therapeutic targets to reverse cancer drug resistance.
Similar content being viewed by others
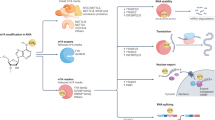
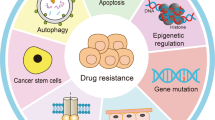
Introduction
Drug resistance in cancer treatment
Cancer remains the leading cause of incidence and mortality worldwide.1,2 The development of cancer is a complex process with significant biological characteristics, such as abnormal cell proliferation and differentiation, a high degree of molecular heterogeneity and epithelial–mesenchymal transition (EMT).3 Because most cancers have progressed to the middle or late stages when diagnosed, molecular targeted drug therapy and chemotherapy are the main treatment options.4 The most common therapeutic drugs include cisplatin, sorafenib, oxaliplatin, 5-fluorouracil, and epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs).5 However, long-term therapies usually lead to acquired drug resistance and poor prognosis. The main underlying mechanisms of drug resistance include: (1) drug efflux and alterations in drug metabolism; (2) alterations of drug targets; (3) DNA damage repair (DDR); (4) deregulation of apoptosis and autophagy; (5) resistance-promoting adaptive responses; (6) alterations in tumor microenvironment; and (7) epigenetic changes.6,7
Epigenetics refers to a “heritable” phenomenon in which the phenotype changes are independent of DNA sequence. Epitranscriptomics, also called “RNA epigenetics,” is a branch of epigenetics and refers RNA editing and noncoding RNA regulations. Epitranscriptomics plays essential roles in alternative splicing, nuclear export, transcript stability, and translation of RNAs.8,9 Epiproteomics is the posttranslational modifications (PTMs) that involve histone acetylation, SUMOylation, phosphorylation, and ubiquitination.10,11,12 The PTMs might regulate various biological processes via modulating chromosomal structures or regulating the binding of chromatin.13 Recent studies in RNA and protein modifications mainly focus on evaluating drug response to screening drugs suitable for individual patients or as the molecular targets to pioneer new ways of cancer treatment.14,15,16,17 In this review, we will discuss the role of posttranscriptional and PTM in cancer drug resistance and therapeutic targets.
mRNA modification in cancer drug resistance
In this section, we focus on the mechanism of cancer drug resistance in three different types of RNA modifications: alternative splicing, adenosine-to-inosine (A-to-I) modification, and mRNA methylation (Fig. 1 and Table 1).

mRNA modification in cancer drug resistance. a Schematic representation of examples of alternative splicing patterns causing cancer drug resistance, including skipping of one exon, skipping of multiple exons, mutually exclusive exons, and exon inclusion. b Schematic representation of A-to-I RNA editing mediated drug-resistance-related functional consequences including structure modification of targeted protein, target escape from silencing of miRNA, off-target effects of miRNA, pre-miRNA degradation, aberrant splicing of targeted mRNA. c Schematic representation of m6A modification network in targeted genes causing cancer drug resistance. In the nucleus, m6A is deposited in nascent pre-mRNA by a “writer” multiprotein complex (i.e., METTL3, METTL14, and other related protein) and removed by “eraser” demethylases (i.e., FTO and ALKBH5). In the cytoplasm, the m6A modifications are recognized by “reader” proteins, resulting in stabilization or decay or enhanced translation. Specific examples of each mRNA modification event discussed in the text are shown
Alternative splicing and cancer drug resistance
Alternative splicing is a process by which introns are differentially removed from a single precursor mRNA (pre-mRNA) to generate multiple mature mRNA products.18,19 More than 95% of human genes are transcribed into pre-mRNAs that undergo alternative splicing.20,21,22 Since alternative splicing represents a frequent mechanism underlying the expansion of transcriptomes and proteomes in higher eukaryotes, it plays numerous critical roles in both normal and disease processes. Global analysis has revealed at least 15,000 cancer specific splice variants in 27 types of cancers,20 indicating that alternative splicing is a significant mechanism contributing to the progression of cancer development, including cell proliferation, apoptosis, invasion, metastasis, angiogenesis, and drug resistance.21 Thus, it is becoming increasingly clear that alternative splicing regulates many of the biological and pathological processes. Therefore, alternative splicing could be potential targets for the development of new cancer therapeutics.22 To illustrate the alternative splicing patterns and programs that cancer cells apply to gain drug resistance, we describe below an exemplary set of functionally important alternative splicing events.
Exon skipping is one of the most important alternative splicing processes in drug-resistant cancer cells. In leukemia, the enzyme folylpolyglutamate synthetase (FPGS) is responsible for the intracellular retention of folates and antifolates by polyglutamylation. Aberrant splicing of FPGS induced skipping of exon 12 and generated a nonfunctional FPGS enzyme, which leads to reduced retention of antifolates and causes cancer cells resistant to folate antagonist methotrexate.23 In patients with B-cell malignancies treated with adoptive T cells expressed chimeric antigen receptors against CD19 (CART-19), the expression of alternatively spliced CD19 isoform lacking exon 2 caused failure of initiation of CART-19-mediated cancer cell death.24 TGF-β-activated kinase 1 (TAK1) promoted TGF-β-induced apoptosis in response to TGF-β activation. However, TAK1 variable exon 12 exerted opposite function that constitutively supported TGF-β-induced EMT and activated nuclear factor-kappa B (NF-κB) signaling pathway, eventually causing chemotherapeutic resistance.25 Breast and ovary cancer cells can overcome deleterious germline mutations in BRCA1 (the gene encoding breast cancer type-1 susceptibility protein) by alternative splicing. Among the splicing products, BRCA1-Δ11q retains residual activity, triggering resistance to cisplatin and poly ADP-ribose polymerase (PARP) inhibitors.26
In order to escape from drug mediated apoptosis, targeted genes would undergo multiple exons skipping to delete the specific domains targeted by cancer drugs. BRAF is an oncogene belonging to RAS/MAPK signaling pathway, which controls several important cellular functions including proliferation and migration. About 90% of melanomas harbor BRAF V600E mutation, which leads to the constitutive activation of RAS/MAPK signaling pathway and malignant cell proliferation. Vemurafenib is a potent RAF kinase inhibitor with remarkable clinical activity against BRAF (V600E) melanoma. However, patients rapidly develop resistance to vemurafenib treatment. Mechanistically, patients harboring isoform BRAF3-9 (Δ exons 4–8) or BRAF2-6 (Δ exons 3–5) that could eliminate RAS-binding domains often develop drug resistance.27,28 Advanced prostate cancer is commonly treated with drugs that inhibit androgen biosynthesis or antagonize the interaction between androgen and androgen receptor (AR). AR splice variant 7, which lacked the ligand-binding domain (exon 4–8), was constitutively resistant to androgen-targeted therapies.29
Mutually exclusive exons represent a rare subtype of RNA splicing. However, once it occurs, the cells harboring the spliced product with drug-resistant function would be evolutionally selected and accumulated resulting in cancer drug resistance. B-cell CLL/lymphoma 2 (BCL-2)-like 11 (BIM), a pro-apoptotic member of the BCL-2 family, is required for TKIs to induce apoptosis in kinase-driven cancers. A polymorphism switched BIM splicing from exon 3 to exon 4 would result in deletion of pro-apoptotic BCL-2-homology domain 3 (BH3) and confer intrinsic TKI resistance in both CML and EGFR NSCLC cells. Patients with this mutant protein had a poorer response to tyrosine kinase inhibitors than individuals without the polymorphism.30 In addition to exon removal and switching, intron retention is another mechanism that cancer cells applied for drug resistance. Interferon (IFN) treatment is effective in hematological malignancies through mediating cell apoptosis. Signal transducer and activator of transcription 2 (STAT2) is a transcription factor that contributes to the activation of IFN responsive genes. However, cancer cells frequently develop a new splice variant of STAT2 that contains intron 19 and a premature stop codon, leading to resistance to apoptosis induced by IFN and a number of chemotherapeutic agents (camptothecin, staurosporine, and doxorubicin (DOX)).31
A-to-I modification and cancer drug resistance
In eukaryotes, A-to-I editing in double-stranded RNA is one of the most prevalent RNA modifications. This process involves hydrolytic deamination of adenosine, catalyzed by the adenosine deaminase acting on RNA (ADAR) family members (ADAR1, ADAR2, and ADAR3).32 The newly generated inosine base is interpreted by the ribosome as guanosine, and this event could occur in protein-coding region during mRNA translation, leading to altered protein products,33,34 noncoding regions, such as introns, 5′ and 3′ untranslated regions (UTRs), and repetitive sequences, such as human Alu elements.35 Deregulation of ADAR1 has emerged as a dominant driver of cancer progression and therapeutic resistance.36 The transition from pre-malignant progenitor to therapy resistant cancer stem cell (CSC) is often accompanied by aberrant ADAR1 activation.37 The most common mechanism involved in A-to-I editing induced drug resistance during therapeutic treatment is protein mutation. In multiple myeloma, ADAR1 enhanced Alu-dependent editing and transcriptional activity of GLI1, a Hedgehog (Hh) signaling pathway transcriptional activator and self-renewal agonist, leading to an R701G amino acid change, which stabilized GLI1 transcriptional activity by preventing the binding of a critical Hh signaling pathway negative regulator, and resulted in promotion of immunomodulatory drug resistance.38 Importantly, inactivation of ADAR1 reduced the resistance to blockade and overcame the resistance to immunotherapy.39
In the process of microRNAs (miRNAs) maturation, the stem-loop secondary structure adopted by primary transcripts of miRNA genes (pri-miRNAs) and miRNA precursors (pre-miRNAs) enable the interactions between the A-to-I editing machinery and the miRNA biogenesis pathway.40 ADARs can suppress miRNA maturation, which is another mechanism of gene regulation causing drug resistance.41 In leukemia, ADAR1-mediated miRNA editing impaired let-7 biogenesis and enhanced the self-renewal of leukemic stem cell (LSC). A small-molecule antagonizes ADAR1 on LSC self-renewal in stromal co-cultures restored let-7 biogenesis.42 Dihydrofolate reductase (DHFR) plays a key role in folate metabolism in cancers, and is a target of chemotherapeutic agents including methotrexate and pemetrexed. Upregulation of ADAR1 induced the expression of DHFR by editing the miR-25-3p, a miRNA targeting DHFR, which could enhance cellular proliferation and resistance to methotrexate.43 The 3’UTRs are important regions for posttranscriptional regulation mediated by miRNA. Modification of 3’ UTR sequence plays crucial role in cancer drug resistance. ADAR1-induced A-to-I editing stabilizes a proto-oncogene, mouse double minute 2 homolog (MDM2). Modulating the 3’ UTR region of MDM2 by ADAR1 can prevent the binding of miR-155, and enhance the reprogramming of progenitor cells into dormant leukemia stem cells.44 In some cases, A-to-I editing could also trigger abnormal splicing leading to drug resistance. A-to-I editing induced mis-splicing of GSK3β (Δ exon 9) in LSC, as a result, β-catenin expression was enhanced, leading to cancer progression and TKI resistance.37,45
mRNA methylation and cancer drug resistance
Over 100 different types of posttranscriptional RNA modifications have been documented among all living organisms so far.46,47 Although the majority of these modifications are found in tRNA and rRNA,48 increasing evidence have shown that the epigenetic modifications in messenger RNA (mRNA) are essential for almost every step of RNA biogenesis, physiology and turnover.49 Among them, N6 -methyladenosine (m6A) is the most prevalent modification that occurs in the mRNAs.50 The modification is catalyzed by the m6A methyltransferase “writers” and then recognized by m6A binding protein “readers”, and the m6A mark can be removed by demethylase “erasers”. The core m6A writer complex includes methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14).51 The m6A erasers include fat mass and obesity-associated (FTO) and AlkB homolog 5 (ALKBH5) demethylases.52,53 The readers were firstly discovered to be YT521-B homology (YTH) domain-containing proteins (YTHDF1, 2, 3 and YTHDC1, 2).51 Soon afterward, more different types of readers were revealed.54,55 Interestingly, they exert diverse functions with different mechanisms.55 For example, YTHDF2 specifically recognizes and destabilizes m6A modified RNAs and re-localizes these RNAs to processing bodies; while YTHDF1 stimulates mRNA translation by interacting with translation initiation factors. The m6A marks are enriched at the RRACH (R = G or A, H = A, C, or U) motif around stop codons, 3’ or 5’ UTRs, and internal long exons.51,56,57 The m6A deposition is the beginning of the journey of methylation regulation. The m6A mRNA might be demethylated by erasers, or exported into cytoplasm and subject to be “read”. The dramatic and dynamic variations of mRNA modifications had been found in every step of biological processing, especially in tumor transformation and cellular reprogramming,58 implying their biological significance in cancer therapeutic treatment.
