
Abstract
The placenta is the single most reliable source for precise information on intrauterine environment, as well as maternal and fetal health. It mediates the physiology of two distinct yet highly interconnected individuals. The pathology that develops in the placenta, and the adaptations the placenta undergoes to mitigate this pathology, may influence the later life health of the mother and baby. Pathological placental examination provides a unique opportunity to explore and understand the intrauterine environment, as well as providing a record of events that may be associated with adverse pregnancy outcomes. A number of placental lesions have been described in association with various neonatal morbidities. The purpose of this review is to summarize the evidence for the association of placental pathologic lesions with neurodevelopmental outcomes infants with specific neonatal morbidities, including (1) neonatal encephalopathy, (2) bronchopulmonary dysplasia, (3) congenital heart diseases, and (4) autism spectrum disorders. For each of these disease processes, we will also propose specific research priorities in future studies. We conclude with a hospital-specific protocol for triaging which placentas should receive histological evaluation as a fundamental first step for the field of neuroplacentology to guide precision-based therapeutic approaches in the affected newborns.
Impact
-
The purpose of this review is to summarize the evidence for placental origins of neonatal diseases.
-
We propose specific research priorities in the field of neuroplacentology in future studies.
-
We also present a targeted hospital-based approach for triaging which placentas should receive histological evaluation.
Similar content being viewed by others
Introduction
The placenta is a unique organ that sits at the fetal–maternal interface. It is the sole source of nutrition and oxygen for the fetus, the barrier against teratogenic or other potential insults, and the provider of a record that reflects the intrauterine environment.1,2 Perhaps the most distinctive feature of the placenta is our ability to study and examine it in its entirety after delivery. However, pathological examination of the placenta has traditionally been an under-utilized aspect of perinatal medicine, even though the specimen is readily available and the costs of routine pathological examination are moderate.3,4 The Stillbirth Collaborative Research Network dramatically demonstrated the overwhelming importance of placental examination in the investigation of stillbirth, far surpassing any other test in usefulness.5 The usefulness of each test was as follows: placental pathology 64.6% (95% confidence interval [CI] 57.9–72.0), fetal autopsy 42.4% (95% CI 36.9–48.4), genetic testing 11.9% (95% CI 9.1–15.3), testing for antiphospholipid antibodies 11.1% (95% CI 8.4–14.4), fetal–maternal hemorrhage 6.4% (95% CI 4.4–9.1), glucose screen 1.6% (95% CI 0.7–3.1), parvovirus 0.4% (95% CI 0.0–1.4), and syphilis 0.2% (95% CI 0.0–1.1).5
It is also becoming increasingly clear that impaired placental functioning can have major implications for the live-born infant. Awareness among pediatricians, however, of the benefit of placental findings for neonatal care is limited. In our opinion, this is a missed opportunity. Information on placental lesions can often be helpful towards explaining an abnormal neonatal outcome and might have consequences for treatment.6
This article aims to provide a review of the literature looking at the relation between placental lesions and neurodevelopmental outcomes infants with specific neonatal in morbidities, including (1) neonatal encephalopathy (NE), (2) bronchopulmonary dysplasia (BPD), (3) congenital heart diseases (CHDs), and (4) autism spectrum disorders (ASDs).
NE in term and near term neonates
NE, resulting from hypoxic–ischemic encephalopathy (HIE), is a fetal disorder due to prenatal hypoxia–asphyxia. It is an important cause of neonatal brain injury, occurs in 1–3 per 1000 term births,7 and is responsible for 23% of neonatal deaths worldwide.8 Survivors of NE are at risk for life-long neurodevelopmental disabilities, such as cerebral palsy (CP), cognitive impairment, hearing disabilities, and cortical vision disabilities.9,10,11 The complex causal pathways underlying NE are poorly understood.12 Potential contributing factors include antenatal conditions such as advanced maternal age, infertility treatment, intrauterine growth restriction, and maternal hypertension, as well as intrapartum complications, such as maternal infection and sentinel events (e.g., placental abruption, uterine rupture).13,14,15,16,17,18,19 A better understanding of the timing and pathophysiology underlying NE could inform new strategies to prevent this condition and could help tailor specific therapies to subsets of infants with NE.
