
Abstract
Background
Preterm birth is the leading cause of mortality and morbidity in young children, with over a million deaths per year worldwide arising from neonatal complications (NCs). NCs are moderately heritable although the genetic causes are largely unknown. Therefore, we investigated the impact of accumulated genetic variation (burden) on NCs in non-Hispanic White (NHW) and non-Hispanic Black (NHB) preterm infants.
Methods
We sequenced 182 exomes from infants with gestational ages from 26 to 31 weeks. These infants were cared for in the same time period and hospital environment. Eighty-one preterm infants did not develop NCs, whereas 101 developed at least one severe complication. We measured the effect of burden at the single-gene and exome-wide levels and derived a polygenic risk score (PRS) from the top 10 genes to predict NCs.
Results
Burden across the exome was associated with NCs in NHW (p = 0.05) preterm infants suggesting that multiple genes influence susceptibility. In a post hoc analysis, we find that PRS alone predicts NCs (AUC = 0.67) and that PRS is uncorrelated with GA (\(\widehat {\it{\rho }}\) = 0.05; p = 0.53). When PRS and GA at birth are combined, the AUC is 0.87.
Conclusions
Our results support the hypothesis that genetic burden influences NCs in NHW preterm infants.
Similar content being viewed by others
Introduction
Preterm birth is the leading cause of mortality and morbidity in young children (age <5 years) worldwide, with more than a million deaths per year arising from neonatal complications (NCs).1,2,3 Therefore, to improve the health outcomes of preterm infants in the era of precision medicine and to reduce medical costs, we need to develop better predictive models for NCs. One attractive approach is to combine well-established risk factors (e.g., gestational age (GA) at birth) with genetic risk factors (e.g., exonic mutations) into a single predictor of NC.
Based on evidence from twin studies, we know that genes play a role in individual NC like bronchopulmonary dysplasia (BPD) and retinopathy of prematurity (ROP). In particular, Bhandari et al.4 estimated the heritability of BPD at 53% in a multicenter twin study using logistic regression with mixed effects; and in a retrospective twin study of ROP, Bizzaro et al. estimated the heritability at 70%.5 However, pinpointing the specific genes that account for the impact of sex and race on susceptibility for NCs has been much more difficult.
Epidemiological studies demonstrate that NCs may also be influenced by GA, sex, and race. For example, Draper et al.6 showed that GA is negatively correlated with risk for NC. In addition, Trembath et al.7 and Peelen et al.8 showed that preterm males have increased risk for NCs relative to preterm females. Finally, Loftin et al.9 and Ryan et al.10 showed that, conditional on GA, non-Hispanic White infants have increased risk relative to non-Hispanic Black infants.
The objective of this study is to test the hypothesis that, among preterm infants, the accumulation of genetic variation across coding regions of the genome (i.e., the exome) influences risk for NCs. As a corollary, we sought to determine whether any observed sex and race disparities in NCs relate to the burden as defined by the gene-specific accumulation of minor alleles found by whole-exome sequencing (WES). Our study was partially motivated by several promising examples in the study of complex traits for adult diseases and morbidities (e.g., schizophrenia, Parkinson’s disease, and obesity), where investigators have found associations with the accumulation of minor alleles.11,12,13 Lastly, in a post hoc analysis, we compared the predictive power of a burden-based polygenic risk score (PRS) and a composite biomarker that combines PRS and GA into a single predictor of NCs. Overall, we (1) demonstrate that NCs are influenced by the accumulation of minor alleles found by WES in non-Hispanic White preterm infants, (2) did not detect an effect of burden on NCs in non-Hispanic Black preterm infants, (3) confirm the effects of previously reported traditional risk factors (e.g., GA) and show that the impact of minor allele accumulation is independent of GA (at least within the GA range studied), and (4) show that susceptibility to NCs can be accurately predicted by a composite biomarker that combines GA and PRS into a single predictor of NCs.
