
Abstract
Nitric oxide (NO) has critical roles in a wide variety of key biologic functions and has intricate transport mechanisms for delivery to key distal tissues under normal conditions. However, NO also plays important roles during disease processes, such as hypoxia–ischemia, asphyxia, neuro-inflammation, and retinopathy of prematurity. The effects of exogenous NO on the developing neonatal brain remain controversial. Inhaled NO (iNO) can be neuroprotective or toxic depending on a variety of factors, including cellular redox state, underlying disease processes, duration of treatment, and dose. This review identifies key gaps in knowledge that should prompt further investigation into the possible role of iNO as a therapeutic agent after injury to the brain.
Impact
-
NO is a key signal mediator in the neonatal brain with neuroprotective and neurotoxic properties.
-
iNO, a commonly used medication, has significant effects on the neonatal brain.
-
Dosing, duration, and timing of administration of iNO can affect the developing brain.
-
This review article summarizes the roles of NO in association with various disease processes that impact neonates, such as brain hypoxia–ischemia, asphyxia, retinopathy of prematurity, and neuroinflammation.
-
The impact of this review is that it clearly describes gaps in knowledge, and makes the case for further, targeted studies in each of the identified areas.
Similar content being viewed by others


Introduction
There has been significant progress to uncover the role of nitric oxide (NO) in physiological and pathological conditions in many tissues and organs. As reviewed in Part 1 of this two-part review, NO is produced by NO synthases (NOS) and activates intracellular signaling pathways. The most well-known target of NO is the reaction with the iron atom of guanylyl cyclase to activate it and produce cyclic guanosine monophosphate (cGMP) that promotes the vasodilatory functions of NO. However, the biological properties of NO affect multiple other cellular functions and go beyond the activation of cGMP.
One such role of NO is the regulation of distal hemodynamics and vascular tone, an effect that is mediated by the ability of NO to react with iron to form nitrosonium (NO+) ions. In brief, the key iron atom that can react with NO and participate in S-nitrosothiols (R-SNO) formation is typically bound to proteins. The most abundant of these proteins is Hgb. Hgb contains both iron and a thiol group and hence is ideal for transport and regulation of NO. In mammals, NO reacts with a highly conserved residue of cysteine at the 93 position of the beta-chain of Hgb (Cysβ93) to form S-nitroso-Hgb (SNO-Hgb).1 The formation of SNO-Hgb is the main transporter of NO to tissues. Changes in partial pressure of oxygen can cause allosteric changes in Hgb, which results not only in well-described changes of its affinity to carbon dioxide, but also in the release of NO and vasodilatation. Thus, SNO-Hgb has the ability to play the role of NO carrier or donor depending on the redox state of the red blood cell.
Inhaled NO (iNO) can act as a donor of NO in biological systems and transfer from the lung to the brain. iNO can be either neuroprotective or neurotoxic depending on a variety of factors, including cellular redox state, underlying disease processes, duration of treatment, and dose. NO plays significant roles during disease processes, such as hypoxia–ischemia, asphyxia, neuro-inflammation, and retinopathy of prematurity, which associate with significant morbidity in the neonatal population. In the second part of this review, we present the effects of iNO under abnormal conditions and we present key gaps in knowledge that should prompt further investigation into the possible role of iNO as a therapeutic agent after injury to the brain.
iNO and hypoxia–ischemia
Does iNO interfere with the endogenous NOS system?
Physiologically, endogenous NO is produced by the actions of two enzymes, namely neuronal NOS (nNOS), which is activated by N-methyl-d-aspartate (NMDA) receptors in a phasic, acute mode2 and endothelial NOS (eNOS), expressed in the endothelium of blood vessels, which is responsible for steady, low concentrations of NO.3,4 Indeed, both steady-state eNOS and acute, phasic nNOS are required for optimal neuronal function, plasticity, and memory.3
Brain hypoxia–ischemia increases NOS activity and NO brain content. In rodents, the magnitude of increase of in vivo NO production can be up to 1000 times above the baseline and occurs within minutes after brain ischemia induced by middle cerebral artery occlusion.5 Experiments with electron paramagnetic resonance spectroscopy provided evidence that ischemic injury increases the production of NO in parallel to the duration of brain ischemia, and increases even further after reperfusion.6,7,8 A 2-h occlusion of the middle cerebral artery (MCA) in rats followed by reperfusion resulted in increased SNO-Hgb in the jugular blood, as measured by electron spin resonance spectroscopy, as early as 30 min of recirculation.9 The messenger RNA (mRNA) for nNOS increased as early as 15 min after ischemia in rodents with increasing NOS activity in the first hour of the experiment.10 Interestingly, in a study of prolonged in vivo ischemia, eNOS mRNA decreased with time, while nNOS mRNA increased. Further, nNOS mRNA containing neurons were resistant to ischemia.11
In newborn pigs, NOS activity either remained unchanged after 1 h of hypoxia12 or decreased throughout the brain except in the cortex,13 but increased after reoxygenation.13 Two hours of focal ischemia in mice, followed by reperfusion induced a rapid decrease in nNOS activity followed by increase in inducible NOS (iNOS) activity.14 The observed differences in NOS activation patterns after brain hypoxia between rodent and swine animal models have not been adequately explained. Human NOS activation after hypoxia might resemble the swine pattern, given the significant homology between swine and human proteins.15,16
Whether iNO associates, blocks, or induces endogenous brain NOS is difficult to assess since this varies with the redox status of any given cell. Currently, there are no data to investigate the effect of iNO on cerebral NOS, especially after prolonged use in the immature neonatal brain. Interestingly, developmental increases in NO-mediated protein modifications (S-nitrosocysteine and 3-nitrotyrosine) occur in an animal model of chronic lung disease (CLD), and iNO increased NO metabolites, such as nitrate, nitrite, SNO, and S-nitrotyrosine, without affecting the endogenous activity of NOS.17
The application of iNO after brain hypoxia is complicated by the non-homogeneous changes of NOS modifications in various areas of the brain. For example, in newborn pigs exposed to hypoxia–ischemia followed by reperfusion, a significant decrease in NOS activity occurs in all parts of the brain except the cortex in the acute phase. Upon reperfusion, NOS activity increases most prominently in the thalami.13 Using a modified magnetic resonance technique to quantify NO radicals, areas that had high NOS activity, such as the cortex, hippocampus, hypothalamus, amygdala, and the substantia nigra, had higher levels of NO radical trapping.18 The occlusion of the right middle cerebral artery in a rodent animal model was associated with histopathologic findings in the hippocampus, as well as increased tissue concentrations of NO and its derivatives.19 Brain nitrite was significantly increased in the right versus left cortex after right middle cerebral artery occlusion, with a return to baseline by 60 min after hypoxia. Cortical cGMP was also increased after occlusion. NOS activity increased ~10-fold from baseline 10 min after occlusion in the right cortex and decreased markedly thereafter. Collectively, these studies show that even if NO derived from iNO could be homogeneously distributed in the brain, the effects would be variable given the different distribution and activation of the endogenous NOS enzymes, especially during hypoxia. We conclude that there are currently no data that define the role of iNO in the regulation of endogenous brain NOS.
