
Abstract
New information is emerging concerning the influence of environmental factors (e.g., viruses, pollutants, nutrients) on fetal lung development and the prenatal modulation of cellular and molecular effectors essential to the control of airway function, which may shed new light into the pathogenesis of chronic obstructive pulmonary disease in childhood. In particular, recent studies have shown that nanosize biological and inorganic particles (e.g., respiratory viruses and pollutants) are able to spread hematogenously across the placenta from mother to offspring and interfere with lung development during critical “windows of opportunity”. Furthermore, the nutritional balance of maternal diet during pregnancy can affect postnatal lung structure and function. Adverse prenatal environmental conditions can predispose to increased airway reactivity by inducing aberrant cholinergic innervation of the respiratory tract, enhanced contractility of the airway smooth muscle, and impaired innate immunity. Such changes can persist long after birth and might provide a plausible explanation to the development of chronic airway dysfunction in children, even in the absence of atopic predisposition. Insight into maternal−fetal interactions will contribute to a better understanding of the pathogenesis of highly prevalent diseases like bronchiolitis and asthma, and may lead to more precise preventative and therapeutic strategies, or new indications for existing ones.
Similar content being viewed by others


Introduction
The most prevalent respiratory disorders in infants and children, i.e., bronchiolitis, non-atopic wheezing, and atopic asthma, involve complex interactions between microbes, environmental and nutritional conditions, and the cells responsible for immune and inflammatory responses in the respiratory tract. For many years, the placenta was perceived as a perfectly secure gate between the mother and the fetus, protecting the latter from environmental influences because it was impenetrable to most physical, chemical, and biological agents. Hence, most previous studies exploring the effects of environmental conditions as early determinants of airway disease were based on the assumption that the placenta was shielding the fetus from any adverse interaction, keeping the growing lungs “healthy” until birth.
However, there is mounting evidence that confutes this paradigm and provides robust evidence that maternally derived inflammation caused by environmental exposures during pregnancy might indeed impact postnatal lung development. Most of the studies published so far have focused on the prenatal origins of bronchopulmonary dysplasia (BPD), and it is now widely accepted that placental inflammation (e.g., chorioamnionitis), chemical exposures (e.g., nicotine), maternal disease (e.g., preeclampsia), and intrauterine growth restriction can interfere with fetal lung development and predispose to premature birth, thereby increasing the risk of BPD.1 Prenatal infections of the respiratory tract, particularly with Ureaplasma species ascending from the maternal genitourinary tract, have also been associated with adverse pregnancy outcomes and BPD.2
More recent studies from our laboratories suggest that prenatal environmental exposures can interfere with fetal lung development selectively and without causing preterm delivery and the profound structural changes typical of BPD. Specifically, our data indicate that common respiratory infections, exposure to particulate pollution, and maternal diet during pregnancy can modify gene expression in the developing lungs, leading to lasting changes in innervation patterns and smooth muscle contractility, as well as both innate and adaptive immunity. Indeed, the first “window of opportunity” to interact with the outside environment is during prenatal life, when biological, chemical, and physical factors have a stronger impact on fetal development and play a critical role for lifelong homeostasis. This review discusses some of the existing evidence that transplacental transmission of environmental factors to the human fetus has important and long-lasting influence on lung development (Fig. 1). The premise is that such evidence is likely to lead to radically new prophylactic and therapeutic strategies aimed at protecting the unborn child from previously unknown threats, thereby limiting subsequent respiratory pathology during childhood.