Methods
Study design, patient population, and samples
This study was approved by the Institutional Review Board at Nationwide Children’s Hospital (NCH). The study utilized the Perinatal Research Repository (PRR), which is a data and biospecimen repository of preterm neonates admitted to the neonatal intensive care unit (NICU) at NCH. Parents of infants eligible for inclusion in the PRR (i.e., infants <37 completed weeks of gestation) were approached to provide written informed consent for their participation and for the participation of their child. Once consent was obtained, blood or buccal swabs were obtained for DNA extraction. Samples were processed for DNA extraction at the NCH Biopathology Center and stored at −80 °C until analysis. Clinical data were abstracted from the electronic medical record upon death or discharge by research personnel who were not directly involved in the present study.
Of the infants enrolled in PRR, eligible preterm infants for this study were singletons with GA at birth between 26 and 31 weeks, inclusive. Infants with known chromosomal abnormalities or congenital anomalies were a priori excluded. GA range was chosen on the rationale that, for infants with GA <26 weeks, extreme immaturity could potentially overwhelm any genetic component. Conversely, severe NCs are far less common in infants born at ≥32 weeks’ gestation. Because both outcomes tend to reduce statistical power, restricting GA to the range of 26–31 weeks would likely provide the greatest power to detect genetic factors influencing NC.
Clinical phenotype of relevant NCs and study groups
Among eligible infants with samples and data available as of December 2015, we defined as “susceptible” (SUS) those infants who were diagnosed at death or discharge with at least one of the of the following severe short-term neonatal outcomes.
-
BPD was defined as a requirement for oxygen and/or positive airway pressure at 36 weeks postmenstrual age.14
-
NEC was defined as Bell’s stage ≥2.15
-
ROP was defined as stage ≥2 according to the ICROP.16
-
Severe IVH was defined as grade ≥3 according to the Papile classification.17
We defined “resilient” (RES) as those infants who had none of the NCs listed above. For each group, we recorded the following: GA at birth, the birth weight, Apgar scores (at 1 and 5 min), race, sex, delivery route, any exposure to antenatal steroids, whether surfactant was given, and maternal characteristics initiating birth.
We used Wilcoxon rank-sum tests to compare quantitative variables and chi-squared tests to compare categorical variables. A p value ≤ 0.05 was considered significant.
Sequencing
WES was performed on each infant using the SureSelectXT Target Enrichment System for Illumina Paired End Sequencing Protocol (Agilent Technologies, CA). DNA libraries were captured and enriched for exons using the SureSelect Clinical Research Exome Version 1 Kit (Agilent). Paired-end 96 base pair reads were generated for exome-enriched libraries sequenced across eight Illumina HiSeq 2500 runs. Samples were sequenced to an average of 76× depth of coverage, with a minimum depth of 50× targeted region coverage.
Following sequencing, primary data analysis consisted of using Illumina’s Real-Time Analysis software to perform base calling and quality scoring from the raw intensity files. The resulting base call format files were then converted and demultiplexed using Illumina’s bcl2fastq2 Conversion Software into the standard FASTQ file format appropriate for secondary analysis.
Secondary analysis was performed using Churchill, a pipeline developed in house for the discovery of human genetic variation that implements a best practice workflow for variant discovery and genotyping.18 Churchill utilizes the Burrows–Wheeler Aligner to align sequence data to the GRCh37/hg19 reference genome. Duplicate sequence reads were removed using PicardTools (version 1.104). Local realignment was performed on the aligned sequence data using the Genome Analysis Toolkit (version 3.3-0) Churchill’s own deterministic implementation of base quality score recalibration was used. The GATK’s HaplotypeCaller was used to call variants. All analysis was performed by uploading FASTQ files to GenomeNext LLC, which automated execution of the Churchill pipeline for the entire dataset. Resulting VCF files were downloaded from GenomeNext for subsequent analysis. We used GRCh37/hg19 from the University of California at Santa Cruz database19,20,21 and the 1000 Genome project phase 322 for reference human genome annotation.
