
Abstract
Background
Loss-of-function variants in MID1 are the most common cause of Opitz G/BBB syndrome (OS). The interpretation of intronic variants affecting the splicing is a rising issue in OS.
Methods
Exon sequencing of a 2-year-old boy with OS showed that he was a carrier of the de novo c.1286–10G>T variant in MID1. In silico predictions and minigene assays explored the effect of the variant on splicing. The minigene approach was also applied to two previously identified MID1 c.864+1G>T and c.1285+1G>T variants.
Results
Minigene assay demonstrated that the c.1286–10G>T variant generated the inclusion of eight nucleotides that predicted generation of a frameshift. The c.864+1G>T and c.1285+1G>T variants resulted in an in-frame deletion predicted to generate a shorter MID1 protein. In hemizygous males, this allowed reclassification of all the identified variants from “of unknown significance” to “likely pathogenic.”
Conclusions
Minigene assay supports functional effects from MID1 intronic variants. This paves the way to the introduction of similar second-tier investigations in the molecular diagnostics workflow of OS.
Impact
-
Causative intronic variants in MID1 are rarely investigated in Opitz syndrome.
-
MID1 is not expressed in blood and mRNA studies are hardly accessible in routine diagnostics.
-
Minigene assay is an alternative for assessing the effect of intronic variants on splicing.
-
This is the first study characterizing the molecular consequences of three MID1 variants for diagnostic purposes and demonstrating the efficacy of minigene assays in supporting their clinical interpretation.
-
Review of the criteria according to the American College of Medical Genetics reassessed all variants as likely pathogenic.
Similar content being viewed by others
Introduction
Opitz G/BBB syndrome (OS) (MIM 145410) is a rare and genetically heterogeneous condition primarily affecting midline structures, and including an X-linked form due to hemizygous variants in the Midline 1 (MID1) gene, and an autosomal-dominant form. Some cases are caused by heterozygous variants in Sperm Antigen with Calponin Homology and Coiled-Coil Domains (SPECC1L) gene.1,2,3 Main clinical manifestations of OS include hypertelorism/telecanthus, cleft lip and/or palate, laryngo-tracheo-esophageal abnormalities, congenital heart defects, anorectal anomalies, hypospadias, and specific malformations of the central nervous system.3,4,5,6,7,8,9 Variable expression occurs even among members of the same family. In the X-linked form, heterozygous females sometimes present with hypertelorism and other anomalies.7
MID1, also termed TRIM18, encodes an E3 ubiquitin ligase that belongs to the Tripartite Motif (TRIM) family.10 MID1 contains an N-terminal TRIM module composed of three domains (Really Interesting New Gene-RING finger, two B-box domains and a coiled-coil region)10 followed by a C-terminal subgroup one signature (COS) domain, a Fibronectin type III repeat (FN3), and a SPla and the RYanodine Receptor (SPRY) associated with a PRY domain.1,10,11 MID1 is a central player in several cellular processes and, through its RING domain, controls the stability and/or the activity of protein phosphatase 2A (PP2A).11 The MID1/PP2A complex regulates the mammalian Target Of Rapamycin Complex 1 (mTORC1) signaling pathway that, in turn, affects multiple cellular functions, including cell metabolism, growth, translation and turnover of proteins, autophagy, and cytoskeleton organization. Accordingly, MID1 mutated fibroblasts show increased levels of PP2A, which leads to a general hypo-phosphorylation of microtubule-associated proteins11 and decreased mTORC1 formation,12 with consequent alterations of the downstream phosphatidylinositol 3-kinase (PI3K/AKT) and extracellular signal-regulated kinase (Ras/ERK) pathways.13
MID1 directly associates with microtubules14 through its COS domain, and regulates their dynamics in defined cell cycle phases.15,16,17 MID1-deprived HeLa cells display division defects, including cytokinetic arrest and delayed or aborted abscission, which induce cell bi-nucleation or death.15 The MID1-dependent control of microtubule dynamics regulates other cellular processes, such as cell adhesion and cell migration, in which microtubules are involved.18,19
