
Abstract
Background
The aim of this study was to investigate the effect of coenzyme Q10 (CoQ10), a commonly used nutritional supplement, on intracranial aneurysm (IA) initiation and progression in a mouse model, as well as the mechanism.
Methods
Hydrogen peroxide (H2O2) was used to treat mouse-derived vascular smooth muscle cells (VSMCs) to induce oxidative injury, followed by incubation with CoQ10. In the mouse IA model established by elastase injection, CoQ10 was orally administered at 10 mg/kg every other day for 14 days, during which the incidence of IA, rupture rate, symptom-free survival, and systolic blood pressure were recorded.
Results
CoQ10 promoted the expression of nuclear factor erythroid 2-related factor 2 and antioxidant enzymes. In H2O2-treated VSMCs, reactive oxygen species and cell apoptosis were reduced by CoQ10. In IA mice, CoQ10 treatment decreased the rupture rate of IA, improved the symptom-free survival, and reduced systolic blood pressure. Macrophage infiltration and expression of pro-inflammatory cytokines in the cerebral arteries were mitigated by CoQ10 treatment.
Conclusions
CoQ10 is effective in reducing oxidative stress in VSMCs, thereby attenuating IA formation and rupture in mice. CoQ10 also alleviates inflammation and restores normal phenotypes of VSMCs in the cerebral arteries. Our data suggest that CoQ10 is a potentially effective drug for managing IA.
Impact
-
To investigate the effect of CoQ10, a commonly used nutritional supplement, on IA initiation and progression in a mouse model, as well as the mechanism.
-
CoQ10 promoted the expression of Nrf2 and antioxidant enzymes. In H2O2-treated VSMCs, ROS and cell apoptosis were reduced by CoQ10.
-
CoQ10 is effective in reducing oxidative stress in VSMCs, thereby attenuating IA formation and rupture in mice.
Similar content being viewed by others


Introduction
Intracranial aneurysms (IAs) affect ~2–5% of the population, and the rupture of IA, as a leading cause of subarachnoid hemorrhage, often results in neurological failure or mortality. Microsurgery and endovascular interventions have made significant advances in treating IA, but morbidity and mortality rates remain formidably high.1 Consequently, there is an urgent need to develop noninvasive pharmacological therapies to prevent the progression and rupture of IA.
Efforts to elucidate the pathogenesis of IA have identified a number of targets for intervention, including IA-associated oxidative stress, inflammation, and vascular muscle cell dysfunction.2,3 It has been shown that sustained inflammation of the arterial vascular wall caused by hemodynamic stress is an important contributing factor of endothelial dysfunction, changes in the vascular smooth muscle cell (VSMC) phenotype, extracellular matrix remodeling, and cell death, which ultimately lead to IA formation.3 These cellular processes are manifested by degeneration, dilation, and eventual rupture of the vessel wall. The inflammatory response associated with IA formation is accompanied by the production of a large number of reactive oxygen species (ROS),4 which in turn further enhance inflammation. Consequently, strategies to alleviate inflammation and oxidative stress are desperately needed for the treatment of IA.
Coenzyme Q10 (CoQ10), also known as ubiquinone, is a naturally occurring fat-soluble vitamin-like substance found in the mitochondria, cell membranes, and the blood. CoQ10 is a cofactor mainly used as a mitochondrial enzyme that phosphorylates adenosine triphosphate (ATP) and mediates cellular metabolism. In its reduced form, CoQ10 serves as a free radical scavenger and protects membranes and proteins from damage induced by lipid peroxidized lipoproteins and ROS.5 In addition, it has been shown that CoQ10 activates the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway,6 which governs the expression of a cascade of genes to regulate the physiological and pathophysiological outcomes of oxidant exposure. Nrf2 activation is mediated by inducing antioxidant-reactive elements to reduce oxidative stress, which promotes the expression of several downstream detoxifying enzymes.7 Nrf2 is a recently emerged regulator of cellular resistance to oxidants, and the ability of CoQ10 in promoting Nrf2 expression makes CoQ10 a potential drug to treat IA.