Assessment of genetic burden
Burden can be tested on multiple levels (i.e., at the level of individual genes, the whole exome, and across a selected set of selected genes).
Single-gene burden
We defined single-gene burden as the total count of minor alleles across a given region that includes 7.5 kilobases upstream and downstream flanking sequences. Gene regions were further filtered to remove genes with excessive amounts of missing data (i.e., genes with >90% missing sequence data), variants with extremely low reads (i.e., number of reads <1), and variants with extremely high reads (i.e., number of reads >2.5 standard deviations from the median). These analyses were conducted for each infant and for all genes in the human genome.
However, because our sequence data are organized around the count of alternate alleles at polymorphic sites, and because alternate alleles are not necessarily minor alleles, computing the single-gene burden is not trivial. Specifically, we scored the number of minor alleles at each site as follows:
-
0 if the site is homozygous for the alternate allele and the alternate allele is major (i.e., no minor alleles are present).
-
1 if the site is heterozygous (site carries exactly 1 minor allele).
-
2 if the site is homozygous for the alternate allele and the alternate allele is minor (i.e., both alleles present are minor).
Note, in our data, sites which are homozygous for the reference allele are not recorded. That said, if we index infants by i, genes by j, and polymorphic sites by k, then the single-gene burden of the jth gene in the ith infant is Bij ≡ \(\mathop {\sum }\nolimits_k {\mathrm{M}}_{ijk}\). For each gene, we use a logistic regression (with GA as covariate) to test single-gene burden for association to NC; SUS is coded as 1 (i.e., high risk) and RES is coded as 0 (i.e., low risk). To correct for the number of multiple tests, we implemented a Bonferroni procedure.
Exome-wide burden
In contrast to our single-gene test (described above), we also assessed the joint effect of all genes by looking for an excess of low p values (i.e., p < 0.05) among the observed burden p values. (Note, burden p values were corrected for heteroscedasticity23,24). Because burden is discrete, the distribution of burden p values is not uniform under the null hypothesis of “no association.” Therefore, to determine the statistical significance of the observed burden p values, we performed a permutation test.25,26 Specifically, we first computed the observed area under the curve (AUC) from the cumulative distribution of the burden p values. Then we permuted preterm infant status (e.g., RES and SUS) 10,000 times to obtain a permutation distribution for AUC. Because a large observed AUC is evidence for an excess of low p values, we used the proportion of permutations with AUC larger than (or equal to) the observed AUC to estimate the permutation p value.
Burden-based PRSs
Using the results of our whole-exome association study (WEAS) of NCs in non-Hispanic Whites, we derived a PRS based on burden. Specifically, for the ith infant we computed the PRS27 as \(\mathop {\sum }\nolimits_i \widehat {\beta _{ik}}B_{ik}\), where for the kth gene, \(\widehat {\beta _k}\) is the estimated coefficient for burden and Bik is the observed burden in the kth infant. The summation is taken over the burden of ten genes showing the strongest evidence for association. We then used the “ROCR” package in R28 to compute the Receiver Operating Characteristic (ROC) curve29 and the corresponding AUC for PRS alone. For comparison, we also computed the ROC curve and AUC for GA alone and for our proposed composite biomarker, where the estimated coefficients from a logistic regression were used to combine PRS and GA into a single predictor of NC. Then we averaged the AUC over a tenfold cross-validation procedure30 to mitigate the negative effects of over-fitting. Finally, we used the average AUC and 95% confidence interval (CI) to compare the predictive power of each predictor.