Flow chart illustrating the proposed pathogenetic model. In this model, respiratory viruses like RSV spread hematogenously from the pregnant mother’s respiratory tract and target the developing fetal lungs. In addition to the tolerogenic effect deriving from exposure of the preimmune fetus to viral antigens, the severity of this intrauterine infection can be modulated by other extrinsic factors like maternal caloric intake and exposure to particulate pollution during pregnancy. Complex interactions between genetic, epigenetic, and environmental cues shape all structural components of the fetal airways during a “window of opportunity” of critical importance for organ development and lifelong host-microbial and immune homeostasis. As a result, the infant’s airways are predisposed to exaggerated inflammation and bronchospasm, which leads to persistent airway hyperreactivity, recurrent airflow obstruction, and progressive structural remodeling
Vertical transmission of respiratory viruses
Recent studies show that we may come down with our first “cold” before we are even born, and this can play a critical role in modulating the structure and function of the respiratory system. The respiratory syncytial virus (RSV)—a pleiomorphic paramyxovirus with average diameter of 50−250 nm—is the most common cause of acute lower respiratory infections (ALRI) in young children worldwide, and 33 million occurrences of RSV-related ALRI in 2015 resulted in 3 million hospitalizations and 60,000 in-hospital deaths of children younger than 5 years.3 Until our 2013 publication proposing for the first time that RSV could spread hematogenously from the infected airways of a pregnant mother to the lungs of her unborn child via the placenta,4 it was universally accepted that fetal lungs were protected from maternal respiratory infections during gestation. Since then, our research has provided proof-of-concept that this central axiom of respiratory medicine (the “sterile womb dogma”) is incorrect, first using cellular and animal models4,5,6 and more recently also with human studies.7
The most important discoveries in this area stem from our original observation that RSV could spread outside of the respiratory tract and seed nonpulmonary tissues, particularly the human bone marrow mesenchymal stem cells,8 which implies hematogenous dissemination of replicating virions. Based on this knowledge, we hypothesized—and later confirmed in a rat model—that RSV is able to spread across the placenta from the respiratory tract of the mother to the fetus (Fig. 2), and it can also be detected postnatally in the lungs throughout development and into adulthood.4 Importantly, vertical RSV infection was associated with dysregulation of critical neurotrophic pathways—particularly the nerve growth factor (NGF)/TrkA receptor axis—during fetal development, leading to aberrant cholinergic innervation of the respiratory tract and increased airway reactivity after postnatal reinfection with RSV.

Propagation of vertically transmitted RSV. Co-cultures of human airway epithelial cells with extracts of whole fetuses delivered from rat dams inoculated with a recombinant RSV-A2 strain expressing the red fluorescent protein gene (rrRSV), or with sterile medium. After 48 h of incubation, the cells were then stained with a polyclonal antibody against the nerve growth factor (NGF). The micrographs show red fluorescence in the cytoplasm of cells exposed to fetal extracts from RSV-infected dams, confirming the presence of actively replicating infectious virus associated with markedly increased green NGF immunoreactivity. As RFP expression reflects virus infection and production of the transgene rather than virus spread and plaque formation, this methodology is more accurate than the standard plaque assay to assess RSV infectivity. Magnification = ×60. Methods are outlined in an online Supplemental file
A subsequent study provided further evidence that pups born to RSV-infected mothers exhibit selective impairment of innate antiviral immunity and persistent airway dysfunction after postnatal reinfections with the same virus.5 The most significant effects measured after a primary early-life infection were a sharply decreased CD8+ T-cell response combined with virtual suppression of key Th1-type cytokines like interferon gamma (IFN-γ) and interleukin (IL)-2; a large increase in the expression of the prototypical neurotrophin NGF; and increased pre- and post-synaptic innervation of the airway smooth muscle combined with intrinsic hypercontractility delaying its return to resting tone. These changes persisted after secondary reinfections and might provide a plausible explanation to the development of chronic airway dysfunction in a subpopulation of children with a history of severe RSV infections in infancy. We also developed novel pharmacologic strategies aiming at selectively and “physiologically” prevent or treat the effects of RSV infection with minimal side effects for mother and fetus, using selected microRNAs (miRNAs) to inhibit viral replication9 or limiting the virus’ ability to penetrate the airway epithelial barrier by increasing cAMP expression with forskolin.10
Concerning the molecular mechanisms involved, a recent study has shown that human bronchial epithelial cells (HBECs) from asthmatic children exhibit higher basal transient receptor potential cation channel subfamily V member 1 (TRPV1) expression and activity compared to nonasthmatic controls and, even more importantly, that RSV infection of HBECs from asthmatic children—but not from adults—leads to a sharp increase in TRPV1 activation.11 TRPV1 belongs to a family of proteins originally characterized in neuronal cells as Ca2+-permeable, nonselective cation channels that sense physical and chemical stimuli such as noxious heat, acid pH, and chemical irritants.12 Subsequent studies have localized TRPV1 expression to nonneuronal cells like bronchial epithelial cells,13,14,15,16,17 where it plays an important pathophysiologic role through stimulation of cough reflex,18 increased mucus production,19 airway smooth muscle contraction,20 and increased vascular permeability.21 These new findings raise the possibility that newborn lungs might be preprogrammed to develop cough, wheezing and inflammation once re-exposed to RSV independently from canonical allergic mechanisms, and also imply that TRPV1 inhibition could lead to novel pharmacological strategies able to reduce the impact of RSV infections in asthmatic and nonasthmatic children.