Placental gas exchange and nutrient delivery are vital to the health of the fetus both during fetal development and during the labor and delivery process. Therefore, placental abnormalities may confer susceptibility to NE and asphyxial brain injury. Epidemiological studies have shown that placental pathology is associated with NE, and when present, it doubles the risk of NE.20 Several placental findings have been reported in term infants diagnosed with NE, including acute histologic chorioamnionitis,21,22 placental vascular abnormalities or infarction,23,24,25 and chronic villitis of unknown origin.22,26
Acute histologic chorioamnionitis
In earlier studies of infants with NE, acute chorioamnionitis with fetal inflammatory response was found in nearly one-third of the cohort (n = 93), whereas, only 5% of term controls showed similar histopathology (n = 816).27 Subsequent reports27 have also associated severe chorioamnionitis with funisitis with NE, especially if fetal thrombotic vasculopathy was also present.23,28 In our own study of 86,274 neonates, published in 2015, we found a total of 120 with perinatal acidosis and NE, the severity of which was associated with acute histologic chorioamnionitis with or without fetal inflammatory response.22 Similar results were reported by Owen et al. in a population-based study,29 where chorioamnionitis was found to be more prevalent in those with CP who had NE as newborns, compared with those with CP who had not manifested NE. Others have shown that high rates of placental inflammation are often found in infants with apparent perinatal asphyxia.21,26
Chronic villitis
One major contributor of NE is chronic villitis, which is commonly considered a non-infectious inflammatory process. This entity is defined by chronic inflammatory T cells of maternal origin.30 In the majority of placental pathologic specimens, no infective organism is recognized and the condition is known as villitis of unknown etiology (VUE). VUE is thought to arise from a breakdown in the maternal tolerance for the foreign fetoplacental cells and is observed in approximately 10% of term placentas.30 High-grade VUE is a risk factor for NE in many recent studies.26,27,28 VUE is also associated with brain lesions that are commonly attributed to asphyxial birth events,26 as well as a multitude of other morbidities, including fetal growth restriction, nonreassuring fetal heart rate patterns, need for emergent surgical delivery, severe acidosis in the absence of a sentinel event, CP, and even intrauterine fetal demise.31 We have also noted that diffuse/patchy chronic villitis (high-grade villitis) is associated with abnormal neurodevelopmental outcome following hypothermia treatment in neonates with moderate-to-severe NE22 (Table 1). Findings from our studies22 and others21 suggest that cooling is less likely to improve neurodevelopmental outcomes in neonates with inflammatory placental lesions and/or thrombotic vasculopathy.
Summary and future directions
Placental lesions are common in NE, suggesting that clinical HIE is, in many cases, associated with inflammation with or without antenatal infection and/or thrombotic vasculopathy.32 Therapeutic hypothermia was designed as an intervention for acute perinatal asphyxia. It is highly likely that infants with inflammation of the placental and/or thrombotic vasculopathy and NE represent a subgroup of vulnerable infants less likely to benefit from hypothermia.33 Neuroprotective medications under investigation, including erythropoietin and melatonin, have strong anti-inflammatory properties, which may contribute to their efficacy.34 Evidence of maternal and/or placental inflammation with or without infection will be important in determining their anti-inflammatory potential and in elucidating a possible mechanism for their therapeutic effect. Furthermore, in animal models, new treatments, including growth factors, neurosteroids, and specific anti-inflammatory medications, ought to be assessed in current experimental models of hypoxia and/or ischemia that mimic human NE. Animal models of inflammation combined with hypoxic injury35 are likely to better reproduce the complexity of human NE.
In the future, human trials of therapeutic hypothermia plus neuroprotective medications should include placental histology, molecular markers of placental infection, and bacterial culture data, as well as maternal history. These data would be useful to test the hypothesis that severe inflammation of the placenta with fetal inflammatory response is associated with NE and should be considered for secondary analyses of those trials. This would facilitate determination of infectious vs. non-infectious inflammation as contributors to NE. Placental pathology may play a key role in determining which infants are most likely to benefit from cooling. Specifically, placental lesions with evidence of fetal inflammatory response (e.g., funisitis and vasculitis) are known to be highly associated with NE and may provide targets for future cooling-plus trials. Development of rapid point of care placental pathology reporting will be essential to facilitate therapeutic decision-making in a timely manner.
BPD in preterm infants
BPD occurs in an estimated 45% of infants born at <29 weeks gestational age (GA), making it the most common respiratory morbidity in surviving extremely low birth weight infants (ELGANs).36 Those with BPD are at risk for significant morbidities, including neurodevelopmental impairment, chronic cardiopulmonary disease, growth failure, deficits of hearing and vision, and shortened lifespan.37 Risk factors for BPD are numerous and include fetal growth restriction, lack of exposure to antenatal steroids, histopathologic evidence of chorioamnionitis, sepsis, prolonged need for mechanical ventilation and supplemental oxygen, and male sex, as well as GA and birth weight.37 Placental pathology is associated with morbidities and mortality in preterm infants, but there is conflicting evidence regarding the association between specific placental lesions and BPD.38,39,40,41 Studies in this field have many limitations such as small sample size and varying definitions of placental pathologies. This results in heterogeneous classifications of placental pathology and complicates interpretation. One of the most common placental findings associated with a neonatal outcome is that between acute histologic chorioamnionitis and BPD.