The biological significance of mRNA methylation is recently uncovered, however, the mechanism of regulation of m6A modification in drug resistance remains poorly understood. Most studies focused on the regulation of the transcripts that participated in the maintenance and modulation of the stemness and self-renewing CSCs that are thought to be responsible for complex tumor heterogeneity, cancer progression and therapeutic resistance.59,60,61 SOX2 is one of the major regulators in tumor initiation and cancer stem-cell functions.62 One Study showed that m6A writer METTL3 interacted with the 3′UTR of SOX2 mRNA and lead to methylation and stabilization of SOX2 mRNA in glioma stem-like cells (GSCs). The enhanced expression of METTL3 increased SOX2 expression, which maintained the stemness and radioresistance of GSCs.63 Interestingly, METTL3 is a multifunctional protein that not only has the activity of transmethylation, but can also regulate the mRNA translation in cytoplasm. In lung cancer, METTL3 promoted translation of oncogenic mRNA, epidermal growth factor receptor (EGFR), independent of its catalytic activity. METTL3 shuttled from nucleus to cytoplasm and interacted with ribosomes. Such interactions promoted EGFR mRNA translation leading to cell proliferation, survival, and invasion of cancers.64
The process of “reading” and “erasing” of m6A methylation marks are essential for regulation of genes that are responsible for drug resistance. In acute myeloid leukemia (AML), YTHDF2 is overexpressed and is required for disease initiation. Deletion of reader YTHDF2 compromises LSC development and propagation by increasing the half-life of tumor necrosis factor receptor 2 (TNFR2). Importantly, YTHDF2 is not essential for normal hematopoietic stem cells, indicating that YTHDF2 is a unique therapeutic target which specifically inhibits LSCs.65 In cervical cancer, FTO was found to induce DNA repair activity and drug resistance to chemoradiotherapy by increasing β-catenin mRNA through m6A methylation.66
Recently, inhibition of m6A eraser was found to be a potential strategy for overcoming drug resistance. Inhibition of m6A demethylases FTO and ALKBH5 was found to effectively overcome PARP inhibitor resistance in BRCA-mutated epithelial ovarian cancers.67 Mechanistically, deletion of m6A erasers increased FZD10 mRNA m6A modification and led to stabilization of FZD10 and upregulation of the Wnt/beta-catenin signaling pathway.67
In some cases, METTL3 promotes drug resistance through various pathways simultaneously. For example, METTL3 increased the mRNA of YAP, an effector of Hippo signaling pathway, leading to promotion of castration resistance in direct and indirect manners. On one hand, METTL3 prevented the degradation of YAP mRNA caused by miR-1914-3p via increasing the m6A modification of a noncoding RNA MALAT1 which was an RNA sponge of miR-1914-3p. On the other hand, METTL3 promoted YAP mRNA translation by recruiting YTHDF1/3 and EIF3b to the translation initiation complex machinery.68 Interestingly, a recent study revealed a crucial role of the m6A regulator in response to immunotherapy. Anti-PD-1 checkpoint blockade therapy had been proven as an important therapy for melanoma. However, more than 50% of patients didn’t show a durable response to immunotherapy. Inhibition of FTO increased m6A methylation in PD-1, leading to PD-1 mRNA decay through the m6A reader YTHDF2, eventually, sensitized melanoma to anti-PD-1 treatment. This study provides evidence suggesting that targeting m6A methylation regulator is a novel strategy for sensitizing immunotherapy.69
Noncoding RNA modification in cancer drug resistance
The process of eukaryotic transcription refers to the genetic information being transformed from DNA to RNA.70 Subsequently, the message RNA (coding RNA) is translated into protein and the noncoding RNA performs function of posttranscriptional modification. The linear noncoding RNAs can be divided into three categories: miRNA, siRNA, and piRNA with the lengths of <50 nt; rRNA, tRNA, and snRNA with the lengths from 50 to 500 nt; and Long noncoding RNA (LncRNA) with the length of more than 500 nt.71 LncRNA can directly interact with target genes to activate or inhibit the expression of target genes. In addition, it can also act as competitive endogenous RNA (ceRNA) to interact with miRNA and participate in the regulation of gene expression.72,73 Because of the similar structures between lncRNA and mRNA, miRNA may negatively regulate lncRNA expression through a mechanism similar to mRNA.74 Unlike other linear RNAs, circular RNAs are closed circular structures, which lack the 5′ and 3′ ends and their expressions are more stable. Functional studies have revealed that circRNAs can release the inhibitory effect of miRNA on its target genes by acting as miRNA sponge.75,76 Currently, noncoding RNAs have attracted a lot of attention as potential targets of drug resistance in cancers due to their functions in cell proliferation, metastasis, and EMT (Fig. 2).77,78,79,80 In this section, we will focus on the role of miRNAs, LncRNAs, and circRNAs in chemoresistance.

