
Abstract
Necrotizing enterocolitis (NEC) is a devastating condition affecting up to 5% of neonatal intensive care unit (NICU) admissions. Risk factors include preterm delivery, low birth weight, and antibiotic use. The pathogenesis is characterized by a combination of intestinal ischemia, necrosis of the bowel, reperfusion injury, and sepsis typically resulting in surgical resection of afflicted bowel. Targeted medical therapy remains elusive. Chondroitin sulfate (CS) holds the potential to prevent the onset of NEC through its anti-inflammatory properties and protective effect on the gut microbiome. The purpose of this review is to outline the many properties of CS to highlight its potential use in high-risk infants and attenuate the severity of NEC. The purpose of this review is to (1) discuss the interaction of CS with the infant microbiome, (2) review the anti-inflammatory properties of CS, and (3) postulate on the potential role of CS in preventing NEC.
Impact
-
NEC is a costly medical burden in the United States.
-
Breast milk is the best preventative measure for NEC, but not all infants in the NICU have access to breast milk.
-
Novel therapies and diagnostic tools are needed for NEC.
-
CS may be a potential therapy for NEC due to its potent anti-inflammatory properties.
-
CS could be added to the formula in an attempt to mitigate breast milk disparities.
Similar content being viewed by others


Introduction
Necrotizing enterocolitis (NEC) is responsible for 2–5% of all neonatal intensive care unit (NICU) admissions worldwide.1 The majority of cases involve preterm infants, with an incidence rate of 7–9% in the very low birth weight (VLBW) population.2,3 Targeted medical treatment for NEC is limited and many infants often require surgery. Due to the rapid progression of the disease, infants with NEC are often diagnosed when the disease is advanced and requires surgery, thereby increasing morbidity, mortality, and cost burden.4,5 Despite extensive research, there has been no significant advances in treating NEC over the past 30 years.
Preventative treatment for most diseases requires an adept understanding of its etiology and pathophysiology. Proposing an effective solution for NEC is difficult as its pathophysiology remains poorly understood. There are several theories concerning the pathophysiology of NEC. One theory is that unfavorable intestinal bacterial colonization, called dysbiosis, disrupts the mucosal lining triggering an acute inflammatory response allowing bacteria to translocate from the gut to the bloodstream. This process is associated with dysfunction of intestinal microcirculation, hypoxia, and eventual necrosis and perforation of the bowel.
Preterm infants are especially susceptible to NEC for a variety of reasons. The preterm immune system is immature and more likely to mount an excessive inflammatory response towards invasive gut flora.6 Compounding this, preterm infants have higher intestinal permeability and are at increased risk of the translocation of bacteria.7 Newborns also have decreased mucosal defenses against pathogens, including a deficiency of immunoglobulin A (IgA) and lactoferrin, making them more susceptible to inflammatory disease.8
Fortunately, there is one well-established method to reduce the chance of newborns developing NEC: breast milk. It has been well studied that the administration of human breast milk (HM) provides a dose-dependent protective effect against NEC, reducing incidence by as much as 4%.9,10 HM also decreases rates of mortality and other conditions, such as retinopathy, sepsis, and bronchopulmonary dysplasia.11,12 Preterm babies fed exclusively formula have a 6–10 times higher rate of NEC occurrence compared to the HM-fed group.13 The mechanism behind such a protection is multivariate. HM has been shown to alter the composition of the gut microbiome as well as alter gene expression in enterocytes.14,15 Many compounds derived from HM have shown immune-modulating activity (Table 1).16
While HM, particularly maternally derived HM, is very effective at reducing risk for NEC, there are several disparities in its availability to infants in NICU’s across the United States. Only 72% of infants in the NICU actually receive HM, leaving a significant number relying on formula feeds.5,17 These disparities are further associated with race, ethnicity, socioeconomic status, language, and sexual orientation of the parents.18
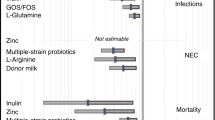
Choosing the most efficacious compounds in HM to fortify formula has great potential for reducing the incidence of NEC (Fig. 1). HM contains many protective factors, including secretory IgA, lactoferrin, and various oligosaccharides, that are not in formulas.3 One such oligosaccharide that is gaining interest is the glycosaminoglycan chondroitin sulfate (CS). Traditionally used to slow the progression of osteoarthritis (OA), isolated CS is a growing candidate to reduce intestinal inflammation and combat dysbiosis in preterm infants or those with inflammatory disease.19,20,21 The purpose of this review is to (1) discuss the interaction of CS with the infant microbiome, (2) review the anti-inflammatory properties of CS, and (3) postulate on the potential role of CS in preventing NEC as a supplement in formula feeds.