Maternal vascular malperfusion (MVM)
MVM has also been reported to be associated with BPD and pulmonary artery hypertension in extremely preterm infants.42,43,44 In one recent studying using the Redline classification of placental pathology, Mestan et al.44 showed that MVM was associated with BPD and pulmonary arterial hypertension (PAH) in a large cohort of preterm infants. They conducted a 5-year retrospective study of premature infants born at ≤28 weeks gestation. Among 283 births, 121 had MVM, of which 67 (55%) developed BPD and 24 (20%) had PAH. Among the common neonatal complications of extreme prematurity, BPD was the only outcome that was increased with MVM (P < 0.001). After adjustment for birth weight, fetal growth restriction, preeclampsia, and other factors, infants with MVU were more likely to develop BPD (adjusted odds ratio (OR) = 2.6; 95% CI = 1.4, 4.8). Certain MVU sublesions (fibrinoid necrosis/acute atherosis and distal villous hypoplasia/small terminal villi) were increased with pulmonary hypertension (PH; P < 0.001).44,45,46 There is evidence from animal studies that fetal stressors that adversely affect pulmonary vascular growth, such as in utero hypoxia–ischemia, may have important contributions in later onset neonatal chronic lung disease. An important example of this is the sheep model of chronic fetal hypoxia, in which Rozance et al.47 have shown that uteroplacental insufficiency leads to the classic histologic lung findings of BPD (e.g., disrupted alveolar development and pulmonary vascular remodeling). Interestingly, the pulmonary vascular components of their model closely resemble the placental histologic description of MVM.47
Multiple placental pathologies
Complex placental histopathology (e.g., multiple lesions and/or pathologies) and their association with neonatal morbidities have not been studied extensively. In one study, multiple placental pathologic lesions were associated with idiopathic intrauterine growth retardation.48 Our group categorized placental lesions into none, 1, or ≥2 (multiple lesions) and reported for the first time the number of placental pathologic lesions with the BPD and/or death in extremely preterm infants.49 Acute chorioamnionitis was the most common pathologic lesion, occurring in nearly half of this cohort of neonates, with fetal vasculitis occurring in one-third. MVM was present in 20% of the study population. Large numbers of placentas were small for GA (SGA; 20%). Sixteen percent of the placentas were large for GA (LGA), and 77% of them showed histological signs of chorioamnionitis. In infants with multiple placental pathologic lesions (34%), the most common combination was that of LGA + chorioamnionitis [n = 30, 12%], followed by SGA + chorioamnionitis [n = 15, 6%] and MVM + chorioamnionitis [n = 11, 5%] (Fig. 1). The presence of multiple lesions was significantly associated with subsequent death and/or the occurrence of BPD. After controlling for confounding factors, the association with the presence of moderate–severe BPD (OR: 3.9; 95% CI: 1.5–10; P < 0.01) remained significant. The association of BPD with multiple placental lesions may suggest that there are additive effects between vascular and inflammatory processes, which contribute to the development of BPD. Therefore, we propose that multiple placental insults occurring throughout pregnancy impart a greater risk of developing of BPD than isolated events.

MC chorioamnionitis without fetal vasculitis, FC chorioamnionitis with fetal vasculitis, MVU maternal vascular underperfusion, FTV fetal thrombotic vasculopathy, HGV high-grade villitis, VE villous edema, SGA small for gestational age placenta, LGA large for gestational age placenta. The most common pathological lesion in the placentas in our patient population was histological acute chorioamnionitis (n = 119, 49%) and maternal vascular underperfusion (including SGA placentas) (n = 96, 40%). The most common complex placental pathological lesions (multiple lesions) were a combination of histological acute chorioamnionitis with LGA placentas (n = 30, 12%), followed by histological acute chorioamnionitis with SGA placentas (n = 15, 6%) and acute chorioamnionitis with MVU (n = 11, 5%). Chronic villitis and villous edema is seen in very small percentage of placentas (<5%).
Future directions
The high morbidity and mortality of BPD-PH in the preterm population warrant concerted efforts in identification of therapeutic options. The presence of various placental pathologies such as MVM, fetal thrombotic vasculopathy, chronic villitis, villous edema, and acute chorioamnionitis should be included in future predictive models of BPD. Preclinical and prospective clinical studies should be used to determine the impact of placental lesions and their role in development of neonatal morbidities, including BPD.
ASDs in preterm infants
Children born significantly preterm are at greater risk of ASDs.50,51,52,53,54 The prevalence of ASD in children born <29 weeks GA (ELGANs) is 7.1% relative to the 1.5% risk of ASD in the general U.S. population.54,55 The risk of ASD decreases with increasing GA,54,56 from 15.0% for 23–24 weeks, 6.5% for 25–26 weeks, to 3.4% for 27 weeks GA.54 Low GA is a marker of the immaturity and vulnerability of the central nervous system, as well as other physiological systems, that function to protect the developing brain and, when perturbed, put the developing fetus at risk.57,58,59 Thus the exposures that promote preterm birth might also promote ASD. Preterm delivery is often induced by maternal–fetal infection/inflammation.60,61,62 Early parturition by aberrant inflammation has a role in linking maternal–fetal infection/inflammation and preterm birth. In addition to driving preterm birth, exposure to maternal–fetal infection/inflammation is also a leading factor in the presence and severity of brain injury/neurodevelopmental disturbance in the infant.63,64,65
Changes in placental histology may impact placental function and fetal outcomes.66,67 As such, the placenta is uniquely positioned to directly influence fetal programming. Importantly, sex-specific vulnerabilities in the ability of the placenta to respond to insults may explain why the preponderance of ASD cases are male.68 However, the placenta remains an underutilized source of information about exposures during pregnancy. Interestingly, many of the known risk factors for ASD are associated with evidence of placental inflammation or placental histopathology. For example, preterm birth, maternal obesity, elevated cytokines in amniotic fluid, pre-gestational diabetes, and maternal infection during pregnancy are all risk factors for ASD that are also associated with placental inflammation or other placental histopathology.69,70,71,72,73,74,75
Histologic chorioamnionitis and MVM
In a recent matched case–control study with 55 cases of ASD and 99 matched controls, it was reported that histologic evidence of placental inflammatory lesions, as well as MVM pathology, was associated with increased risk of ASD.76 In another study, it was noted that placentas of children with ASD had a 40% reduction in chorionic surface vascular branch points, with 2 fewer branch generations and diminished expansion of the surface vessels to the chorionic plate border.77 However, these reports were limited to infants born term and near term. In a recent study,78 the authors explored the relationship of placental pathology with the incidence of ASD in preterm infants. They noted that histological chorioamnionitis and preterm birth increased the odds of ASD.