What is the role of the iNOS and does inhaled NO affect its expression or function?
In the central nervous system (CNS), iNOS (or NOS2) is not present at baseline, but is expressed under stress. The primary physiological role of iNOS is the destruction of pathogens as part of the innate immune response.20 iNOS is regulated at the transcriptional or post-transcriptional level after cellular activation by a variety of danger signals using intracellular signaling pathways, such as nuclear factor kappa B (NF-κB) and activator of transcription 1a.21 These transcription factors bind to the promoter of the iNOS gene and induce its expression. As a result, the effect of iNOS is time limited, persisting for several hours after the initial insult has been removed as the enzyme is fully degraded.22 iNOS is induced in microglia23 and is associated with late and persistent activation of these cells after hypoxia–ischemia in the brain.24 For these reasons, iNOS is considered to link hypoxic brain injury with neuroinflammation.
NO produced by iNOS can bind to iron or sulfur atoms and deactivate a variety of enzymes, or form peroxynitrite and induce DNA damage.25,26 NO is also a physiologic regulator of oxidative phosphorylation. Excessive NO production competes with oxygen and inhibits mitochondrial respiration by blocking the complex IV of cytochrome oxidase.27,28
In the brain, there are several protective mechanisms that prevent the excessive activation of iNOS. Availability of l-arginine,29 activation of argininosuccinate synthase (which converts l-citrulline back to l-arginine and hence maintains substrate balance),30 presence of particular cytosolic proteins that can deactivate iNOS, such as Karilin,31 or absence of tetrahydrobiopterin (an enzymatic cofactor),32 are some of the mechanisms that can decrease excessive iNOS activation.
iNOS differs from eNOS and nNOS not only in the calcium sensitivity, but also in the cytosolic location (nNOS and iNOS are cytosolic enzymes versus eNOS, which is largely membrane associated) and the molecular mass (similar to eNOS, but smaller than nNOS).33 iNOS, although previously thought not to require calcium for activation, in fact requires lower concentrations of calcium due to its structural co-localization with calmodulin (a calcium-binding intermediate messenger).34 All synthases have similar structures and include both an oxygenase domain (which binds heme and l-arginine) and a reductase domain (which binds FAD and NADPH), while requiring dimerization for full function. In the case of iNOS there are only small structural differences compared to the other types of NOS,35 which makes the development of specialized inhibitors challenging.36,37,38
Administration of the iNOS inhibitor aminoguanidine39 reduces brain injury, indicating that excessive NO production exerts cytotoxic effects in the developing brain, while inhibition of iNOS using 2-iminobiotin prevented activation of caspase-3, a key step of intrinsic apoptotic pathway, after hypoxic brain injury.40
Interestingly, NO itself can deactivate iNOS in a non-reversible manner via feedback mechanisms. NO donors (such as GSNO (S-nitroso-glutathione)) inhibit, while NO scavengers (such as Hgb) increase iNOS activity.41 Smith et al.42 proposed that the mechanism of iNOS deactivation occurs via S‐nitrosation and iNOS dimer dissociation. Despite the control of iNOS by NO, an anticipated effect of iNO on this pathway has not been proven so far.
In summary, after hypoxia ischemia, iNOS becomes activated in the microglia, results in excess NO production, and is associated with several deleterious effects on the developing brain. Use of iNO during the late phases after hypoxia ischemia could decrease the activation of iNOS. The presented data of irreversible inhibition of iNOS after NO use is of unclear clinical significance and needs to be studied further.
Can iNO prevent excitotoxicity after brain hypoxia–ischemia?