Infant gut microbiome
To better understand the relationship between CS and the intestinal microbiome, it is best to discern the gut flora of healthy, full-term infants first. Each person has a uniquely structured composition of organisms that play a personalized role in digestion, immune activity, and neurologic signaling.22 This system coalesces from the beginning of development, but only forms a true ecosystem after birth. The gut microbiome of a healthy infant undergoes three distinct stages of evolution starting at 3 months of age. According to data from over 900 children, large amounts of Bifidobacterium are a hallmark of the first year of life and are heavily influenced by HM feeding. This is supported by other studies showing the colonization of anaerobes like Bifidobacterium and Bacteroides as important in the first weeks of life.23 The first days to weeks of life are of particular interest, as NEC most often occurs during these times.24 Fortunately, controlled environments, such as the NICU, foster predictable patterns of infant gut colonization, thus providing insights into factors at play during this time.25 These factors include antibiotic use, mode of childbirth, and feeding patterns.26
Despite the complexity, two major themes persist. The first is diversity. Many studies have demonstrated a strong correlation between the range of microbial species and disease. Alpha diversity, characterized by the number of taxonomic groups and distribution of the abundances of the groups, summarizes the structure and richness of a microbial community.27 Loss of alpha diversity in adult gut flora has been seen in inflammatory bowel disease, obesity, and colitis, as well as neurological disorders like Alzheimer disease.28,29,30 NEC and celiac disease are also associated with lower alpha diversity.31,32,33 Contrary to these results, some studies have found no significant difference in species diversity in the stool of NEC and control patients.34,35 This can be explained by the fact that not all NEC patients harbor a dysbiotic gut. A preterm infant with a microbial profile of that of a “healthy” infant may still develop NEC if insult to the bowel comes in the form of ischemia or inflammation. Loss of alpha diversity may only increase the risk of NEC, not cause it. However, a plethora of data support diversity as a protective factor.1,31,36,37 Whether too much diversity is detrimental is also not understood. Until more data emerges, it is likely best to consider the loss of alpha diversity as a risk factor for dysbiosis and NEC pathogenesis.
The second theme in the microbiome is balance. In most cases, a higher ratio of commensal bacteria protects the bowel by preventing pathogenic organisms from attaching to intestinal endothelium and causing inflammation.38,39 If a large amount of non-commensal species overtakes the gut, the balance may tip in favor of NEC pathogenesis. For example, infants born vaginally are predominantly colonized by Firmicutes, such as Lactobacillus spp., characteristic of the mother’s vaginal canal and stool. Once born, babies fed with HM commonly host large amounts of Lactobacillus and Bifidobacteria that play multiple roles in preventing dysbiosis.40 Formula-fed infants demonstrate increased levels of Escherichia coli, Clostridium difficile, and Bacteroides fragilis. If this effect was profound enough, it may disrupt the balance in the infant microflora. Such a scenario may explain the higher incidence of NEC in formula-fed infants.41 Paradoxically, studies show that formula infants have a more diverse microbiome, which is unexpected as formula feeding is associated with worse health outcomes.42,43 Perhaps, excessive diversity can negatively impact the balance of commensal to pathogenic flora in the gut. However, these findings are controversial.44 The detrimental effects of formula feeding may be better explained by the hyperosmolarity of formula regimens that damage the endothelial lining and microcirculation of the gut, likely offsetting any beneficial effect of increased diversity.45
Dysbiosis and preterm infants
There is no single pathogen that can cause dysbiosis, as the microflora is a complex ecosystem. Even though the triggers are multifactorial and the pathogenesis can be variable, dysbiosis frequently involves loss of diversity or balance in the microflora.1,31,46,47 It typically displays a disproportionate colonization of invasive lipopolysaccharide (LPS)-containing Gram-negative species that trigger inflammatory cascades and can prevent the growth of important obligate anaerobes like Bifidobacterium or protective species like Bacteroides.48 In particular, dysbiosis is strongly linked with large increases in the number of Gram-negative bacteria as infants age. For example, the most commonly seen species in stools of NEC infants are E. coli, Salmonella enterolitica, and various members of the Enterobacteriaceae and Pseudomonades families.49,50,51 This is further supported by a case study finding that the relative amounts of Gram-negative Proteobacteria, such as Klebsiella, were larger compared to the amounts of Firmicutes, such as Lactobacillus.52 Until our understanding of the role of the microbiome in the infant gut increases, attenuating dysbiosis in NEC infants may be best approached from a broad view of the entire gut profile rather than focusing on targeting specific microbes.
Because undergoing preterm delivery is such a strong risk factor for NEC, the microbiome profile of such infants is of great interest as it fits many patterns key to dysbiosis. The flora of preterm infants typically harbors a larger ratio of Proteobacteria compared to term infants.23 An overabundance of Staphylococci bacteria has been documented in VLBW preterm infant stool as well.53,54,55 Furthermore, one study found a reduced percentage of Bacteroidaceae and increased the percentage of Lactobacillaceae in preterm infant stool compared to that of term infants.56 These findings may be explained by extensive antibiotic use in preterm infants. Commonly used antibiotics for neonates are ampicillin and gentamicin. Despite their usefulness in fighting infection, prolonged use of such drugs has been correlated with a two-fold increase in NEC incidence.57 Evidence shows that antibiotic use in preterm infants lowers the bacterial diversity necessary to keep Proteobacteria or invasive Gram-negative populations in check.48,58 Excessive administration of these drugs may tip the balance of the microflora ecosystem to an unfavorable state conducive to NEC. Perhaps, a simultaneous look into reducing antibiotic use and CS therapy will prove fruitful.