The functions of noncoding RNAs in cancer drug resistance. LncRNA can directly interact with target genes, or act as ceRNA to interact with miRNA to participate in gene expressions; circRNAs can act as “miRNA sponge” to release the inhibitory effect of miRNA on its target genes. The noncoding RNAs could be potential targets of drug resistance in cancers due to their functions in cell proliferation, metastasis, and EMT
miRNAs and cancer drug resistance
Recent studies have shown that miRNAs are involved in cisplatin (DDP)-mediated cancer drug resistance. Ubiquitin-conjugating enzyme E2C (UBE2C) is an active proto-oncogene and highly expressed in DDP-resistant NSCLC cells. miR-495 targeted 3′UTR of UBE2C, which acted as a transcriptional factor and downregulated the expression of cancer drug resistance associated genes such as ABCG2 and ERCC1, thus reversing DDP resistance in NSCLC cells via reducing EMT, cell migration, and invasion.81 Another miRNA, miR-146b could bind to protein tyrosine phosphatase 1B, inhibit the EMT process, and reduce cisplatin resistance in human lung adenocarcinoma (LUAD) cells.82 Analysis of miRNA expression profiles and experiments in cisplatin-resistant and -sensitive ovarian cancer cells showed that miR-141 was overexpressed in cisplatin-resistant cells. miR-141 could directly target KEAP1, an oxidative stress regulator, and induced cisplatin resistance in ovarian cancer cells by activating NF-κB pathway.83 Exosomes are vesicles with a diameter of 40–100 nm. They play important roles in regulating tumor microenvironment, metastasis, and drug resistance by promoting transportation of mRNAs and miRNAs.84,85 The cancer-associated fibroblasts derived exosomal miR-196a accelerated head and neck cancer cell proliferation and cisplatin resistance through downregulating expression of target genes: CDKN1B and ING5.86 miR-936 suppressed cell proliferation, migration in glioma87, and non-small cell lung cancer,88 and induced drug resistance to cisplatin and DOX via targeting G protein-coupled receptor 78 (GRP78) in laryngeal squamous cell carcinoma cells.89
In addition, other EGFR-TKIs involved in drug resistance through regulating miRNAs expressions. Notch receptor 3 (NOTCH3) is highly expressed in LUAD and gefitinib-resistant cells. MiR-150 decreased IC50 of gefitinib, downregulated the expressions of target gene NOTCH3, which was positively correlated with collagen 1A1 expression, providing a potential therapeutic target for LUAD treatment.90 Bone morphogenetic protein 4 (BMP4) accelerates cancer cell energy metabolism and is upregulated in the EGFR-TKI-resistant cells. However, low-expression of miR-139-5p was found in TKI-resistant cells and combination of miR-139-5p and yuanhuadine significantly suppressed BMP4 expression and tumor growth in the resistant NSCLC cells and cell-derived xenograft (CDX) mouse model.91 Microarray analysis revealed that miR-214-3p was significantly decreased in multidrug resistant cells. MiR-214-3p directly targeted ABCB1 and XIAP, promoted cell apoptosis, and sensitized retinoblastoma cells to multiple chemotherapeutic drugs. Overexpression of ABCB1 or XIAP could reverse chemoresistance induced by miR-214-3p.92 Tao et al. showed that miR-451a promoted the sensitivity of lung cancer cells to DOX via targeting c-myc to reduce expression of N-cadherin and Vimentin and enhance expression of E-cadherin.93 Han et al. revealed that miR-552 could promote the self-renewal, tumorigenesis. and sorafenib resistance via activating protein kinase B (AKT) phosphorylation in liver tumor-initiating cells (T-ICs).94
LncRNAs and cancer drug resistance
A number of studies have identified the function of lncRNAs in cancer drug resistance via various methods. A study using CRISPR activation of lncRNA system was developed to target 14,701 lncRNA genes in cytarabine-resistant acute myeloid leukemia and found that lncRNA GAS6-AS2 promoted cytarabine resistance via activating GAS6/TAM signaling pathway.95 Another study using whole-exome sequencing and transcriptional profiling in cetuximab-resistant cells in three-dimensional culture showed that lncRNA MIR100HG-derived miR-100 and miR-125b were overexpressed in cetuximab-resistant head and neck squamous cell carcinoma and colorectal cancer cells.96 RNA sequencing of drug-resistant and drug-sensitive NSCLC cells revealed that lncRNA ATP2B1 (or lncRNA HUWE1)-miR-222-5p-TAB could be the potential ceRNA regulatory network, which involved in drug resistance in NSCLC cells.97 LncRNA MSTRG51053.2 may act as the ceRNA for miR-432-5p in cisplatin resistance via targeting target genes such as MGST1, MGST3, GST-ω1, and ABCG2 in NSCLC cells.98 LncRNA MALAT1-miR-22-3p-ZFP91 axis could promote oxaliplatin resistance in gastric cancer cells.99 LncRNA KCNQ1OT1 induced the chemoresistance to DOX in acute myeloid leukemia by targeting TSPAN3 through sponging miR-193a-3p.100
Autophagy is a process which can be divided into three phases: (1) cells engulf and encapsulate cytoplasmic proteins or organelles into vesicles; (2) vesicles fuse with lysosomes to form autophagy lysosomes; and (3) autophagy lysosomes degrade the contents, recycle amino acids, fatty acids, and nucleotides.101,102 Eventually, autophagy achieves the goal of renewal of damaged cell organelles, misfolded proteins, and provides nutrients and energy for cells.103,104 Some studies have shown that lncRNAs accelerate cancer drug resistance via modulating the expressions of autophagy-associated genes. Knockdown of LncRNA-HOTAIR could promote the sensitivity of crizotinib in NSCLC cells via suppressing autophagy and ULK1 phosphorylation.105 LncRNA LINC00160 upregulated the expressions of autophagy-associated proteins such as LC3I/II and ATG5, and then induced autophagy and drug resistance in hepatocellular carcinoma cells via miR-132-PIK3R3 axis.106 LncRNA MALAT1 induced autophagy and facilitated the resistance to cisplatin in gastric cancer via miR-30b-ATG5107 or miR-23b-3p-ATG12108 or in hepatocellular carcinoma cells via HIF-2α-MALAT1-miR-216b.109 The lncRNA SNHG family has recently been implicated in the modulation of drug resistance. SNHG6-miR-26a-5p-ULK1 axis could promote colorectal cancer cell resistance to 5-fluorouracil through inducing autophagy.110 Another member of SNHG family, lncRNA SNHG1 also contributed to sorafenib resistance by activating AKT pathway and inducing autophagy via sponging miR-21 in hepatocellular carcinoma cells.111 In addition, lncRNA SNHG14 induced the expressions of autophagy-related proteins such as RAB5A and ATG4D via sponging miR-101 and then enhanced the gemcitabine resistance in pancreatic cancer (Fig. 3).112

An illustration of the process of autophagy and the roles of lncRNAs in drug resistance via autophagy. The cells engulf and encapsulate cytoplasmic proteins or organelles into vesicles, and then vesicles fuse with lysosomes to form autophagy lysosomes, subsequently, autophagy lysosomes degrade the contents, recycle amino acids, fatty acids, and nucleotides. LncRNA MALAT1, and SNHG family could facilitate drug resistance via inducing autophagy and activating expressions of autophagy-related proteins
CircRNAs and cancer drug resistance
Circular RNA can relay the inhibitory effect of miRNA on its target genes by acting as miRNA sponge. There is a growing evidence that circRNAs accelerate cancer drug resistance.113 CircAKT3 is highly expressed in cisplatin-resistant gastric cancer cells. Huang et al. showed that circAKT3 upregulated PIK3R1 expression via sponging miR-198 and promoted cisplatin resistance in gastric cancer.114 CircPAN3 induced DOX resistance in acute myeloid leukemia via regulating autophagy-associated AMPK/mTOR signaling pathway and protein expressions of LC3I/II and p62115 or miR-153-5p/miR-183-5p-XIAP axis.116 Hsa_circ_0079662 interacted with hsa-miR-324-5p as the ceRNA and enhanced the resistance to oxaliplatin via TNF-α pathway in human colon cancer.117 Hsa_circ_0060060 accelerated expressions of autophagy marker LC3 and p62 through miR-144-3p/TGF-α axis and promoted cisplatin resistance in papillary thyroid carcinoma and anaplastic thyroid carcinoma cells.118 In addition, circCELSR1 was upregulated in paclitaxel-resistant ovarian cancer cells. Inhibition of circCELSR1 enhanced paclitaxel-induced cytotoxicity via upregulating FOXR2 expression or acting as the sponge for miR-1252 and increased cell apoptosis.119 Dong et al found that circ_0076305 was upregulated in NSCLC and promoted cisplatin resistance in NSCLC by upregulating STAT3 via targeting miR-296-5p.120
However, some studies indicated that circRNAs increased cancer drug sensitivity. For example, Li et al. showed that circ_0002483 enhanced paclitaxel sensitivity in NSCLC by targeting GRB2, FOXO1, and FOXO3 via miR-182-5p.121 Sang et al. found that Hsa_circ_0025202 could inhibit tumor progression and enhance the sensitivity of cancer cells to tamoxifen in breast cancer via targeting miR-182-5p/FOXO3a axis.122 Moreover, Liang et al. indicated that decreased expression of circKDM4C in breast cancer suppressed DOX resistance through miR-548p/PBLD pathway.123 The roles and the molecular mechanisms of noncoding RNAs in cancer drug resistance are outlined in Table 2.
Protein modification in cancer drug resistance
PTM refers to the enzymatic modification after the biosynthesis of proteins, which is crucial for regulating and maintaining the functions of proteins. A large portion of human proteins have gone through at least one round of PTM after being synthesized. The PTM status of human proteins retrieved from Uniprot database (www.uniprot.org) is summarized and illustrated in Fig. 4. Among different types of PTMs, phosphorylation is the most common one, with 7977 human proteins containing 40,694 phosphorylation sites, and serine is the most common phosphorylated amino acid. Among these 7977 proteins, the top-five-enriched GO biological processes are “organelle organization,” “cell localization,” “regulation of cellular component organization,” “positive regulation of metabolic process,” and “establishment of localization in cell.” Acetylation ranks the second, with 3379 human proteins containing 6604 acetylation sites, and lysine is the most preferred acetylation amino acids. Among these 3379 proteins, the top-five-enriched GO biological processes are “organelle organization,” “cellular catabolic process,” “mRNA metabolic process,” “catabolic process,” and “organic substance catabolic process.” Ubiquitination ranks the third, with 1025 proteins being ubiquitinated. Among these 1025 proteins, the top-five-enriched GO biological processes are “cellular protein modification process,” “protein modification process,” “macromolecule modification,” “protein modification by small protein conjugation or removal,” “positive regulation of metabolic process.” Methylation ranks the fourth, with 966 proteins containing 1216 methylation sites, and asparagine is the most ordinary methylation amino acid. Among these 966 proteins, the top-five-enriched GO biological processes are “mRNA metabolic process,” “organelle organization,” “small GTPase-mediated signal transduction,” “mRNA processing,” and “Ras protein signal transduction.” Glycosylation ranks the fifth, with 811 proteins containing 4534 glycosylation sites, and asparagine is the most preferred glycosylation amino acid. Among these 811 proteins, the top-five-enriched GO biological processes are “biological adhesion,” “cell adhesion,” “extracellular structure organization,” “immune system process,” and extracellular matrix organization.” The mechanisms of PTMs in chemoresistance can either be direct, in which the modifications disrupt binding sites; or indirect, in which upstream modifications lead to pathway blockage. In this section, we will briefly discuss the role of PTM in chemoresistance from the perspectives of drug inactivation/efflux, drug target modifications, DDR, cell death resistance, EMT and metastasis. Some of the reported protein targets causing chemoresistance are listed in Table 3.

The PTM status of human proteins. All data is retrieved from Uniprot database and updated as of 2015-05. The number of proteins with different types of PTMs are illustrated in the barplot (left); the percentages of amino acids modified in each type of PTMs are illustrated in the circle plots (right)
Drug inactivation
Certain drugs need metabolic modification to become their active forms, and some cancer cells have developed the ability to modify/shut down these activation processes or to use other processes to deactivate the active forms of drugs. For example, capecitabine, under the brand name Xeloda, is a widely used chemotherapeutic drug in the treatment of breast cancer, gastric cancer, and colorectal cancer.124 Inside the human body, capecitabine needs to be metabolized into 5-FU, a thymidylate synthase inhibitor which interrupts the process of pyrimidine thymidine synthesis.125 This process is mediated through three enzymes, carboxylesterases (CES), cytidine deaminase (CDA), and thymidine phosphatase (TYMP), as detailed in Fig. 5a. TYMP is a dual-role enzyme in cancer development and treatment: on one side, after phosphorylated by protein kinase C, TYMP is capable of converting doxifluridine (5-dFUR), an intermediate product of capecitabine, to its active form, 5-FU, which is effective in cancer treatments;126 on the other side, TYMP has been found as a pro-oncogene in many studies, where it is overexpressed in many types of cancers, and the overexpression of TYMP promotes tumor angiogenesis and inhibits apoptosis.127 As for CES and CDA, although there havn’t been any direct evidence, the changes of catalytic activity of CES and CDA genes in some cancer patients with SNPs might be related to PTMs.128,129 Aside from mutations, CES is also positively regulated by p53, a well-known tumor repressor, which is often mutated or posttranslational modified in cancer cells (will be discussed in the following context),130 hence the downregulation of CES expression in cancers can result in chemoresistance.