NO participates in NMDA-mediated cytotoxicity, possibly via the formation of ROS and via inhibition of energy production at the mitochondria level. Glutamate, the main excitatory amino acid implicated in excitotoxicity, acts mainly on NMDA, and also on alpha-3-amino-hydroxy-5-methyl-4-isoxazole (AMPA), kainate, and metabotrobic receptors. Physiologically nNOS, the main source of NO in CNS, is coupled to the NMDA receptor by the scaffolding protein PSD95 via calcium-dependent processes.43,44 In addition to NMDA, AMPA also triggers endogenous NO formation and hence cGMP production, although this link is less well described.45
Excessive NMDA activation is linked to downstream activation of nNOS and further production of NO, but also increases oxidative phosphorylation rates and superoxide production. Excess superoxide can react with NO to produce peroxynitrite, which, in turn, is directly toxic to mitochondrial proteins of the electron transport chains, and attacks protective enzymes, such as superoxide dismutase, causing a vicious cycle.46,47 Although peroxynitrite is produced initially inside the mitochondria, it can translocate to the nucleus and cause DNA fragmentation and initiate apoptotic and necrotic mechanisms, via activation of caspases, production of poly (ADP-ribose) polymerase 1,48 depletion of energy intermediates,49 and release of apoptosis-inducing factor.50
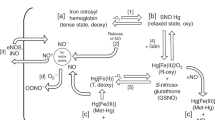
From the biochemical standpoint, iNO has significant effects on the brain after hypoxia–ischemia (Fig. 1). Pretreatment with iNO for 24 h had a protective effect, as shown by histopathology of the cortex and periventricular white matter of rats whose brains were injected with NMDA receptor activators such as NMDA and ibotenate, which activates NMDA and metabotrobic receptors, or S-Willardiine, which activates AMPA and kainate receptors. The effects were also dose dependent. Further, iNO-treated rats had less astrogliosis as quantified by GFAP, and microglial activation, using a macrophage marker, ED-1. In S-Willardiine-induced brain damage, there was a protective effect of iNO only in the cortex and not in the white matter. In NMDA- and ibotenate-treated animals, the protective effect of iNO was global, but there was no effect if iNO was applied after the injury. In fact, iNO-treated animals showed a downregulation of all types of NMDA receptors.51

Nitric oxide from the lung exposed to iNO can be carried in the red blood cells using the R-SNO system of hemoglobin and diffused into the cell [1]. NO also can be endogenously produced via the NOS system [2]. Intracellularly, NO can target various pathways with different biologic effects. For example, NO reactions with superoxide produces peroxynitrite and interfere with electron transport chain integrity with subsequent necrosis [3]. The presence of NO in the cell also results in modulation of intracellular signals (such as Ras/Erk, activation of c-Jun and c-Fos, or HSP70 and bcl-2). The balance of these signals can pivot between survival or programmed cell death [4]. VEGF activation is also be influenced by NO [5]. Another interesting interaction is the association with NMDA receptors. The redox status of the cell favoring the formation of nitrosonium ion might decrease the activity of NMDA-inducing neuroprotection [6]. NO also decreases the cAMP-mediated phosphorylation of CREB, which binds to CRE represented by a palindromic sequence (TGACGTCA) [7]. Decreased activity of CRE as well the corresponding genes downregulate the formation of an array of NMDA receptor subunits [9]. Under conditions of stress, NMDA in turn can be activated and increase pCREB via increased intracellular calcium levels [8].
NO has been associated with improved tissue tolerance to ischemia, also known as preconditioning. iNO affects pathways involving the cAMP response element (CRE) and the corresponding binding protein CREB. CRE/CREB are sensors of intracellular danger signals and are characterized by their ability to modulate an array of genes in the brain that affect memory, learning, and neuron survival.52 Many of these effects observed in hypoxia–ischemia occur via the phosphorylation of CREB mediated by NMDA receptors and associated changes in cytosolic calcium. There is evidence that CREB activation is involved with ischemic preconditioning53 and inhibition of CREB-mediated gene transcription interferes with the development of ischemia tolerance.54 NO has also been implicated in Ras activation associated with NMDA. NO can activate the Ras and extracellular-regulated kinase (Erk) pathways in neurons, as well as protein kinase C in myocardium,55 and offers resistance to injury.56 In turn, Erk activation can prevent apoptosis. Further, iNO in rats decreases Akt, phosphorylated Akt, and pCREB protein.51
The neuroprotective effect of iNO, when associated with NMDA, depends on the redox state of the microenvironment. Lipton et al.57 suggested that nitrosonium ion (NO+) plays an important role in the neuroprotective effects of NO. Downregulation of NMDA receptor activity is achieved by a reaction with a thiol group at the receptor’s modulatory site. This reaction is not mediated by NO itself, but occurs under conditions of S-nitrosylation of the NMDA receptor thiol in the presence of NO+. Under conditions of cellular stress, which typically deplete the thiol pool, the reaction is minimal or reversed. Under such conditions, NO can also participate in S-nitrosylation of a variety of other proteins including p21 ras,58 and caspase-3.59
Thus far, there are limited data addressing the use of iNO with therapeutic hypothermia as synergistic therapy for hypoxic–ischemic injury. NO is involved in the beneficial biochemical effects that are observed with therapeutic hypothermia especially in the early phases after brain hypoxia, although the evidence is not strong.60 In animal studies, therapeutic hypothermia decreases the accumulation of excitatory amino acids, suppresses NO production, and superoxide formation.61,62,63 In an animal model of MCA occlusion, increases in markers of NO synthesis (nitrite, cGMP levels) and NOS activity were abolished with hypothermia.64 Interestingly, in a cardiac arrest/CPR model using wild-type and eNOS knockout mice followed by hypothermia, the deficiency of eNOS abolished the beneficial effects of hypothermia after CPR and iNO at high doses improved survival in hypothermia-treated mutant mice.65
In summary, iNO affects intracellular mechanisms after hypoxia–ischemia via the NMDA receptor, a key mediator of excitotoxicity. These observations are tempered by the need for pretreatment with iNO, which limits its application in clinical settings. There are limited data for any synergism between iNO and hypothermia neuroprotection.