CS as a preventative therapy
CS has many intriguing properties, particularly in the extracellular matrix. As a derivative of the glycosaminoglycan family of connective tissue structures, it is found naturally in humans in HM, cartilage, nervous structures, and various organs playing both structural and physiologic roles.59 CS is a polymer of alternating and repeating sequences of glucuronic acid and galactosamine typically sulfated at the C4 and C6 position. Like other glycosaminoglycans, this structure enables endogenous CS to bind various core proteins forming proteoglycans. This formation is not simply structural, as CS-derived proteoglycans appear to regulate cell signaling in different pathways and locations throughout the body.60,61,62
CS separates itself from other glycosaminoglycans and oligosaccharides as a candidate for formula supplementation in a few ways. One way is in the diversity of its biologic activity. In addition to sharing probiotic and anti-inflammatory properties with other compounds in HM, CS appears unique in its propensity to increase sulfate-reducing bacteria populations.21,63 CS also holds various antioxidant and antithrombotic properties.64,65,66,67 This is important, as the concentration of CS in HM appears to change over time. Preterm mothers harbor more CS in their milk than term mothers.68,69 Perhaps, there is an evolutionary advantage to having more CS available in HM for mothers caring for preterm infants.
Another way CS differentiates itself is its extensive clinic use as an anti-inflammatory agent in OA. CS has been shown to upregulate hyaluronic acid and type 2 collagen synthesis in human chondrocytes, prevent apoptosis of chondrocytes in animal models, and regulate chondrocyte signaling.70 However, its anti-inflammatory properties are of most interest for CS’s potential use for NEC patients. Several OA-related animal studies have shown its propensity to limit the synthesis of pro-inflammatory mediators like nitric oxide, prostaglandin E2, and nuclear factor-kB (NF-kB)-mediated products70 Similarly, serum levels of cytokines interleukin-1 (IL-1), tumour necrosis factor-α (TNFα), and IL-6 were reduced in rats given varying doses of oral CS.71 Immune dysregulation and intestinal inflammation is part of the pathophysiology of NEC, and these anti-inflammatory properties of CS may provide benefits in a NEC animal model.
Traditionally, oral CS is used for OA patients. Its pharmacokinetic properties and bioavailability have been studied extensively in the OA model. It is administered orally at doses from 800 to 1200 mg/day and is primarily absorbed by the distal gastrointestinal (GI) tract in the paracellular pathway.70,72 It is absorbed in relatively small amounts in the small intestine likely through endocytosis. CS is not significantly metabolized in the stomach or small intestine, but is mostly metabolized distal to the ileum.72 Estimates on the exact bioavailability of oral CS range from 10 to 20%, with the majority of the absorbed CS being in the form of depolymerized derivatives70,73 While there is some debate on the exact bioavailability, it is known that peak concentrations occur at 2–6 h after dosage. CS is not metabolized by the cytochrome P450 system, it follows first-order kinetics, and is a long-acting drug, with a slow onset of action and biologic effect that can accumulate over the course of months.70,73 The half-life of CS in humans is 15 h and steady-state concentrations have been achieved after 4 days of oral therapy. It is important to note that clinical trials of CS in OA patients have reported no major side effects nor elevations of serum enzymes.59 The FDA rates CS as “Generally Recognized as Safe.” A few separated case reports have linked liver toxicity to glucosamine and CS therapy, but it remains unclear whether CS was a causal agent. CS is well tolerated, with few reported cases of an immune-allergenic response.59
Since NEC primarily affects the small intestine, a question remains regarding CS’s therapeutic potency if it is not absorbed and metabolized significantly until the distal GI tract. One possible explanation is that CS may function through bacterial pathways as intestinal flora metabolize CS throughout the GI tract.21 A second theory is that CS needs to be absorbed systemically before it can act to reduce inflammation in the small intestine. Another major concern for CS supplementation is the effect of the pasteurization process on the biologic components of donor and formula feeds. Some data suggest that pasteurization reduces levels of key components of HM, such as IgA, growth factors, and other enzymes.74 Fortunately, the times and temperatures involved appear unable to degrade CS or reduce concentrations of glycosaminoglycans.75 CS-supplemented feeds can theoretically still be pasteurized.
CS and the microbiome
Given that the pathophysiology of NEC involves intestinal dysbiosis and decrease alpha diversity, medications that promote a diverse and balanced microbiome have preventative potential. For CS to work, it must be absorbed by resident microflora as well as enterocytes. Studies on antibiotic use demonstrate varying levels of CS fractional absorption, thus there must be some interplay between CS, the microflora, and the body.21 The exact details of how CS influences the microbiome are not yet known. However, CS appears to work through three mechanisms: (1) promoting the growth of commensal genera in the microflora to maintain diversity and balance, (2) decreasing the invasion of pathogenic bacteria across the gut wall, and (3) increasing relative abundance of sulfate-reducing bacteria.