Multiple placental pathologic lesions
ASD is ~7-fold more prevalent in extremely preterm infants when compared to the general population.54 The association between increased risk of ASD and lower GA is independent of neurodevelopmental impairments.54 In a recently published matched case–control study,79 we explored the relationship between placental pathologic lesions and ASD in preterm infants ≤28 weeks GA. The major result of the study was that children with ASD had a twofold greater incidence of multiple placental pathologies as compared to matched controls [11/16 (69%) vs.16/48 (33%), respectively; P = 0.01].79 Particularly, the combination of acute chorioamnionitis with fetal vasculitis and LGA placentas was associated with the future diagnosis of ASD in this distinctive patient population. However, single placental pathologies were not associated with ASD [5/16 (31%) vs. 21/48 (43%), respectively; P = 0.1].79
Changes in placental histopathology may increase the risk of ASD via different pathways, depending on the timing of exposure. For example, chronic placental inflammatory processes that manifest early in pregnancy may alter placental angiogenesis and may have downstream effects on fetal angiogenesis and neurogenesis.80 Chronic inflammation affects angiogenesis, which is a process that is associated with pregnancy complications.81 Alternatively, it is possible that placental pathologies that manifest during the third trimester or close to the time of delivery may have direct effects on the rapidly developing fetal brain by altering cytokine flux and distribution.82,83,84 These examples suggest that the type of placental histopathology present at the time of delivery broadly reflects the timing (i.e., GA) of the exposure during pregnancy.85
Summary and future directions
Recent data suggest that placental pathology findings may be valuable for identifying newborns at high risk of developing ASD and could potentially shed light on intrauterine risk factors increasing the risk of ASD in preterm infants. Future clinical and mechanistic studies should be designed to provide further insight and inform clinical risk assessment and prevention strategies for reducing ASD among high-risk children.
CHD and stroke
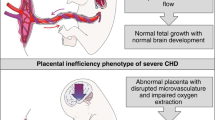
Now that survival from CHD has increased significantly with advanced surgical techniques and medical care, a new focus has been placed on optimizing neurologic outcomes.86 Currently, survivors of CHD have a 75% risk of experiencing developmental delay.87 Severe forms of CHD impart multiple potential causative factors for developmental problems: prolonged postnatal hypoxia while awaiting surgical repair, cardiopulmonary bypass, postnatal recovery with its complications, etc. However, prenatal imaging demonstrates brain growth disturbance in fetal CHD as early as the second trimester,88 as well as delayed brain maturation and white matter abnormalities, among others.89 There is new growing evidence that placental function is abnormal in pregnancies complicated by fetal CHD with reduced oxygen saturation in the umbilical vein in fetal CHD measured using phase-contrast magnetic resonance imaging (MRI) and T2 mapping90 and differences in blood-oxygen-level-dependent signal MRI,91 as well as a larger size for a given fetal weight than controls.92 Emerging evidence is linking abnormal placental hemodynamics with the smaller brain size and delayed brain maturation in fetuses with CHD.90
Placental pathologic lesions
Histologic studies demonstrate the microstructural abnormalities that likely contribute to the impaired hemodynamic function of the placenta in these pregnancies. Common lesions reported in CHD placentas include thrombosis, infarction, chorangiosis, and hypomature villi.93 Placental histology in neonates with hypoplastic left heart syndrome (HLHS) demonstrates villous hypomaturity with increased syncytial nuclear aggregates, indicating failed branching of the villous tree, as well as decreased terminal villi. Increased leptin expression in the HLHS placenta is thought to be an attempt to compensate for the vascular immaturity. Leptin is produced by the placenta as a pro-angiogenic hormone with levels directly correlating with placental weight.94 Likewise, in placentas from fetuses with transposition of the great arteries there are higher rates of chorangiosis and villous hypomaturity compared to other forms of CHD.95 Coupled with imaging findings of larger placenta for a given fetal size, these results indicate a phenotype of placental inefficiency.