Approximately one hundred MID1 variants have been reported in OS (https://databases.lovd.nl/shared/genes/MID1).1,5,18,19,20 Disease-causing variants are distributed along the entire gene without recognized mutational hot-spots; they belong to all known molecular classes with an excess of predicted null alleles. Occasionally, OS is caused by whole gene or intragenic large deletions.9 The molecular repertoire of MID1 disease-causing variants is consistent with a loss-of-function mechanism.5 As often occurs in Mendelian disorders due to loss-of-function mechanisms, the interpretation of intronic variants affecting splicing is a potential concern in X-linked OS. In agreement with the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) standards, functional studies are a powerful tool for investigating the pathogenicity potential of intronic variants. However, MID1 is only expressed in non-accessible tissues and its mRNA is not detected in peripheral blood. Therefore, in vitro analyses, such as the minigene assay, may represent a good alternative to assess the splicing effect of candidate intronic variants in MID1, as recently introduced by the ACMG.18
Here, exome sequencing (ES) identified a de novo c.1286–10G>T variant of uncertain significance (VUS) in MID1. Minigene assay demonstrated its effect on splicing and the introduction of a premature termination codon (PTC). The same approach was used to characterize the effect at the RNA level of two previously published MID1 c.864+1G>T and c.1285+1G>T variants. Both resulted in an in-frame deletion. For all studied variants, the application of the minigene assay improved their clinical interpretation according to ACMG/AMP criteria.
Materials and methods
Family enrollment, genomic DNA extraction, and quantification
Individual 1 and his unaffected parents were routinely followed in the Division of Medical Genetics at Fondazione IRCCS-Casa Sollievo della Sofferenza (San Giovanni Rotondo, Italy). Individuals 2 and 3 and their relatives were previously reported.9 The families provided written informed consent to molecular testing and to the full content of this publication. This study is in accordance with the 1984 Helsinki declaration and its subsequent versions and received IRB approval at Fondazione IRCCS-Casa Sollievo della Sofferenza (approval no. 2021/13/CE). Genomic DNAs were extracted from peripheral blood leukocytes by using Bio Robot EZ1 (Quiagen, Solna, Sweden), according to the manufacturer’s instructions. The DNA was quantified with Nanodrop 2000 C spectrophotometer (Thermo Fisher Scientific, Waltham, MA).
Exome sequencing
Individual 1’s DNA was analyzed by ES using the SureSelect Custom Constitutional Panel 17 Mb (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s instructions. This is a combined shearing-free transposase-based library prep and target-enrichment solution, which enables comprehensive coverage of a comprehensive set of OMIM genes. Sequencing was performed on a NextSeq 500 platform (Illumina, San Diego, CA) by using MidOutput flow cells (300 cycles), with a minimum expected coverage depth of 100×. The average coverage obtained was 111× and a minimum coverage of 20× in 96.4% of analyzable regions. FastQC files were checked and trimmed, mapped reads recalibrated and processed, and variants annotated by the Alissa Align & Call bioinformatics pipeline (Agilent Technologies, Santa Clara, CA). Annotated variants were then filtered and interpreted with an internally implemented variant triage system by Alissa Interpret pipeline (Agilent Technologies, Santa Clara, CA). Variants were first prioritized according to the following criteria: (i) variant absent in allele frequency population databases; (ii) variant reported in allele frequency population databases, but with a minor allele frequency (MAF) lower than 0.01; (iii) nonsense/frameshift variant in genes previously described as disease-causing by haploinsufficiency or loss of function; (iv) missense variant with a REVEL score ≥0.70; (v) variant affecting canonical splicing sites (i.e., ±1 or ±2 positions); (vi) variant predicted and/or annotated as pathogenic/deleterious in Clinical Variations (ClinVar) database and/or Leiden Open Variation Database (LOVD) without evidence of conflicting interpretation. For autosomal recessive genes, only homozygous or double heterozygous variants were selected. For autosomal dominant genes, assuming a de novo origin of the disease (parents unaffected), variants with a MAF between 0.01 and 0.001 were labeled as “low priority”. Given the phenotypic characteristics of the proband, a virtual gene panel was generated by the use of the following Human Phenotype Ontology terms: hypertelorism (HP:0000316), hypospadias (HP:0000047) and abnormality of the posterior cranial fossa (HP:0000932). Selected variants were interpreted according to guidelines from the ACMGG/AMP.19