The proof-of-concept formulated with cellular and animal models has been translated by systematic studies conducted over several years confirming that RSV crosses the uterine-placental interface in humans and infects the developing fetal lungs by vertical transmission. In particular, we have published the first documented case of RSV infection caused by prenatal transmission of this virus from a mother infected during pregnancy to her newborn son.7 In this newborn showing symptoms consistent with congenital viral pneumonia, microbiological tests revealed high serum titers of anti-RSV antibodies combined with presence of RSV RNA in blood samples obtained with sterile procedure immediately after birth. Serologic tests for RSV were also positive in the mother and correlated with a history of respiratory symptoms during gestation. Simultaneously, vertical transmission of RSV in humans was reported by another group of investigators in Australia in 26 of 45 (58%) cord blood samples tested by droplet digital (dd)PCR, a next-generation PCR technique with greater analytical sensitivity compared to conventional real-time PCR due to an enhanced ability to read nucleic acid at a single molecule level.22 Together, these data suggest that:
-
(a)
Albeit the clinical manifestations of RSV are predominantly circumscribed to the respiratory system, transient RSV viremia during pregnancy allows transmission through the placenta and subsequent access to the developing fetal lungs, which might be modulated by concurrent environmental factors and can be targeted by pharmacological interventions.
-
(b)
In utero RSV infection of the fetus during its preimmune development, i.e., before the establishment of immunological self-tolerance, induces selective tolerance towards viral antigens. Consequently, newborns originally exposed in utero may be failing to recognize the virus as pathogenic and nonself during an early-life reinfection, which explains previous epidemiologic reports of weak anti-RSV Th1 immunity and persistent post-RSV airway dysfunction in childhood.
-
(c)
Although RSV-exposed fetal lungs may not show overt structural changes, more subtle changes in critical molecular mechanisms may have persistent functional consequences, in experimental models as well as in humans. Specifically, viral transmission and replication in the developing lung tissues of the fetus may modulate expression and function of ion channels and receptors that are essential to the control of postnatal respiratory physiology, thereby predisposing humans to airway hyperreactivity during infancy, childhood, and perhaps adulthood.
Exposure to environmental pollution in utero
Over the last several decades, a growing body of evidence has linked the exposure to ambient air pollution with respiratory morbidity and mortality. A recent analysis conducted by the World Health Organization (WHO) underscored that children are more at risk from air pollution than adults, revealing that over 90% of children in the world breathe polluted air that can increase the risk for asthma and other chronic diseases.23 Exposure to pollution is associated with more than one in four deaths among children younger than 5 years old, and approximately 600,000 children in this age range die every year from respiratory infections attributable to pollution. Data also show that 93% of the global pediatric population is exposed to levels of ambient fine particulate matter (FP, ≤2.5 μm in diameter) above WHO guidelines. Notably, another very recent study revealed that even short-term exposure to FP pollution in young age increases the risk of ALRI, and in particular increases the susceptibility to RSV infection and its clinical severity.24
Several studies from our lab have provided mechanistic evidence that exposure of the airways to particles promotes RSV replication and enhances the magnitude of RSV-induced airway inflammation and hyperreactivity by altering local autonomic innervation, reducing innate antiviral defense, and increasing epithelial permeability. These effects are size-dependent and most severe after exposure to the ultrafine particulate fraction (UFP, ≤100 nm in diameter). Owing to their smaller size and higher surface area, these virus-sized particles exhibit high deposition in the lower respiratory tract, evade phagocytosis, translocate to the systemic circulation, and frequently act as carriers for co-pollutants of industrial and biological origin (e.g., gases, chemicals, bacteria, viruses).25 Yet, environmental UFP levels are not regulated other than being loosely included under FP—of which UFP are only a small fraction—in great part because of the lack of studies differentiating the effects associated with UFP exposures from those related to other particle size fractions and gaseous co-pollutants.26
Among nanoparticulate pollutants, Titanium(IV) oxide (TiO2)—an inorganic pigment used in a wide array of industrial products and prototypic in size to ultrafine ambient air particles—have drawn special attention because they have widespread use in commercial applications27 (paints, photocatalysts,28 antibacterial films or sprays,29 sunscreens and cosmetics), and cause inflammation and injury of the lower airways in a dose-dependent fashion.25,30,31 We first reported that in vivo exposure of newborn and weanling rats to inhaled TiO2-UFP upregulates lung expression of NGF and brain-derived neurotrophic factor (BDNF), which are key regulatory agents of neuronal development and responsiveness and play an important role in the pathophysiology of childhood asthma.32 This mechanism leads to the development of airway hyperreactivity and inflammation only in young rats, whereas exposure to the same concentration of TiO2-UFP does not influence airway function or structure in older adult animals, once again supporting the existence of a critical window of vulnerability during the early stages of lung development.