iNO and asphyxia
Administration of iNO offers neuroprotection to asphyxiated newborn pigs after severe meconium aspiration syndrome. Aaltoren et al.66 allocated newborn pigs to groups with and without concurrent asphyxia, as well as with or without instillation of meconium intratracheally (MAS). Examination of the brains in the MAS group showed that asphyxia resulted in neuronal injury in the cortical, cerebellar, and hippocampal hilar regions. In a subsequent study, meconium-affected newborn pigs were treated with 20 p.p.m. of iNO starting 30 min after the insult. The extent of brain injury was analyzed by histology, and quantifying brain tissue lipid peroxidation products, reduced glutathione (GSH), myeloperoxidase activity, and oxidized DNA. While iNO did not change the systemic or carotid hemodynamics in this model, treatment was associated with reduced neuronal injury, diminished oxidized DNA in the hippocampus, and decreased GSH in the cortex, but without any significant change in lipid peroxidation or myeloperoxidase activity.67
The effect of NO on cerebral autoregulation in lieu of an abnormal partial pressure of carbon dioxide (pCO2) is quite controversial. NOS inhibitors generally attenuate the increase of CBF that occurs with CO2 in a dose-dependent manner.68,69 Other studies, however, do not show the NO dependency of hypercarbia-induced vasodilatation.70 In experiments with graded hypercapnia in rats, the higher the pCO2, the less likely that the vasodilatation was NO mediated.71 NO-mediated regulation of CO2 vasodilatation occurs between 60 and 100 Torr, but very high pCO2 (>100 Torr) are NO independent. Further, increased pCO2 is associated with perivascular acidosis and smooth muscle relaxation, and NO appears to participate as an intermediate resulting in these two outcomes.71
The changes in autoregulation and vasodilatation, as well changes in the metabolism of cells that occur with iNO, could explain the associated changes seen in cerebral electrical activity. There are limited data on the effects of iNO on the electrical activity of the brain, especially in newborns with hypoxia–ischemia. In a Pediatric ICU setting, it was noted that critically sick infants treated for congenital heart disease or severe respiratory distress developed electroencephalographic (EEG) changes after iNO treatment. The EEG abnormalities reported included background slowing, low voltage, burst suppression, and sharp waves independent of age and NO indication.72 However, in preclinical studies, results are conflicting. In a neonatal sheep model, six lambs treated with iNO (2–60 p.p.m.) for Group B Streptococcus-induced pulmonary hypertension had serial assessments of their electrocortical activity and indices of antioxidative capacity and lipid peroxidation, but none of these parameters changed significantly.73 A case report on an adult patient who developed lower motor neuron disease resulting in paralysis after 2 weeks of treatment with iNO suggests a potential neurotoxic effect.74 It is obvious that more studies are needed on the exact effects of iNO in the brain electrical activity, whether it can induce seizures, and the duration and clinical significance of these effects.
Physiological NO mediates carbon dioxide-related vasodilation. Although iNO affects metabolism and cerebral blood flow, there is no solid evidence of its effect on EEG.
The role of iNO in retinopathy of prematurity
NO strongly promotes vascular growth. Vascular endothelial growth factor (VEGF), an angiogenic factor that affects cell survival, proliferation, and migration, activates eNOS to stimulate the production of endogenous NO.75,76,77 The effects of VEGF in stimulating angiogenesis and increasing vascular permeability are mediated by NO.78,79 VEGF receptor engagement increases intracellular calcium by triggering a signaling cascade that results in Src kinase-mediated activation of phospholipase C-gamma (PLC-γ), with parallel activation of the phosphoinositide 3 kinase/Akt pathway system80,81,82 (Fig. 2). Akt phosphorylates serine 1177 on eNOS and this post-translational modification activates the enzyme to catalyze the production of NO.83,84 PLC-γ also activates diacylglycerol, which also upregulates eNOS, albeit more slowly. Hypoxic conditions upregulate VEGF by the activation of hypoxia-inducible factor-1.85 VEGF effects on vascular tone in coronary arteries is also mediated by intracellular calcium and NOS.86

Akt phosphorylates eNOS and this modification activates the production of NO acutely. PLC-γ activates diacylglycerol, which also upregulates eNOS slowly. Hypoxic conditions upregulate VEGF by the activation of HIF-1. NO can activate the production of VEGF playing a role of proximal regulator and distal effector in this pathway.
Insulin-like growth factor-1 (IGF-1) and VEGF play important roles in normal development of retinal vessels. Preterm newborns have decreased baseline IGF-1 and ability to produce new IGF-1. These low concentrations of IGF-1 are responsible for the disruption of retinal vessel growth. VEGF production is regulated by oxygen and is a major factor in the development of retinopathy of prematurity (ROP). The relative hyperoxia that occurs after birth suppresses the production of VEGF, resulting in an avascular retina, while subsequently, intermittent hypoxia drives VEGF synthesis in combination with IGF-1 to cause abnormal neovascularization.87,88
The effects of iNO on ROP are controversial. In a systematic review, 14 randomized controlled trials of iNO therapy in preterm infants were included and noted a reduction of 7% in the combined outcome of death or CLD. iNO did not affect other complications of prematurity, such as severe ROP.89 In a recent prospective study, which included infants with gestational age <32 weeks and/or birth weight <1500 g, ROP developed in 22%. In addition to well-established risk factors (gestational age, birth weight, prolonged stay, and prolonged mechanical ventilation), treatment with iNO was found to be an independent risk factor for ROP.90 Askie et al.91 performed individual-patient data analysis in which raw data from 3298 individual participants of 12 randomized controlled trials were included. They did not find evidence that iNO therapy had a significant effect on the primary end points of death, CLD, or ROP. The association between iNO and ROP found in the study from the Netherlands could be explained from the fact that infants exposed to iNO had a higher disease severity and higher initial dosing of iNO was used. The mechanisms behind the possible association of iNO therapy and ROP are not well established. Use of iNO is associated with fluctuations in arterial oxygen and weaning from iNO is not well standardized, especially in extremely immature babies.92,93
iNO may have an important role in the development of ROP, but the effects of iNO on ROP are controversial and need further evaluation.