Sanger sequencing and variant validation
The candidate variant identified by ES was confirmed by Sanger sequencing using individual 1’s and his parents’ DNA. Exon 8 of MID1 (NM_000381) was amplified by polymerase chain reaction (PCR) using a set of primers located at least 60 nucleotides from the exon–intron boundaries. Primer sequences are reported in Table S1. The amplified product was subsequently purified using ExoSAP-IT PCR Product Cleanup Reagent (Thermo Fisher Scientific, Waltham, MA) and sequenced using BigDye Terminator v1.1 sequencing kit (Thermo Fisher Scientific, Waltham, MA). The fragment obtained was purified using DyeEx plates (Qiagen, Tübingen, Germany), resolved on ABI Prism 3130 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA) and analyzed using the Sequencher software (Gene Codes, Ann Arbor, MI). The identified variant was also confirmed on an independent DNA extraction. Nucleotide variant nomenclature follows the format indicated in the Human Genome Variation Society (HGVS, http://www.hgvs.org) recommendations. Nucleotide numbers are derived from the cDNA sequence of MID1 (GenBank accession no. NM_000381.1); +1 corresponds to the A of the ATG initiation translation codon.
Splicing prediction
The molecular effects of identified splice site variants were predicted on the basis of in silico splice predictors, including NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and Berkeley Drosophila Genome Project (BDGP, http://www.fruitfly.org/seq_tools/splice.html).
Minigene generation
The in vitro splicing assay was carried out using a pSPL3 exon-trapping vector provided by Tompson and Young.20 Briefly, the pSPL3 vector contains a small artificial gene composed of an SV40 promoter, an exon–intron–exon sequence containing functional splice donor and acceptor sites, and a late polyadenylation signal. A multiple cloning site, including XhoI and BamHI restriction sites, is located within the intronic region between vector exons V1 and V2. For MID1 c.1286–10G>T, wild-type MID1 exon 8 was directly amplified by PCR from control DNA with specific primers containing additional XhoI (forward) and BamHI (reverse) restriction sites. The PCR reaction amplified the entire exon 8 sequence plus an additional 466 bp (5′) and 224 bp (3′) of the flanking intronic regions. For MID1 c.864+1G>T, a DNA fragment of 890 bp, including the MID1 exon 4, 471 bp (5′), and 313 bp (3′) of the flanking intronic regions, was amplified from control DNA with specific primers. For MID1 c.1285+1G>T, an 850 bp genomic DNA fragment that comprises MID1 exon 7 and its flanking sequences (456 bp at the 5′ and 253 bp at the 3′) was amplified by PCR with primers containing the appropriate restriction enzyme sites from control DNA. After PCR amplification, PCR products were purified and subjected to restriction enzyme digestion and then ligated into a pSPL3 exon-trapping vector. All constructs were Sanger sequenced to verify the correctness of the wild-type DNA fragments. The three pSPL3 constructs containing the MID1 variants were generated from the respective wild-type vectors by using the QuickChange II site-directed mutagenesis kit (Stratagene, San Diego, CA) and a Pfu Taq polymerase (Promega Corporation, Madison, WI), according to the manufacturer’s instructions. The constructs were verified by Sanger sequencing. All primer sequences are listed in Table S1.
Cell cultures and transfections
Human embryonic kidney (HEK) 293 cell lines were maintained in Dulbecco’s Modified Eagle Medium/Nutrient D-MEM with Glutamax (Thermo Fisher Scientific, Waltham, MA) plus 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and 1% penicillin and streptomycin (P/S, 100 U/ml and 100 μg/ml, respectively) (Thermo Fisher Scientific, Waltham, MA). Cell cultures were grown in a 5% CO2 incubator at 37 °C. HEK293 cells were plated in a six-well dish at a density of 4.0 × 105 cells and then transfected with the generated pSPL3 vectors using Lipofectamine 3000 reagent (Thermo Fisher Scientific, Waltham, MA), following the manufacturer’s instructions.