Results
Clinical characteristics of the study groups
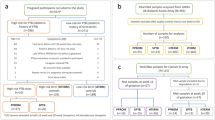
The workflow of the subjects with inclusion and exclusion criteria is summarized in Fig. 1. There were 287 eligible babies of which 94 fulfilled SUS criteria and 125 were classified as RES, after excluding for multiple births. Of these 219 infants, high-quality exome sequencing data was successfully generated on 209 newborns (SUS: n = 90, RES: n = 119). In anticipation of future meta-analyses, we did not exclude eligible candidates based on race or sex. However, owing to analytical limitations of understanding the clinical significance of burden in admixed populations with small sample sizes, we restricted our analyses to non-Hispanic White (n = 131) and non-Hispanic Black (n = 51) infants. Two infants in our study died as a result of their complications. Table 1 summarizes the clinical outcomes, and Table 2 summarizes the clinical characteristics of the SUS and RES groups limited to non-Hispanic Whites and non-Hispanic Blacks.

From a total sample of 287 preterm infants, only 182 were retained for the final analysis. Exclusion criteria included: DNA control check, multiple births, and unknown race of the preterm infants.
As anticipated, the SUS group had a much lower birth weight (p < 0.001) and GA (p < 0.001) at birth. The SUS group also had more (p < 0.05) male newborns than did the RES group. In addition, the Apgar scores at 1 and 5 min also were statistically different (p < 0.009 and p < 0.03, respectively) between the two groups of infants, with SUS group having somewhat lower scores. There was no significant difference in delivery route (p < 0.38), in antenatal steroids (p < 0.20), or surfactant administration (p < 0.96) between the two groups.
Confirmation of known risk factors in our study groups
Several authors have previously reported associations between NCs and GA,31 between NC and sex,1,32 and between NCs and race.32,33 Therefore, we sought to confirm these findings in our sample of 182 preterm infants. First, we found that GA is associated with NCs in non-Hispanic Whites [odds ratio (OR)GA = 0.43; 95% CI: (0.31, 0.56)] and non-Hispanic Blacks [ORGA = 0.42; 95% CI: (0.24, 0.63)]. Second, we tested for an association between sex and NCs with GA as a covariate. Although the evidence for association was not statistically significant, the effect of male sex may be stronger in non-Hispanic Blacks [ORSex = 3.6; 95% CI: (0.84, 17.3)] than in non-Hispanic Whites [ORSex = 1.9; 95% CI: (0.77, 4.83)].
Within each race, male preterm infants appear to have higher risk of NCs than female preterm infants. Third, we found suggestive evidence for an association between race and NCs with GA as a covariate (ORRace = 2.2; 95% CI: (0.98, 5.22)], implying that non-Hispanic White preterm infants may be more susceptible to NCs than non-Hispanic Black preterm infants.
Exome-wide burden associates with NCs in non-Hispanic White infants
Using the human genome reference annotations (GRCh37/hg19) from the University of California at Santa Cruz database19,20,21 we identified a total of 27,939 gene regions. After filtering the WES data, we retained a total of 23,854 gene regions for our analysis of data from non-Hispanic White infants and 20,232 gene regions for analysis of non-Hispanic Black infants. On average, each infant provided sequence data at about 273,168 variants.
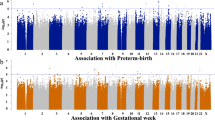
In our study, burden was not statistically significant for any gene at the exome-wide level in either cohort (i.e., non-Hispanic White and Black preterm infants; Fig. 2). However, after permuting infant status (i.e., SUS and RES) 10,000 times and using AUC as our test statistic, we did find an excess of low p values across all genes in non-Hispanic Whites (p = 0.05; Fig. 3). The excess of low p values suggests that burden influences NCs through multiples genes of small effect, especially since no single gene was statistically significant at the exome-wide level.

After correcting for multiple tests, no single gene in non-Hispanic White (left) and non-Hispanic (right) preterm infants is statistically significant at the exome-wide level (dashed line).

The distribution of p values in NHWs (n = 75 SUS, n = 56 RES) shows a statistically significant excess of low with p values (p = 0.05), suggesting that genetic burden influences neonatal complications in NHWs. By contrast, the distribution of p values in NHBs (n = 25 SUS, n = 26 RES) is inconclusive (p = 0.4).