We then explored the interactions between TiO2-UFP and human RSV in vitro using primary HBECs (Fig. 3), and confirmed that TiO2 exposure followed by RSV infection enhance synergistically the synthesis of NGF, along with concomitant upregulation of its high-affinity receptor TrkA.33 In turn, NGF-TrkA signaling upregulates the gene expression of beclin-1, a mammalian homolog of yeast Atg6/Vps30 that is at the heart of a regulatory complex for the class III PI3K/hVps34, and whose activity is essential during autophagosome formation.34,35 The resulting antiapoptotic effect augments RSV replication by allowing host cells to better adapt to the stress of viral invasion and survive while the virus completes its replication cycle.

Pre-exposure to ultrafine particles increases RSV replication. Human bronchial epithelial cells were exposed to 10 μg/ml of TiO2-UFP for 24 h and then infected with rrRSV at 0.5 MOI for 24 h. Quantitative PCR analysis (left panel) showed increased RSV copy numbers in cells exposed to TiO2-UFP. Data are shown as mean ± SD (n = 5 experiments); *p < 0.05 compared to nonexposed RSV-infected cells. Also, confocal microscopy of human bronchial epithelial cells pre-exposed to TiO2-UFP before infection with rrRSV showed increased red fluorescent protein expression (right panel). Methods are outlined in an online Supplemental file
While there is some information about the postnatal respiratory effects of UFP exposure in children,36 very little is known about the impact of their transplacental migration on fetal lung development. The need to fill this information gap is rapidly growing because, while humans have always been exposed to combustion-derived airborne nanoparticles, today’s broad use of nanotechnology has increased exponentially the production of engineered nanomaterials for use in domestic and biomedical applications to which pregnant women are frequently exposed. There is solid evidence that the transport of particles across the human placenta is exquisitely size-dependent.37 Indeed, during the late phase of pregnancy, particles with a diameter of 50 nm cross the mature human placenta rather quickly, whereas their transfer rate declines sharply at 80 nm, and there is minimal or no permeability beyond ~250 nm.38 During early and mid-pregnancy, however, the layer between maternal and fetal circulation is thicker and the number of fetal capillaries is lower, thus permeability is strictly limited to the UFP nanosize range.39 There is also evidence suggesting the actual size of hemochorial placental pores is only ~10 nm,40 which implies that even most UFP cross the placenta using as of yet unidentified active transport system(s). TiO2-UFP transfer has been confirmed also in an engineered 3D placental-barrier-on-chip microdevice.41
Importantly, in vivo exposure of pregnant rats to TiO2-UFP or other nanosize particles by intratracheal instillation was found by our lab and others to result in detection and localization of the same particles in the placenta and fetal lungs, and is associated to changes in RNA transcription and epigenetic histone modifications,42 as well as long-lasting impairment of lung development in the progeny.43 The need for mechanistic research on the effects of perinatal UFP exposure was recently emphasized by the first population-based study showing that maternal UFP exposure during the second trimester of pregnancy—corresponding to the pseudoglandular and canalicular phases of lung development—is associated with an increased risk of developing asthma in children by age 6, independent of other pollutants including FP.44
Beside size and chemical composition, another important determinant of particulate exposure is the climate, as the prolonged thermal inversions commonly occurring in winter months can raise dramatically the concentrations of FP/UFP in the air we breathe. These wintertime inversions usually coincide worldwide with the RSV epidemic season.45 Thus, because nanosize metallic and biological (e.g., viruses) particles can be transmitted from a pregnant mother to her progeny via the placental circulation, it is reasonable to believe that during winter months millions of pregnant women living in urban areas and exposed to peaks of particulate pollution while being infected by respiratory viruses are simultaneously or sequentially affected by such agents, which may result in synergistic pathological responses affecting the fetus. Yet, little is known about the consequences of concurrent maternal exposure to specific pollutants and infections for fetal lung development, and we hope additional research in this area will become a priority for funding agencies.