Similarly, placental pathology could provide a link in the causal pathway of perinatal stroke (with or without an associated cardiac defect). A recent systematic review of ten studies reported that placental abnormalities were more common among children with perinatal stroke than in the control population with thromboinflammatory process representing the most frequent abnormality.96 However, the results should be interpreted cautiously considering the low frequency of placental examination and lack of uniform pathology reporting.
Summary and future directions
Evidence is accumulating that the placenta in fetal CHD differs from normal in both its structure and function. The data linking the placenta and fetal brain development in CHD are abundant. However, many obstacles still exist in further defining this link. Given the infrequency of the severe forms of CHD, most studies on placental pathology in CHD necessitate grouping of CHD subtypes, which complicates efforts to develop a deeper understanding of placental maladaptation. In addition, placental pathologic examination is not routinely performed in pregnancies complicated by fetal CHD. The first priority is to develop robust protocols to examine placentas from pregnancies complicated by fetal CHD along with larger, multisite data-sharing consortiums to enhance sample sizes of specific cardiac lesions for future investigations. Lastly, the timing of brain dysmaturation in fetuses with CHD requires further study. We should consider more careful imaging assessment of brain and placenta by MRI in severe forms of fetal CHD. Evaluation of placental growth by MRI for comparison to published normative data may prove useful.97 Closer and more sophisticated monitoring would improve our understanding of when potential placental interventions may need to be implemented to impact neurodevelopment.
A targeted hospital-based approach for placental pathologic examination
Taken together, data presented in this review stress the need for precision in diagnostic criteria, terminology, and stratification of placental abnormalities, as well as standardized collection. At Parkland Hospital, a large county hospital with ~14,000 births/year, we have developed a protocol for triaging which placentas should receive histological evaluation. Several obstetric and fetal conditions had been identified at our hospital as warranting the attendance at delivery of a specialized pediatric resuscitation team. These same high-risk deliveries receive routine histological examination of the placenta. These deliveries included: (1) fetuses <36 weeks gestation, (2) estimated fetal weight <2000 g, (3) emergency forceps, (4) thick meconium, (5) nonreassuring fetal heart rate, (6) fetuses of insulin-dependent diabetic mothers, (7) presence of life-threatening fetal anomalies, (8) fetal hydrops, (9) diagnosis of chorioamnionitis with meconium and fetal heart rate abnormalities, (10) infant depression at birth requiring bag mask ventilation for >15 s, and (11) any other situation that the obstetrician determined to warrant the resuscitation team. We follow the systematic classification for harmonization of reporting.98
Conclusions
This review emphasizes the need of placental collection and evaluation for all clinical trials on perinatal brain injury. In addition to insights from histopathology, proteomic and epigenetic analyses of placenta may be especially useful for assessing the impact of injury in the developing brain. An effort to retain the placenta for future histological, biochemical, and molecular analyses should be considered. However, such implementation and change in practice comes with challenges. For placental pathology to truly enter the domain of precision medicine, histological information or molecular biomarkers will need to be rapidly available, with information gathered in real time to facilitate diagnosis and urgent treatment.99
We hope that the review will stimulate future clinical and mechanistic studies, which may provide further insight and inform clinical risk assessment and prevention strategies for reducing long-term neurodevelopmental sequela among high-risk children. It is important to note that, while placental lesions until this point have been considered individually, the concurrent finding of multiple independent placental lesions could be one of the strongest demonstrated associations with adverse neurologic outcomes.23,28 In addition, the severity and timing of the different lesions are important. The synergistic effect of multiple placental lesions is maximal when they are severe and develop at different times.23 One final important caveat: while many correlations between placental lesions and adverse neonatal outcomes have been outlined in this review, to make the jump to causation one must carefully consider and synthesize (1) the clinical scenario, including the presence or absence of a sentinel event; (2) the nature of the lesion(s) identified; and (3) the severity and/or multiplicity of the lesions. Despite these challenges, it is clear that perinatal, childhood, and long-term neurodevelopmental outcomes are linked with placental histology across the spectrum of perinatal brain injury.
References
Bonnin, A. & Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 197, 1–7 (2011).
Robbins, J. R. & Bakardjiev, A. I. Pathogens and the placental fortress. Curr. Opin. Microbiol. 15, 36–43 (2012).
Keating, S. T. & El-Osta, A. Epigenetics and metabolism. Circ. Res. 116, 715–736 (2015).
Nelissen, E. C. et al. Epigenetics and the placenta. Hum. Reprod. Update 17, 397–417 (2011).
Page, J. M. et al. Diagnostic tests for evaluation of stillbirth: results from the Stillbirth Collaborative Research Network. Obstet. Gynecol. 129, 699–706 (2017).
Ravishankar, S. & Redline, R. W. The placenta. Handb. Clin. Neurol. 162, 57–66 (2019).
Yates, L. et al. Influenza A/H1N1v in pregnancy: an investigation of the characteristics and management of affected women and the relationship to pregnancy outcomes for mother and infant. Health Technol. Assess. 14, 109–182 (2010).
Black, R. E. et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375, 1969–1987 (2010).