RNA extraction and reverse transcription
Total RNA was extracted with the RNeasy Mini Kit 48 h after transfection (Qiagen, Tübingen, Germany), treated with RNase-free DNase (Qiagen, Tübingen, Germany), quantified by Nanodrop (Thermo Fisher Scientific, Waltham, MA), and reverse-transcribed with the QuantiTect Reverse Transcription Kit (Qiagen, Tübingen, Germany) according to the manufacturer’s protocol. The cDNA was PCR amplified using pSPL3 vector-specific primers corresponding to sequences in vector exons V1 and V2 (V1—forward and V2—reverse). The pSPL3 vector with no genomic DNA insert generated a single reverse-transcribed PCR product of 257 bp from vector exons V1 and V2. The primers used for cDNA amplification are provided in Table S1. The amplified fragments were visualized on a 1% agarose gel, purified, and subsequently Sanger sequenced.
Results
Clinical report
Individual 1 was a 2-year-old boy, the only child of apparently unrelated and healthy parents. Family history was unremarkable for congenital anomalies and genetic disorders. Amniocentesis was performed for advanced maternal age and showing 46,XY. Second trimester ultrasound screening revealed a dilated cisterna magna likely due to a Blake’s pouch cyst. This finding was confirmed by fetal magnetic resonance imaging (MRI). The proband was born at 35 weeks due to spontaneous rupture of the membranes. Birth parameters were: length 45 cm (8th centile adjusted by gestational age), weight 2850 g (55th centile), head circumference 32 cm (16th centile), and Apgar score 8 (1 min)/8 (5 min). At birth, clinical hypertelorism, bitemporal depression and penoscrotal hypospadias were noted. Brain MRI performed shortly after birth confirmed the Blake’s pouch cyst and showed a small cerebellar venous malformation. Heart ultrasound was normal, while abdominal ultrasound revealed minimal kidney asymmetry. Lactation, nutrition and dentition were normal. He held his head up at 1 month, sat alone at 8 months, walked alone at 14 months and said first words at 10 months. Somatic growth was always within normal ranges. The patient underwent surgical correction of the penoscrotal hypospadias at 13 months without complications. At 1 year of age, he underwent a third brain MRI which revealed a dilated cisterna magna, thinning of the corpus callosum, a small venous malformation of the right cerebellar hemisphere and cyst of the septum pellucidum (Fig. 1a–c). No convulsions noted. Sleep electroencephalogram was normal at 1 year. At 2 years of age, height was 94.5 cm (97th centile), weight 15 kg (97th centile) and head circumference 48.5 cm (50th centile). Facial features included clinical hypertelorism with an internal intercanthal distance of 36 mm (+2 SD) and external intercanthal distance of 90 mm (+2 SD), prominent nasal bridge, a short nose, low-set ears. Intraoral cavity examination revealed bifid uvula. Shawl scrotum and surgical residues of the urologic intervention were also evident.

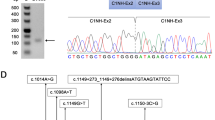
a–c Brain magnetic resonance imaging at 2 years showing dilated cisterna magna (asterisk) and thin corpus callosum (a), a small cerebellar venous malformation (arrow; b), and cyst of the septum pellucidum (asterisk; c). d Sanger sequencing chromatogram of both unaffected parents (I-1 and I-2) and proband (Individual 1, II-1). The MID1 c.1286–10G>A variant is indicated by a yellow bar on the proband’s chromatogram. e Family pedigree includes two unaffected parents and one affected son indicated with a filled symbol. f Minigene assay: (Upper panel) Electropherogram showing DNA sequencing analysis of PCR product amplified with primers targeting exon 8 and flanking intronic regions of MID1. Nucleotide sequences are provided. (Middle panel) Analysis of the c.1286–10G>A variant using minigene construct. The position of variant site and fragment containing exon 8 and its adjacent introns are indicated: the dark gray box indicates exon 8, the light gray boxes indicate vector exons V1 and V2, black lines between exons indicate introns, upper dashed lines indicate normal splicing product, instead, bottom dashed lines represent the exon skipping event. Analysis of mRNA from transfected HEK293 cells via RT-PCR (on the gel, pSPL3: empty vector; WT: wild type; MUT: mutated) and (Lower panel) Sanger sequencing.