In the current study, we carefully investigated the potential and mechanism of CoQ10 in reducing IA formation and rupture. We first constructed a hydrogen peroxide (H2O2)-induced oxidation of VSMC damage model, and explored if CoQ10 treatment effectively activated the Nrf2 signaling pathway and activated antioxidant-related enzyme expression. Subsequently, we constructed a mouse model of IA by stereotaxically injecting 2.5 μL elastase at the right basal cistern, followed by induction of systemic hypertension by administering angiotensin II, a method that has been widely used to induce IA in mice and shown to recapitulate human IAs.8,9 We evaluated whether CoQ10 reduced IA incidence and rupture and improved the survival of mice. The effects of CoQ10 in regulating inflammation were assessed by the amount of infiltrated macrophages in the cerebral arteries, as well as the expression of pro-inflammatory cytokines. In addition, whether CoQ10 restored the normal phenotypes of VSMCs in IA was explored. Our data could shed light on the pharmacological effects of CoQ10 in alleviating IA.
Materials and methods
Cell culture
VSMCs were collected from mice using a previous protocol.10 Briefly, thoracic aortas from mice were cut into pieces of 1–2 mm in length after removing the adventitia and endothelium, which were then placed in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% fetal bovine serum. VSMCs shed from explants were harvested and cultured in DMEM (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. Cells between passages 3–7 were used. For treatment with H2O2, VSMCs were cultured in a medium containing 0.5 mM H2O2 for 12 h. CoQ10 was added to incubate cells at the final concentration of 20 μM according to a previous study.11
Western blot
Cells and tissues were homogenized in the lysis buffer. To extract nuclear protein from cells, the Nuclear Extract Kit (Abcam, Cambridge, MA) was used and the remaining cytoplasmic proteins were also collected. Protein was loaded in 10% gel for sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to nitrocellulose membranes and incubated in 0.5% bovine serum albumin (BSA) to block nonspecific binding. Primary antibodies (acquired from Cell Signaling, Danvers, MA) were added to the membranes and incubated at 4 °C overnight. Horseradish peroxidase-conjugated secondary antibodies were then added to the membrane and incubated for 4 h at 4 °C. The amount of protein was quantified using the NIH ImageJ software and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression.
Quantitative real-time PCR
Quantitative real-time PCR was conducted to measure the levels of Nrf2, glutamate-cysteine ligase catalytic subunit (GCLC), NAD(P)H quinone dehydrogenase 1 (NQO1), superoxide dismutase 1 (SOD-1), vascular platelet-derived growth factor-B (PDGF-B), endothelial growth factor (VEGF), smooth muscle α-actin (SMA), and smooth muscle 22α (SM22α). Total RNA was extracted from VSMCs or cerebral arteries using the Trizol agent (Invitrogen, Waltham, MA), and then reverse-transcribed into DNA. Real-time PCR was performed using the SYBR Green Kit (Thermo Fisher Scientific, Waltham, MA) and a thermocycler (Eppendorf, Germany). The primers used in this study were designed and synthesized by GenePharma (Shanghai, China). GAPDH was used as an internal control to normalize the expression of other genes. The following primer sequences were used: Nrf2: forward, ACCACCGCCAGAAAGAG and reverse, ACGGGCAGGTTGATTAT. GCLC: forward, GCWGCYTCACCCTTCTACA and reverse, RTAAAACATGCCCTCCWGGAC. NQO1: forward, ACGGCGATGAGACGATG and reverse, CTGAAACCCGCAGAAGT. SOD-1: forward, CCTCGGCAACGTGACTGCTA and reverse, ACTTGGCTATTCCGATGACACC. PDGF-B: forward, GCTCTTCCTGTCTCTCTG-3 and reverse: GGTCACTCAGCATCTCATA-3. VEGF: forward, CCTCCGAAACCATGAACTTT and reverse, TTCTTTGGTCTGCATTCACATT. SMA: forward, AGTCGCCATCAGGAACCTCGAG‐3 and reverse, ATCTTTTCCATGTCGTCCCAGTTG. SM22α: forward, TTCTGCCTCAACATGGCCAAC and reverse, CACCTTCACTGGCTTGGATC. GAPDH: forward, TGCCTCCTGCACCACCAACT and reverse, CGCCTGCTTCACCACCTTC.