Effect of maternal nutrition on fetal lungs
Intrauterine exposure to imbalanced maternal nutrition may cause a shift in the trajectory of structural and functional airway development toward a hyperreactive phenotype. As we had shown in a previous population-based study significant correlation between childhood asthma and early abnormalities of lipid and glucose metabolism,46 we sought to determine in a rat model whether maternal nutrition in pregnancy affects postnatal metabolic and respiratory outcomes in the offspring.47 On gestation day 1, dams were switched from standard chow to either high-fat hypercaloric diet (HFD) or control diet. Terminal experiments were performed on newborn and weanling offspring of dams fed the study diet during gestation and lactation, and on adult offspring maintained on the same diet as their mother. Pups born from HFD dams developed early metabolic abnormalities which persisted throughout development.
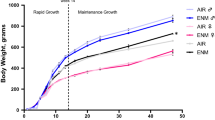
Cytokine expression analysis of lung tissues from pups born to HFD dams revealed a strong proinflammatory pattern. Also gene expression of neurotrophic factors and receptors was upregulated in the same lungs, and this was associated to higher respiratory system resistance and lower compliance at baseline, as well as hyperreactivity to aerosolized methacholine (Fig. 4). Notably, HFD dams delivered pups prone to develop more severe disease after postnatal RSV infection, due to impaired antiviral defense. Thus, maternal nutrition in pregnancy is not only an independent determinant of airway inflammation and hyperreactivity in offspring, but also increases the risk of severe RSV bronchiolitis independently from the pre-pregnancy nutrition of the mother or postnatal nutrition of the offspring. Finally, a very recent study found that nutritional factors—specifically the intake of omega-3 and omega-6 fatty acids—can modulate the effect of particulate matter exposure on lung function,48 contributing additional evidence to the evolving concept that the intersection and interaction of infectious, environmental, and dietary factors may be essential determinants of asthma incidence and morbidity.

Effect of maternal diet on offspring susceptibility to RSV. After 5 days of infection at 2 weeks of age, RSV copy number (top left panel) and change in NGF expression (bottom left panel) in lung tissues of weanlings born from dams fed high-fat hypercaloric diet (HFD) during pregnancy was on average threefold higher than in age-matched weanlings born from dams fed control diet. Also, the increase of respiratory system resistance (Rrs) in response to aerosolized methacholine challenge (right panel) was significantly larger in infected HFD weanlings than in infected controls. Data are expressed as the mean ± SEM (n = 7 rats per group); *p < 0.05 compared to age-matched controls born to dams fed with control diet. Methods are outlined in an online Supplemental file
All these findings support the principles summarized in the “Fetal Programming Hypothesis” (Fig. 5), originally formulated by Dr. David Barker.49 Dr. Barker believed that public healthcare was failing because of its focus on treatment rather than prevention, and that the bulk of available resources should rather be dedicated to the protection of pregnant women. Indeed, chronic diseases like obesity, diabetes and asthma have become epidemic and their management in adulthood is escalating the costs of healthcare to proportions unsustainable for any world economy. If we want to prevail in the war against chronic diseases that so far have eluded any therapeutic strategy, it is essential to recognize that the months spent in our mothers’ womb may be the most consequential of our lives, and identify the intrauterine and early-life events shaping organ development to prevent or redirect dysfunctional phenotypes before they result in actual disease. Securing a balanced and healthy diet, clean air to breathe, and viral prophylaxis to all pregnant women and their offspring during early childhood should be the first and most important step in this direction.