The mechanisms of PTMs in cancer cell chemoresistance. a The inactivation of Capecitabine through the regulation of CES, CDA, TYMP, and DPD enzymes. b The drug efflux process mediated by ABC transporter proteins and the modifications of these transporters. c The modification of ERBB receptors through mutations and PTMs resulting in multiple drug resistance. d DNA damage repair system in cancer cells could also result in drug resistance, and this process is mediated through the repression of ATM and ATR, as well as p53 proteins, and induction of specialized DNA polymerases, such as Poly beta, kappa and zeta. e The repression of apoptosis in cancer cells, which is mainly achieved through the inhibition of p53 via either mutation or PTMs. The overexpression of MCL-1/BCL-2 and repression of BAX/BAK proteins also contribute to this process. f The repression of autophagy in cancer cells. MTORC1 is triggered through PI3K-AKT pathways, which further inhibits the phosphorylation of ULK1, and impedes the autophagy process. g The activation of EMT in cancer cells. EMT process is triggered through multiple signaling pathways including TGFβR and WntR, which activate SNAIL and TWIST transcription factors. These EMT-TFs repress the expression of E-cadherin and promote the expression of N-cadherin, vimentin, and fibronectin, which further promote EMT process
Aside from impeding the activation process, deactivation also contributes to drug resistance. For example, dihydropyrimidine dehydrogenase (DPD) is a key enzyme in pyrimidine catabolism,131 and is capable of reducing the pyrimidine double bonds of 5-FU, and converting it to dihydrofluorouracil. This process deactivates 5-FU and also results in chemoresistance. In certain type of cancer (e.g., head and neck squamous cell carcinoma), DPD is overexpressed, and overexpression of DPD is one of the main reasons causing 5-FU resistance.132
Drug efflux
Efflux is the process of moving a variety of compounds out of cells, which is mediated by some ATP-binding cassette (ABC) transporters, which are also named multiple drug resistance (MDR) proteins. Among them, P-glycoprotein, encoded by ABCB1 (MDR1), is one of the most widely studied transporters.133 P-glycoprotein is highly expressed in many drug-resistant tumors and is involved in the efflux of many anticancer drugs such as anthracycline, daunorubicin, epipodophyllotoxins, and others (Fig. 5b).134,135,136 The regulation of P-glycoprotein is explored in many studies: Takada et al. showed that the expression of P-glycoprotein was positively regulated by MAPFK signaling pathways in human breast cancer;137 Henrique et al. found that ABCB1 was epigenetically regulated through posttranslational histone modification in prostate cancer;138 Xie et al. revealed that Pim-1 could phosphorylate P-glycoprotein, which protects P-glycoprotein from degradation and enables its glycosylation.139 Overexpression of Pim-1 in many human cancers indirectly contributes to P-glycoprotein-mediated drug-efflux and chemoresistance.140 The Pim-1 inhibitor SGI-1776 was reported to overcome P-gp-mediated drug resistance.141
In terms of 5-FU, although there have been activation and deactivation processes in many cancer types, drug efflux also contributes to the chemoresistance (Fig. 5b). 5-FU and its downstream product fluorodeoxyuridylate (FdUMP) can be pumped out of cells through several transporters such as ABCC3/4/5/11.142,143,144 PTMs play important roles in these ABC transporter-mediated drug resistance, e.g., phosphorylation directly affects the efficiency of transporters;145 glycosylation influences the stability of transporters and protects them from proteases.146
Drug target modification
Most of the cancer drugs aim for some specific proteins, causing structural changes of targets, leading to the blockage of certain pathways, and resulting in death of cancer cells. To resist these effects, many cancer cells alter the target proteins either by decreasing/halting their expression or modifying their structures to hinder the binding process (Fig. 5c). For example, EGFR, one of the most intensively studied receptor-tyrosine kinases (RTKs), was found to have a variety of mutations and PTMs that resulted in multiple chemoresistance in many different types of cancers: T790M mutation was directly related to gefitinib and erlotinib resistance in non-small cell lung cancer;147 lack of glycan sialyation due to K521 polymorphism resulted in cetuximab resistance in head and neck cancers;148 methylation of R1175 by PRMT5 modulated EGF-induced phosphorylation at W1173, which further promotes ERK activation in breast cancer;149 SRC kinase-mediated EGFR ubiquitination, and degradation in colorectal cancer.150 Aside from EGFR, other ErbB family members also showed alterations in many cancers. Phosphorylation by PKB inhibited the activation of HER2 in breast cancer and resulted in resistance to herceptin;151,152 neural precursor cell expressed and developmentally downregulated 4, an E3 ubiquitin ligase, mediated the degradation of HER3 in prostate cancer through ubiquitination.153
Cancer cells could also use alternative pathways to bypass or compensate for drug actions. For example, VanMeter et al. revealed an alternative mechanism of downstream protein activation, which is independent of EGFR in non-small cell lung cancer;154 Sergina et al. showed that overexpression of HER3 could lead to a compensatory shift in phosphorylation-dephosphorylation equilibrium, resulting in gefitinib resistance;155 Canfield et al. found that HER4 was upregulated in HER2+ breast cancer with lapatinib resistance, and knockdown of HER4 significantly decreased ATK phosphorylation levels, indicating the bypassing receptor function of HER4 in lapatinib-resistant cells.156
DNA damage repair (DDR)
Many cancer drugs can cause direct or indirect DNA damages, and the removal of these DNA lesions leads to cell survival. DDR system could induce cell-cycle arrest, leading to DNA repair, which protects the cell or leads to cell apoptosis and results in cell death (Fig. 5d). Increasing DNA repair and damage tolerance as well as evading apoptosis were the potential mechanisms of chemotherapy resistance in cancer cells.157 After chemotherapeutic treatment, DDR could induce a rapid but faulty repair mechanism, and the tolerance of DNA damages were achieved through several specialized DNA polymerases, such as poly beta, kappa, and zeta, that had low fidelity in DNA duplication and resulted in mutations.158,159 The overexpression of these specialized polymerases in chemoresistant cancers has been revealed in many studies. For example, elevated expression of Poly beta had been found in drug-resistant cancer cells,160 and knockdown of Poly beta by siRNA resensitized cancer cells to cisplatin;159 upregulated poly kappa had also been examined in lung cancer161 and inactivation of p53 promoted the expression of poly kappa.162 The genomic instability caused by tolerance of mutations was one of the main features of cancer.163
Ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) kinases are activated by stresses of DNA double strand breaks and single strand breaks. Activation of ATM/ATR induces cell-cycle arrest (through CHK1 and CHK2) and cell apoptosis (through p53). In many cancer cells, the expression of ATM/ATR is downregulated164 and the activity of ATM/ATR is also reduced by upregulating WIP1 phosphatase, which dephosphorylates ATM/ATR and its substrates, e.g., p53.165 Interestingly, in certain types of cancers, where ATM/ATR is uncoupled from cell-cycle arrest and apoptosis, the overexpression of ATM/ATR has been found in many studies, which proves the importance of ATM/ATR in chemoresistance.166
Cell death resistance
A significant hallmark of cancer cells is the ability of resisting cell death,167 hence evading apoptosis and autophagy is one of the most important abilities of cancer cells. In normal cells, apoptosis is induced either through extrinsic or intrinsic signaling pathways. In cancer cells, the components in both extrinsic and intrinsic signaling pathways are either mutated or mis-regulated, therefore apoptosis process is impeded (Fig. 5e). For example, extrinsic apoptosis pathways were triggered by surface death receptors such as FAS, DR4/5, and in many cancers those death receptors were often mutated or PTM modified,168 that greatly impeded apoptosis process. Moreover, some decoy receptors such as TRID and TRUNDD were overexpressed in certain cancers,169 which repressed extrinsic signaling pathway-induced apoptosis. Intrinsic signaling pathways are mainly induced by p53, which is often mutated or modified in cancer cells, as reviewed in Mansoori et al.,170 resulted in inhibiting the intrinsic apoptosis process from very beginning. Aside from p53 mutation, p65 subunit of nuclear factor-kappa B, is one of the regulators of tumorigenesis, and the suppression of p65 signaling could enhance the DOX-induced apoptosis in cervical cancer.171 MCL-1/BCL-2, the anti-apoptotic proteins, was found to be overexpressed in many types of cancers.172,173 BAX, the pro-apoptotic protein, could be phosphorylated by AKT at residue 184 in breast cancer, which prevented it from entering into mitochondria, resulting in chemoresistance.174 Caspase 3, the executioner of apoptosis, was phosphorylated by p38-MAPK, which was negatively regulated by TYMP, and the overexpression of TYMP in many cancers helped cancer cell to evade apoptosis and contributed to chemoresistance.127 Jaime-Sánchez et al.175 showed that EL4 cells overexpressing Bcl-XL or DNC3 (a dominant negative form of caspase 3) proteins exhibited multidrug resistance such as DOX, and these EL4 cells could be eliminated by antigen-specific primed cytotoxic T cells. Recently, Hu et al.176 have showed that culling-ring ubiquitin ligase 4 (CRL4) could regulate the expression of BIRC3 (one of the inhibitors of apoptosis proteins) through STAT3 pathway, and BIRC3 is associated with cisplatin-resistance in ovarian cancer cells, suggesting the potential functional role of CRL4 and BIRC3 as novel therapeutic targets for cisplatin-resistant patients.