Flow cytometry
To detect ROS levels, cells were incubated with 2′,7′-dichlorofluorescein diacetate (DCFH-DA, Beyotime, China) for 30 min at 37 °C. After washing with phosphate-buffered saline three times, flow cytometry was performed to quantify positively stained cells. Cells were stained with Annexin V-FITC/PI solution (Thermo Fisher) for 15 min, followed by flow cytometry analysis to detect apoptotic cells.
Animals
All animal protocols were approved by the Ethics Committee of Quanzhou First Hospital Affiliated to Fujian Medical University. C57BL/6J mice (8–10 weeks of age, purchased from Cyagen Biosciences Inc., Suzhou, China) were stereotaxically injected with 2.5 μL elastase at the right basal cistern. Angiotensin II was immediately administered to induce systemic hypertension. The tail-cuff method was used to measure systolic blood pressure. CoQ10 was administered orally at the dose of 10 mg/kg. In the sham group, the mice underwent stereotaxis injection of an equal volume of saline. In the vehicle group, the IA mice were orally administered with saline. The survival of mice was monitored for 3 weeks, and mice were perfused and pathologically examined for incidence of IA and rupture. Entire cerebral arteries with or without aneurysm induction were collected for examination.
Immunofluorescence imaging
Immunofluorescence imaging of ROS production was performed on cells stained with DCFH-DA using confocal laser fluorescence microscopy (FV1000, Olympus, Tokyo, Japan). The IA lesions were cryosectioned at a thickness of 5 μm and blocked with 1% BSA. Primary antibodies specific for F4/80 and α-SMA, and AlexaFluor488- or AlexaFluor594-conjugated secondary antibodies were then sequentially added to the sections. All antibodies were acquired from Cell Signaling (Danvers, MA). DAPI (Sigma Aldrich, St. Louis, MO) was used to stain the nuclei. Confocal laser fluorescence microscopy was conducted to acquire images of the sections and images were processed by ImageJ.
Statistical analysis
All data were presented as mean ± standard deviation (SD) based on at least three independent biological repeats. GraphPad Prism 7 was used for statistical analysis. Kaplan–Meier curve was analyzed using log-rank test to determine statistical significance between survival curves. Statistical difference was determined using one-way or repeated-measures analysis of variance followed by a post hoc test.
Results
CoQ10 treatment activates Nrf2 signaling in VSMCs with oxidative damage
To induce oxidative damage, the VSMCs harvested from mice were treated with 5 mM H2O2 for 12 h. The levels of Nrf2, SOD-1, GCLC, and NQO1 in the cells pretreated with CoQ10 for 12 h before adding H2O2 were compared to those of cells without H2O2 treatment (control) or only H2O2 treatment (vehicle). As shown in Fig. 1, Western blot analysis showed that both nuclear Nrf2 and cytosolic Nrf2 became downregulated in VSMCs after H2O2 treatment (Fig. 1a–c), but pretreatment of VSMCs with CoQ10 restored the levels of nuclear (p < 0.01, compared to the vehicle group) and cytosolic (p < 0.05, compared to the vehicle group) Nrf2. Since Nrf2 plays an important role in regulating the resilience of cells to oxidative stress, the restoration of Nrf2 suggested that the damage induced by H2O2 treatment was alleviated. Furthermore, we also analyzed the levels of antioxidant enzymes, including SOD-1, GCLC, and NQO1, which showed that, while the vehicle group demonstrated higher levels of SOD-1, GCLC, and NQO1, the CoQ10 group demonstrated substantially higher upregulation of these enzymes (~8-fold upregulation for SOD-1, ~5-fold upregulation for GCLC, and ~4-fold upregulation for NQO1, all p < 0.001 compared to the vehicle group) (Fig. 1d–f).