Evaluating various predictors of NC
We compared the ROC curves of PRS alone and our composite biomarker (PRS+GA) in terms of AUC (Fig. 4). Recall that our composite biomarker combines GA and PRS into a single predictor of NC, where PRS is computed from the top ten genes (Table 3) of the exome-wide analysis for non-Hispanic White infants. However, this comparison does not account for over-fitting. To compare the predictive power of PRS alone and PRS+GA without over-fitting, we averaged the AUC of each predictor in a tenfold cross-validation procedure (see “Methods”). The average AUC for PRS alone was 0.67 (p < 0.003) and increased to 0.87 (p < 0.001) when GA was combined with PRS (Table 4). Note that the average AUC based on PRS+GA is significantly larger than the average AUC based on PRS alone (p = 0.0012). Moreover, for each gene in the PRS gene set, the average burden was higher among SUS preterm infants than among RES preterm infants (data not shown). This suggests that large values of PRS are associated with increased risk of NC. Interestingly, PRS and GA were not correlated [\(\hat \rho\) = 0.05; p = 0.53]. This may explain why the predictive power of our composite biomarker, which combines PRS and GA into a single predictor of NC, is so high compared to the predictive power of PRS alone.

After correcting for over-fitting, the average AUC of PRS+GA dropped to 87%, and the average AUC of PRS alone dropped to 67%. The difference in predictive power between PRS+GA and PRS alone is significant (p = 0.0012).
Discussion
Preterm infants are at increased risk for NC, and as such, predicting the health outcomes of preterm infants could have a tremendous impact on their outcomes, and furthermore could be extremely useful in the development of preventative therapies by identifying high-risk populations. In this study, we confirm that GA is an important predictor of NC, and we find suggestive evidence for an effect of race. Our data also indicate that male sex may play a stronger role in non-Hispanic Blacks than in non-Hispanic Whites. Furthermore, we show that the combination of traditional risk factors and genetic risk factors can substantially improve prediction. In what follows, we discuss each of these findings, their implications, and any relevant limitations of our study.
Genetic predictors of NC
We demonstrate that in aggregate (i.e., across genes) the accumulation of variants found by WES is positively correlated with NC, but as with most whole-exome association studies, there are potential limitations. First, there is always the question of how should one summarize variation across potentially overlapping, nested, and alternatively spliced genomic regions. Here we took the simplest possible approach and assessed burden at the gene level, where the start and end positions of each gene were given by the UCSC GRCh38/hg38 annotation file (see “Data description” for more details). Second, because exons are highly conserved, exonic variation in human populations is typically low.34 As such, WEAS often require large sample sizes to detect an association.35 Because it is often difficult to collect large samples of preterm infants,36 most WEAS studies of preterm infants are underpowered. For example, given the size of the genetic effects that we see in our non-Hispanic White infants, our non-Hispanic Black sample (n = 51) is probably too small to detect an association at either the single-gene level or at the exome-wide level.
Similarly, in non-Hispanic Whites, we investigated the role of intronic variants by comparing genes with “little or no” evidence for association (i.e., p values > 0.1) to genes with “suggestive” evidence for association (i.e., p values < 0.1). In genes with “little or no” evidence, 87% of all variants were intronic, whereas in genes with “suggestive” evidence 89% of all variants were intronic. While intronic variation could play an important role in the genetics of NCs, we would need many more preterm infants to ensure that the observed increase of 2% is, in fact, statistically significant.
In a post hoc analysis, we tested our newly discovered PRS gene set, which contains ten genes showing the largest evidence for association. There is almost always some difficultly in deciding exactly how many genes to include in a PRS risk score.27,37 Nevertheless, we decided to use the top ten genes because the degree of over-fitting seemed acceptably small (data not shown). To further mitigate the potentially negative effects of over-fitting, we chose to implement a tenfold cross-validation procedure. Interestingly, our PRS does not appear to be correlated with GA, which likely stems from the fact that our logistic regression included GA as a covariate, but may also suggest that NCs and prematurity have different genetic etiologies.