Autophagy suppresses tumor cells via lysosomal degradation pathway, and this process is mediated through ULK1 complex and VPS34 complex. ULK1 functions in complex with ATG13 and FIP2000,177 which further phosphorylates AMBRA1 and BECN1 and leads the translocation of VPS34 complex to ER and initiates autophagy process.178 In cancer cells, MTORC1 is activated through the PI3K-AKT pathway. Activated MTORC1 phosphorylates ULK1 on S758, which inhibits the function of ULK1 and hence impedes autophagy process. Moreover, mutations in MTOR, as well as upstream or downstream signaling components, conferring constitutive activation of MTOR signaling have also been reported in many studies.179,180
EMT and cancer metastasis
EMT refers to the process of converting epithelial cells into mesenchymal cells. Tumor metastasis is the process of primary tumor cells entering the bloodstream or lymph vessels and settling at other sites. Classically, EMT is considered as a promoter of metastasis, during which cancer cells acquire mobility and the capacity to migrate away from the primary site.181 EMT is triggered and regulated by a complex network as summarized in Thiery and Sleeman182 and executed by EMT transcription factors (EMT-TFs) consisted of SNAIL and TWIST family members.183 These EMT-TFs repress the expression of E-cadherin, which is a glycoprotein helping in epithelial cell anchorage, and stimulate cells to gain mesenchymal markers, such as N-cadherin, vimentin, and fibronectin.
One of the main mechanisms of EMT-mediated chemoresistance is through E-cadherin. Thomson et al.184 found that NSCLC cells with E-cadherin expression showed greater sensitivity to EGFR kinase inhibition (such as erlotinib), and cells overexpressed vimentin/fibronectin after EMT were insensitive to EGFR inhibition. Many EMT-TFs, such as SNAIL1, SNAIL2, and TWIST, possess the ability of repressing the expression of E-cadherin. The activity of these TFs is mainly regulated through a number of PTMs. For example, Snail was phosphorylated by GSK3β, which led to its cytosolic localization and β-Trcp-mediated ubiquitination.185 SNAIL was phosphorylated by LATS2 and PAK1, which promoted its nuclear transport and increased its stability when GSK3β was inhibited by RTK or WntR signaling pathways.186,187 Similarly, TWIST was acetylated by p300/CBP-associated factor, which regulated its intracellular location and transcriptional activity in urothelial cancer cells.187
Besides E-cadherin, EMT-TFs could induce chemoresistance through other intermediates. For example, TWIST could transcriptionally upregulate the expression of AKT2, which induced paclitaxel resistance in breast cancer, and this resistance was reduced with the silence of AKT2;188 TWIST could also repress the expression of estrogen receptor-α (ER) together with histone deacetylase 1 in breast tumors, which might contribute to the hormone-resistance in ER-negative breast cancer.189
Epitranscriptomic and epiproteomic modifications as therapeutic targets
Epitranscriptomic modifications
The roles of mRNA and noncoding RNA modification in drug resistance indicated that epitranscriptomic modifications could be the potential therapeutic targets in cancers (Fig. 6). Knockdown of METTL3 in pancreatic cancer showed significantly increased sensitivity to drugs such as 5-fluorouracil, cisplatin, gemcitabine, and irradiation.190 The downregulation of m6A demethylases FTO and ALKBH5 increased FZD10 m6A methylation and reduce sensitivity to PARPi in BRCA-mutated epithelial ovarian cancers.67 Zhu et al. showed that expression of miR-506-3p was decreased in gefitinib-resistant PC-9R cells, and miR-506-3p mimic could reverse the resistance of NSCLC cells to gefitinib through targeting YAP1.191 MiR-1269b directly targeted PTEN to activate PI3K/AKT signaling pathway and miR-1269b inhibitor could overcome cisplatin resistance in human NSCLC cells, speculating that miR-1269b could be a potential target for treatment of NSCLC.192 MiR-186 decreased TWIST1 expression via reversing mesenchymal-to-epithelial transition phenotype and resensitized ovarian cancer cells to cisplatin.193 Another miRNA, miR-20b could target ADAM9 to decrease the 5-FU resistance in colon cancer.194 MiR-153 directly targeted ABCE1 to inhibit gefitinib resistance in lung cancer, which may provide a therapeutic target to reverse the resistance to gefitinib in the future.195

Epitranscriptomic and epiproteomic modifications could be the potential therapeutic targets in cancers. The critical genes and proteins in both modifications could reverse the resistance of cancer cells to chemotherapeutic drugs such as 5-fluorouridine, EGFR-TKI, and cisplatin
LncRNA CCAT1 was overexpressed in esophageal cancer and its knockdown significantly repressed the tumor growth and promoted the sensitivity of cisplatin via miR-143/PLK1/BUBR1 axis, which suggested that CCAT1 could be a potential therapeutic target to overcome the cisplatin resistance in esophageal cancer.196 The depletion of lncRNA UCA1 attenuated the activity of Wnt/β-catenin signaling pathway and increased the tamoxifen sensitivity in breast cancer.197 Knockdown of lncRNA XIST could sensitize colorectal cancer to DOX via miR-124/SGK1 axis, which indicated that lncRNA XIST could be a potential therapeutic target to overcome chemoresistance in colorectal cancer.198 Silencing to lncRNA SNHG15, a bifunctional MYC-regulated noncoding RNA, could inhibit the cancer progression in CRC cells and promote the sensitivity of CRC cells to 5-FU, indicating that lncRNA SNHG15 could be a potential prognostic marker for chemotherapy.199 Expression of circRNA_101505 was downregulated in cisplatin-resistant hepatocellular carcinoma cells, and cisplatin toxicity was enhanced by circRNA_101505 sponging miR-103/NOR1 pathway.200
Epiproteomic modifications
Various studies have indicated that targeting epiproteomic modifications can ameliorate drug resistance in cancers (Fig. 6). Histone methyltransferase SMYD2 promoted tumor progression in renal cell carcinoma, and combination treatment with SMYD2 inhibition and anticancer drugs significantly reduced the tumor volumes and weights, suggesting that SMYD2 could be a potential target for RCC treatment.201 PI3K/mTOR inhibitor and EGFR repression played the coordinated role in animal survival and gefitinib-targeted therapy in malignant glioma.202 NGI-1, an inhibitor of oligosaccharyltransferase could block the interaction between MET and EGFR, resulting in increasing sensitivity to gefitinib and osimertinib in EGFR mutated NSCLC cells.203 AM-1-124 specifically targeted STAT3 and downregulated STAT3 phosphorylation overcame drug resistance in TKI-resistant chronic myeloid leukemia cells.204 TC-N19, which is the dual inhibitor of EGFR and cMET, degraded both proteins via ubiquitin proteasome pathway and overcame gefitinib resistance in NSCLC cells.205 Qi et al. found that the phosphorylation of ERK increased in gefitinib-resistant NSCLC cells and the inhibition of ERK phosphorylation reversed gefitinib resistance via suppressing autophagy in lung cancer.206 Knockdown of ubiquitin-specific peptidase 8 (USP8) decreased the phosphorylation of EGFR, c-MET, ERBB2, and ERBB3, and a synthetic USP8 inhibitor displayed a smaller tumor size and a reversed gefitinib resistance in H1975 CDX model.207 WZB117, a specific inhibitor of Glut1, significantly increased the 5-FU resistance and could be used as the potential treatment in patients with 5-FU-resistant colon cancers.208 Protein tyrosine phosphatase receptor J (PTPRJ) was downregulated in human cervical tumor tissues and inhibition of PTPRJ could have promoted the resistance to 5-FU through activating JAK1/STAT3 pathway.209 Histone methyltransferase NSD2 mediated BCL-2 and SOX2 H3K36me2 modification and activated the levels of p-ERK and p-AKT in osteosarcoma. Knockdown of NSD2 induced cell apoptosis and led to the enhancing sensitivity of osteosarcoma to cisplatin.210 WP1130, the USP9x inhibitor, induced the degradation of transcription factor PBX1 and accelerated cell apoptosis in prostate cancer, which provided a new idea for prostate cancer treatment.211
Conclusions and future perspectives
Epigenetic alternations in epitranscriptomic and epiproteomic modifications play critical roles in cancer treatment and drug resistance. Future researches on the function of epigenetics in vivo and its effect on conquering drug resistance are warranted. For example, even though emerging evidences have indicated that m6A regulators play important roles in cancer drug resistance by modulating the epitranscriptome, the research on m6A methyltransferase family is mainly focused on METTL3 and METTL14, and the underlying molecular mechanisms of other m6A methyltransferase members in drug resistance need to be further investigation. Recently, miRNAs have been discovered in exosomes, a structure that contained abundant genetic information and widely distributed in various body fluids. The potential advantages of miRNAs in exosomes imply that the roles in cancer drug resistance are worthy explored. The mutations related to cancer drug resistance have been identified, such as EGFR T790M and C797S. However, the relationship between mutations and protein expressions is not fully consistent. Temporal proteomic is being used as an emerging technology to target drug resistance and researchers showed that the combination of KRASi and HSP90 inhibitor (17-AAG) or cell-cycle inhibitor (CDK4/6i) could block cell growth and inhibit cancer drug resistance,212,213 which suggested that proteomic can be used as an effective treatment strategy for overcoming cancer drug resistance. Overall, targeting epigenetic alternations may improve cancer treatment and provide new approaches in overcoming drug resistance.
The dynamic intratumoral heterogeneity and the increased clonal repopulation are the main causes of cancer acquired resistance to platinum-based chemotherapy. Single-cell RNA-seq (scRNA-seq), the technology which allows transcriptomic analysis in individual cells, can dissect the heterogeneity and subpopulations in tumor microenvironment during cancer drug resistance.214 More attempts have been made to reveal the mechanism of drug resistance in cancers by scRNA-seq.215,216,217 For example, scRNA-seq from paclitaxel-sensitive and -resistant esophageal squamous cell carcinoma (ESCC) identified the subpopulations of paclitaxel-resistant ESCC cells, and research on the mechanism revealed the carfilzomib, a proteasome inhibitor, could reverse the paclitaxel resistance via activating the HIF-1 pathway.218 The transcriptome mapping of cisplatin-resistant tumor cells by scRNA-seq uncovered a novel gene COX7B, and inhibition of COX7B reduced the sensitivity of cisplatin, which provided the valuable insights into chemosensitivity in cancers.219
An era of single-cell omics has arrived, and the future clinical applications based on epitranscriptomics and epiproteomics are very promising: (i) single-cell multiple omics sequencing technique can be used widely to analyze the transcriptome, proteome, epitranscriptome, and epiproteome simultaneously at the single-cell level in drug resistance cancer cells, which allows us to reveal the unknown mechanisms and targets;220,221 (ii) personalized single-cell sequencing provides comprehensive clues to optimize the therapeutic strategy against relapsing cancers;222 and (iii) application of single-cell sequencing on tumor liquid biopsy can surveil and prevent the drug-resistant events during therapeutic treatment.223
References
Choi E. K. et al. Body mass index and 20 specific cancers: re-analyses of dose-response meta-analyses of observational studies. Ann. Oncol. 29, 749–757 (2018).