Molecular findings in Individual 1
ES of Individual 1’s DNA revealed the unpublished c.1286–10G>A variant located in intron 7 of MID1. No further rare variants were detected in the remaining high-priority genes. The c.1286–10G>A variant was absent in major databases, including dbSNP, ExAC, 1000 Genomes, and gnomAD. The variant was confirmed by Sanger sequencing of patient’s DNA and was not detected in the patient’s parents, and therefore was assumed as de novo (Fig. 1d, e). In silico analysis predicted that the variant would likely lead to the use of an alternative cryptic splice site nearby, potentially resulting in inclusion of intronic nucleotides causing a frameshift with a consequent PTC. As MID1 is not physiologically expressed in the blood, to validate in silico analysis, we employed a minigene-based splice approach (Fig. 1f). The minigenes which carried MID1 wild-type sequence and c.1286–10G>A variant were transfected into HEK293 cell line. Reverse transcriptase-PCR (RT-PCR) analysis performed on total RNA extracted from both transfected cells detected a ~420 bp band. Sanger sequencing of the PCR products confirmed the inclusion of eight nucleotides of intron 7 in the mutant MID1 mature RNA (r.1285_1286insCTTTGCAG). The aberrantly spliced transcript is not present in the expressed sequence tag database (EST, https://www.ncbi.nlm.nih.gov/dbEST/index.html). The CTTTGCAG insertion was predicted to generate a frameshift which leads to a truncated MID1 protein lacking the C-terminal residues [p.(Ser429Thrfs*13)] (Fig. S1a). The generated PTC was located in exon 8 at a distance >50 nucleotides upstream of the 3’-most exon-exon junction, a condition that generally triggers nonsense-mediated mRNA decay (NMD). The novel variant was submitted to LOVD (individual ID #00391499). According to the ACMG guidelines and further recommendations for the application of functional data,18 the variant was classified as likely pathogenic by the attribution of the following criteria: PS3_moderate, PM2_moderate, PM6_moderate and PP4_supporting (Table 1).
Splicing study of the c.864+1G>T and c.1285+1G>T variants
We functionally characterized two other MID1 canonical splicing variants, c.864+1G>T and c.1285+1G>T, previously identified in two sporadic male patients affected by OS (Individuals 2 and 3). Segregation study showed that both variants were inherited from the unaffected mothers of the two patients.9 These variants were expected to disrupt the intron 4 and 7 canonical splice donor site, respectively. In order to explore the molecular events associated with these variants, we used the minigene approach (Fig. 2). pSPL3 vectors harboring the wild-type and mutant sequence of MID1 exons 4 or 7 and flanking introns were transfected into HEK293 cell line. Concerning the c.864+1G>T variant, RT-PCR analysis detected a 365 bp band in cells transfected with wild-type region and a 257 bp band in the cells expressing the mutant plasmid (Fig. 2a). Direct sequencing of the amplified products revealed that the larger fragment contained exon 4 as expected. In contrast, the shorter fragment lacked exon 4 (Fig. 2a). The exon 4 skipping event may lead to the synthesis of a shorter MID1 transcript (r.757_864del) which encodes a smaller protein, p.(Asn254_Val289del), defective of 36 residues (aa 253–289) of the coiled-coil domain. Regarding the c.1285+1G>T variant, analysis of the splicing products from the minigene assay, revealed that cells transfected with the wild-type vector yielded the expected 403 bp band (Fig. 2b). In contrast, cells transfected with the mutant vector yielded a band at 257 bp. Sequencing of both fragments confirmed the predicted splicing events (Fig. 2b). The deletion of exon 7 generated a shorter transcript (r.1142_1285del) which encodes a MID1 protein that lacks 49 (381–429 residues) C-terminal residues [p.(Ala381_Ser429delinsGly)]. Both the variants were submitted to LOVD (individual ID #00391500 and #00391501, respectively). According to the ACMG guidelines and further recommendations for the application of functional data18 and PVS1,21 these variants were classified as likely pathogenic by attribution of the following criteria: PS3_moderate, PM2_moderate, PM4_moderate, and PP4_supporting (Table 1). PVS1 is the only criterion designated as very strong strength level for pathogenicity in the ACMG guideline. PVS1 can be attributed to a null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a gene where loss of function is a known mechanism of disease.18