iNO and inflammatory systemic responses
Generally, iNO can exert pro-and anti-inflammatory actions as shown in several preclinical lung studies. For example, when macrophages from mouse lungs were subjected to variable NO concentrations (10–100 p.p.m. for up to 48 h), there was an increased inflammatory response, especially to high concentrations of NO.94 In lung extracts from lambs exposed to iNO at high (20 p.p.m.) or low (5 p.p.m.) doses, although high-dose iNO affected hemodynamics, low doses decreased activation of neutrophils by 80%.95 High-dose iNO prevented neutrophil migration and leak caused by intratracheal administration of interleukin-1 (IL-1) in rats96 and macrophage accumulation after complement fragment instillation in the lungs of rabbits, but did not affect bone marrow neutrophil production.97 Newborn pigs given 50 p.p.m. of iNO concurrently with hyperoxia was associated with higher neutrophil apoptosis,98 while iNO attenuated pulmonary inflammation in a neonatal rat hyperoxia model with downregulation of a variety of genes involved in inflammation (IL-6, cytokine-induced neutrophilic chemoattractant-1), coagulation, fibrinolysis, and cell cycle regulation (p21).99
Overall, the data from these studies show that iNO affects the immune system, again in a dose-dependent manner. Lower doses decrease neutrophil and macrophage activation, whereas longer duration and higher doses of iNO have a pro-inflammatory effect. Circulating neutrophils can contribute to the development of brain injury. The number of neutrophils in the lung following iNO may also be decreased by induction of programmed cell death.98 Under normal conditions, iNO can downregulate lung-derived cytokines and free radical production, which could lead to a decrease in brain injury.100 iNO can also induce neuroprotection through several other mechanisms, including the regulation of circulating leukocytes.101 Rats with severe intrauterine growth retardation induced by protracted gestational hypoxia leading to diffuse white matter injury associated with severe neuroinflammation were subjected to low-dose iNO (5 p.p.m.). This iNO exposure during the first postnatal week significantly decreased cell death and microglial activation, caused oligodendroglia cell proliferation (via upregulation of cyclin-dependent kinase inhibitor-p27kip), and improved myelination.102 In experiments that investigated the effect of allicin in traumatic brain injury (TBI), the Akt/eNOS pathway mediated a significant anti-inflammatory activity.103 Low-dose iNO also affects the developing brain white matter, inflammation, and repair.104
Another aspect of neuroinflammation involves the actions of glucocorticoid receptors (GR). iNO increases GR expression after various insults, including sepsis and TBI. In a newborn pig model of sepsis, Da et al.105 demonstrated that endotoxin infusion markedly reduced GR expression in target organs. However, NO upregulated GRs and decreased the inflammatory response as measured by NF-κB and tumor necrosis factor-α. The iNO was initiated at 30 p.p.m. after the insult, with effects seen only in combination with glucocorticoids. None of the interventions were effective alone. Simultaneous administration of steroids and NO also preserved normal histology in target organs.
iNO affects local and systemic inflammatory responses. NO decreases microglial activation and white matter injury after hypoxia–ischemia possibly via downregulation of systemic or lung-derived cytokines and modulation of GRs.
Implications and conclusions
The evidence that iNO can be neuroprotective in perinatal brain injury remains inconclusive. The protection conferred on the ischemic brain by NO is possibly linked to vasodilation, which improves or diverts cerebral blood flow during an acute insult. Biochemically, NO, by inducing nitrosylation of metal containing enzymes, can decrease the generation of reactive oxygen species and the resulting oxidative stress. In addition, iNO can scavenge radicals, converting them into biologically active S-nitrosothiols. iNO may also contribute to neuroprotection after ischemia in the developing brain depending on its concentration and the timing of exposure after the insult. Implementation of iNO during the reperfusion period might exacerbate brain injury. iNO exposure at low doses promotes vascular collaterals and reduces brain damage, but these data derive from adult animal studies of arterial stroke rather than global ischemia. iNO can alter autoregulation and might have direct beneficial effects on apoptosis and excitotoxicity mainly via NMDA receptor modulation. Low doses of iNO given for a prolonged time reduces neuroinflammation and white matter injury, while high doses might be proinflammatory with limited data on its effects on endogenous NO production.
Clinically, it is challenging to identify the transition of brain ischemia to reperfusion. There is a dearth of preclinical studies to address the effects of iNO during therapeutic hypothermia following brain injury and no studies of large animal translational models to mirror human physiology. Such preclinical studies are necessary to delineate the effects of iNO in combination with hypothermia. Studies evaluating NIRS and other real-time neurophysiological monitoring to track the reperfusion injury during NO administration could be useful for titration and identification of neonates that might benefit from iNO to prevent brain injury.
References
Jia, L., Bonaventura, C., Bonaventura, J. & Stamler, J. S. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380, 221–226 (1996).
Hardingham, N., Dachtler, J. & Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell Neurosci. 7, 190 (2013).
Hopper, R. A. & Garthwaite, J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J. Neurosci. 26, 11513–11521 (2006).
Garthwaite, J. NO as a multimodal transmitter in the brain: discovery and current status. Br. J. Pharm. 176, 197–211 (2019).
Malinski, T., Bailey, F., Zhang, Z. G. & Chopp, M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 13, 355–358 (1993).