CS induces the growth of commensal microflora
Evidence suggests that CS exposure promotes growth of certain species of gut-protective bacteria. This effect can be seen with both isolated CS and HM, which contains CS. A review by Shmagel et al.21 analyzed eight studies performed on the effects of CS and glucosamine administration on the microbiome in both mouse and human models. They found “moderate-quality” evidence that CS exposure increased the relative abundance of genus Bacterioides, an important player in the early colonization of the infant gut. Two of the analyzed studies using the mouse model noted an increase in the Desfulfovibrio piger population, a sulfate-reducing bacteria. In terms of overall gut diversity, they found mixed results.21 Similar conclusions were found in the study by Liu et al.76 They found that long-term CS administration decreased blood LPS levels, reduced prevalence of Proteobacteria in stool, and increased intestinal Bacteroides populations. These findings were supported by another study carried out by Ford et al.36 Rather than isolated CS, Ford et al.36 investigated the effects of HM on the microbiome of VLBW infants. Perhaps, the most interesting aspect of their work was their separation of maternal HM and donor HM. The stool samples from infants receiving majorly maternal HM had increased gut diversity of Bacteroides and Bifidobacterium populations compared to the donor HM group. If such is the case, then increasing the supply of readily available maternal HM for VLBW infants may improve patient outcomes and reduce the cost of care. By extension, if the effect of HM depends on its source (maternal or donor), then structural or physiological properties of CS between sources may slightly differ as well. This has not been studied, but is supported by the fact that CS from different species, like shark or bovine, are absorbed at different rates in humans.59 Further inquiry may unveil interesting conclusions. Taking these findings into account, increasing the abundance of genera, such as Bacteroides, may appear counterintuitive to an effort of promoting diversity. However, such genera are typically less populous in NEC infants34,77 CS exposure may restore a healthy balance in microflora lacking Bacteroides or Bifidobacterium. Such a restoration may lower the relative abundance of invasive Gram-negative bacteria, potentially reducing the risk of NEC development.
CS limits translocation of gut bacteria
Another potential beneficial property of CS is its ability to limit translocation and invasion of bacteria into the bloodstream. Burge et al.78 used an in vitro model with T84 cells formed in a monolayer and treated with CS. The cells were then exposed to E. coli and evaluated for inflammatory markers, cell viability, and transposition of the bacteria across the monolayer. Bacterial translocation was measured using trans-epithelial electrical resistance to gauge tight junction integrity. They described a concentration-dependent effect, with no loss of cell function at CS concentrations up to 750 μg/ml. At the same amount, there was a 75% decrease in bacterial translocation and invasion across the cell monolayer.78 Translating the value 750 μg/ml to clinical terms is difficult, as ingesting a prescribed dosage of CS will not necessarily lead to similar intestinal concentrations. The gut is not a simple monolayer of cells. For context, peak serum concentrations of oral CS typically fall within the range of 2–12 μg/ml for OA patients.70,73 The side-effect profile of CS minimizes any adverse outcomes or potential cellular damage if intestinal concentrations do exceed the 750 μg/ml threshold. Most importantly, these findings by Burge et al.78 were consistent with the anti-inflammatory properties of CS.
Increasing the thickness or number of barriers will also prevent translocation of pathogenic species. This can be best understood by considering that chondroitin is a component of mucin. Mucin acts as a natural physical barrier to luminal bacteria and is secreted by goblet cells in the intestinal tract. Increasing the supply of CS in the environment will theoretically allow for increased production of mucin.21 Many members of the genus Bacteroides are known to metabolize dietary glycans like CS in the gut.79 If starved, these bacteria will break down mucin and induce inflammation and eventually translocation of bacteria.80
Anti-inflammatory properties of CS
The pathophysiology of NEC is extremely complex and involves more than the microbiome. Inflammation plays a central role. While the etiology behind the inflammation is still unclear, how the immature innate immune system increases susceptibility to NEC has been studied. It has been reported that immature intestine has underdeveloped expression of IkB, a mediator that normally limits NF-kB.6 In theory, this would lead to exaggerated NF-kB-related cascades and higher levels of pro-inflammatory markers, such as IL-1, IL-6, IL-8, and TNFα. This is supported by a study analyzing human enterocytes. It was found that immature cells secreted a larger amount of IL-8 in response to stimulation compared to mature cells.81 These findings make sense in the context of Toll-like receptors (TLRs) expression, specifically TLR4. LPS from pathogenic bacteria activate TLR4, which in turn induces the activity of NF-kB. . Previous studies concluded that TLR4-deficient mice experienced decreased amounts of enterocyte apoptosis and TNFα levels in a NEC model compared to wild-type counterparts. These studies also found that NEC severity was attenuated in TLR4-deficient mice.82 Thus, reducing pro-inflammatory markers in preterm infants may be useful in preventing NEC pathogenesis. While research into this approach was limited, there are some studies investigating the reduction of the aberrant inflammatory response in NEC.11,82 Compounds like glucocorticoids and anti-cytokines have been used for prevention of NEC, but hold concerns regarding side-effect profiles.8 CS presents as a safe alternative for its use for NEC. The exact profiling of the anti-inflammatory properties of CS remains elusive, but mounting evidence unveils many mechanisms behind them. These mechanisms include its inhibition of NF-kB activity and its regulation of immune mediator cells like macrophages and mast cells. (Fig. 2)

Chondroitin sulfate works to decrease pro-inflammatory mediators while working to modulate the intestinal microbiome.