Race and sex as predictors of NCs
To date, the strongest predictors for NCs are GA [Manuck (2016)],38 birth weight,39,40 sex,41 and race.10 While the evidence for an association between race and NCs is only suggestive in our data (p = 0.06), when we imputed the percentage of African ancestry in each preterm infant from the available exome sequence data, we found that imputed African ancestry was negatively correlated with risk for NCs (p = 0.049; data not shown). This suggests that preterm infants of African descent may be less likely to develop NCs. Furthermore, our results (and the results of Morse et al.32) suggest that the effect of sex may depend on race. Lastly, our PRS gene set predicted NCs poorly in non-Hispanic Black infants. Here the PRS was computed from the PRS gene set (and the corresponding regression coefficients) identified in non-Hispanic Whites and from the observed burden of non-Hispanic Black infants. Although the relatively small number of non-Hispanic Black infants in our study (n = 51) could explain our inability to detect an association, another possible explanation is that NCs in non-Hispanic Blacks and non-Hispanic Whites are influenced by different genes.
Study limitations
To the best of our knowledge, this is the first-ever WES study of NC, and as such, there are limitations that we describe below. First, although we have “lumped” several complications together into a single trait (i.e., NC), the genetic heterogeneity in our sample of preterm infants is mitigated (to some degree) by the fact that more than 90% of our SUS infants have BPD (see Table 1). Second, our study is in some sense a study of extreme phenotypes because we have RES (resilient) infants that are very preterm (i.e., GA between 26 and 31 weeks); and relative to a standard case-control design, using extreme phenotypes in this way increases our power to detect genetic factors associated with NC.
Candidate genes and improved biomarkers for NC
Among the ten genes composing our PRS score for NC, the top score was assigned to RANBP2 (Ran-binding protein-2), a protein located on the cytoplasmic surface of the nuclear pore complex that plays a role in intracellular trafficking.42 Although additional studies need to investigate the mechanistic role of RANBP2 in the pathogenesis of NCs, there is some biologically plausibility based on what is known so far about this protein. First, the mouse RANBP2 knockout is embryonic lethal suggesting that RANBP2 plays an important role in development. And studies on conditional knockout mice linked the cause of lethality to inadequate nuclear import, although whether this affected a broad spectrum of proteins or a small subset remains to be determined.43 Second, several RANBP2 mutations in children are known to cause acute necrotizing encephalopathy, a disorder where previously normal children develop encephalopathy in response to a common viral infection. Therefore, the possible involvement of RANBP2 in a broader adaptive response such as the ones required by premature newborns in the context of a NICU environment needs to be studied.44
Another gene among the top ten with direct biological plausibility is GUCY1A3. This gene encodes for the α1 subunit of the soluble guanylyl cyclase (sGC) enzyme, and it is important in the nitric oxide/cGMP signaling pathway—a pathway that regulates sensitivity to nitric oxide. Note that nitric oxide is an established mediator of newborn lung development, and when inhaled, it has been proposed to exert therapeutic benefit to prevent BPD. While randomized clinical trials have yielded conflicting results on the protective effect of inhaled nitric oxide in the general population, there is the suggestion of a possible subgroup benefit in non-white infants. Furthermore, a polymorphism in GUCY1A3 has been previously associated with decreased risk for pulmonary hypertension in a high-altitude population.45
Although we limited our candidate gene discussion to RANBP2 and GUCY1A3, future mechanistic investigations of NCs should be extended to other genes as well, especially because genetic burden could have additive effects in genes with seemingly unrelated function. Furthermore with regard to BPD specifically, it would be interesting to determine whether the accumulation of minor alleles in GUCY1A3 modulates the therapeutic response to inhaled nitric oxide.46
Interestingly, while the burden-based PRS predicts NCs in non-Hispanic Whites, it is a poor predictor of GA. This implies (and our AUC analyses confirm) that our composite predictor of NC—which combines information from both PRS and GA—should perform better than a predictor based on PRS alone. Overall, we believe that our composite predictor could facilitate the design of individualized treatments for preterm infants that, in turn, could substantially improve health outcomes and reduce hospitalization costs.