(Upper panel) Electropherogram showing DNA sequencing analysis of PCR product amplified with primers targeting exons 4 a and 7 b and flanking intronic regions of MID1. Nucleotide sequences are provided. (Middle panel) Analysis of the c.864+1G>T and c.1285+1G>T variants using minigene construct (a, b). The position of variant site and fragment containing exons 4 and 7 and their adjacent introns are indicated: the dark gray boxes indicates exons 4 and 7, the light gray boxes indicate vector exons V1 and V2, black lines between exons indicate introns, upper dashed lines indicate normal splicing product, instead, bottom dashed lines represent the exon skipping event. Analysis of mRNA from transfected HEK293 cells via RT-PCR (on the gel, pSPL3: empty vector; WT: wild type; MUT: mutated) and (Lower panel) Sanger sequencing.
Discussion
Deleterious variants occurring in MID1 are the most common cause of OS and loss-of-function is the leading molecular pathogenesis. Accordingly, most nonsense and frameshift variants, as well as GT-AG 1–2 splice site variants predicted to undergo NMD, can be interpreted as (likely) pathogenic given the attribution of PVS1 with a high strength level (i.e., strong or very strong). Nevertheless, other types of non-coding variants, such as canonical splice site variants with a downgraded PVS1 strength level (i.e., moderate or supporting) and variants falling in other intronic sequences, need further molecular proof of their functional consequence to escape the default classification of “variants of unknown significance”. For this reason, intronic variants are typically under-reported in MID1-related OS.
In the present study, we first investigated the effect of the novel MID1 c.1286–10G>T variant on RNA splicing by minigene assay. The preferred method to study the effect of variants on pre-mRNA splicing is the analysis of patient-derived mRNA obtained from blood or other key tissue(s). However, in many cases, the candidate gene is not physiologically expressed in available patient’s tissues and/or routine clinical activities hamper multiple sampling. In these scenarios, the minigene assay results are relatively fast and accurate for characterizing potential splicing aberrations. Minigenes typically contain a genomic fragment from the gene of interest that includes the exon(s) and partial or entire flanking intronic regions, and are able to express wild-type or aberrant pre-mRNAs by transient transfection, thus, providing a rapid assay for evaluating the transcriptional effect of intronic variants.20 This approach can present some limitations, as it is an in vitro assay that requires the construction of an artificial hybrid gene and that may not detect tissue specific mis-splicing. The minigene-based system has been extensively employed to functionally characterize variants in a number of disease-associated genes, such as BRCA1,22 BRCA2,23 CFT,24 and CHD7.25 Recently,26 the use of a minigene assay was reported as an efficient tool to characterize mRNA splicing profiles of 29 intronic and exonic NF1 variants. Additionally, minigene-based system can be used to analyze heterozygous variants as it permits the study of mRNA generated from a single allele (wild type or mutant), unlike classical cDNA analysis that usually does not distinguish the transcripts derived from each allele, unless a polymorphism can be exploited.
Our approach indicated that the MID1 c.1286–10G>A variant alters mRNA by activating a novel acceptor splice site that induces the expression of an alternative splice form that contains a PTC. The PTC is located in exon 8 at a distance more than 50–55 nucleotides upstream of the last exon–exon junction, a condition that generally triggers NMD.27 According to the rules of NMD, the MID1 mutant transcripts should be targets of NMD and this would confirm the MID1 loss of hemizygosity as a key molecular mechanism of disease.