CS limits NF-kB activity
NF-kB plays a central role in the inflammatory pathway of the immune system by mediating the secretion of acute-phase reactants like IL-1, IL-6, and TNFα.83 The therapeutic potential of CS therefore hinges on its ability to limit NF-kB to reduce the severity of NEC. Research into this primarily involves the use of CS for OA. Stabler et al.84 investigated the capacity for CS to limit NF-kB expression in THP-1 macrophages. They cultured macrophages and treated them with the NF-kB activators, such as LPS and hyaluronic acid, to induce an inflammatory response. They concurrently exposed the macrophages to varying levels of CS. Results showed that CS reduced the release of IL-1 and blocked NF-kB expression from activated macrophages compared to controls. Another study investigated the capacity for CS to limit inflammatory markers in healthy patients. Navarro et al.85 conducted a randomized, double-blind control trial with a patient cohort screened for medical concerns. Experimental groups received oral capsules of combined CS and glucosamine. The results revealed a significant drop in serum C-reactive protein in the experimental group compared to placebo. Using KEGG (Kyoto Encyclopedia of Genes and Genomes) and Gene Ontology databases, Navarro et al.85 also conducted gene-set enrichment pathway analysis and found a significant reduction in cytokine–cytokine receptor interaction, JAK/STAT (Janus kinase-signal transducer and activator of transcription) signaling, and intestinal IgA production. A study by Campo et al.86 supported these findings. Using mouse articular chondrocytes stimulated by LPS to induce an inflammatory state, they found that CS blunted the response by reducing TNFα, IL-1, IL-6, and interferon-γ (IFNγ) levels. Data from Chan et al.87 concluded that CS supplementation, in combination with glucosamine, also reduced IL-1-induced gene expression of NF-kB, prostaglandin E, inducible nitric oxide synthase, and cyclooxygenase-2 in a bovine cartilage model.
A potential drawback for CS to antagonize NF-kB activity is its potential to sequester TGF-β2 in the gut lumen. TGF- β2 is an immune modulator that limits macrophage expression of cytokines through the NF-kB pathway. A study by Namachivayam et al.88 used various techniques to measure TGF-β2 bioactivity in the presence of different compounds derived from HM. They found that CS could bind milk-borne TGF-β2 and limit its ability as an anti-inflammatory molecule. When the HM was treated with chondroitinase, the bioactivity of TGF-β2 increased. Also, evidence suggests that isolated CS cannot directly trigger or interact with TLRs.89 These revelations appear concerning for the potential of CS to limit NF-kB activity. However, the study by Namachivayam et al.88 had a small sample size and only focused on aqueous factors of HM, but not the fat compartment, which contains significant amounts of TGF-B2. Since CS is mainly degraded in the large intestine, trapped TGF-β2 can still act distally.70 CS potentially limits NF-kB from a multifaceted approach as discussed earlier. It is likely that any pro-inflammatory aspects of CS are outweighed by the many anti-inflammatory ones. Further inquiry may dissolve any ambiguity.
CS and immune cells
The capacity for CS to regulate the activity of various immune cells holds much interest. Tan et al.90 measured the effect of CS on the expression of inflammatory mediators in macrophages derived from mouse bone marrow. Results concluded that CS reduced the expression of IL-6 and TNFα by macrophages activated by LPS and INFy. The expression of IL-10 increased. This suggests that CS alters the phenotype of macrophages away from an inflammatory role towards an anti-inflammatory state conducive to wound healing. The macrophages pretreated with CS prior to exposure of LPS or INFγ had the strongest decrease in inflammatory gene expression.90 If these findings are replicated in the gut, then treating preterm or VLBW infants prophylactically with CS may reduce the risk of developing NEC. Tan et al.90 concluded that these effects were in part due to suppression of NF-kB activity in macrophages.
CS also appeared to modulate the activity of mast cells. Gross and Theoharides91 investigated the effect of CS on human mast cell secretion of TNF and C-X-C motif chemokine ligand 8 (CXCL8). They concluded that mast cells pretreated with CS and then activated with IL-33 exhibited a significant reduction of TNF and CXCL8 secretion. Further inquiry revealed that CS did not alter gene expression of TNF and CXCL8, decrease IL-33 surface receptor localization, nor block mast cell degranulation. They did find that CS was endocytosed by mast cells in a calcium-independent manner. Gross and Theoharides91 concluded that CS must act through an unknown intracellular mechanism to limit the secretion of TNF and CXCL8. Whether the pathway involved NF-kB or something else was unknown. Current understanding of the pathogenesis of NEC does not involve mast cells. However, their role in other intestinal disorders like inflammatory bowel disease opens the possibility of an undiscovered function in NEC patients.92
Conclusion
While our knowledge on pathogenesis of NEC is incomplete, reducing inflammation and preventing dysbiosis may reduce the severity of NEC and could reduce mortality in preterm and low birth weight infants. CS is an intriguing candidate for NEC therapy due to its safe side-effect profile, its anti-inflammatory properties, and its favorable influence on the microbiome.
References
Gupta, A. & Paria, A. Etiology and medical management of NEC. Early Hum. Dev. 97, 17–23 (2016).
Hull, M. A. et al. Mortality and management of surgical necrotizing enterocolitis in very low birth weight neonates: a prospective cohort study. J. Am. Coll. Surg. 218, 1148–1155 (2014).
Knell, J., Han, S. M., Jaksic, T. & Modi, B. P. Current status of necrotizing enterocolitis. Curr. Probl. Surg. 56, 11–38 (2019).
Li, Q. Y. et al. Differences in the clinical characteristics of early- and late-onset necrotizing enterocolitis in full-term infants: a retrospective case–control study. Sci. Rep. 7, 43042 (2017).
Boundy, E. O., Perrine, C. G., Nelson, J. M. & Hamner, H. C. Disparities in hospital-reported breast milk use in neonatal intensive care units—United States, 2015. Morb. Mortal. Wkly Rep. 66, 1313–1317 (2017).
Nanthakumar, N. et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS ONE 6, e17776 (2011).