Shankaran, S. et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 366, 2085–2092 (2012).
Perez, A. et al. Long-term neurodevelopmental outcome with hypoxic-ischemic encephalopathy. J. Pediatr. 163, 454–459 (2013).
Pin, T. W., Eldridge, B. & Galea, M. P. A review of developmental outcomes of term infants with post-asphyxia neonatal encephalopathy. Eur. J. Paediatr. Neurol. 13, 224–234 (2009).
ACOG. (ed) Neonatal Encephalopathy and Neurologic Outcome (AAP, Washington, DC, 2014).
Badawi, N. et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ 317, 1554–1558 (1998).
Badawi, N. et al. Antepartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ 317, 1549–1553 (1998).
West, C. R. et al. Antenatal antecedents of moderate or severe neonatal encephalopathy in term infants-a regional review. Aust. NZ J. Obstet. Gynaecol. 45, 207–210 (2005).
Nelson, K. B. et al. Antecedents of neonatal encephalopathy in the Vermont Oxford Network Encephalopathy Registry. Pediatrics 130, 878–886 (2012).
Hayes, B. C. et al. A case-control study of hypoxic-ischemic encephalopathy in newborn infants at >36 weeks gestation. Am. J. Obstet. Gynecol. 209, 29.e1–29.e19 (2013).
Martinez-Biarge, M. et al. Antepartum and intrapartum factors preceding neonatal hypoxic-ischemic encephalopathy. Pediatrics 132, e952–e959 (2013).
Okereafor, A. et al. Patterns of brain injury in neonates exposed to perinatal sentinel events. Pediatrics 121, 906–914 (2008).
Zhang, P. & Benirschke, K. Placental pathology casebook: serpentine aneurysms of the placenta with fetal consequences. J. Perinatol. 20, 63–65 (2000).
Wintermark, P. et al. Placental pathology in asphyxiated newborns meeting the criteria for therapeutic hypothermia. Am. J. Obstet. Gynecol. 203, 579.e1–579.e9 (2010).
Mir, I. N. et al. Placental pathology is associated with severity of neonatal encephalopathy and adverse developmental outcomes following hypothermia. Am. J. Obstet. Gynecol. 213, 849.e1–849.e7 (2015).
Redline, R. W. & O'Riordan, M. A. Placental lesions associated with cerebral palsy and neurologic impairment following term birth. Arch. Pathol. Lab. Med. 124, 1785–1791 (2000).
Redline, R. W. Severe fetal placental vascular lesions in term infants with neurologic impairment. Am. J. Obstet. Gynecol. 192, 452–457 (2005).
Vik, T. et al. The placenta in neonatal encephalopathy: a case-control study. J. Pediatr. 202, 77.e3–85.e3 (2018).
Harteman, J. C. et al. Placental pathology in full-term infants with hypoxic-ischemic neonatal encephalopathy and association with magnetic resonance imaging pattern of brain injury. J. Pediatr. 163, 968.e2–995.e2 (2013).
McDonald, D. G. et al. Placental fetal thrombotic vasculopathy is associated with neonatal encephalopathy. Hum. Pathol. 35, 875–880 (2004).
Hayes, B. C. et al. The placenta in infants >36 weeks gestation with neonatal encephalopathy: a case control study. Arch. Dis. Child. Fetal Neonatal Ed. 98, F233–F239 (2013).
Owen, M. et al. Brain volume and neurobehavior in newborns with complex congenital heart defects. J. Pediatr. 164, 1121.e1–1127.e1 (2014).
Redline, R. W. Villitis of unknown etiology: noninfectious chronic villitis in the placenta. Hum. Pathol. 38, 1439–1446 (2007).
Greer, L. G. et al. An immunologic basis for placental insufficiency in fetal growth restriction. Am. J. Perinatol. 29, 533–538 (2012).
Jacobs, S. E. et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. CD003311 (2013).
McIntyre, S. et al. Does aetiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy influence the outcome of treatment?. Dev. Med. Child Neurol. 57(Suppl 3), 2–7 (2015).
Ofek-Shlomai, N. & Berger, I. Inflammatory injury to the neonatal brain - what can we do? Front. Pediatr. 2, 30 (2014).
Bolouri, H. et al. Innate defense regulator peptide 1018 protects against perinatal brain injury. Ann. Neurol. 75, 395–410 (2014).
Stoll, B. J. et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993-2012. JAMA 314, 1039–1051 (2015).
Jensen, E. A. & Schmidt, B. Epidemiology of bronchopulmonary dysplasia. Birth Defects Res. A Clin. Mol. Teratol. 100, 145–157 (2014).
Sarno, L. et al. Histological chorioamnionitis and risk of pulmonary complications in preterm births: a systematic review and meta-analysis. J. Matern. Fetal Neonatal Med. https://doi.org/10.1080/14767058.2019.1689945 (2019).
Thomas, W. & Speer, C. P. Chorioamnionitis is essential in the evolution of bronchopulmonary dysplasia-the case in favour. Paediatr. Respir. Rev. 15, 49–52 (2014).