Bray F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Saitoh M. Involvement of partial EMT in cancer progression. J. Biochem. 164, 257–264 (2018).
Ayati A. et al. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 99, 103811 (2020).
Sio, T. T., Ko, J., Gudena, V. K., Verma, N. & Chaudhary, U. B. Chemotherapeutic and targeted biological agents for metastatic bladder cancer: a comprehensive review. Int J. Urol. 21, 630–637 (2014).
Holohan, C., Van Schaeybroeck, S., Longley, D. B. & Johnston, P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013).
Liu K. et al. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 19, 54 (2020).
Kang, K. A. & Hyun, J. W. Oxidative stress, Nrf2, and epigenetic modification contribute to anticancer drug resistance. Toxicol. Res. 33, 1–5 (2017).
Dominissini, D. & Rechavi, G. Epitranscriptome regulation. Nat. Struct. Mol. Biol. 28 (2018).
Stram, A. R. & Payne, R. M. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol. Life Sci. 73, 4063–4073 (2016).
Rape M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 19, 59–70 (2018).
Han, Z. J., Feng, Y. H., Gu, B. H., Li, Y. M. & Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 52, 1081–1094 (2018).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Kelly, A. D. & Issa, J. J. The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 42, 68–77 (2017).
Ciesielski O. et al. The epigenetic profile of tumor endothelial cells. Effects of combined therapy with antiangiogenic and epigenetic drugs on cancer progression. Int J. Mol. Sci. 21, 2606 (2020).
Izzo, S., Naponelli, V. & Bettuzzi, S. Flavonoids as epigenetic modulators for prostate cancer prevention. Nutrients 12, 1010 (2020).
Chiappinelli, K. B., Zahnow, C. A., Ahuja, N. & Baylin, S. B. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 76, 1683–1689 (2016).
Oltean, S. & Bates, D. O. Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318 (2014).
Scotti, M. M. & Swanson, M. S. RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32 (2016).
Kahles A. et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224.e6 (2018).
Sveen, A., Kilpinen, S., Ruusulehto, A., Lothe, R. A. & Skotheim, R. I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427 (2016).
El Marabti, E. & Younis, I. The cancer spliceome: reprograming of alternative splicing in cancer. Front. Mol. Biosci. 5, 80 (2018).
Stark, M., Wichman, C., Avivi, I. & Assaraf, Y. G. Aberrant splicing of folylpolyglutamate synthetase as a novel mechanism of antifolate resistance in leukemia. Blood 113, 4362–4369 (2009).
Sotillo E. et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 5, 1282–1295 (2015).
Tripathi, V., Shin, J. H., Stuelten, C. H. & Zhang, Y. E. TGF-beta-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 38, 3185–3200 (2019).
Wang Y. et al. The BRCA1-Delta11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 76, 2778–2790 (2016).
Shi H. et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93 (2014).
Salton M. et al. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 6, 7103 (2015).
Antonarakis E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038 (2014).
Ng K. P. et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 18, 521–528 (2012).
Du Z. et al. Interferon-resistant Daudi cell line with a Stat2 defect is resistant to apoptosis induced by chemotherapeutic agents. J. Biol. Chem. 284, 27808–27815 (2009).
Bass B. L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846 (2002).
Xu, L. D. & Ohman, M. ADAR1 editing and its role in cancer. Genes 10, 12 (2018).
Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349 (2010).
Levanon E. Y. et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005 (2004).
Jiang, Q., Crews, L. A., Holm, F. & Jamieson, C. H. M. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 17, 381–392 (2017).
Han L. et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 28, 515–528 (2015).
Lazzari E. et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 8, 1922 (2017).
Ishizuka J. J. et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019).
Yang W. et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 13, 13–21 (2006).
Negi V. et al. Altered expression and editing of miRNA-100 regulates iTreg differentiation. Nucleic Acids Res. 43, 8057–8065 (2015).
Zipeto M. A. et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 19, 177–191 (2016).
Nakano, M., Fukami, T., Gotoh, S. & Nakajima, M. A-to-I RNA editing up-regulates human dihydrofolate reductase in breast cancer. J. Biol. Chem. 292, 4873–4884 (2017).
Jiang Q. et al. Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell 35, 81–94.e7 (2019).
Abrahamsson A. E. et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc. Natl Acad. Sci. U. S. A. 106, 3925–3929 (2009).
Boccaletto P. et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307 (2018).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Li, X., Xiong, X. & Yi, C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14, 23–31 (2016).
Roignant, J. Y. & Soller, M. m(6)A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 33, 380–390 (2017).
Yue, Y., Liu, J. & He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 (2015).
Dominissini D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Jia G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Zheng G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Panneerdoss S. et al. Cross-talk among writers, readers, and erasers of m(6)A regulates cancer growth and progression. Sci. Adv. 4, eaar8263 (2018).
Liao, S., Sun, H. & Xu, C. YTH domain: a family of N(6)-methyladenosine (m(6)A) readers. Genom. Proteom. Bioinform. 16, 99–107 (2018).
Meyer K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Meyer K. D. et al. 5’ UTR m(6)A promotes cap-independent translation. Cell 163, 999–1010 (2015).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322 (2020).
Zhang C. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. U. S. A. 113, E2047–E2056 (2016).
Cui Q. et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634 (2017).
Zhang S. et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606.e6 (2017).
Boumahdi S. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250 (2014).
Visvanathan A. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 37, 522–533 (2018).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016).
Paris J. et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell 25, 137–148.e6 (2019).
Zhou S. et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol. Carcinog. 57, 590–597 (2018).
Fukumoto T. et al. N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 79, 2812–2820 (2019).
Jin D. et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J. Hematol. Oncol. 12, 135 (2019).
Yang S. et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 10, 2782 (2019).
Crick F. Central dogma of molecular biology. Nature 227, 561–563 (1970).
Anastasiadou, E., Jacob, L. S. & Slack, F. J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18 (2018).
Chen, L., Zhou, Y. & Li, H. LncRNA, miRNA and lncRNA-miRNA interaction in viral infection. Virus Res. 257, 25–32 (2018).
Beermann, J., Piccoli, M. T., Viereck, J. & Thum, T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol. Rev. 96, 1297–1325 (2016).
Huang Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J. Cell Mol. Med. 22, 5768–5775 (2018).
Kristensen L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet 20, 675–691 (2019).
Panda A. C. Circular RNAs act as miRNA sponges. Adv. Exp. Med. Biol. 1087, 67–79 (2018).
Leonetti A. et al. MicroRNAs as a drug resistance mechanism to targeted therapies in EGFR-mutated NSCLC: Current implications and future directions. Drug Resist. Updat. 42, 1–11 (2019).
Wei L. et al. Noncoding RNAs in gastric cancer: implications for drug resistance. Mol. Cancer 19, 62 (2020).
Bhat A. A. et al. Role of non-coding RNA networks in leukemia progression, metastasis and drug resistance. Mol. Cancer 19, 57 (2020).
Jiang W. et al. Long non-coding RNAs as a determinant of cancer drug resistance: towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updat. 50, 100683 (2020).
Guo J. et al. The miR 495-UBE2C-ABCG2/ERCC1 axis reverses cisplatin resistance by downregulating drug resistance genes in cisplatin-resistant non-small cell lung cancer cells. EBioMedicine 35, 204–221 (2018).
Han, Q., Cheng, P., Yang, H., Liang, H. & Lin, F. miR-146b reverses epithelial-mesenchymal transition via targeting PTP1B in cisplatin-resistance human lung adenocarcinoma cells. J. Cell Biochem. 11 (2019).
van Jaarsveld M. T. et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene 32, 4284–4293 (2013).
Zhang H. D. et al. Exosome: a novel mediator in drug resistance of cancer cells. Epigenomics 10, 1499–1509 (2018).
Mashouri L. et al. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 18, 75 (2019).
Qin X. et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 20, 12 (2019).
Wang D. et al. MicroRNA-936 induces cell cycle arrest and inhibits glioma cell proliferation by targeting CKS1. Am. J. Cancer Res. 7, 2131–2143 (2017).
Zhou, X. & Tao, H. Overexpression of microRNA-936 suppresses non-small cell lung cancer cell proliferation and invasion via targeting E2F2. Exp. Ther. Med. 16, 2696–2702 (2018).
Lin X. J. et al. miR-936 suppresses cell proliferation, invasion, and drug resistance of laryngeal squamous cell carcinoma and targets GPR78. Front. Oncol. 10, 60 (2020).
Zhang Y. et al. NOTCH3 overexpression and posttranscriptional regulation by miR-150 were associated with EGFR-TKI resistance in lung adenocarcinoma. Oncol. Res. 27, 751–761 (2019).
Bach D. H. et al. BMP4 upregulation is associated with acquired drug resistance and fatty acid metabolism in EGFR-mutant non-small-cell lung cancer cells. Mol. Ther. Nucleic Acids 12, 817–828 (2018).
Yang, L., Zhang, L., Lu, L. & Wang, Y. miR-214-3p regulates multi-drug resistance and apoptosis in retinoblastoma cells by targeting ABCB1 and XIAP. Onco Targets Ther. 13, 803–811 (2020).
Tao L. et al. MiR-451a attenuates doxorubicin resistance in lung cancer via suppressing epithelialmesenchymal transition (EMT) through targeting c-Myc. Biomed. Pharmacother. 125, 109962 (2020).
Han T. et al. miR-552 regulates liver tumor-initiating cell expansion and sorafenib resistance. Mol. Ther. Nucleic Acids 19, 1073–1085 (2020).
Bester A. C. et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell 173, 649–664.e20 (2018).
Lu Y. et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat. Med. 23, 1331–1341 (2017).
Kong X. et al. Analysis of lncRNA, miRNA and mRNA-associated ceRNA networks and identification of potential drug targets for drug-resistant non-small cell lung cancer. J. Cancer 11, 3357–3368 (2020).
Zhang J. et al. A novel long non-coding RNA, MSTRG.51053.2 regulates cisplatin resistance by sponging the miR-432-5p in non-small cell lung cancer cells. Front. Oncol. 10, 215 (2020).