A vascular smooth muscle cell (VSMC) oxidative damage model was developed by incubating VSMC with hydrogen peroxide (H2O2, 0.5 mM) for 12 h. VSMC was pretreated with CoQ10 (20 μM) for 12 h before stimulation with H2O2. The Nrf2 signaling was detected. a–c Western blot analysis of Nrf2 in the nuclear and cytoplasm fractions. *P < 0.05 and **p < 0.01, compared with the vehicle-treated group. d–f mRNA expressions of antioxidant enzymes, including superoxide dismutase 1 (SOD-1), glutamate-cysteine ligase catalytic subunit (GCLC), and NAD(P)H quinone dehydrogenase 1 (NQO1). g Mice vascular smooth muscle cells (VSMCs) were treated with CoQ10 (20 μM) for 12 h and the levels of Nrf2 were detected by Western blot. ***P < 0.001, compared with the vehicle-treated group.
To validate the role of CoQ10 in the modulation of Nrf2 signaling, VSMCs without H2O2 treatment were incubated with CoQ10 (20 μM) for 12 h, and the level of Nrf2 was examined, and we found that CoQ10 could significantly enhance the nuclear/cytosolic Nrf2, confirming CoQ10 could effectively increase Nrf2 expression (Fig. 1g).
CoQ10 reduces production of ROS and improves cell viability of H2O2-treated VSMCs
We next assessed whether the CoQ10-enhanced antioxidative capacity of VSMCs could reduce the production of ROS and improve cell viability. Flow cytometry (Fig. 2a) and immunofluorescence staining (Fig. 2b) suggested that while ROS production was higher in cells treated with H2O2 (vehicle group), pretreatment with CoQ10 significantly reduced the ROS production. Annexin V-FITC/PI staining flow cytometry also suggested that while H2O2 treatment led to increased apoptotic rate, CoQ10 treatment markedly reduced apoptosis induced by H2O2 (p < 0.001 vs. vehicle group, Fig. 2c, d). Furthermore, the expression of Nrf2 was knocked down by small interfering RNAs, and we found that CoQ10 showed a slight effect on the cell apoptosis of H2O2-treated mice VSMCs compared to when Nrf2 was downregulated (Fig. 2c, d). These evidences suggested that CoQ10 protected VSMCs from apoptosis and mitigated production of ROS.

The intracellular productions of ROS in vehicle or CoQ10-treated VSMC were assessed by flow cytometry (a) and confocal microscopy (b). c Apoptosis of VSMCs cells was examined by flow cytometry after staining with PI and Annexin V-FITC. VSMCs were transfected with Lipofectamine 2000/siNrf2 for Nrf2 downregulation. d Percentage of apoptotic VSMC cells based on the FACS analysis. ***P < 0.001, compared with the vehicle-treated group.
CoQ10 administration prevents IA rupture in a mouse model
To evaluate the effects of CoQ10 in reducing IA formation and rupture in vivo, we constructed an IA mouse model by injecting elastase into the basal cistern, and CoQ10 was administered orally every other day for 14 days. The normal mice (control group) and IA mice without CoQ10 treatment (vehicle group) were used as controls. Incidence of IA formation, rupture, symptom-free survival, and systolic blood pressure was monitored every week to day 21. Compared to the vehicle group, CoQ10 group demonstrated decreased incidence of IA (27 vs. 29, p < 0.05, Fig. 3a) and rupture rate (33.3% vs. 82.7%, p < 0.01, Fig. 3b). The symptom-free survival of the CoQ10 group was also significantly improved compared to the vehicle group (p < 0.01, Fig. 3c). Despite significantly increased systolic pressure, CoQ10 treatment effectively lowered systolic blood pressure compared to the vehicle group at 14 days post IA (p < 0.01, Fig. 3d).