Ma, B. et al. Microbial biomarkers of intestinal barrier maturation in preterm infants. Front. Microbiol. 9, 2755 (2018).
Choi, Y. Y. Necrotizing enterocolitis in newborns: update in pathophysiology and newly emerging therapeutic strategies. Korean J. Pediatr. 57, 505–513 (2014).
Miller, J. et al. A systematic review and meta-analysis of human milk feeding and morbidity in very low birth weight infants. Nutrients 10, 707 (2018).
Meinzen-Derr, J. et al. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J. Perinatol. 29, 57–62 (2009).
Herrmann, K. & Carroll, K. An exclusively human milk diet reduces necrotizing enterocolitis. Breastfeed. Med. 9, 184–190 (2014).
Hair, A. B. et al. Beyond necrotizing enterocolitis prevention: improving outcomes with an exclusive human milk-based diet. Breastfeed. Med. 11, 70–74 (2016).
Lucas, A. & Cole, T. J. Breast milk and neonatal necrotising enterocolitis. Lancet 336, 1519–1523 (1990).
Sitarik, A. R. et al. Breast milk transforming growth factor beta is associated with neonatal gut microbial composition. J. Pediatr. Gastroenterol. Nutr. 65, e60–e67 (2017).
Guner, Y. S. et al. P-glycoprotein induction by breast milk attenuates intestinal inflammation in experimental necrotizing enterocolitis. Lab. Invest. 91, 1668–1679 (2011).
Institute of Medicine of the National Academies. Infant Formula: Evaluating the Safety of New Ingredients (The National Academies Press, Washington, 2004).
Profit, J. et al. Racial/ethnic disparity in NICU quality of care delivery. Pediatrics 140, e20170918 (2017).
Sigurdson, K., Morton, C., Mitchell, B. & Profit, J. Correction: disparities in NICU quality of care: a qualitative study of family and clinician accounts. J. Perinatol. 38, 1123 (2018).
Simental-Mendia, M. et al. Effect of glucosamine and chondroitin sulfate in symptomatic knee osteoarthritis: a systematic review and meta-analysis of randomized placebo-controlled trials. Rheumatol. Int. 38, 1413–1428 (2018).
Hori, Y. et al. Effects of chondroitin sulfate on colitis induced by dextran sulfate sodium in rats. Jpn J. Pharmacol. 85, 155–160 (2001).
Shmagel, A. et al. The effects of glucosamine and chondroitin sulfate on gut microbial composition: a systematic review of evidence from animal and human studies. Nutrients 11, 294 (2019).
Franzosa, E. A. et al. Identifying personal microbiomes using metagenomic codes. Proc. Natl Acad. Sci. USA 112, E2930–E2938 (2015).
Staude, B. et al. The microbiome and preterm birth: a change in paradigm with profound implications for pathophysiologic concepts and novel therapeutic strategies. Biomed. Res. Int. 2018, 7218187 (2018).
Gonzalez-Rivera, R., Culverhouse, R. C., Hamvas, A., Tarr, P. I. & Warner, B. B. The age of necrotizing enterocolitis onset: an application of Sartwell’s incubation period model. J. Perinatol. 31, 519–523 (2011).
La Rosa, P. S. et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl Acad. Sci. USA 111, 12522–12527 (2014).
Yang, I. et al. The infant microbiome: implications for infant health and neurocognitive development. Nurs. Res. 65, 76–88 (2016).
Willis, A. D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 10, 2407 (2019).
Mosca, A., Leclerc, M. & Hugot, J. P. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front. Microbiol. 7, 455 (2016).
Minter, M. R. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 6, 30028 (2016).
Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
McMurtry, V. E. et al. Bacterial diversity and Clostridia abundance decrease with increasing severity of necrotizing enterocolitis. Microbiome 3, 11 (2015).
Stewart, C. J. et al. The preterm gut microbiota: changes associated with necrotizing enterocolitis and infection. Acta Paediatr. 101, 1121–1127 (2012).
Sanchez, E. et al. Reduced diversity and increased virulence—gene carriage in intestinal enterobacteria of coeliac children. BMC Gastroenterol. 8, 50 (2008).
Neu, J. & Pammi, M. Pathogenesis of NEC: impact of an altered intestinal microbiome. Semin. Perinatol. 41, 29–35 (2017).
Brower-Sinning, R. et al. Mucosa-associated bacterial diversity in necrotizing enterocolitis. PLoS ONE 9, e105046 (2014).
Ford, S. L. et al. Improved feeding tolerance and growth are linked to increased gut microbial community diversity in very-low-birth-weight infants fed mother’s own milk compared with donor breast milk. Am. J. Clin. Nutr. 109, 1088–1097 (2019).
Dobbler, P. T. et al. Low microbial diversity and abnormal microbial succession is associated with necrotizing enterocolitis in preterm infants. Front. Microbiol. 8, 2243 (2017).
Houghteling, P. D. & Walker, W. A. Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J. Pediatr. Gastroenterol. Nutr. 60, 294–307 (2015).
Gareau, M. G., Sherman, P. M. & Walker, W. A. Probiotics and the gut microbiota in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 7, 503–514 (2010).
Lin, H. C. et al. Oral probiotics prevent necrotizing enterocolitis in very low birth weight preterm infants: a multicenter, randomized, controlled trial. Pediatrics 122, 693–700 (2008).