Fetal Programming Hypothesis. Prenatal events associated with environmental agents able to cross the placenta can modify the trajectory of structural and functional fetal development and generate a pathological neonatal phenotype. Such a phenotype will have dysfunctional interactions with the outside environment later in life, increasing the risk of disease. Postnatal events, such as re-exposure to infections, pollution, or imbalanced diet during childhood, could further shift the positioning of the adult phenotype, thereby exacerbating the mismatch between phenotype and environment and causing more severe or persistent disease
Conclusions
This article reviews some of the growing evidence that prenatal exposure to respiratory viruses, particulate pollution, and nutritional imbalance can affect lung development in utero and have—at least in animal models—persistent effects on airway function and structure throughout life. Although seemingly different, all environmental exposures discussed in this review share direct proinflammatory effects on fetal lung tissues, mediated at least in part by common molecular signaling pathway. Furthermore, nanosized pollution and hypercaloric diet share the synergism with concurrent RSV infection because both of them interfere with innate antiviral immunity. These concepts are a logical evolution of the Barker’s Fetal Programming hypothesis—also known as Developmental Origins of Health and Disease (DOHaD)—applied to the respiratory system, and highlight the critical role that infection prevention, clean air, and healthy diet can play during pregnancy in protecting the respiratory health of the progeny. A better understanding of the long-term consequences of prenatal environmental exposures on human lungs—which are currently being investigated as part of a project recently funded by the National Heart, Lung, and Blood Institute (NHLBI)—will open the way to clinical trials exploring whether maternal prevention and/or early life treatment can adequately protect against prenatal agents and reduce their postnatal sequelae. Indeed, selective pharmacological modulation of lung development might become one day a completely new strategy for more precise and effective management of the most common respiratory diseases in children, like bronchiolitis and asthma.
References
Jobe, A. H. Mechanisms of lung injury and bronchopulmonary dysplasia. Am. J. Perinatol. 33, 1076–1078 (2016).
Viscardi, R. M. & Kallapur, S. G. Role of Ureaplasma respiratory tract colonization in BPD pathogenesis: current concepts and update. Clin. Perinatol. 42, 719–738 (2015).
Shi, T., McAllister, D. A. O’Brien, K. L., Simoes, E. A., Madhi, S. A. et al. RSV Global Epidemiology Network. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390, 946–958 (2017).
Piedimonte, G., Walton, C. & Samsell, L. Vertical transmission of respiratory syncytial virus modulates pre- and postnatal innervation and reactivity of rat airways. PLoS ONE 8, e61309 (2013).
Brown, P. M., Harford, T. J., Agrawal, V., Belkadi, A., Yen-Lieberman, B. et al. Prenatal exposure to respiratory syncytial virus alters postnatal immunity and airway smooth muscle contractility during early-life reinfections. PLoS ONE 12, e0168786 (2017).
Piedimonte, G. & Perez, M. K. Alternative mechanisms for respiratory syncytial virus (RSV) infection and persistence: could RSV be transmitted through the placenta and persist into developing fetal lungs? Curr. Opin. Pharm. 16, 82–88 (2014).
Manti, S., Cuppari, C., Lanzafame, A., Salpietro, C., Leonardi, S. et al. Vertical transmission of respiratory syncytial virus infection in humans. Pediatr. Pulmonol. 52, E81–84 (2017).
Rezaee, F., Gibson, L. F., Piktel, D., Othumpangat, S. & Piedimonte, G. Respiratory syncytial virus infection in human bone marrow stromal cells. Am. J. Respir. Cell Mol. Biol. 45, 277–286 (2011).
Othumpangat, S., Walton, C. & Piedimonte, G. MicroRNA-221 modulates RSV replication in human bronchial epithelial cells by targeting NGF expression. PLoS ONE 7, e30030 (2012).
Rezaee, F. et al. cAMP-dependent activation of protein kinase A attenuates respiratory syncytial virus-induced human airway epithelial barrier disruption. PLoS ONE 12, e0181876 (2017).
Harford, T. J., Rezaee, F., Scheraga, R. G., Olman, M. A. & Piedimonte, G. Asthma predisposition and respiratory syncytial virus infection modulate transient receptor potential vanilloid 1 function in children’s airways. J. Allergy Clin. Immunol. 141, 414–416 (2018).