a Incidence of unruptured and ruptured aneurysms, b rupture rate, and c symptom-free curve of mice in different groups. Mice that did not have aneurysms were excluded from this analysis. **P < 0.01 compared with the vehicle-treated group. d Systolic blood pressure. **P < 0.01, compared with the vehicle-treated group.
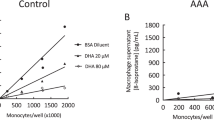
CoQ10 inhibits macrophage infiltration in IA lesions and lowers expression of pro-inflammatory cytokines
Since dysregulated inflammation is one of the contributing factors of IA formation and progression, we harvested IA lesions and performed immunofluorescence staining of F4/80 to assess the level of macrophage infiltration (Fig. 4a) and used Western blot analysis to quantify the expression of F4/80, interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α) proteins (Fig. 4b–e). As expected, elastase treatment resulted in increased macrophage infiltration in the smooth muscles, but CoQ10 mitigated macrophage infiltration (Fig. 4a), which was verified by Western blot analysis of F4/80 expression (Fig. 4b). Furthermore, CoQ10 pronouncedly inhibited pro-inflammatory cytokines (Fig. 4c–e). Hence, CoQ10 effectively alleviated inflammation in the cerebral arteries.

a Macrophages infiltrating in IA lesions from mice were visualized by immunostaining for F4/80 (macrophage, green, AlexaFluor488), colocalized with α-SMA (smooth muscle cells, red, AlexaFluor594) and DAPI (nucleus, blue). b The levels of F4/80 in IA lesions were examined by western blot. Expression of IL-6 (c), IL-1β (d), and TNF-α (e) in cerebral arteries from treated mice were examined by real-time PCR (up) and western blot (low). **P < 0.01 and ***p < 0.01, compared with the vehicle-treated group.
CoQ10 restores normal physiology of VSMCs in the cerebral arteries
To further elucidate the effects of CoQ10 in alleviating IA formation and rupture, we explored whether the normal phenotypes of VSMCs in the cerebral arteries could be restored by CoQ10 treatment. As shown in Fig. 5a, b, while the expression of VEGF and PDGF-B was markedly upregulated, suggesting augmented angiogenesis, CoQ10 treatment significantly downregulated the levels of VEGF and PDGF-B. Furthermore, while SM22α and αSMA levels were downregulated in the vehicle group, suggesting an impaired contractile phenotype, CoQ10 treatment upregulated their levels as well (Fig. 5c, d). These data indicated that CoQ10 potently restored the normal phenotypes of VSMCs in the cerebral arteries.

Expressions of vascular endothelial growth factor (VEGF) (a) and platelet-derived growth factor-B (PDGF-B) (b) in cerebral arteries in the mRNA level were examined by real-time PCR. VEGF and PDGF-B are two main angiogenic growth factors. **P < 0.01, compared with vehicle-treated group. Expressions of smooth muscle 22α (SM22α) (c) and smooth muscle α-actin (αSMA) (d) in cerebral arteries in the mRNA level were examined by real-time PCR. SM22α and αSMA are two specific markers of VSMC with contractile phenotype. **P < 0.01 and ***p < 0.001, compared with the vehicle-treated group.