Hoyos, A. B. Reduced incidence of necrotizing enterocolitis associated with enteral administration of Lactobacillus acidophilus and Bifidobacterium infantis to neonates in an intensive care unit. Int. J. Infect. Dis. 3, 197–202 (1999).
Dieterich, C. M., Felice, J. P., O’Sullivan, E. & Rasmussen, K. M. Breastfeeding and health outcomes for the mother-infant dyad. Pediatr. Clin. N. Am. 60, 31–48 (2013).
Thompson, A. L., Monteagudo-Mera, A., Cadenas, M. B., Lampl, M. L. & Azcarate-Peril, M. A. Milk- and solid-feeding practices and daycare attendance are associated with differences in bacterial diversity, predominant communities, and metabolic and immune function of the infant gut microbiome. Front. Cell Infect. Microbiol. 5, 3 (2015).
Ho, N. T. et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. 9, 4169 (2018).
Steele, J. R., Meskell, R. J., Foy, J. & Garner, A. E. Determining the osmolality of over-concentrated and supplemented infant formulas. J. Hum. Nutr. Diet. 26, 32–37 (2013).
Kriss, M., Hazleton, K. Z., Nusbacher, N. M., Martin, C. G. & Lozupone, C. A. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Curr. Opin. Microbiol. 44, 34–40 (2018).
Wang, Z. et al. Characteristic dysbiosis of gut microbiota of Chinese patients with diarrhea-predominant irritable bowel syndrome by an insight into the pan-microbiome. Chin. Med. J. 132, 889–904 (2019).
Tanaka, S. et al. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 56, 80–87 (2009).
Mai, V. et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE 6, e20647 (2011).
Underwood, M. A. & Sohn, K. The microbiota of the extremely preterm infant. Clin. Perinatol. 44, 407–427 (2017).
Coggins, S. A., Wynn, J. L. & Weitkamp, J. H. Infectious causes of necrotizing enterocolitis. Clin. Perinatol. 42, 133–154, ix (2015).
Warner, B. B. et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: a prospective case-control study. Lancet 387, 1928–1936 (2016).
de la Cochetiere, M. F. et al. Early intestinal bacterial colonization and necrotizing enterocolitis in premature infants: the putative role of Clostridium. Pediatr. Res. 56, 366–70. (2004).
Aujoulat, F. et al. Temporal dynamics of the very premature infant gut dominant microbiota. BMC Microbiol. 14, 325 (2014).
Romano-Keeler, J. et al. Distinct mucosal microbial communities in infants with surgical necrotizing enterocolitis correlate with age and antibiotic exposure. PLoS ONE 13, e0206366 (2018).
Arboleya, S. et al. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J. Pediatr. 166, 538–544 (2015).
Kuppala, V. S., Meinzen-Derr, J., Morrow, A. L. & Schibler, K. R. Prolonged initial empirical antibiotic treatment is associated with adverse outcomes in premature infants. J. Pediatr. 159, 720–725 (2011).
Greenwood, C. et al. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J. Pediatr. 165, 23–29 (2014).
Volpi, N. Chondroitin sulfate safety and quality. Molecules 24, 1447 (2019).
Kastana, P. et al. Insight into the role of chondroitin sulfate E in angiogenesis. FEBS J. 286, 2921–2936 (2019).
Dyck, S. et al. Perturbing chondroitin sulfate proteoglycan signaling through LAR and PTPsigma receptors promotes a beneficial inflammatory response following spinal cord injury. J. Neuroinflamm. 15, 90 (2018).
Uhlin-Hansen, L., Eskeland, T. & Kolset, S. O. Modulation of the expression of chondroitin sulfate proteoglycan in stimulated human monocytes. J. Biol. Chem. 264, 14916–14922 (1989).
Pichette, J., Fynn-Sackey, N. & Gagnon, J. Hydrogen sulfide and sulfate prebiotic stimulates the secretion of GLP-1 and improves glycemia in male mice. Endocrinology 158, 3416–3425 (2017).
Liu, X. et al. Antithrombotic activities of fucosylated chondroitin sulfates and their depolymerized fragments from two sea cucumbers. Carbohydr. Polym. 152, 343–350 (2016).
Egea, J., Garcia, A. G., Verges, J., Montell, E. & Lopez, M. G. Antioxidant, antiinflammatory and neuroprotective actions of chondroitin sulfate and proteoglycans. Osteoarthr. Cartil. 18, S24–S27 (2010).
Pai, V. C. et al. The chondroitin sulfate moiety mediates thrombomodulin-enhanced adhesion and migration of vascular smooth muscle cells. J. Biomed. Sci. 25, 14 (2018).
Zancan, P. & Mourao, P. A. Venous and arterial thrombosis in rat models: dissociation of the antithrombotic effects of glycosaminoglycans. Blood Coagul. Fibrinolysis 15, 45–54 (2004).
Coppa, G. V. et al. Glycosaminoglycan content in term and preterm milk during the first month of lactation. Neonatology 101, 74–76 (2012).
Coppa, G. V. et al. Composition and structure elucidation of human milk glycosaminoglycans. Glycobiology 21, 295–303 (2011).
Henrotin, Y., Mathy, M., Sanchez, C. & Lambert, C. Chondroitin sulfate in the treatment of osteoarthritis: from in vitro studies to clinical recommendations. Ther. Adv. Musculoskelet. Dis. 2, 335–348 (2010).