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Inoue, K. et al. Functional vanilloid receptors in cultured normal human epidermal keratinocytes. Biochem. Biophys. Res. Commun. 291, 124–129 (2002).
Scotland, R. S., Chauhan, S., Davis, C., De Felipe, C., Hunt, S. et al. Vanilloid receptor TRPV1, sensory C-fibers, and vascular autoregulation: a novel mechanism involved in myogenic constriction. Circ. Res. 95, 1027–1034 (2004).
Seki, N. et al. Expression and localization of TRPV1 in human nasal mucosa. Rhinology 44, 128–134 (2006).
Zhang, F., Yang, H., Wang, Z., Mergler, S., Liu, H. et al. Transient receptor potential vanilloid 1 activation induces inflammatory cytokine release in corneal epithelium through MAPK signaling. J. Cell Physiol. 213, 730–739 (2007).
Reilly, C. A. et al. Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol. Sci. 73, 170–181 (2003).
Groneberg, D. A., Niimi, A., Dinh, Q. T., Cosio B., Hew M. et al. Increased expression of transient receptor potential vanilloid-1 in airway nerves of chronic cough. Am. J. Respir. Crit. Care Med. 170, 1276–1280 (2004).
Yu, H., Li, Q., Zhou, X., Kolosov, V. P. & Perelman, J. M. Transient receptor potential vanilloid 1 receptors mediate acid-induced mucin secretion via Ca2+ influx in human airway epithelial cells. J. Biochem. Mol. Toxicol. 26, 179–186 (2012).
Lin, R. L., Hayes, D. Jr. & Lee, L. Y. Bronchoconstriction induced by hyperventilation with humidified hot air: role of TRPV1-expressing airway afferents. J. Appl. Physiol. 106, 1917–1924 (2009).
Hu, D. E., Easton, A. S. & Fraser, P. A. TRPV1 activation results in disruption of the blood-brain barrier in the rat. Br. J. Pharm. 146, 576–584 (2005).
Fonceca, A. M., Chopra, A., Levy, A., Noakes, P. S., Poh, M. W. et al. Infective respiratory syncytial virus is present in human cord blood samples and most prevalent during winter months. PLoS ONE 12, e0173738 (2017).
World Health Organization. Ambient Air Pollution: A Global Assessment of Exposure and Burden of Disease (2016).
Horne, B. D., Joy, E. A., Hofmann, M. G., Gesteland, P. H., Cannon, J. B. et al. Short-term elevation of fine particulate matter air pollution and acute lower respiratory infection. Am. J. Respir. Crit. Care Med. 198, 759–766 (2018).
Oberdorster, G. Pulmonary effects of inhaled ultrafine particles. Int. Arch. Occup. Environ. Health 74, 1–8 (2001).
Baldauf, R. W., Devlin, R. B., Gehr, P., Giannelli, R., Hassett-Sipple, B., et al. Ultrafine particle metrics and research considerations: review of the 2015 UFP workshop. Int. J. Environ. Res. Public Health 13, 1054 (2016).
Xia, T., Kovochich, M., Brant, J., Hotze, M., Sempf, J., et al. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 6, 1794–1807 (2006).
Sun, D., Meng, T. T., Loong, T. H. & Hwa, T. J. Removal of natural organic matter from water using a nano-structured photocatalyst coupled with filtration membrane. Water Sci. Technol. 49, 103–110 (2004).
Shieh, K. J. et al. Antibacterial performance of photocatalyst thin film fabricated by defection effect in visible light. Nanomedicine 2, 121–126 (2006).
Chen, D. L. & Schuster, D. P. Imaging pulmonary inflammation with positron emission tomography: a biomarker for drug development. Mol. Pharm. 3, 488–495 (2006).
Grassian, V. H., O’Shaughnessy, P. T., Adamcakova-Dodd, A., Pettibone, J. M. & Thorne, P. S. Inhalation exposure study of titanium dioxide nanoparticles with a primary particle size of 2 to 5 nm. Environ. Health Perspect. 115, 397–402 (2007).
Scuri, M., Chen, B. T., Castranova, V., Reynolds, J. S., Johnson, V. J. et al. Effects of titanium dioxide nanoparticle exposure on neuroimmune responses in rat airways. J. Toxicol. Environ. Health 73, 1353–1369 (2010).