Discussions
CoQ10 is a common fat-soluble micronutrient found in the human body and is a coenzyme for mitochondrial oxidative phosphorylation. A variety of human malignancies are characterized by CoQ10 deficiency, which is concomitant with dysregulated oxidative stress and inflammatory responses. Hence, supplementation of CoQ10 has been shown as an effective pharmacological approach, whereby CoQ10 is exploited as an antioxidant to neutralize free radicals. Besides, CoQ10 was also shown to promote the expression of antioxidant enzymes and activate antioxidative signaling pathways, such as Nrf2. In heart failure, the amount of CoQ10 in the myocardial tissues decreases, and supplementation of CoQ10 counteracts the loss of antioxidants and reduces ROS, protecting plasma lipoproteins and cell membranes.12 In preeclampsia, CoQ10 also adopts an antioxidant role and alleviates hypertension to improve preeclamptic symptoms in women. In IA, there is also a disturbed balance between oxidation and antioxidation in the body, and the protective function of the body’s antioxidant enzymes is compromised. The applications of CoQ10 in other brain diseases, including ischemia-induced injury,13 brain atrophy,14 amyloid deposition,15 neurological disorders,16 and so on, have also been explored. However, to our best knowledge, the pharmacological efficacy of CoQ10 in IA has not been investigated, and our study has for the first time clarified that orally administered CoQ10 confers protection against IA in the elastase-induced IA mouse model. Our results are in agreement with the effects of CoQ10 in regulating vascular endothelial function.17,18,19
To investigate the antioxidative effects of CoQ10 in alleviating injury of vascular endothelial cells, we treated VSMCs with H2O2 and assessed whether incubating VSMCs with CoQ10 could activate the Nrf2 signaling and promote the expression of antioxidative enzymes, including SOD-1, GCLC, and NQD-1, as well as attenuating ROS production and cell apoptosis. Indeed, our results indicated that CoQ10 conferred significant protection for VSMCs against oxidative injury by upregulating the Nrf2 signaling pathway and antioxidant enzymes. The role of CoQ10 in protecting against H2O2-induced cell injury has been investigated using similar approaches in bone marrow-derived stem cells (BMSCs), where CoQ10 was shown to mitigate apoptosis of transplanted BMSCs in the spinal cord by activating the Nrf2 signaling,19 and therefore enhancing regenerative efficacy in spinal cord injury. To exclude the possibility that the CoQ10-induced upregulation of Nrf2 is a result of improvement from H2O2-induced cellular damage, VSMCs without H2O2 treatment were incubated with CoQ10. Indeed, we found that CoQ10 could significantly enhance the nuclear/cytosolic Nrf2, confirming that CoQ10 could effectively increase Nrf2 expression. Here, we performed in vitro studies on VSMCs, since IA is characterized by remodeling of vascular endothelial cells,20 and the molecular changes induced by CoQ10 in VSMCs could be representative of pharmacological effects of CoQ10 in cerebral arteries in IA. A number of previous studies have also employed VSMCs as the cells of interest to test the efficacies of pharmacological compounds or genes to treat IA.21,22,23
We verified that orally administered CoQ10 reduced the incidence and rupture rate of IA in mice. The IA mouse model was constructed by injecting elastase into the basal cistern, which is a common method to establish IA in animals24,25 that recapitulates pathogenesis of IA in humans. In this model, elastase attenuates elastic lamina in the vascular endothelium, which is also a pathological manifestation in human aneurysms. Here, we showed that the decreased IA formation and rupture concurred with significantly improved symptom-free survival and reduced systolic blood pressure in IA mice. The improved mouse survival was accompanied by the CoQ10-reduced inflammation in IA mice, as evidenced by the marked reduction in macrophage infiltration in the cerebral arteries and pronouncedly lower levels of pro-inflammatory cytokines including IL-6, IL-1β, and TNF-α. Macrophage infiltration is a pivotal factor in the pathogenesis and progression of IA,26 since macrophage and macrophage-mediated cytokine production play an important role in inflammation and hemodynamically induced vascular remodeling that leads to the development, growth, and rupture of IA. The effects of CoQ10 in inhibiting macrophage infiltration and production of pro-inflammatory cytokines also underscore its clinical utility in attenuating oxidative stress and inflammation.