Korotkyi, O. et al. Effect of chondroitin sulfate on blood serum cytokine profile during carrageenan-induced edema and monoiodoacetate-induced osteoarthritis in rats. Rev. Recent Clin. Trials 14, 50–55 (2019).
Barthe, L. et al. In vitro intestinal degradation and absorption of chondroitin sulfate, a glycosaminoglycan drug. Arzneimittelforschung 54, 286–292 (2004).
du Souich, P. Absorption, distribution and mechanism of action of SYSADOAS. Pharm. Ther. 142, 362–374 (2014).
Nutrition ECo, Arslanoglu, S. et al. Donor human milk for preterm infants: current evidence and research directions. J. Pediatr. Gastroenterol. Nutr. 57, 535–542 (2013).
Burge, K., Bergner, E., Gunasekaran, A., Eckert, J. & Chaaban, H. The role of glycosaminoglycans in protection from neonatal necrotizing enterocolitis: a narrative review. Nutrients 12, 546 (2020).
Liu, F. et al. Chondroitin sulfate disaccharides modified the structure and function of the murine gut microbiome under healthy and stressed conditions. Sci. Rep. 7, 6783 (2017).
Rotimi, V. O. & Duerden, B. I. The bacterial flora of neonates with congenital abnormalities of the gastro-intestinal tract. J. Hyg. 88, 69–81 (1982).
Burge, K. Y., Hannah, L., Eckert, J. V., Gunasekaran, A. & Chaaban, H. The protective influence of chondroitin sulfate, a component of human milk, on intestinal bacterial invasion and translocation. J. Hum. Lact. 35, 538–549 (2019).
Tuncil, Y. E. et al. Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. mBio 8 (2017).
Salyers, A. A., Vercellotti, J. R., West, S. E. & Wilkins, T. D. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol. 33, 319–322 (1977).
Claud, E. C. et al. Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc. Natl Acad. Sci. USA 101, 7404–7408 (2004).
Nanthakumar, N. N., Fusunyan, R. D., Sanderson, I. & Walker, W. A. Inflammation in the developing human intestine: a possible pathophysiologic contribution to necrotizing enterocolitis. Proc. Natl Acad. Sci. USA 97, 6043–6048 (2000).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Stabler, T. V., Huang, Z., Montell, E., Verges, J. & Kraus, V. B. Chondroitin sulphate inhibits NF-kappaB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthr. Cartil. 25, 166–174 (2017).
Navarro, S. L. et al. Randomized trial of glucosamine and chondroitin supplementation on inflammation and oxidative stress biomarkers and plasma proteomics profiles in healthy humans. PLoS ONE 10, e0117534 (2015).
Campo, G. M. et al. Glycosaminoglycans modulate inflammation and apoptosis in LPS-treated chondrocytes. J. Cell. Biochem. 106, 83–92 (2009).
Chan, P. S., Caron, J. P. & Orth, M. W. Short-term gene expression changes in cartilage explants stimulated with interleukin beta plus glucosamine and chondroitin sulfate. J. Rheumatol. 33, 1329–1340 (2006).
Namachivayam, K. et al. Transforming growth factor-beta2 is sequestered in preterm human milk by chondroitin sulfate proteoglycans. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G171–G180 (2015).
Frey, H., Schroeder, N., Manon-Jensen, T., Iozzo, R. V. & Schaefer, L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 280, 2165–2179 (2013).
Tan, G. K. & Tabata, Y. Chondroitin-6-sulfate attenuates inflammatory responses in murine macrophages via suppression of NF-kappaB nuclear translocation. Acta Biomater. 10, 2684–2692 (2014).
Gross, A. R. & Theoharides, T. C. Chondroitin sulfate inhibits secretion of TNF and CXCL8 from human mast cells stimulated by IL-33. Biofactors 45, 49–61 (2019).
De Winter, B. Y., van den Wijngaard, R. M. & de Jonge, W. J. Intestinal mast cells in gut inflammation and motility disturbances. Biochim. Biophys. Acta 1822, 66–73 (2012).
Green Corkins, K. & Shurley, T. What’s in the bottle? A review of infant formulas. Nutr. Clin. Pract. 31, 723–729 (2016).
Vandenplas, Y., Zakharova, I. & Dmitrieva, Y. Oligosaccharides in infant formula: more evidence to validate the role of prebiotics. Br. J. Nutr. 113, 1339–1344 (2015).
Acknowledgements
This work was supported by K08DK113226 from the National Institutes of Health, the Koret Foundation, The George H. Clowe’s Memorial Research Career Development Award, and the Department of Surgery at the Indiana University School of Medicine.
Author information
Authors and Affiliations
Contributions
T.A.K. drafted the manuscript, made critical revisions, and approved the final submission. B.D.H., A.R.P., H.L., W.C.S., and T.A.M. all made critical revisions to the manuscript and approved its final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Patient consent
This was not a clinical trial and human research was not conducted. Thus, consent was not required
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Knowles, T.A., Hosfield, B.D., Pecoraro, A.R. et al. It’s all in the milk: chondroitin sulfate as potential preventative therapy for necrotizing enterocolitis. Pediatr Res 89, 1373–1379 (2021). https://doi.org/10.1038/s41390-020-01125-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01125-7
This article is cited by
-
Necrotizing enterocolitis: current understanding of the prevention and management
Pediatric Surgery International (2024)