Conclusions
This work demonstrates clearly our ability to predict NCs among non-Hispanic White preterm infants using information from both genetic risk factors (e.g., burden) and traditional risk factors (e.g., GA). Provided that future studies of NCs continue to collect genetic data on preterm infants from under-represented populations—where the chance of preterm birth is higher—then more efficacious composite biomarkers could be developed and implemented for members of these extremely vulnerable and historically understudied populations.
References
Glass, H. C. et al. Outcomes for extremely premature infants. Anesth. Analg. 120, 1337–1351 (2015).
Liu, L. et al. Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the sustainable development goals. Lancet 388, 3027–3035 (2016).
Mathews, T. J. & MacDorman, M. F. Infant mortality statistics from the 2007 period linked birth/infant death data set. Natl Vital Stat. Rep. 59, 1–30 (2011).
Bhandari, V. et al. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics 117, 1901–1906 (2006).
Bizzarro, M. J. et al. Genetic susceptibility to retinopathy of prematurity. Pediatrics 118, 1858–1863 (2006).
Draper, E. S., Manktelow, B., Field, D. J. & James, D. Prediction of survival for preterm births by weight and gestational age: retrospective population based study. BMJ 319, 1093–1097 (1999).
Trembath, A. & Laughon, M. M. Predictors of bronchopulmonary dysplasia. Clin. Perinatol. 39, 585–601 (2012).
Peelen, M. J. et al. Impact of fetal gender on the risk of preterm birth, a national cohort study. Acta Obstet. Gynecol. Scand. 95, 1034–1041 (2016).
Loftin, R., Chen, A., Evans, A. & DeFranco, E. Racial differences in gestational age-specific neonatal morbidity: further evidence for different gestational lengths. Am. J. Obstet. Gynecol. 206, 259.e251–256 (2012).
Ryan, R. M. et al. Black race is associated with a lower risk of bronchopulmonary dysplasia. J. Pediatr. 207, 130.e2–135.e2 (2019).
He, P. et al. Accumulation of minor alleles and risk prediction in schizophrenia. Sci. Rep. 7, 11661 (2017).
Still, C. D. et al. High allelic burden of four obesity SNPs is associated with poorer weight loss outcomes following gastric bypass surgery. Obesity (Silver Spring) 19, 1676–1683 (2011).
Zhu, Z. et al. Enrichment of minor alleles of common SNPs and improved risk prediction for Parkinson’s disease. PLoS ONE 10, e0133421 (2015).
Jobe, A. H. & Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 163, 1723–1729 (2001).
Bell, M. J. et al. Epidemiologic and bacteriologic evaluation of neonatal necrotizing enterocolitis. J. Pediatr. Surg. 14, 1–4 (1979).
International Committee for the Classification of Retinopathy of Prematurity. The International Classification of Retinopathy of Prematurity revisited. Arch. Ophthalmol. 123, 991–999 (2005).
Papile, L. A., Burstein, J., Burstein, R. & Koffler, H. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. J. Pediatr. 92, 529–534 (1978).
Kelly, B. J. et al. Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol. 16, 6 (2015).
Haeussler, M. et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858 (2019).
Rosenbloom, K. R. et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 41, D56–D63 (2013).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Barton, S. J. et al. Correction of unexpected distributions of P values from analysis of whole genome arrays by rectifying violation of statistical assumptions. BMC Genomics 14, 161 (2013).
Mackinnon, J. G. & White, H. Some heteroskedasticity-consistent covariance-matrix estimators with improved finite-sample properties. J. Econ. 29, 305–325 (1985).
Ludbrook, J. & Dudley, H. Why permutation tests are superior to t and F tests in biomedical research. Am. Stat. 52, 127–132 (1998).
Bush, W. S. & Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 8, e1002822 (2012).
Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 9, e1003348 (2013).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Hajian-Tilaki, K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Casp. J. Intern. Med. 4, 627–635 (2013).
Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Ser. B Stat. Methodol. 36, 111–147. (1974).
Ward, R. M. & Beachy, J. C. Neonatal complications following preterm birth. BJOG 110(Suppl 20), 8–16 (2003).
Morse, S. B. et al. Racial and gender differences in the viability of extremely low birth weight infants: a population-based study. Pediatrics 117, e106–e112 (2006).
Schieve, L. A. & Handler, A. Preterm delivery and perinatal death among black and white infants in a Chicago-area perinatal registry. Obstet. Gynecol. 88, 356–363 (1996).
Irimia, M. et al. Widespread evolutionary conservation of alternatively spliced exons in Caenorhabditis. Mol. Biol. Evol. 25, 375–382 (2008).
Hong, E. P. & Park, J. W. Sample size and statistical power calculation in genetic association studies. Genomics Inf. 10, 117–122 (2012).
Torgerson, D. G. et al. Ancestry and genetic associations with bronchopulmonary dysplasia in preterm infants. Am. J. Physiol. Lung Cell. Mol. Physiol. 315, L858–L869 (2018).
Choi, S. W., Heng Mak, T. S. & O’Reilly, P. F. A guide to performing Polygenic Risk Score analyses. bioRxiv 416545. https://doi.org/10.1101/416545 (2018).
Manuck, T. A. et al. Preterm neonatal morbidity and mortality by gestational age: a contemporary cohort. Am. J. Obstet. Gynecol. 215, 103.e101–103 e114 (2016).
Gooden, M., Younger, N. & Trotman, H. What is the best predictor of mortality in a very low birth weight infant population with a high mortality rate in a medical setting with limited resources? Am. J. Perinatol. 31, 441–446 (2014).
Abolfotouh, M. A., Al Saif, S., Altwaijri, W. A. & Al Rowaily, M. A. Prospective study of early and late outcomes of extremely low birthweight in Central Saudi Arabia. BMC Pediatr. 18, 280 (2018).
Zisk, J. L. et al. Do premature female infants really do better than their male counterparts? Am. J. Perinatol. 28, 241–246 (2011).
Yokoyama, N. et al. A giant nucleopore protein that binds Ran/TC4. Nature 376, 184–188 (1995).
Hamada, M. et al. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J. Cell Biol. 194, 597–612 (2011).
Neilson, D. E. et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am. J. Hum. Genet. 84, 44–51 (2009).
Wilkins, M. R. et al. alpha1-A680T variant in GUCY1A3 as a candidate conferring protection from pulmonary hypertension among Kyrgyz highlanders. Circ. Cardiovasc. Genet. 7, 920–929 (2014).
Hasan, S. U. et al. Effect of inhaled nitric oxide on survival without bronchopulmonary dysplasia in preterm infants: a randomized clinical trial. JAMA Pediatr. 171, 1081–1089 (2017).
Acknowledgements
WES was performed by the Biomedical Genomics Core in The Research Institute at Nationwide Children’s Hospital. We thank the participants and their families for their involvement in this study, the Ohio Perinatal Research Network (OPRN) for recruiting preterm infants, and Dr. Will C. Ray for his help in editing the manuscript. We are also grateful to Andrew M Corris, Senior Licensing Associate, who provided professional advice concerning the patentability of various aspects of this research. This work was supported by the Abigail Wexner Research Institute at Nationwide Children’s Hospital and the Center for Clinical and Translational Science CTSA Grant UL1TR002733.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data. W.C.L.S. and K.M.G. drafted the article, and P.W., B.K., M.K., I.A.B., and L.D.N. revised it critically for important intellectual content. All authors gave final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Stewart, W.C.L., Gnona, K.M., White, P. et al. Prediction of short-term neonatal complications in preterm infants using exome-wide genetic variation and gestational age: a pilot study. Pediatr Res 88, 653–660 (2020). https://doi.org/10.1038/s41390-020-0796-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-0796-7