Lastly, we also showed that CoQ10 restored the normal phenotypes of VSMCs in vivo, as suggested by the reduced levels of VEGF and PDGF and increased levels of SM22α and SMAα in VSMCs harvested from the cerebral arteries of IA mice. These data suggest that CoQ10 inhibits the expression of angiogenic growth factors, thereby mitigating angiogenesis, which is a contributing factor to vascular modeling.27 CoQ10 also promotes the contractile phenotypes of VSMCs since the rupture of IA is associated with decreased elasticity of vasculature.28,29
Our study has several limitations. First, although we showed marked effects of CoQ10 in reducing symptoms of IA using a dose of 10 mg/kg/day, it is possible that a higher dose of CoQ10 could result in better therapeutic outcomes and therefore a dose-escalation study would be of great benefit. Second, further studies on the precise molecular targets of CoQ10 in vivo to regulate macrophage infiltration and production of pro-inflammatory cytokines are needed.
In summary, we herein evaluate the efficacies of CoQ10 in alleviating oxidative damage induced by H2O2 in VSMCs, suggesting that CoQ10 activates the Nrf2 signaling, promoting the expression of antioxidative enzymes while reducing ROS production and cell apoptosis. We also demonstrate that orally administered CoQ10 confers significant protection in IA mice against IA rupture by improving mouse survival. Meanwhile, systolic blood pressure, macrophage infiltration, and pro-inflammatory cytokine production are reduced by CoQ10. CoQ10 also restores normal phenotypes of VSMCs by decreasing angiogenic factors and increasing contractile phenotypes. Together, our data provide evidence supporting the utility of CoQ10 in treating IA. Further clinical studies are warranted to corroborate the benefit of CoQ10 intake in IA patients.
References
Toth, G. & Cerejo, R. Intracranial aneurysms: review of current science and management. Vasc. Med. 23, 276–288 (2018).
Penn, D. L., Witte, S. R., Komotar, R. J. & Connolly, Jr. E. S. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J. Clin. Neurosci. 21, 28–32 (2014).
Hashimoto, T., Meng, H. & Young, W. L. Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol. Res. 28, 372–380 (2006).
Zou, L., Hou, Y., Yu, B., Li, S. & Du, Y. The effect of intravascular interventional embolization and craniotomy on MMP-2, MMP-9 and caspase3 in serum of intracranial aneurysm patients. Exp. Ther. Med. 16, 4511–4518 (2018).
Santoro, M. M. The antioxidant role of non-mitochondrial CoQ10: mystery solved! Cell Metab. 31, 13–15 (2020).
Li, L. et al. Protective effects of coenzyme Q10 against hydrogen peroxide-induced oxidative stress in PC12 cell: the role of Nrf2 and antioxidant enzymes. Cell. Mol. Neurobiol. 36, 103–111 (2016).
Mohammadzadeh, M. et al. Nrf-2 overexpression in mesenchymal stem cells reduces oxidative stress-induced apoptosis and cytotoxicity. Cell Stress Chaperones 17, 553–565 (2012).
Nuki, Y. et al. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension 54, 1337–U1108 (2009).
Strange, F., Gruter, B. E., Fandino, J. & Marbacher, S. Preclinical intracranial aneurysm models: a systematic review. Brain Sci. 10, 134 (2020).
Zhu, Q. et al. Intermedin reduces neointima formation by regulating vascular smooth muscle cell phenotype via cAMP/PKA pathway. Atherosclerosis 266, 212–222 (2017).
Inui, M. et al. Mechanisms of inhibitory effects of CoQ(10) on UVB-induced wrinkle formation in vitro and in vivo. Biofactors 32, 237–243 (2008).
Singh, U., Devaraj, S. & Jialal, I. Coenzyme Q10 supplementation and heart failure. Nutr. Rev. 65, 286–293 (2007).
Li, H., Klein, G., Sun, P. & Buchan, A. M. CoQ10 fails to protect brain against focal and global ischemia in rats. Brain Res. 877, 7–11 (2000).