The impact of NMD on the whole transcripts carrying a PTC may vary among different individuals and tissues,28,29,30 and this phenomenon can explain significant clinical variability within and between families carrying the same deleterious allele. For example, it is quite common for rare truncating variants of the dystrophin gene falling after the 70th exon and, therefore, near to the 3’ of the gene which can result in extremely variable phenotypes. Concerning MID1, at the moment, we do not have enough information for discriminating where the boundary determining the chance of escaping NMD is located. Our data showed that the c.1286–10G>A splicing variant generates a premature stop codon in the third last exon. If we assume that the inclusion of the premature stop codon is near the 3’ end, it could be enough to partially escape NMD leaving a residual amount of truncated protein could be synthetized and act as a hypomorphic allele due to the lack of the C-terminal residues. In fact, MID1 is an X-linked gene and our proband is hemizygous for the mutated allele. In this allelic state, theoretically, even a small amount of encoded protein could restore the phenotype if the truncated protein does not cause significant cell damage. To further corroborate the potential pathogenic effect of the c.1286–10G>T variant, we performed an expression study on ARPE-19 cells (Supplementary Data). Altogether, these findings allowed reclassifying the DNA substitution from “variant of unknown significance” (one moderate and one supporting criteria) to “likely pathogenic” (three moderate and one supporting criteria) (Table 1).
We also applied the minigene assay on two previously published MID1 canonical splice site variants, i.e., c.864+1G>T and c.1285+1G>T, which are considered unaffected by NMD in AutoPVS1 (http://autopvs1.genetics.bgi.com/) (and, therefore, received a downgraded PVS1). The minigene analysis demonstrated that the c.864+1G>T variant affects pre-mRNA splicing by causing the skipping of exon 4. The aberrant transcript was predicted to lead to the synthesis of a shorter protein, p.(Asn254_Val289del), defective of 36 residues of the coiled-coil domain. The MID1 c.1285+1G>T nucleotide substitution resulted in the skipping of exon 7. This alternative event would cause the translation of a MID1 protein which lacks 49 residues of the FN3 domain, p.(Ala381_Ser429delinsGly). We hypothesized that both MID1 variants, which cause an in-frame deletion, could have a deleterious effect on its product due to the loss of a large portion of protein crucial for MID1 dimerization and subcellular localization. Nevertheless, it remains to be determined if the variants have a similar or rather different effect in their natural expression context. Accordingly, the minigene assay definitely excluded the insertion of a PTC (i.e., removal of PVS1_moderate), and added a moderate criterion for the in-frame deletion in non-repetitive sequences (i.e., PM4_moderate) and a second moderate criterion for functional data (i.e., PS3_moderate), which allowed the definition of “likely pathogenic” for both variants (Table 1).
In conclusion, this study demonstrated the power of minigene assay in refining clinical interpretation of intronic variants in MID1. It is a rapid, efficient and cost-effective tool with increasing clinical application especially for variants in genes that present a limited or absent expression in peripheral blood. The dramatic increase of inconclusive reports with VUS in next-generation sequencing diagnostics should prompt laboratories to develop complementary techniques supporting genomic data with functional information to improve their diagnostic yield.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Quaderi, N. A. et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 17, 285–291 (1997).
Kruszka, P. et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J. Med. Genet. 52, 104–110 (2015).
Robin, N. H. et al. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat. Genet. 11, 459–461 (1995).
Gaudenz, K. et al. Opitz G/BBB syndrome in Xp22: mutations in the MID1 gene cluster in the carboxy-terminal domain. Am. J. Hum. Genet. 63, 703–710 (1998).
Cox, T. C. et al. New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome. Hum. Mol. Genet. 9, 2553–2562 (2000).
Pinson, L. et al. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J. Med. Genet. 41, 381–386 (2004).