Villamor-Martinez, E. et al. Association of chorioamnionitis with bronchopulmonary dysplasia among preterm infants: a systematic review, meta-analysis, and metaregression. JAMA Netw. Open 2, e1914611 (2019).
Ericson, J. E. & Laughon, M. M. Chorioamnionitis: implications for the neonate. Clin. Perinatol. 42, 155–165 (2015).
Lee, H. J. et al. Chorioamnionitis, respiratory distress syndrome and bronchopulmonary dysplasia in extremely low birth weight infants. J. Perinatol. 31, 166–170 (2011).
Pappas, A. et al. Chorioamnionitis and early childhood outcomes among extremely low-gestational-age neonates. JAMA Pediatr. 168, 137–147 (2014).
Mestan, K. K. et al. Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta 35, 570–574 (2014).
Kunjunju, A. M. et al. Bronchopulmonary dysplasia-associated pulmonary hypertension: clues from placental pathology. J. Perinatol. 37, 1310–1314 (2017).
Yallapragada, S. G. et al. Placental villous vascularity is decreased in premature infants with bronchopulmonary dysplasia-associated pulmonary hypertension. Pediatr. Dev. Pathol. 19, 101–107 (2016).
Thorn, S. R. et al. The intrauterine growth restriction phenotype: fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin. Reprod. Med. 29, 225–236 (2011).
Salafia, C. M. et al. Placental pathology of idiopathic intrauterine growth retardation at term. Am. J. Perinatol. 9, 179–184 (1992).
Mir, I. N. et al. Impact of multiple placental pathologies on neonatal death, bronchopulmonary dysplasia, and neurodevelopmental impairment in preterm infants. Pediatr. Res. 87, 885–891 (2019).
Hack, M. et al. Behavioral outcomes of extremely low birth weight children at age 8 years. J. Dev. Behav. Pediatr. 30, 122–130 (2009).
Johnson, S. et al. Autism spectrum disorders in extremely preterm children. J. Pediatr. 156, 525–31 e2 (2010).
Pinto-Martin, J. A. et al. Prevalence of autism spectrum disorder in adolescents born weighing <2000 grams. Pediatrics 128, 883–891 (2011).
Treyvaud, K. et al. Psychiatric outcomes at age seven for very preterm children: rates and predictors. J. Child Psychol. Psychiatry 54, 772–779 (2013).
Joseph, R. M. et al. Prevalence and associated features of autism spectrum disorder in extremely low gestational age newborns at age 10 years. Autism Res. 10, 224–232 (2017).
Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators & Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 63, 1–21 (2014).
Kuzniewicz, M. W. et al. Prevalence and neonatal factors associated with autism spectrum disorders in preterm infants. J. Pediatr. 164, 20–25 (2014).
Dammann, O. & Leviton, A. Brain damage in preterm newborns: might enhancement of developmentally regulated endogenous protection open a door for prevention?. Pediatrics 104(Pt 1), 541–550 (1999).
Leviton, A. et al. The wealth of information conveyed by gestational age. J. Pediatr. 146, 123–127 (2005).
Sanders, E. J. & Harvey, S. Peptide hormones as developmental growth and differentiation factors. Dev. Dyn. 237, 1537–1552 (2008).
Goldenberg, R. L. et al. Epidemiology and causes of preterm birth. Lancet 371, 75–84 (2008).
Nadeau, H. C., Subramaniam, A. & Andrews, W. W. Infection and preterm birth. Semin. Fetal Neonatal Med. 21, 100–105 (2016).
Wu, H. C. et al. Subclinical histologic chorioamnionitis and related clinical and laboratory parameters in preterm deliveries. Pediatr. Neonatol. 50, 217–221 (2009).
Leviton, A. et al. The risk of neurodevelopmental disorders at age 10years associated with blood concentrations of interleukins 4 and 10 during the first postnatal month of children born extremely preterm. Cytokine 110, 181–188 (2018).
Anblagan, D. et al. Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 6, 37932 (2016).
Malaeb, S. & Dammann, O. Fetal inflammatory response and brain injury in the preterm newborn. J. Child Neurol. 24, 1119–1126 (2009).
Kramer, B. W. et al. Decreased expression of angiogenic factors in placentas with chorioamnionitis after preterm birth. Pediatr. Res. 58, 607–612 (2005).
Roescher, A. M. et al. Placental pathology, perinatal death, neonatal outcome, and neurological development: a systematic review. PLoS ONE 9, e89419 (2014).
Rosenfeld, C. S. Sex-specific placental responses in fetal development. Endocrinology 156, 3422–3434 (2015).
Atladottir, H. O. et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 40, 1423–1430 (2010).
Chess, S. Autism in children with congenital rubella. J. Autism Child Schizophr. 1, 33–47 (1971).
Guinchat, V. et al. Pre-, peri- and neonatal risk factors for autism. Acta Obstet. Gynecol. Scand. 91, 287–300 (2012).
Challier, J. C. et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29, 274–281 (2008).
Roberts, K. A. et al. Placental structure and inflammation in pregnancies associated with obesity. Placenta 32, 247–254 (2011).