Sato, S., Tominaga, T., Ohnishi, T. & Ohnishi, S. T. EPR spin-trapping study of nitric oxide formation during bilateral carotid occlusion in the rat. Biochim. Biophys. Acta 1181, 195–197 (1993).
Tominaga, T., Sato, S., Ohnishi, T. & Ohnishi, S. T. Potentiation of nitric oxide formation following bilateral carotid occlusion and focal cerebral ischemia in the rat: in vivo detection of the nitric oxide radical by electron paramagnetic resonance spin trapping. Brain Res. 614, 342–346 (1993).
Sato, S., Tominaga, T., Ohnishi, T. & Ohnishi, S. T. Electron paramagnetic resonance study on nitric oxide production during brain focal ischemia and reperfusion in the rat. Brain Res. 647, 91–96 (1994).
Kumura, E. et al. Nitrosyl hemoglobin production during reperfusion after focal cerebral ischemia in rats. Neurosci. Lett. 177, 165–167 (1994).
Zhang, Z. G. et al. Upregulation of neuronal nitric oxide synthase and mRNA, and selective sparing of nitric oxide synthase-containing neurons after focal cerebral ischemia in rat. Brain Res. 654, 85–95 (1994).
Guo, Y. et al. Regulation of cerebellar nitric oxide production in response to prolonged in vivo hypoxia. J. Neurosci. Res. 49, 89–97 (1997).
Groenendaal, F. et al. Cytosolic and membrane-bound cerebral nitric oxide synthase activity during hypoxia in cortical tissue of newborn piglets. Neurosci. Lett. 206, 121–124 (1996).
Jiang, K. et al. Effect of hypoxia and reoxygenation on regional activity of nitric oxide synthase in brain of newborn piglets. Neurosci. Lett. 206, 199–203 (1996).
Grandati, M. et al. Calcium-independent NO-synthase activity and nitrites/nitrates production in transient focal cerebral ischaemia in mice. Br. J. Pharm. 122, 625–630 (1997).
Rushworth, S. A., Bravery, C. A. & Thompson, S. High sequence homology between human and pig CD40 with conserved binding to human CD154. Transplantation 69, 936–940 (2000).
Gonzalez-Ramon, N. et al. The major acute phase serum protein in pigs is homologous to human plasma kallikrein sensitive PK-120. FEBS Lett. 371, 227–230 (1995).
Munson, D. A. et al. Pulmonary and systemic nitric oxide metabolites in a baboon model of neonatal chronic lung disease. Am. J. Respir. Cell. Mol. Biol. 33, 582–588 (2005).
Kuppusamy, P. et al. Three-dimensional imaging of nitric oxide production in the rat brain subjected to ischemia–hypoxia. J. Cereb. Blood Flow Metab. 15, 899–903 (1995).
Kader, A., Frazzini, V. I., Solomon, R. A. & Trifiletti, R. R. Nitric oxide production during focal cerebral ischemia in rats. Stroke 24, 1709–1716 (1993).
Chakravortty, D. & Hensel, M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 5, 621–627 (2003).
Kleinert, H., Schwarz, P. M. & Forstermann, U. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 384, 1343–1364 (2003).
MacMicking, J., Xie, Q. W. & Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 (1997).
Nomura, Y. & Kitamura, Y. Inducible nitric oxide synthase in glial cells. Neurosci. Res. 18, 103–107 (1993).
Vannucchi, M. G. et al. Expression of neuronal and inducible nitric oxide synthase in neuronal and glial cells after transient occlusion of the middle cerebral artery. Neuroscience 136, 1015–1026 (2005).
Fernhoff, N. B., Derbyshire, E. R. & Marletta, M. A. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc. Natl Acad. Sci. USA 106, 21602–21607 (2009).
Burney, S. et al. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424, 37–49 (1999).
Moncada, S. & Erusalimsky, J. D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell. Biol. 3, 214–220 (2002).
Brown, G. C. & Borutaite, V. Nitric oxide, cytochrome c and mitochondria. Biochem. Soc. Symp. 66, 17–25 (1999).
Manner, C. K., Nicholson, B. & MacLeod, C. L. CAT2 arginine transporter deficiency significantly reduces iNOS-mediated NO production in astrocytes. J. Neurochem. 85, 476–482 (2003).
Kawahara Kea Co-induction of argininosuccinate synthetase, cationic amino acid transporter-2, and nitric oxide synthase in activated murine microglial cells. Brain Res. Mol. Brain Res. 90, 165–173 (2001).
Youn, H. et al. Under-expression of Kalirin-7 Increases iNOS activity in cultured cells and correlates to elevated iNOS activity in Alzheimer’s disease hippocampus. J. Alzheimers Dis. 12, 271–281 (2007).
Cho, H. J. et al. Inducible nitric oxide synthase: identification of amino acid residues essential for dimerization and binding of tetrahydrobiopterin. Proc. Natl Acad. Sci. USA 92, 11514–11518 (1995).
Alderton, W. K., Cooper, C. E. & Knowles, R. G. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357, 593–615 (2001).
Clapham, D. E. Calcium signaling. Cell 131, 1047–1058 (2007).
Fischmann, T. Oea Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat. Struct. Biol. 6, 233–242 (1999).
Ratovitski EAea Kalirin inhibition of inducible nitric-oxide synthase. J. Biol. Chem. 274, 993–999 (1999).
Garvey EPea Potent and selective inhibition of human nitric oxide synthases. Inhibition by non-amino acid isothioureas. J. Biol. Chem. 269, 26669–26676 (1994).
Cinelli, M. A., Do, H. T., Miley, G. P. & Silverman, R. B. Inducible nitric oxide synthase: regulation, structure, and inhibition. Med. Res. Rev. 40, 158–189 (2020).