Zhang, Z., Li, M. & Zhang, Z. lncRNA MALAT1 modulates oxaliplatin resistance of gastric cancer via sponging miR-22-3p. Onco Targets Ther. 13, 1343–1354 (2020).
Sun, H., Sun, Y., Chen, Q. & Xu, Z. LncRNA KCNQ1OT1 contributes to the progression and chemoresistance in acute myeloid leukemia by modulating Tspan3 through suppressing miR-193a-3p. Life Sci. 241, 117161 (2020).
Zhang S. F. et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 11, 225–238 (2015).
Sisinni L. et al. Endoplasmic reticulum stress and unfolded protein response in breast cancer: the balance between apoptosis and autophagy and its role in drug resistance. Int J. Mol. Sci. 20, 857 (2019).
Li Y. J. et al. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 36, 52 (2017).
Smith, A. G. & Macleod, K. F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 247, 708–718 (2019).
Yang Y. et al. Silencing of LncRNA-HOTAIR decreases drug resistance of non-small cell lung cancer cells by inactivating autophagy via suppressing the phosphorylation of ULK1. Biochem. Biophys. Res. Commun. 497, 1003–1010 (2018).
Zhang W. et al. Long non-coding RNA LINC00160 functions as a decoy of microRNA-132 to mediate autophagy and drug resistance in hepatocellular carcinoma via inhibition of PIK3R3. Cancer Lett. 478, 22–33 (2020).
Zhang, Y. F., Li, C. S., Zhou, Y. & Lu, X. H. Propofol facilitates cisplatin sensitivity via lncRNA MALAT1/miR-30e/ATG5 axis through suppressing autophagy in gastric cancer. Life Sci. 244, 117280 (2020).
YiRen H. et al. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol. Cancer 16, 174 (2017).
Yuan P. et al. The HIF-2alpha-MALAT1-miR-216b axis regulates multi-drug resistance of hepatocellular carcinoma cells via modulating autophagy. Biochem. Biophys. Res. Commun. 478, 1067–1073 (2016).
Wang X. et al. LncRNA SNHG6 promotes chemoresistance through ULK1-induced autophagy by sponging miR-26a-5p in colorectal cancer cells. Cancer Cell Int. 19, 234 (2019).
Li W. et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 38, 183 (2019).
Zhang, X., Zhao, P., Wang, C. & Xin, B. SNHG14 enhances gemcitabine resistance by sponging miR-101 to stimulate cell autophagy in pancreatic cancer. Biochem. Biophys. Res. Commun. 510, 508–514 (2019).
Tang, Q. & Hann, S. S. Biological roles and mechanisms of circular RNA in human cancers. Onco Targets Ther. 13, 2067–2092 (2020).
Huang X. et al. Circular RNA AKT3 upregulates PIK3R1 to enhance cisplatin resistance in gastric cancer via miR-198 suppression. Mol. Cancer 18, 71 (2019).
Shang J. et al. CircPAN3 contributes to drug resistance in acute myeloid leukemia through regulation of autophagy. Leuk. Res. 85, 106198 (2019).
Shang J. et al. CircPAN3 mediates drug resistance in acute myeloid leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp. Hematol. 70, 42–54.e3 (2019).
Lai M. et al. Hsa_circ_0079662 induces the resistance mechanism of the chemotherapy drug oxaliplatin through the TNF-alpha pathway in human colon cancer. J. Cell Mol. Med. 24, 5021–5027 (2020).
Liu F. et al. Circular RNA EIF6 (Hsa_circ_0060060) sponges miR-144-3p to promote the cisplatin-resistance of human thyroid carcinoma cells by autophagy regulation. Aging 10, 3806–3820 (2018).
Zhang S. et al. circCELSR1 (hsa_circ_0063809) contributes to paclitaxel resistance of ovarian cancer cells by regulating FOXR2 expression via miR-1252. Mol. Ther. Nucleic Acids 19, 718–730 (2020).
Dong Y. et al. Circ_0076305 regulates cisplatin resistance of non-small cell lung cancer via positively modulating STAT3 by sponging miR-296-5p. Life Sci. 239, 116984 (2019).
Li X. et al. Hsa_circ_0002483 inhibited the progression and enhanced the Taxol sensitivity of non-small cell lung cancer by targeting miR-182-5p. Cell Death Dis. 10, 953 (2019).
Sang Y. et al. circRNA_0025202 regulates tamoxifen sensitivity and tumor progression via regulating the miR-182-5p/FOXO3a axis in breast cancer. Mol. Ther. 27, 1638–1652 (2019).
Liang Y. et al. circKDM4C suppresses tumor progression and attenuates doxorubicin resistance by regulating miR-548p/PBLD axis in breast cancer. Oncogene 38, 6850–6866 (2019).
Walko, C. M. & Lindley, C. Capecitabine: a review. Clin. Ther. 27, 23–44 (2005).
Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003).
Li, W. & Yue, H. Thymidine phosphorylase: a potential new target for treating cardiovascular disease. Trends Cardiovasc Med. 28, 157–171 (2018).
Elamin, Y. Y., Rafee, S., Osman, N., KJ, O. B. & Gately, K. Thymidine phosphorylase in cancer; enemy or friend? Cancer Microenviron. 9, 33–43 (2016).
Wang D. et al. Human carboxylesterases: a comprehensive review. Acta Pharm. Sin. B 8, 699–712 (2018).
Frances, A. & Cordelier, P. The emerging role of cytidine deaminase in human diseases: a new opportunity for therapy? Mol. Ther. 28, 357–366 (2020).
Ozaki, T. & Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 3, 994–1013 (2011).
Chung T. et al. Dihydropyrimidine dehydrogenase is a prognostic marker for mesenchymal stem cell-mediated cytosine deaminase gene and 5-fluorocytosine prodrug therapy for the treatment of recurrent gliomas. Theranostics 6, 1477–1490 (2016).
Shimada S. et al. Dihydropyrimidine dehydrogenase overexpression correlates with potential resistance to 5-fluorouracil-based treatment in head and neck squamous cell carcinoma. Transl. Cancer Res. 7, 411–419 (2018).
Katayama, K., Noguchi, K. & Sugimoto, Y. Regulations of P-Glycoprotein/ABCB1/MDR1 in human cancer cells. N. J. Sci. 2014, 476974 (2014).
Kartner, N., Shales, M., Riordan, J. R. & Ling, V. Daunorubicin-resistant Chinese hamster ovary cells expressing multidrug resistance and a cell-surface P-glycoprotein. Cancer Res. 43, 4413–4419 (1983).
Mickisch, G. H., Pai, L. H., Gottesman, M. M. & Pastan, I. Monoclonal antibody MRK16 reverses the multidrug resistance of multidrug-resistant transgenic mice. Cancer Res. 52, 4427–4432 (1992).
Hamada, H. & Tsuruo, T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc. Natl Acad. Sci. U. S. A. 83, 7785–7789 (1986).
Takada, T., Suzuki, H., Gotoh, Y. & Sugiyama, Y. Regulation of the cell surface expression of human BCRP/ABCG2 by the phosphorylation state of Akt in polarized cells. Drug Metab. Dispos. 33, 905–909 (2005).
Henrique R. et al. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genom. 14, 898 (2013).
Xie, Y., Burcu, M., Linn, D. E., Qiu, Y. & Baer, M. R. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol. Pharm. 78, 310–318 (2010).
Bachmann, M. & Möröy, T. The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol. 37, 726–730 (2005).
Natarajan K. et al. The Pim kinase inhibitor SGI-1776 decreases cell surface expression of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and drug transport by Pim-1-dependent and -independent mechanisms. Biochem. Pharm. 85, 514–524 (2013).
Nambaru P. K. et al. Drug efflux transporter multidrug resistance-associated protein 5 affects sensitivity of pancreatic cancer cell lines to the nucleoside anticancer drug 5-fluorouracil. Drug Metab. Dispos. 39, 132–139 (2011).
Fukuda, Y. & Schuetz, J. D. ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem. Pharm. 83, 1073–1083 (2012).
Oguri T. et al. MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol. Cancer Ther. 6, 122–127 (2007).
Zhang, Z., Liu, F. & Chen, J. Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 170, 483–491.e8 (2017).
Czuba, L. C., Hillgren, K. M. & Swaan, P. W. Post-translational modifications of transporters. Pharm. Ther. 192, 88–99 (2018).
Kuang Y. et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin. Cancer Res. 15, 2630–2636 (2009).
Braig F. et al. Cetuximab resistance in head and neck cancer is mediated by EGFR-K(521) polymorphism. Cancer Res. 77, 1188–1199 (2017).
Hsu J. M. et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 13, 174–181 (2011).
Lu Y. et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 67, 8240–8247 (2007).
Molina M. A. et al. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res 61, 4744–4749 (2001).
Gijsen M. et al. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 8, e1000563 (2010).
Huang Z. et al. The E3 ubiquitin ligase NEDD4 negatively regulates HER3/ErbB3 level and signaling. Oncogene 34, 1105–1115 (2015).
VanMeter A. J. et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGPR) phosphorylation events associated with mutated EGFR compared with wild type. Mol. Cell Proteom. 7, 1902–1924 (2008).
Sergina N. V. et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445, 437–441 (2007).
Canfield K. et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 14, 648–655 (2015).
Salehan, M. R. & Morse, H. R. DNA damage repair and tolerance: a role in chemotherapeutic drug resistance. Br. J. Biomed. Sci. 70, 31–40 (2013).
Zhang Y. et al. Human DNA polymerase kappa synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res. 28, 4147–4156 (2000).
Albertella, M. R., Lau, A. & O’Connor, M. J. The overexpression of specialized DNA polymerases in cancer. DNA Repair 4, 583–593 (2005).
Topalis, D., Gillemot, S., Snoeck, R. & Andrei, G. Distribution and effects of amino acid changes in drug-resistant α and β herpesviruses DNA polymerase. Nucleic Acids Res. 44, 9530–9554 (2016).
Wang J.O. et al. DNA polymerase kappa, implicated in spontaneous and DNA damage-induced mutagenesis, is overexpressed in lung cancer. Cancer Res. 61, 5366–5369 (2001).
Wang Y. et al. Elevated expression of DNA polymerase kappa in human lung cancer is associated with p53 inactivation: Negative regulation of POLK promoter activity by p53. Int J. Oncol. 25, 161–165 (2004).