So, J. et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. Am. J. Med. Genet. A 132A, 1–7 (2005).
Mnayer, L., Khuri, S., Merheby, H. A., Meroni, G. & Elsas, L. J. A structure-function study of MID1 mutations associated with a mild Opitz phenotype. Mol. Genet. Metab. 87, 198–203 (2006).
Ferrentino, R., Bassi, M. T., Chitayat, D., Tabolacci, E. & Meroni, G. MID1 mutation screening in a large cohort of Opitz G/BBB syndrome patients: twenty-nine novel mutations identified. Hum. Mutat. 28, 206–207 (2007).
Reymond, A. et al. The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001).
Trockenbacher, A. et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 29, 287–294 (2001).
Liu, E., Knutzen, C. A., Krauss, S., Schweiger, S. & Chiang, G. G. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Natl Acad. Sci. USA 108, 8680–8685 (2011).
Carracedo, A. & Pandolfi, P. P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27, 5527–5541 (2008).
Short, K. M. & Cox, T. C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 281, 8970–8980 (2006).
Gholkar, A. A. et al. The X-linked-intellectual-disability-associated ubiquitin ligase Mid2 interacts with astrin and regulates astrin levels to promote cell division. Cell Rep. 14, 180–188 (2016).
Zanchetta, M. E., Napolitano, L. M. R., Maddalo, D. & Meroni, G. The E3 ubiquitin ligase MID1/TRIM18 promotes atypical ubiquitination of the BRCA2-associated factor 35, BRAF35. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1844–1854 (2017).
Zanchetta, M. E. & Meroni, G. Emerging roles of the TRIM E3 ubiquitin ligases MID1 and MID2 in cytokinesis. Front. Physiol. 10, 274 (2019).
Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Tompson, S. W. & Young, T. L. Assaying the effects of splice site variants by exon trapping in a mammalian cell line. Bio Protoc. 7, e2281 (2017).
Abou, Tayoun et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Steffensen, A. Y. et al. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur. J. Hum. Genet. 22, 1362–1368 (2014).
Fraile-Bethencourt, E. et al. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 13, e1006691 (2017).
Giorgi, G. et al. Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin. Chem. Lab. Med. 53, 1719–1723 (2015).
Villate, O. et al. Functional analyses of a novel splice variant in the CHD7 gene, found by next generation sequencing, confirm its pathogenicity in a Spanish patient and diagnose him with CHARGE Syndrome. Front. Genet. 9, 7 (2018).
Morbidoni, V. et al. Hybrid minigene assay: an efficient tool to characterize mRNA splicing profiles of NF1 variants. Cancers 13, 999 (2021).
Hentze, M. W. & Kulozik, A. E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96, 307–310 (1999).
Linde, L. et al. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur. J. Hum. Genet. 15, 1156–1162 (2007).
Lejeune, F. Nonsense-mediated mRNA decay, a finely regulated mechanism. Biomedicines 10, 141 (2022).
Sarkar, H. et al. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum. Mol. Genet. 28, 1865–1871 (2019).
Acknowledgements
The authors thank the families for their kind availability in sharing the findings within the scientific community. We acknowledge S.W. Tompson (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison) for providing pSPL3 vector.
Funding
This work was supported by the Ricerca Corrente 2018–2020 Program from the Italian Ministry of Health and Regione Puglia.
Author information
Authors and Affiliations
Contributions
L.M., G.M. and M.C. conceived and designed the work that led to the submission, acquired data, and played an important role in interpreting the results. F.R., M.M., G.N. and C.F. conducted functional studies. L.B. and S.M. analyzed the exome-sequencing data. L.M., G.M. and M.C. wrote the manuscript. All authors revised the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
COMPETING interests
The authors declare no competing interests.
Ethics approval and consent to participate
Written informed consent was obtained from all individuals for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Micale, L., Russo, F., Mascaro, M. et al. Opitz syndrome: improving clinical interpretation of intronic variants in MID1 gene. Pediatr Res 93, 1208–1215 (2023). https://doi.org/10.1038/s41390-022-02237-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-02237-y