Tsuji, M. et al. Protective effect of aminoguanidine on hypoxic–ischemic brain damage and temporal profile of brain nitric oxide in neonatal rat. Pediatr. Res. 47, 79–83 (2000).
Zhu, C. et al. Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor in neonatal rat cerebral hypoxia–ischaemia. J. Neurochem. 90, 462–471 (2004).
Assreuy, J., Cunha, F. Q., Liew, F. Y. & Moncada, S. Feedback inhibition of nitric oxide synthase activity by nitric oxide. Br. J. Pharm. 108, 833–837 (1993).
Smith, B. C., Fernhoff, N. B. & Marletta, M. A. Mechanism and kinetics of inducible nitric oxide synthase auto-S-nitrosation and inactivation. Biochemistry 51, 1028–1040 (2012).
Christopherson, K. S., Hillier, B. J., Lim, W. A. & Bredt, D. S. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 274, 27467–27473 (1999).
Aarts, Mea Treatment of ischemic brain damage by perturbing NMDA receptor–PSD-95 protein interactions. Science 298, 846–850 (2002).
Giesen, J. et al. AMPA induces NO-dependent cGMP signals in hippocampal and cortical neurons via L-type voltage-gated calcium channels. Cereb. Cortex 30, 2128–2143 (2020).
Dawson, V. L. & Dawson, T. M. Deadly conversations: nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 36, 287–294 (2004).
Ischiropoulos, H. & Beckman, J. S. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J. Clin. Invest. 111, 163–169 (2003).
Eliasson, M. Jea Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 3, 1089–1095 (1997).
Berger, N. A. & Berger, S. J. Metabolic consequences of DNA damage: the role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 38, 357–363 (1986).
Loeffler Mea Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 15, 758–767 (2001).
Pansiot, J. et al. Neuroprotective effect of inhaled nitric oxide on excitotoxic-induced brain damage in neonatal rat. PLoS ONE 5, e10916 (2010).
Lonze, B. E. & Ginty, D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 (2002).
Lee, H. T. et al. cAMP response element-binding protein activation in ligation preconditioning in neonatal brain. Ann. Neurol. 56, 611–623 (2004).
Rybnikova, E. et al. Preconditioning induces prolonged expression of transcription factors pCREB and NF-kappa B in the neocortex of rats before and following severe hypobaric hypoxia. J. Neurochem. 106, 1450–1458 (2008).
Ping, P. et al. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ. Res. 84, 587–604 (1999).
Gonzalez-Zulueta, M. et al. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc. Natl Acad. Sci. USA 97, 436–441 (2000).
Lipton, S. A. et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364, 626–632 (1993).
Lander, H. M., Jacovina, A. T., Davis, R. J. & Tauras, J. M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 271, 19705–19709 (1996).
Mannick, J. B. et al. S-nitrosylation of mitochondrial caspases. J. Cell Biol. 154, 1111–1116 (2001).
Drury, P. P., Gunn, E. R., Bennet, L. & Gunn, A. J. Mechanisms of hypothermic neuroprotection. Clin. Perinatol. 41, 161–175 (2014).
Thoresen, M. et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. NeuroReport 8, 3359–3362 (1997).
McManus, T. et al. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. J. Neurochem. 91, 327–336 (2004).
Lei, B., Adachi, N. & Arai, T. The effect of hypothermia on H2O2 production during ischemia and reperfusion: a microdialysis study in the gerbil hippocampus. Neurosci. Lett. 222, 91–94 (1997).
Kader, A. et al. Effect of mild hypothermia on nitric oxide synthesis during focal cerebral ischemia. Neurosurgery 35, 272–277 (1994). discussion 277.
Kida, K. et al. Beneficial effects of nitric oxide on outcomes after cardiac arrest and cardiopulmonary resuscitation in hypothermia-treated mice. Anesthesiology 120, 880–889 (2014).
Aaltonen, M. et al. Meconium aspiration induces neuronal injury in piglets. Acta Paediatr. 94, 1468–1475 (2005).
Aaltonen, M. et al. Inhaled nitric oxide treatment inhibits neuronal injury after meconium aspiration in piglets. Early Hum. Dev. 83, 77–85 (2007).
Iadecola, C. Does nitric oxide mediate the increases in cerebral blood flow elicited by hypercapnia? Proc. Natl Acad. Sci. USA 89, 3913–3916 (1992).
Niwa, K., Lindauer, U., Villringer, A. & Dirnagl, U. Blockade of nitric oxide synthesis in rats strongly attenuates the CBF response to extracellular acidosis. J. Cereb. Blood Flow Metab. 13, 535–539 (1993).
Adachi, T., Inanami, O. & Sato, A. Nitric oxide (NO) is involved in increased cerebral cortical blood flow following stimulation of the nucleus basalis of Meynert in anesthetized rats. Neurosci. Lett. 139, 201–204 (1992).
Iadecola, C. & Zhang, F. Nitric oxide-dependent and -independent components of cerebrovasodilation elicited by hypercapnia. Am. J. Physiol. 266, R546–R552 (1994).
Moenkhoff, M. et al. Electroencephalogram changes during inhalation with nitric oxide in the pediatric intensive care patient—a preliminary report. Crit. Care Med. 26, 1887–1892 (1998).
Lopes Cardozo, R. H. et al. Inhalation of nitric oxide: effect on cerebral hemodynamics and activity, and antioxidant status in the newborn lamb. Biol. Neonate 69, 284–292 (1996).
Tsai, G. E. & Gastfriend, D. R. Nitric oxide-induced motor neuron disease in a patient with alcoholism. N. Engl. J. Med. 332, 1036 (1995).