Othumpangat, S., Gibson, L. F., Samsell, L. & Piedimonte, G. NGF is an essential survival factor for bronchial epithelial cells during respiratory syncytial virus infection. PLoS ONE 4, e6444 (2009).
Liang, X. H., Jackson, S. Seaman, M., Brown, K., Kempkes, B., et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 (1999).
Yue, Z., Jin, S., Yang, C., Levine, A. J. & Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl Acad. Sci. USA 100, 15077–15082 (2003).
Heinzerling, A., Hsu, J. & Yip, F. Respiratory health effects of ultrafine particles in children: a literature review. Water Air Soil Pollut. 227, https://doi.org/10.1007/s11270-015-2726-6 (2016).
Kulvietis, V., Zalgeviciene, V., Didziapetriene, J. & Rotomskis, R. Transport of nanoparticles through the placental barrier. Tohoku J. Exp. Med. 225, 225–234 (2011).
Wick, P., Malek, A. Manser, P., Meili, D., Maeder-Althaus, X. et al. Barrier capacity of human placenta for nanosized materials. Environ. Health Perspect. 118, 432–436 (2010).
Enders, A. C. & Blankenship, T. N. Comparative placental structure. Adv. Drug Deliv. Rev. 38, 3–15 (1999).
Fournier, S. B., D’Errico, J. N. & Stapleton, P. A. Engineered nanomaterial applications in perinatal therapeutics. Pharm. Res. 130, 36–43 (2018).
Yin, F. et al. A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier. Toxicol. Vitr. 54, 105–113 (2019).
Stapleton, P. A., Hathaway, Q. A., Nichols, C. E., Abukabda, A. B., Pinti, M. V., et al. Maternal engineered nanomaterial inhalation during gestation alters the fetal transcriptome. Part Fibre Toxicol. 15, 3 (2018).
Paul, E., Franco-Montoya, M. L., Paineau, E., Angeletti, B., Vibhushan, S. et al. Pulmonary exposure to metallic nanomaterials during pregnancy irreversibly impairs lung development of the offspring. Nanotoxicology 11, 484–495 (2017).
Lavigne, E., Donelle, J., Hatzopoulou, M., Van Ryswyk, K., Van Donkelaar, A., et al. Spatiotemporal variations in ambient ultrafine particles and the incidence of childhood asthma. Am. J. Respir. Crit. Care Med. 199, 1487–1495 (2019).
Yusuf, S., Piedimonte, G., Auais, A., Demmler, G., Krishnan, S., et al. The relationship of meteorological conditions to the epidemic activity of respiratory syncytial virus. Epidemiol. Infect. 135, 1077–1090 (2007).
Cottrell, L., Neal, W., Ice, C., Perez, M. K. & Piedimonte, G. Metabolic abnormalities in children with asthma. Am. J. Respir. Crit. Care Med. 183, 441–448 (2011).
Griffiths, P., Walton, C., Samsell, L., Perez, M. K. & Piedimonte, G. Maternal high-fat diet in pregnancy results in metabolic and respiratory abnormalities in offspring. Pediatr. Res. 79, 278–286 (2016).
Brigham, E. P., Woo, H., McCormack, M., Rice, J., Koehler, K. et al. Omega-3 and omega-6 intake modifies asthma severity and response to indoor air pollution in children. Am. J. Respir. Crit. Care Med. 199, 1478–1486 (2019).
Barker, D. J. & Thornburg, K. L. The obstetric origins of health for a lifetime. Clin. Obstet. Gynecol. 56, 511–519 (2013).
Acknowledgements
The authors are very grateful to the National Heart, Lung and Blood Institute of the National Institute of Health and the Cystic Fibrosis Foundation for the generous support of some of the research described in this article over the past 20 years. We also thank Dr. Caroline Smallcombe for helping with the preparation of the figures. This work was supported in part by NIH-NHLBI grant RO1HL061007 (G.P.) and Cystic Fibrosis Foundation grant PIEDIM16G0 (G.P.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
G.P. and T.J.H. co-wrote the manuscript. G.P. responded to the reviewer’s critiques.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Piedimonte, G., Harford, T.J. Effects of maternal−fetal transmission of viruses and other environmental agents on lung development. Pediatr Res 87, 420–426 (2020). https://doi.org/10.1038/s41390-019-0657-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0657-4