Ghidini, A. & Salafia, C. M. Gender differences of placental dysfunction in severe prematurity. BJOG 112, 140–144 (2005).
Limperopoulos, C. et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics 121, 758–765 (2008).
Straughen, J. K. et al. The association between placental histopathology and autism spectrum disorder. Placenta 57, 183–188 (2017).
Salafia, C. et al. Characterization of placental growth as a biomarker of autism/ASD risk. Placenta 33, A16–A16 (2012).
Raghavan, R. et al. Preterm birth subtypes, placental pathology findings, and risk of neurodevelopmental disabilities during childhood. Placenta 83, 17–25 (2019).
Mir, I. N. et al. Autism spectrum disorders in extremely preterm infants and placental pathology findings: a matched case-control study. Pediatr. Res. https://doi.org/10.1038/s41390-020-01160-4 (2020).
Naldini, A. & Carraro, F. Role of inflammatory mediators in angiogenesis. Curr. Drug Targets Inflamm. Allergy 4, 3–8 (2005).
Kwiatkowski, S. et al. A common profile of disordered angiogenic factor production and the exacerbation of inflammation in early preeclampsia, late preeclampsia, and intrauterine growth restriction. PLoS ONE 11, e0165060 (2016).
Elovitz, M. A. et al. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int. J. Dev. Neurosci. 29, 663–671 (2011).
Leitner, K. et al. IL-1 receptor blockade prevents fetal cortical brain injury but not preterm birth in a mouse model of inflammation-induced preterm birth and perinatal brain injury. Am. J. Reprod. Immunol. 71, 418–426 (2014).
Maxwell, J. R. et al. Combined in utero hypoxia-ischemia and lipopolysaccharide administration in rats induces chorioamnionitis and a fetal inflammatory response syndrome. Placenta 36, 1378–1384 (2015).
Redline, R. W. Inflammatory responses in the placenta and umbilical cord. Semin. Fetal Neonatal Med. 11, 296–301 (2006).
Marino, B. S. et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation 126, 1143–1172 (2012).
Mussatto, K. A. et al. Risk and prevalence of developmental delay in young children with congenital heart disease. Pediatrics 133, e570–e577 (2014).
Lauridsen, M. H. et al. Fetal heart defects and measures of cerebral size. J. Pediatr. 210, 146–153 (2019).
Khalil, A. et al. Prevalence of prenatal brain abnormalities in fetuses with congenital heart disease: a systematic review. Ultrasound Obstet. Gynecol. 48, 296–307 (2016).
Sun, L. et al. Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation 131, 1313–1323 (2015).
You, W. et al. Hemodynamic responses of the placenta and brain to maternal hyperoxia in fetuses with congenital heart disease by using blood oxygen-level dependent MRI. Radiology 294, 141–148 (2020).
Andescavage, N. et al. 3-D volumetric MRI evaluation of the placenta in fetuses with complex congenital heart disease. Placenta 36, 1024–1030 (2015).
Schlatterer, S. D. et al. Placental pathology and neuroimaging correlates in neonates with congenital heart disease. Sci. Rep. 9, 4137 (2019).
Hassink, S. G. et al. Placental leptin: an important new growth factor in intrauterine and neonatal development?. Pediatrics 100, E1 (1997).
Rychik, J. et al. Characterization of the placenta in the newborn with congenital heart disease: distinctions based on type of cardiac malformation. Pediatr. Cardiol. 39, 1165–1171 (2018).
Roy, B. et al. The role of the placenta in perinatal stroke: a systematic review. J. Child Neurol. 35, 773–783 (2020).
Leon, R. L., Li, K. T. & Brown, B. P. A retrospective segmentation analysis of placental volume by magnetic resonance imaging from first trimester to term gestation. Pediatr. Radiol. 48, 1936–1944 (2018).
Khong, T. Y. et al. Sampling and definitions of placental lesions: Amsterdam Placental Workshop Group Consensus Statement. Arch. Pathol. Lab. Med. 140, 698–713 (2016).
Lester, B. M. & Marsit, C. J. Epigenetic mechanisms in the placenta related to infant neurodevelopment. Epigenomics 10, 321–333 (2018).
Acknowledgements
This work was supported by NIH Grant 5R01NS102617-03 awarded to L.F.C.
Author information
Authors and Affiliations
Contributions
All authors contributed to drafting the article or revising it critically for important intellectual content and final approval of the submitted version. I.N.M. participated in concept, literature search, and data interpretation; drafted the first version of the manuscript; and finalized the manuscript for submission after comments from the other authors. R.L. and L.F.C. participated in concept, literature search, data interpretation, revision of the manuscript, and finalization of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mir, I.N., Leon, R. & Chalak, L.F. Placental origins of neonatal diseases: toward a precision medicine approach. Pediatr Res 89, 377–383 (2021). https://doi.org/10.1038/s41390-020-01293-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01293-6
This article is cited by
-
Future Prospects for Epigenetics in Autism Spectrum Disorder
Molecular Diagnosis & Therapy (2022)