Murohara, T. et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 101, 2567–2578 (1998).
Lee, P. C. et al. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 277, H1600–H1608 (1999).
Fukumura, D. et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl Acad. Sci. USA 98, 2604–2609 (2001).
Sessa, W. C. et al. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J. Biol. Chem. 270, 17641–17644 (1995).
Shesely, E. G. et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA 93, 13176–13181 (1996).
Brock, T. A., Dvorak, H. F. & Senger, D. R. Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am. J. Pathol. 138, 213–221 (1991).
He, H. et al. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J. Biol. Chem. 274, 25130–25135 (1999).
Papapetropoulos, A., Garcia-Cardena, G., Madri, J. A. & Sessa, W. C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 100, 3131–3139 (1997).
Michell, B. J. et al. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 9, 845–848 (1999).
McCabe, T. J., Fulton, D., Roman, L. J. & Sessa, W. C. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J. Biol. Chem. 275, 6123–6128 (2000).
Kimura, H. & Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim. Pol. 50, 49–59 (2003).
Ku, D. D., Zaleski, J. K., Liu, S. & Brock, T. A. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am. J. Physiol. 265, H586–H592 (1993).
Chan-Ling, T., Gock, B. & Stone, J. The effect of oxygen on vasoformative cell division. Evidence that ‘physiological hypoxia’ is the stimulus for normal retinal vasculogenesis. Invest. Ophthalmol. Vis. Sci. 36, 1201–1214 (1995).
Hartnett, M. E. & Penn, J. S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 367, 2515–2526 (2012).
Donohue, P. K. et al. Inhaled nitric oxide in preterm infants: a systematic review. Pediatrics 127, e414–e422 (2011).
van Sorge, A. J. et al. Nationwide inventory of risk factors for retinopathy of prematurity in the Netherlands. J. Pediatr. 164, 494–498 e491 (2014).
Askie, L. M. et al. Inhaled nitric oxide in preterm infants: an individual-patient data meta-analysis of randomized trials. Pediatrics 128, 729–739 (2011).
Ahearn, J., Panda, M., Carlisle, H. & Chaudhari, T. Impact of inhaled nitric oxide stewardship programme in a neonatal intensive care unit. J. Paediatr. Child Health 56, 265–271 (2020).
Shiraishi, J. et al. Standardization of nitric oxide inhalation in extremely preterm infants in Japan. Pediatr. Int. 61, 152–157 (2019).
Weinberger, B. et al. Inhaled nitric oxide primes lung macrophages to produce reactive oxygen and nitrogen intermediates. Am. J. Respir. Crit. Care Med. 158, 931–938 (1998).
Kinsella, J. P. et al. Effects of inhaled nitric oxide on pulmonary edema and lung neutrophil accumulation in severe experimental hyaline membrane disease. Pediatr. Res. 41, 457–463 (1997).
Guidot, D. M., Hybertson, B. M., Kitlowski, R. P. & Repine, J. E. Inhaled NO prevents IL-1-induced neutrophil accumulation and associated acute edema in isolated rat lungs. Am. J. Physiol. 271, L225–L229 (1996).
Sato, Y. et al. Nitric oxide reduces the sequestration of polymorphonuclear leukocytes in lung by changing deformability and CD18 expression. Am. J. Respir. Crit. Care Med. 159, 1469–1476 (1999).
Ekekezie, I. I. et al. Independent and combined effects of prolonged inhaled nitric oxide and oxygen on lung inflammation in newborn piglets. Biol. Neonate 77, 37–44 (2000).
ter Horst, S. A. et al. Inhaled nitric oxide attenuates pulmonary inflammation and fibrin deposition and prolongs survival in neonatal hyperoxic lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L35–L44 (2007).
Haynes, R. L. et al. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 15, 225–233 (2005).
Weinberger, B., Laskin, D. L., Heck, D. E. & Laskin, J. D. The toxicology of inhaled nitric oxide. Toxicol. Sci. 59, 5–16 (2001).
Pham, H. et al. Impact of inhaled nitric oxide on white matter damage in growth-restricted neonatal rats. Pediatr. Res. 77, 563–569 (2015).
Chen, W. et al. Neuroprotective effect of allicin against traumatic brain injury via Akt/endothelial nitric oxide synthase pathway-mediated anti-inflammatory and anti-oxidative activities. Neurochem. Int. 68, 28–37 (2014).
Charriaut-Marlangue, C. et al. Nitric oxide signaling in the brain: a new target for inhaled nitric oxide? Ann. Neurol. 73, 442–448 (2013).
Da, J., Chen, L. & Hedenstierna, G. Nitric oxide up-regulates the glucocorticoid receptor and blunts the inflammatory reaction in porcine endotoxin sepsis. Crit. Care Med. 35, 26–32 (2007).
Acknowledgements
R.S. holds the “William Buchanan Chair in Pediatrics,” and L.C. is supported by NIH Grant 1R01NS102617-01.
Author information
Authors and Affiliations
Contributions
D.A. contributed to the concept of the paper, wrote the initial and revised drafts of this manuscript, and approved the final manuscript as submitted; R.S. contributed to the conceptualization of the paper, reviewed and revised the manuscript, and approved the final manuscript as submitted; L.C. contributed to the conceptualization of the paper, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Competing interests
R.S. is on the Scientific Advisory Council of Mallinckrodt Pharmaceuticals and had no role in the development of this review. D.A. and L.C. have no conflicts of interest to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Angelis, D., Savani, R. & Chalak, L. Nitric oxide and the brain. Part 2: Effects following neonatal brain injury—friend or foe?. Pediatr Res 89, 746–752 (2021). https://doi.org/10.1038/s41390-020-1021-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-1021-4