Li, G., Jack, C. R., Yang, X. F. & Yang, E. S. Diet supplement CoQ10 delays brain atrophy in aged transgenic mice with mutations in the amyloid precursor protein: an in vivo volume MRI study. Biofactors 32, 169–178 (2008).
Moreira, P. I. et al. CoQ10 therapy attenuates amyloid β-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp. Neurol. 196, 112–119 (2005).
Belousova, M., Tokareva, O. G., Gorodetskaya, E., Kalenikova, E. I. & Medvedev, O. S. Intravenous treatment with coenzyme Q10 improves neurological outcome and reduces infarct volume after transient focal brain ischemia in rats. J. Cardiovasc. Pharmacol. 67, 103–109 (2016).
Gao, L. et al. Effects of coenzyme Q10 on vascular endothelial function in humans: a meta-analysis of randomized controlled trials. Atherosclerosis 221, 311–316 (2012).
Larijani, V. N. et al. Beneficial effects of aged garlic extract and coenzyme Q10 on vascular elasticity and endothelial function: the FAITH randomized clinical trial. Nutrition 29, 71–75 (2013).
Huo, J. et al. Coenzyme Q10 prevents senescence and dysfunction caused by oxidative stress in vascular endothelial cells. Oxid. Med. Cell Longev. 2018, 3181759 (2018).
Meng, H. et al. Complex hemodynamics at the apex of an arterial bifurcation induces vascular remodeling resembling cerebral aneurysm initiation. Stroke 38, 1924–1931 (2007).
Sun, L. et al. MiR-29b downregulation induces phenotypic modulation of vascular smooth muscle cells: implication for intracranial aneurysm formation and progression to rupture. Cell Physiol. Biochem. 41, 510–518 (2017).
Guo, F. et al. Increased apoptosis and cysteinyl aspartate specific protease-3 gene expression in human intracranial aneurysm. J. Clin. Neurosci. 14, 550–555 (2007).
Xu, J. et al. The miR-143/145 cluster reverses the regulation effect of KLF5 in smooth muscle cells with proliferation and contractility in intracranial aneurysm. Gene 679, 266–273 (2018).
Nuki, Y. et al. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension 54, 1337–1344 (2009).
Hosaka, K., Downes, D. P., Nowicki, K. W. & Hoh, B. L. Modified murine intracranial aneurysm model: aneurysm formation and rupture by elastase and hypertension. J. Neurointerv. Surg. 6, 474–479 (2014).
Kanematsu, Y. et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 42, 173–178 (2011).
Kamio, Y. et al. Roles of nicotine in the development of intracranial aneurysm rupture. Stroke 49, 2445–2452 (2018).
Leal, A. G., Mori, Y. T., Nohama, P., de Souza, M. A. Three-dimensional hollow elastic models for intracranial aneurysm clipping election–a case study. In 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), 4137–4140 (IEEE, 2019).
Ruigrok, Y. M., Rinkel, G. J. & Wijmenga, C. Genetics of intracranial aneurysms. Lancet Neurol. 4, 179–189 (2005).
Acknowledgements
This study was supported by Science and Technology Project of Quanzhou City of Fujian Province (2018T007R).
Author information
Authors and Affiliations
Contributions
Data collection and analysis: J.H., H.Z., L.Y., J.Z., and Z.J.; study designed and manuscript writing: J.H. and J.Z. All authors approved the final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, J., Zhang, H., You, L. et al. Coenzyme Q10 inhibits intracranial aneurysm formation and progression in a mouse model. Pediatr Res 91, 839–845 (2022). https://doi.org/10.1038/s41390-021-01512-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-021-01512-8
This article is cited by
-
A Future Blood Test to Detect Cerebral Aneurysms
Cellular and Molecular Neurobiology (2023)
-
Dexmedetomidine alleviates inflammatory response and oxidative stress injury of vascular smooth muscle cell via α2AR/GSK-3β/MKP-1/NRF2 axis in intracranial aneurysm
BMC Pharmacology and Toxicology (2022)