Halazonetis, T. D., Gorgoulis, V. G. & Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 (2008).
Song L. et al. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One 6, e25454 (2011).
Le Guezennec, X. & Bulavin, D. V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 35, 109–114 (2010).
Vadnais C. et al. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res. 40, 4483–4495 (2012).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Leonard, B. C. & Johnson, D. E. Signaling by cell surface death receptors: alterations in head and neck cancer. Adv. Biol. Regul. 67, 170–178 (2018).
Ozören, N. & El-Deiry, W. S. Cell surface death receptor signaling in normal and cancer cells. Semin. Cancer Biol. 13, 135–147 (2003).
Mansoori, B., Mohammadi, A., Davudian, S., Shirjang, S. & Baradaran, B. The different mechanisms of cancer drug resistance: a brief review. Adv. Pharm. Bull. 7, 339–348 (2017).
Wu K. J. et al. Synthesis and evaluation of dibenzothiophene analogues as Pin1 inhibitors for cervical cancer therapy. ACS Omega 4, 9228–9234 (2019).
Campbell, K. J. & Tait, S. W. G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol 8, 180002 (2018).
Wertz I. E. et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114 (2011).
Kale J. et al. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 19, e45235 (2018).
Jaime-Sánchez P. et al. Antigen-specific primed cytotoxic T cells eliminate tumour cells in vivo and prevent tumour development, regardless of the presence of anti-apoptotic mutations conferring drug resistance. Cell Death Differ. 25, 1536–1548 (2018).
Hu X. et al. Cul4 E3 ubiquitin ligase regulates ovarian cancer drug resistance by targeting the antiapoptotic protein BIRC3. Cell Death Dis. 10, 104 (2019).
Ganley I. G. et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 (2009).
Spitzer, R., Cleves, A. E. & Jain, A. N. Surface-based protein binding pocket similarity. Proteins 79, 2746–2763 (2011).
Motono C. et al. SAHG, a comprehensive database of predicted structures of all human proteins. Nucleic Acids Res. 39, D487–D493 (2011).
Pópulo, H., Lopes, J. M. & Soares, P. The mTOR signalling pathway in human cancer. Int J. Mol. Sci. 13, 1886–1918 (2012).
Ribatti, D., Tamma, R. & Annese, T. Epithelial-mesenchymal transition in cancer: a historical overview. Transl. Oncol. 13, 100773 (2020).
Thiery, J. P. & Sleeman, J. P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 (2006).
Brabletz, T., Kalluri, R., Nieto, M. A. & Weinberg, R. A. EMT in cancer. Nat. Rev. Cancer 18, 128–134 (2018).
Thomson S. et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 65, 9455–9462 (2005).
Zhou B. P. et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940 (2004).
Zhang K. et al. Lats2 kinase potentiates Snail1 activity by promoting nuclear retention upon phosphorylation. EMBO J. 31, 29–43 (2012).
Yang Z. et al. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 65, 3179–3184 (2005).
Cheng G. Z. et al. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 67, 1979–1987 (2007).
Vesuna F. et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene 31, 3223–3234 (2012).
Taketo K. et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J. Oncol. 52, 621–629 (2018).
Zhu, J., Tao, L. & Jin, L. MicroRNA5063p reverses gefitinib resistance in nonsmall cell lung cancer by targeting Yesassociated protein 1. Mol. Med Rep. 19, 1331–1339 (2019).
Yang, W., Xiao, W., Cai, Z., Jin, S. & Li, T. miR-1269b drives cisplatin resistance of human non-small cell Lung cancer via modulating the PTEN/PI3K/AKT signaling pathway. Onco Targets Ther. 13, 109–118 (2020).
Zhu X. et al. miR-186 regulation of Twist1 and ovarian cancer sensitivity to cisplatin. Oncogene 35, 323–332 (2016).
Fu Q. et al. miR-20b reduces 5-FU resistance by suppressing the ADAM9/EGFR signaling pathway in colon cancer. Oncol. Rep. 37, 123–130 (2017).
Wang L. et al. MiR-153 inhibits the resistance of lung cancer to gefitinib via modulating expression of ABCE1. Cancer Biomark. 25, 361–369 (2019).
Hu M. et al. lncRNA CCAT1 is a biomarker for the proliferation and drug resistance of esophageal cancer via the miR-143/PLK1/BUBR1 axis. Mol. Carcinog. 58, 2207–2217 (2019).
Liu H. et al. Knockdown of long non-coding RNA UCA1 increases the tamoxifen sensitivity of breast cancer cells through Inhibition of Wnt/beta-catenin pathway. PLoS One 11, e0168406 (2016).
Zhu J. et al. Knockdown of long non-coding RNA XIST inhibited doxorubicin resistance in colorectal cancer by upregulation of miR-124 and downregulation of SGK1. Cell Physiol. Biochem. 51, 113–128 (2018).
Saeinasab M. et al. SNHG15 is a bifunctional MYC-regulated noncoding locus encoding a lncRNA that promotes cell proliferation, invasion and drug resistance in colorectal cancer by interacting with AIF. J. Exp. Clin. Cancer Res. 38, 172 (2019).
Luo Y. et al. CircRNA_101505 sensitizes hepatocellular carcinoma cells to cisplatin by sponging miR-103 and promotes oxidored-nitro domain-containing protein 1 expression. Cell Death Discov. 5, 121 (2019).
Yan L. et al. Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics 9, 8377–8391 (2019).
Klingler S. et al. Development of resistance to EGFR-targeted therapy in malignant glioma can occur through EGFR-dependent and -independent mechanisms. Cancer Res. 75, 2109–2119 (2015).
Lopez Sambrooks C. et al. Oligosaccharyltransferase inhibition overcomes therapeutic resistance to EGFR tyrosine kinase inhibitors. Cancer Res. 78, 5094–5106 (2018).
Ali A. M. et al. Disarming an electrophilic warhead: retaining potency in tyrosine kinase inhibitor (TKI)-resistant CML lines while circumventing pharmacokinetic liabilities. ChemMedChem 11, 850–861 (2016).
Wu, D. W., Chen, T. C., Huang, H. S. & Lee, H. TC-N19, a novel dual inhibitor of EGFR and cMET, efficiently overcomes EGFR-TKI resistance in non-small-cell lung cancer cells. Cell Death Dis. 7, e2290 (2016).
Qi M. et al. ERK inhibition represses gefitinib resistance in non-small cell lung cancer cells. Oncotarget 9, 12020–12034 (2018).
Byun S. et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res 19, 3894–3904 (2013).
Liu W. et al. Overcoming 5-Fu resistance of colon cells through inhibition of Glut1 by the specific inhibitor WZB117. Asian Pac. J. Cancer Prev. 15, 7037–7041 (2014).
Yan C. M., Zhao, Y. L., Cai, H. Y., Miao, G. Y. & Ma, W. Blockage of PTPRJ promotes cell growth and resistance to 5-FU through activation of JAK1/STAT3 in the cervical carcinoma cell line C33A. Oncol. Rep. 33, 1737–1744 (2015).
He C. et al. Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis. 10, 65 (2019).
Liu Y. et al. Inhibition of the deubiquitinase USP9x induces pre-B cell homeobox 1 (PBX1) degradation and thereby stimulates prostate cancer cell apoptosis. J. Biol. Chem. 294, 4572–4582 (2019).
Santana-Codina N. et al. Defining and targeting adaptations to oncogenic KRAS(G12C) inhibition using quantitative temporal proteomics. Cell Rep. 30, 4584–4599.e4 (2020).
An Y. et al. Molecular insights into cancer drug resistance from a proteomics perspective. Expert Rev. Proteom. 16, 413–429 (2019).
Ding, S., Chen, X. & Shen, K. Single-cell RNA sequencing in breast cancer: understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 40, 329–344 (2020).
Mitra A. K. et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 30, 1094–1102 (2016).
Lee M. C. et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc. Natl Acad. Sci. U. S. A. 111, E4726–E4735 (2014).
Kim K. T. et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 16, 127 (2015).
Wu H. et al. Single-cell transcriptome analyses reveal molecular signals to intrinsic and acquired paclitaxel resistance in esophageal squamous cancer cells. Cancer Lett. 420, 156–167 (2018).
Tanaka N. et al. Single-cell RNA-seq analysis reveals the platinum resistance gene COX7B and the surrogate marker CD63. Cancer Med. 7, 6193–6204 (2018).
Hou Y. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016).
Angermueller C. et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016).
Lee H. W. et al. Single-cell RNA sequencing reveals the tumor microenvironment and facilitates strategic choices to circumvent treatment failure in a chemorefractory bladder cancer patient. Genome Med. 12, 47 (2020).
Carter L. et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat. Med. 23, 114–119 (2017).
Acknowledgements
This work was supported by grants from the Guangdong Basic and Applied Basic Research fund project (2019B1515120033), the Science and Technology Foundation of Shenzhen (JCYJ20180305164128430), the International Cooperation Foundation of Shenzhen (GJHZ20180928171602104), the Shenzhen Economic and Information Committee “Innovation Chain and Industry Chain” integration special support plan project (20180225112449943), the Shenzhen Public Service Platform on Tumor Precision Medicine and Molecular Diagnosis.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, H., Liu, D., Dong, S. et al. Epitranscriptomics and epiproteomics in cancer drug resistance: therapeutic implications. Sig Transduct Target Ther 5, 193 (2020). https://doi.org/10.1038/s41392-020-00300-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00300-w
This article is cited by
-
EIF4A3-mediated biogenesis of circSTX6 promotes bladder cancer metastasis and cisplatin resistance
Journal of Experimental & Clinical Cancer Research (2024)
-
Regulation and functions of non-m6A mRNA modifications
Nature Reviews Molecular Cell Biology (2023)
-
Bioinformatics analysis and experimental validation of tumorigenic role of PPIA in gastric cancer
Scientific Reports (2023)
-
A-to-I edited miR-411-5p targets MET and promotes TKI response in NSCLC-resistant cells
Oncogene (2023)
-
Amentoflavone and methyl hesperidin, novel lead molecules targeting epitranscriptomic modulator in acute myeloid leukemia: in silico drug screening and molecular dynamics simulation approach
Journal of Molecular Modeling (2023)
